

ABSTRACT
The increasing number of zoonotic infections caused by influenza A virus (IAV) subtypes of avian origin (e.g., H5N1 and H7N9) in recent years underscores the need to better understand the factors driving IAV evolution and diversity. To evaluate the current feasibility of global analyses to contribute to this aim, we evaluated information in the public domain to explore IAV evolutionary dynamics, including nucleotide substitution rates and selection pressures, using 14 IAV subtypes in 32 different countries over a 12-year period (2000 to 2011). Using geospatial information from 39,785 IAV strains, we examined associations between subtype diversity and socioeconomic, biodiversity, and agricultural indices. Our analyses showed that nucleotide substitution rates for 11 of the 14 evaluated subtypes tended to be higher in Asian countries, particularly in East Asia, than in Canada and the United States. Similarly, at a regional level, subtypes H5N1, H5N2, and H6N2 exhibited significantly higher substitution rates in East Asia than in North America. In contrast, the selection pressures (measured as ratios of nonsynonymous to synonymous evolutionary changes [dN/dS ratios]) acting on individual subtypes showed little geographic variation. We found that the strongest predictors for the detected subtype diversity at the country level were reporting effort (i.e., total number of strains reported) and health care spending (an indicator of economic development). Our analyses also identified major global gaps in IAV reporting (including a lack of sequences submitted from large portions of Africa and South America and a lack of geolocation information) and in broad subtype testing which, until addressed, will continue to hinder efforts to track the evolution and diversity of IAV around the world.
IMPORTANCE In recent years, an increasing number of influenza A virus (IAV) subtypes, including H5N1, H7N9, and H10N8, have been detected in humans. High fatality rates have led to an increased urgency to better understand where and how novel pathogenic influenza virus strains emerge. Our findings showed that mutational rates of 11 commonly encountered subtypes were higher in East Asian countries than in North America, suggesting that there may be a greater risk for the emergence of novel pathogenic strains in East Asia. In assessing the potential drivers of IAV subtype diversity, our analyses confirmed that reporting effort and health care spending were the best predictors of the observed subtype diversity at the country level. These findings underscore the need to increase sampling and reporting efforts for all subtypes in many undersampled countries throughout the world.
INTRODUCTION
Influenza A viruses (IAVs) are found throughout the world and cause frequent epidemics in humans and domestic animal species, including poultry, pigs, and horses (1). The IAV genome consists of eight segments of negative-stranded RNA which code for at least 10 proteins. IAVs are classified on the basis of two highly variable glycoproteins, hemagglutinin (HA) and neuraminidase (NA), expressed inside the host cell and assembled on the surface of the virus particles. Avian IAVs are further classified based on their pathogenicity in poultry, with high-pathogenicity avian influenza (HPAI) virus strains causing severe and often fatal disease and low-pathogenicity avian influenza (LPAI) virus strains causing mild disease in domestic fowl. To date, 18 HA and 11 NA antigenic subtypes of IAV have been identified (2, 3). Over 120 unique HA and NA combinations (e.g., H3N2, H5N1, and H10N8) have been documented. Variation among IAVs is further enhanced by their high mutation rates (due to the presence of an RNA polymerase that lacks proofreading ability) and the ability of coinfecting viruses to exchange segments (reassortment), producing novel strains.
Extensive surveillance has shown aquatic birds, particularly, migrating waterfowl, to be the natural reservoirs for nearly all of the currently recognized IAVs (4, 5). However, the recent identification of several unique HA subtypes (H17 and H18) in multiple bat species suggests that they too may be a potentially important reservoir for diverse IAVs (3, 6). Among humans, only 4 IAV subtypes have been documented to have the ability to maintain sustained human-to-human transmission resulting in multiple worldwide pandemics (1). These include currently circulating subtypes H1N1 and H3N2 and sporadically detected subtype H1N2, as well as subtype H2N2, which was in circulation between 1957 and 1968 but has not been detected in humans since.
In the past 2 decades, an increasing number of other IAV subtypes of avian origin have been detected in humans, particularly in individuals with a recent history of bird or poultry contact. Subtypes H6N1, H7N3, H7N7, H9N2, and H10N7 have been primarily associated with nonfatal disease symptoms, including conjunctivitis and mild acute upper respiratory tract infections in humans (6–9), the only exception being a single fatal human case of H7N7 infection in the Netherlands in 2003 (10). In contrast, H5N1 and H7N9 strains have been associated with alarmingly high levels of mortality among infected people but do not sustain human-to-human transmission (11). Another subtype, H10N8, was recently linked to a case of fatal pneumonia (12).
The substantial increase in the number of publicly available IAV sequences in recent years has given researchers and the public health community new opportunities to study the biology and evolutionary dynamics of this globally significant virus. Most of these studies focused specifically on one of several subtypes of primary concern for humans (H1N1, H3N2, H5N1, and H7N9) or for companion animals, including dogs and horses (H3N8) (12–15). While there have been some efforts to look at a broader range of subtypes, those studies combined sequence data from multiple countries in their analyses, which can be useful for examining global or regional trends in IAV evolution and diversity but gave little insight into what may be occurring at the level of the individual country (16, 17). In contrast, our study utilized over 13,000 full-length hemagglutinin (HA) sequences from 12 high-priority subtypes in 32 different countries in order to identify trends in evolutionary dynamics, including nucleotide substitution rates and selection pressures at the local scale as well as the regional scale. We also constructed predictive regression models that incorporated socioeconomic, biodiversity, and agricultural data to examine the drivers of IAV subtype diversity on the country level. The purpose of the study was to identify countries or regions with increased influenza A virus mutation rates in order to better understand where novel strains may next emerge and to identify data gaps in countries and regions that are due to reporting effort (i.e., the total number of strains reported).
MATERIALS AND METHODS
Data analysis and modeling.
The subtype, location of collection, and animal host data for all HA sequences (both partial and full) of influenza A virus strains (through April 2013) were compiled from submissions to GenBank and the Influenza Research Database (IRD). A preliminary assessment of submissions to the other major influenza virus sequence database, GISAID, did not identify any additional sequences that were not available in either GenBank or the IRD.
These data were used to map the global distribution of subtype and host diversity using ArcGIS version 10.2 (Redlands, CA). We also looked at subtype diversity while controlling for reporting effort (the total number of reported strains) by mapping subtype diversity divided by the number of reported strains for each individual country. To minimize bias in our analysis, we restricted the data set to 88 countries from which 10 or more sequences and at least 2 subtypes had been reported. To adjust for countries with high or low reporting effort, both the subtype diversity and reporting effort data were log transformed prior to mapping. Shapiro-Wilk and skewness/kurtosis tests for normality were used to determine the appropriate transformation (i.e., log) to apply to the data set using Stata version 13.1 (StataCorp, College Station, TX).
In addition to these combined-subtype analyses, we also examined the global diversities of HA subtypes and NA subtypes independently. Using all 56,991 influenza A virus strains associated with (partial and full-length) HA and NA sequences available in the IRD database (up to February 2013), we parsed the name of each strain to determine the year of collection, the geographic location of collection, and host. We matched available the location information associated with each strain name to a database of city, state, province, and country names of cities with airports from the International Air Transport Association, as provided by Diio, Inc., LCC, Reston, VA, USA (Diio Mi Express). We first matched the two-letter state (or province) codes, two-letter country codes, country names, state (or province) names, and city names. If there was one unique match, we considered the strain to be georeferenced. By this method, we were able to geolocate sequences from 39,785 strains.
We summarized geolocated strain data by country and calculated the diversities of the HA subtypes and NA subtypes found within a country. We combined the subtype data with country-level socioeconomic data (health care spending, gross domestic product, corruption indices, human population) from the World Bank, agricultural data (cattle, poultry, swine density) from FAO, and biodiversity data (calculated as species richness of wild birds) from BirdLife. We explored all factorial combinations of these predictors in Poisson and negative-binomial generalized linear models using the R program (version 2.15.1; The R Foundation for Statistical Computing).
IAV subtypes were prioritized for analysis according to the geographical distribution and diversity of their animal hosts. Animal hosts were grouped into 26 “animal host groups” based on taxonomical relatedness or similar ecological niches (i.e., shorebirds) of individual species (see Table S1 in the supplemental material). Due to a lack of available data for certain GenBank or IRD submissions, several fairly broad groups, such as the “passerine” and “wild-bird” groups, were included. Among the 122 currently recognized influenza A virus subtypes, we designated 46 “priority” subtypes based on their presence in 3 or more animal host groups within a single country. Of those 46 priority subtypes, we further designated 21 “high priority” if they were also reported from at least 2 different countries, indicating a wider geographic distribution.
Available full-length hemagglutinin (HA) sequences (n = 13,840) from high-priority subtypes submitted to GenBank between 2000 and 2011 were downloaded. The time period used for our analyses was chosen to maximize the number of IAV sequences available from the greatest number of countries across similar and recent time frames. From an extensive evaluation of sequence databases (GenBank, IRD) across all available years, we found an exponential increase in the number of sequences starting at approximately the year 2000. Due to the limited number of avian origin H3N2 sequences available, only human origin H3N2 sequences were used in the analysis. For H3N8, sequences were separated into equine (H3N8-eq) and avian (H3N8-av) strains, reflecting their historical separation into distinct lineages (18). Some human H5N1 sequences were included in the H5N1 analysis, but, due to their sequence similarity (98% to 100%) to avian H5N1 strains sampled in the same countries, the human strains were all presumed to be of avian origin. In addition, due to the global sweep of the swine origin H1N1 strain through the human population in 2009, we excluded this subtype from our analysis because an accurate assessment of nucleotide substitution rates was not possible for the entire time frame (2000 to 2011) that we wished to analyze, since the pre-2009 human strains were completely replaced by the swine origin strain at that time. Full-length HA coding sequences for 12 of the 14 subtypes were trimmed slightly (to approximately 1,650 bp from 1,702 bp). Due to a limited number of available full-length HA sequences from H3N2 and H3N8 strains, we used the largest available partial sequences, consisting of 729 bp (H3N2) and 855 bp (H3N8), for these two subtypes. Sequences were aligned in the program Geneious version 7.0 (Biomatters, Auckland, New Zealand) using the Muscle alignment algorithm. HA sequences were chosen for analysis because they were the most abundant IAV gene sequences available in public databases. Additionally, a recent evolutionary analysis of multiple IAV subtypes showed that the nucleotide substitution rate of the HA gene was similar to the mean substitution rate of all 8 influenza gene segments, indicating that it can be used as a good approximation for the mean evolutionary rate of the entire genome (16).
Estimating substitution rates.
Overall rates of evolutionary change (numbers of nucleotide substitutions per site per year) were estimated using the program BEAST (version 1.7.5), which employs a Bayesian Markov chain Monte Carlo (MCMC) approach, utilizing the number and temporal distribution of genetic differences among viruses sampled at different times (19). For all data sets, an uncorrelated log-normal relaxed molecular clock model was used as it has been shown to best reflect the complex population dynamics of influenza A virus (12). Statistical uncertainty in the data was reflected in the lower and upper bounds of the highest-probability density (HPD) values, where 95% of the sampled values were located, and in each case, chain lengths were run for sufficient time (up to 30 million generations) to achieve coverage, as assessed using the Tracer program (version 1.5). We limited our analysis to countries from which at least 10 HA sequences were available over a time period of 3 or more years, allowing us to evaluate a total of 14 high-priority subtypes. For computational tractability, data sets for individual countries were limited to the 200 randomly chosen HA sequences which captured most of the available country level data. In fact, for the majority of subtypes, 200 tended to be the upper limit of the number of available sequences for the time period analyzed. Data sets for individual world regions, including East Asia, Southeast Asia, South Asia, West Asia, Africa, Europe, and North America, were limited to 500 randomly chosen HA sequences, which captured most of the available region-level data. World regions were designated according to United Nations regional groupings (https://unstats.un.org/unsd/methods/m49/m49regin.htm). To ensure that the partial data sets accurately reflected the complete data sets, we compared nucleotide substitution rates from both partial and complete data sets from 3 different subtypes (H1N2, H5N1, and H9N2) and found that they differed by less than 5% (data not shown), indicating that the substitution rates calculated from partial data sets closely reflected those calculated from complete data sets. In total, we examined substitution rates for HA genes from 14 high-priority subtypes in 32 different countries.
Measurement of selection pressures.
Using the same HA sequence data sets that were used for calculating nucleotide substitution rates, we measured selection pressures on the HA gene by calculating the numbers of nonsynonymous (dN) and synonymous (dS) nucleotide substitutions per site (dN/dS ratio) using the single-likelihood ancestor counting (SLAC) method found within the HYPHY package (35) and accessed through the Datamonkey interface (http://www.datamonkey.org). A dN/dS ratio of greater than 1 is indicative of positive selection, whereas a ratio of less than 1 indicates purifying or negative selection (20). In order to compare dN/dS ratios among individual subtypes, HA sequences for all 14 subtypes were trimmed to cover the same 729-bp region of the HA gene as was available for the H3N2 strains. This region included nucleotides 191 to 920 within the HA1 subunit of the HA gene. Mean dN/dS ratios between individual subtypes were assessed by a t test using independent samples and the program R (version 2.15.1; The R Foundation for Statistical Computing).
RESULTS
The 46 priority and 21 high-priority influenza A virus subtypes identified on the basis of the criteria defined above are presented in Table 1. The human-adapted influenza virus subtypes H1N1, H1N2, H2N2, and H3N2, as well as the more recent zoonotic avian influenza virus (AIV) subtypes H5N1 and H7N9, were all included within the group of high-priority subtypes. In fact, the identification of H7N9 as “high priority” using our methods helped to substantiate the validity of priority selection, since the classification as “high priority” was based on data collected prior to the first recorded human outbreaks in 2013.
TABLE 1.
Priority and high-priority (shaded) influenza A virus subtypes grouped by HA typea
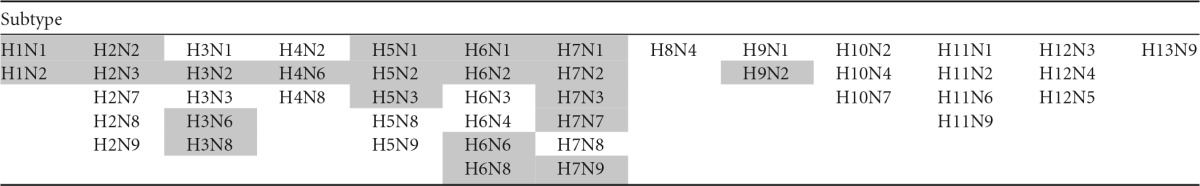
Priority subtypes were designated based on their presence in 3 or more animal host groups within a single country. High-priority subtypes were designated based on their presence in 3 or more animal host groups within a single country and reported in least 2 different countries.
Evaluation of the global distribution by subtype and host diversity indicated that the reported subtype diversity was highest in the United States (n = 100) followed by Canada (n = 65), Japan (n = 49), China (n = 42), and Sweden (n = 40) (Fig. 1A). In contrast, the reported subtype diversity was lowest in Africa (range, 1 to 19) and South America (range, 1 to 10) (Fig. 1A). The diversity of host groups from which influenza A virus subtypes had been reported was greatest in the United States (n = 21) followed by China (n = 19), Thailand (n = 18), South Korea (n = 16), Russia (n = 15), and Hong Kong (n = 15). The hosts in those countries included members of nearly all the host groups (see Table S1 in the supplemental material). Similarly to subtype diversity, reported host diversity was lowest in many parts of Africa and South America, with just one or two hosts (typically humans or farmed poultry) most frequently reported (Fig. 1B). Exceptions included South Africa (n = 11) and Egypt (n = 10) in Africa and Chile (n = 6) and Argentina (n = 5) in South America. Not surprisingly, the countries that had the highest reported diversity of subtypes also had the highest reporting effort (total number of strains reported), while countries that had the lowest reported diversity of subtypes had the lowest reporting effort (r2 = 0.34, P < 0.001). In an attempt to account for reporting bias, we examined the proportion of reported subtype diversity by analyzing the total number of strains reported (Fig. 1C). Controlling for reporting effort showed that, with the exception of Guatemala and Zambia, the greatest reported IAV subtype diversity was observed primarily in northern temperate zone countries, including Russia, Sweden, Norway, Ireland, Hungary, Canada, Netherlands, Switzerland, Czech Republic, Mongolia, and Kazakhstan, as well as in two southern temperate zone countries, Australia and South Africa. Even when only those countries that have conducted broader testing beyond the 2 common human subtypes (H1N1 and H3N2) and the H5N1 subtype (i.e., the countries with 4 or more subtypes reported) were included in the analyses, the same geographical zones were identified (see Fig. S1 in the supplemental material).
FIG 1.

Global distribution of influenza A virus subtypes. (A) Number of unique IAV subtypes (subtype diversity) per country. (B) Number of unique IAV animal host groups with reported IAV sequences per country (see Table S1 in the supplemental material for host group designations). (C) IAV subtype diversity, controlling for reporting effort. Data are presented as the log-normalized proportion of subtype diversity over the number of reported IAV strains per country. Data are based on GenBank and Influenza Resource Database (IRD) submissions as of April 2013. (Maps were created by Kate Thomas with ArcGIS version 10.2 software.)
The best-fit model for predicting HA subtype diversity at the country level included the number of strains reported and health care spending per capita in 2010, both of which were consistently present in all 57 models that were within 2 Akaike's information criterion (AIC) units of the best-fit model for predicting HA subtype diversity, as AIC models within 2 units of the best fit model are generally accepted as having substantial empirical support (21). The best-fit models also included health care spending per capita in 2010 (present in 56 of 57 models), gross domestic product in 2010 (present in 52 of 57 models), pig population density per square kilometer in 2010 (present in 38 of 57 models), and growth rate of pig population density from 1961 to 2010 (present in 37 of 57 models) (Table 2). For NA subtype diversity, the best-fit model included the number of strains reported (consistently present in all 306 models that were within 2 AIC units of the best-fit model), human population growth rate from 1961 to 2010 (present in 186 of the 306 models), and gross domestic product per capita in 2010 (present in 52 of 306 models) (Table 3).
TABLE 2.
Summary of the best model with the lowest AIC value for predicting HA subtype diversity
| Model | AIC value estimate | SE | z value | Pr (>|z|)a | Pb |
|---|---|---|---|---|---|
| Intercept | 0.935 | 0.0696 | 13.433 | <2e-16 | **** |
| Health care spending per capita in 2010 | 0.205 | 0.0405 | 5.074 | 3.89e-07 | **** |
| Gross domestic product in 2010 | −0.0863 | 0.0320 | −2.693 | 0.00709 | *** |
| Pig population density per square kilometer in 2010 | 0.0533 | 0.0290 | 1.84 | 0.06573 | * |
| Growth rate of health care spending from 1995 to 2010 | −0.237 | 0.103 | −2.305 | 0.02115 | ** |
| Growth rate of pig population density from 1961 to 2010 | −0.114 | 0.0734 | −1.558 | 0.1192 | * |
| Loge of no. of strains reported | 0.643 | 0.0715 | 9.004 | <2e-16 | **** |
Pr (>|z|), probability that the z value is greater than the estimate.
*, P < 0.10; **, P < 0.05; ***, P < 0.01; ****, P < 0.001.
TABLE 3.
Summary of the best model with the lowest AIC value for predicting NA subtype diversity
| Predictor | AIC value estimate | SE | z value | Pr (>|z|)a | Pb |
|---|---|---|---|---|---|
| Intercept | 0.779 | 0.0666 | 11.686 | <0.0001 | **** |
| Gross domestic product per capita in 2010 | 0.108 | 0.0513 | 2.10 | 0.0357 | ** |
| Human population growth rate from 1961 to 2010 | −0.160 | 0.0645 | −2.47 | 0.0134 | ** |
| Loge of no. of strains reported | 0.500 | 0.0611 | 8.18 | <0.0001 | **** |
Pr (>|z|), probability that the z value is greater than the estimate.
*, P < 0.10; **, P < 0.05; ***, P < 0.01; ****, P < 0.001.
Due to limited numbers of available sequences, the nucleotide substitution rate analyses for the majority of high-priority subtypes, including H1N2, H3N6, H3N8, H4N6, H5N2, H5N3, H6N1, H6N2, H6N8, H7N3, and H7N7, were restricted to countries in East Asia (China, Hong Kong, Japan, South Korea, and Vietnam), Europe (France, Italy, Russia, Sweden, and the United Kingdom), and North America (the United States and Canada). Analyses of the remaining subtypes, H3N2, H5N1, and H9N2, were much broader, including 32 countries from 7 different world regions. For countries with fewer available sequences and/or fewer sampling years, the variability in the resulting substitution rates was captured in the 95% highest-probability density (HPD) values (akin to 95% confidence intervals) presented in Table S3 in the supplemental material. The 95% HPD values were generally greater for countries with limited sequence data. Because of the limited data for many individual countries, we also analyzed regional data sets, which gave us a greater sample size and similar time spans for comparison. The resulting 95% HPD values for regional data sets are also much closer.
Overall, nucleotide substitution rates (per site per year) ranged from a minimum of 1.43 × 10−3 (H3N8) to a maximum of 11.62 × 10−3 (H7N7). The observed rates for individual subtypes were similar to those reported in previous studies using comparable data sets and analytical techniques (13, 14, 17, 22). Substitution rates in this study differed considerably between individual countries, and although no single country displayed consistently high substitution rates across all subtypes analyzed, predominantly low substitution rates were detected among most of the subtypes from Canada and the United States (Table 4; full details, including 95% HPD values and numbers of sequences analyzed, are presented in Table S3 in the supplemental material). Based on the country-level analyses, it appeared that nucleotide substitution rates for all subtypes, other than H5N3, H7N3, and H7N7, were higher in East Asian countries, including China (8.88 for H5N2), Hong Kong (6.24 for H3N2), Japan (9.98 for H9N2), Mongolia (8.22 for H5N1), South Korea (10.34 for H5N1), and Taiwan (7.81 for H5N2), than in the United States (5.58 for H6N1) and Canada (3.66 for H3N6). A further analysis at the regional level showed that the substitution rates for 3 subtypes (H5N1, H5N2, and H6N2) were significantly higher (no overlap in 95% HPD values) in East Asia (4.24, 5.22, and 5.27, respectively) than in North America (2.26, 2.98, and 2.04, respectively). In contrast, none of the high-priority subtypes analyzed had significantly higher substitution rates in North America than in East Asia (Fig. 2). For H5N1, all of the world regions analyzed, except for West Asia, had significantly higher H5N1 nucleotide substitution rates than North America.
TABLE 4.
Within-country evolutionary rates of influenza A virus HA genes among 14 high-priority subtypes over 12 years (2000 to 2011)
| World region | Country | Within-country evolutionary rate (no. of nucleotide substitutions/site/yr × 10−3) of influenza A virus HA genea |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H1N2 | H3N2 | H3N6 | H3N8-eq | H3N8-av | H4N6 | H5N1 | H5N2 | H5N3 | H6N1 | H6N2 | H6N8 | H7N3 | H7N7 | H9N2 | ||
| East Asia | China | 3.54 | 3.53 | 1.43 | 3.91 | 8.88 | 4.22 | 4.57 | 4.37 | 3.13 | ||||||
| Hong Kong | 3.69 | 6.24 | 4.42 | 10.3 | 5.16 | |||||||||||
| Japan | 4.79 | 3.55 | 5.32 | 3.69 | 3.84 | 7.51 | 6.21 | 3.51 | 9.98 | |||||||
| Mongolia | 8.22 | |||||||||||||||
| South Korea | 3.75 | 3.41 | 5.03 | 1.68 | 3.12 | 10.34 | 7.05 | 6.32 | 6.89 | |||||||
| Taiwan | 4.81 | 4.72 | 7.81 | 4.64 | ||||||||||||
| Southeast Asia | Cambodia | 7.71 | 6.71 | |||||||||||||
| Laos | 4.85 | |||||||||||||||
| Myanmar | 8.89 | |||||||||||||||
| Indonesia | 4.06 | |||||||||||||||
| Thailand | 3.31 | 2.32 | ||||||||||||||
| Vietnam | 4.71 | 4.39 | ||||||||||||||
| South Asia | Bangladesh | 7.71 | 6.49 | |||||||||||||
| India | 4.18 | 8.82 | 5.86 | |||||||||||||
| Pakistan | 1.53 | 7.85 | 5.89 | |||||||||||||
| Iran | 7.31 | 2.95 | ||||||||||||||
| West Asia | Israel | 5.47 | 4.34 | |||||||||||||
| Saudi Arabia | 9.01 | |||||||||||||||
| Turkey | 3.03 | |||||||||||||||
| UAE | 8.78 | |||||||||||||||
| Africa | Egypt | 6.51 | 4.22 | |||||||||||||
| Nigeria | 5.48 | |||||||||||||||
| South Africa | 7.95 | |||||||||||||||
| Europe | France | 6.32 | 3.38 | |||||||||||||
| Germany | 5.81 | |||||||||||||||
| Italy | 6.21 | 3.44 | 4.98 | |||||||||||||
| Romania | 8.86 | |||||||||||||||
| Russia | 6.22 | 4.59 | 5.99 | |||||||||||||
| Sweden | 6.74 | 10.49 | ||||||||||||||
| United Kingdom | 4.15 | 4.59 | 2.46 | |||||||||||||
| North America | Canada | 2.84 | 2.49 | 3.66 | 3.01 | 5.84 | ||||||||||
| United States | 3.35 | 3.45 | 2.42 | 2.92 | 2.43 | 2.99 | 2.26 | 2.38 | 6.12 | 5.58 | 2.04 | 3.23 | 5.16 | 11.62 | 6.51 | |
Boldface indicates significantly higher substitution rates.
FIG 2.

Evolutionary rates of influenza A virus subtypes by region. Evolutionary rates (substitutions per site per year × 103) and 95% highest-probability density (HPD) values of the HA gene for 14 high-priority IAV subtypes from multiple world regions (East [E.] Asia, Southeast[SE.] Asia, South [S.] Asia, West [W.] Asia, Europe, Africa, and North [N.] America) from 2000 to 2011. Up to 500 sequences per region were used in the analysis. H3N8 eq and H3N8 av refer to equine and avian strains of H3N8, respectively.
Analysis of selection pressures across a 729-bp segment (nucleotides 191 to 920) of the HA gene showed that this segment of the HA gene was under the influence of purifying selection (dN/dS = <1) among all subtypes (Table 5). However, several subtypes, including H3N2, H3N8 (equine strains only), and H5N1, exhibited significantly higher dN/dS ratios (P > 0.05) than all other high-priority subtypes except H6N1. This trend was consistent across all of the countries examined.
TABLE 5.
Mean dN/dS ratios of the HA gene (nucleotides 191 to 920) among high-priority IAV subtypes from individual countries during a 12-year time span (2000 to 2011)
| World region and subregion | Country | Mean dN/dS ratioa |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H1N2 | H3N2 | H3N6 | H3N8-eq | H3N8-av | H4N6 | H5N1 | H5N2 | H5N3 | H6N1 | H6N2 | H6N8 | H7N3 | H7N7 | H9N2 | ||
| Asia | ||||||||||||||||
| Eastern | China | 0.20 | 0.51 | 0.50 | 0.34 | 0.20 | 0.31 | 0.26 | 0.21 | 0.18 | ||||||
| Hong Kong | 0.22 | 0.39 | 0.36 | 0.40 | 0.16 | |||||||||||
| Japan | 0.17 | 0.44 | 0.22 | 0.12 | 0.29 | 0.18 | 0.12 | 0.11 | 0.15 | |||||||
| Mongolia | 0.33 | |||||||||||||||
| South Korea | 0.21 | 0.53 | 0.11 | 0.08 | 0.03 | 0.25 | 0.22 | 0.12 | 0.26 | |||||||
| Taiwan | 0.46 | 0.07 | 0.20 | 0.20 | ||||||||||||
| Southeast | Cambodia | 0.41 | 0.19 | |||||||||||||
| Laos | 0.33 | |||||||||||||||
| Myanmar | 0.28 | |||||||||||||||
| Indonesia | 0.28 | |||||||||||||||
| Thailand | 0.35 | 0.35 | ||||||||||||||
| Vietnam | 0.41 | 0.35 | ||||||||||||||
| Southern | Bangladesh | 0.58 | 0.23 | |||||||||||||
| India | 0.49 | 0.25 | 0.13 | |||||||||||||
| Pakistan | 0.45 | 0.25 | 0.12 | |||||||||||||
| Iran | 0.34 | 0.25 | ||||||||||||||
| Western | Israel | 0.61 | 0.19 | |||||||||||||
| Saudi Arabia | 0.15 | |||||||||||||||
| Turkey | 0.15 | |||||||||||||||
| UAE | 0.23 | |||||||||||||||
| Africa | Egypt | 0.62 | 0.43 | |||||||||||||
| Nigeria | 0.25 | |||||||||||||||
| South Africa | 0.56 | |||||||||||||||
| Europe | France | 0.50 | 0.27 | |||||||||||||
| Germany | 0.29 | |||||||||||||||
| Italy | 0.24 | 0.38 | 0.37 | |||||||||||||
| Romania | 0.33 | |||||||||||||||
| Russia | 0.57 | 0.05 | 0.28 | |||||||||||||
| Sweden | 0.64 | 0.12 | ||||||||||||||
| United Kingdom | 0.28 | 0.44 | 0.28 | |||||||||||||
| North America | Canada | 0.38 | 0.08 | 0.07 | 0.16 | 0.11 | ||||||||||
| United States | 0.30 | 0.44 | 0.07 | 0.35 | 0.09 | 0.11 | 0.28 | 0.27 | 0.18 | 0.18 | 0.18 | 0.13 | 0.13 | 0.10 | 0.09 | |
| Global avg | 0.23 | 0.46 | 0.09 | 0.32 | 0.09 | 0.07 | 0.31 | 0.20 | 0.15 | 0.27 | 0.22 | 0.17 | 0.21 | 0.11 | 0.17 | |
Boldface indicates significantly higher dN/dS ratios (P > 0.5).
DISCUSSION
Traditional approaches to studying IAV evolutionary dynamics have typically focused on single subtypes (e.g., H3N2, H3N8, or H5N1) or on analyses that were restricted to coarse geographical scales comparing data from one continent to data from another (13–17). However, in order to identify geographical variations in IAV evolution across a spectrum of subtypes, a more comprehensive approach comparing data from individual countries was needed.
A detailed examination of all publicly available IAV strain data allowed us to prioritize our analyses to a limited set of priority subtypes based on their wider geographic and host distribution. This novel approach identified 21 high-priority subtypes that may be more likely to spread geographically and among different host species. Evolutionary analysis of 14 high-priority subtypes showed that nucleotide substitution rates for all subtypes except H5N3, H7N3, and H7N7 were higher in East Asian countries, including China, Hong Kong, Japan, Mongolia, South Korea, and Taiwan, than in Canada and the United States. However, we did not observe consistently high nucleotide substitution rates across all the subtypes in any single East Asian country, indicating that there was not a specific focal point or evolutionary “hot spot” for all the IAVs analyzed. A regional analysis of nucleotide substitution rates further demonstrated that evolutionary rates for several subtypes, including H5N1, H5N2, and H6N2, were significantly greater in East Asia than in North America. These findings suggested that, among the majority of high-priority subtypes analyzed, novel, potentially pathogenic IAV strains may be more likely to evolve in East Asia. In fact, the majority of emerging IAV strains that have caused disease and mortality in humans in recent years, including those belonging to subtypes H5N1, H7N9, H9N2, and H10N8, were first detected in China and Hong Kong (9, 11, 23). The factors leading to geographical associations with higher substitution rates are not clear, although it is possible that certain practices such as mixed-bird farming and the presence of live-bird markets may serve as drivers for increased substitution rates. It has been shown that the introduction of a novel IAV strain into a bird population can cause the nucleotide substitution rate of that subtype to increase. The introduction of a novel Eurasian lineage of the H6 subtype in North American wild-bird populations in the early 1980s resulted in H6 substitution rates that increased from 2.0 × 10−3 substitutions per site per year to 4.4 × 10−3 substitutions per site per year (4). Similarly, the introduction of a novel H5N2 strain into Mexican poultry farms increased nucleotide substitution rates from 7.0 × 10−3 substitutions per site per year to 28.1 × 10−3 substitutions per site per year (4, 24). Conditions that allow mixing of bird species, such as in live-bird markets, are common throughout East Asia and would likely support the transmission of AIV strains to naive groups of birds. This scenario could in turn lead to increased nucleotide substitution rates such as those observed in the current study.
Another factor that may affect nucleotide substitution rates among influenza viruses is vaccination. While comprehensive application of vaccinations, coupled with careful surveillance and strict biosecurity precautions, has proven to be an effective tool for the control and eradication of IAVs in poultry (25, 26), vaccination programs that are not completely sustained or properly administered have been linked to increased nucleotide substitution rates (22, 27). In Pakistan, for example, a failed attempt in the mid-1990s to eradicate an initial outbreak of HPAI H7N3 in poultry through vaccination efforts resulted in a resurgence of this subtype throughout the following decade (28). Coincidentally, among all of the countries in our study, Pakistan had the highest nucleotide substitution rate for the H7N3 subtype (7.85 × 10−3 substitutions per site per year).
In addition to nucleotide substitution rates, we found that the selection pressures acting on individual subtypes were another important indicator of AIV evolution. The highest dN/dS ratios were detected among H3N2 (consisting of human strains only), H3N8 (equine strains only), and H5N1 (all strains), indicating that nucleotide mutations more frequently resulted in amino acid substitutions among these subtypes. With few exceptions, these trends were consistent among all of the countries analyzed, suggesting that the forces driving selection among IAV subtypes are similar across the globe. One of the major determinants of selection pressure acting on influenza viruses is the host immune response. In our analyses, strains specific to mammals (H3N2 and H3N8-eq) exhibited higher selection pressures than bird-specific strains. A striking example was the difference in mean dN/dS ratios between avian and equine H3N8 strains (0.089 and 0.324, respectively). Within their natural aquatic bird hosts, IAVs are proposed to have reached an “evolutionary stasis” characterized by low rates of evolutionary change, particularly at sites leading to amino acid changes (1). According to this hypothesis, the evolutionary race between virus and host is less intense in avian than in mammalian species, so there is less selective pressure on the virus to maintain amino acid changes that evade host immune responses (29). Selection pressures may also increase following the introduction of a novel IAV subtype into a host population with no prior exposure to that subtype. This was observed following the introduction of the H2N2 subtype into the human population in 1957 (30), as well as with the more recent introduction of HPAI H5N1 virus into naive poultry populations (31). The elevated dN/dS ratios observed among H5N1 subtypes (mean, 0.314; range, 0.15 to 0.61) in our analysis might be a reflection of the rapid spread of this subtype to susceptible host populations throughout much of the globe. Whatever the underlying cause, increased selective pressure acting on a viral population results in a greater accumulation of phenotypic variants, some of which may harbor more-pathogenic properties. Monitoring of selection pressure changes among IAVs worldwide would be a relatively feasible method to help identify emerging, potentially higher-risk strains in the future.
The growing number of unique subtypes detected in humans and poultry in recent years suggests that subtype diversity might be an important factor associated with the emergence of pathogenic IAV strains. After we controlled for reporting effort, our findings showed that subtype diversity was greatest throughout many northern temperate zone countries, including Russia, Sweden, Norway, Ireland, Hungary, Canada, Netherlands, Switzerland, Czech Republic, Mongolia, and Kazakhstan, as well as in two southern temperate zone countries, Australia and South Africa. Much of the observed subtype diversity can be linked to wild migratory birds, in particular, mallards, from which the greatest number of unique subtypes (n = 94) of any bird species have been reported. Extensive breeding and wintering grounds found in temperate zones bring large numbers of migratory birds, often of mixed species, in close contact for large parts of the year, resulting in the maintenance of existing subtypes as well as the establishment of new subtypes through reassortment (32–34). A concerted effort to monitor the IAV subtypes circulating among wild birds in these temperate zones may help to identify emerging subtypes before they make their way into domestic birds.
Assessing the potential drivers of IAV subtype diversity, our analyses confirmed that reporting effort was the best predictor of the observed HA and NA subtype diversity at the country level. This finding underscores the need to increase sampling and reporting efforts for all subtypes in many undersampled countries throughout the world. Notably, for the 143 countries with available sequence data (including partial sequences), 47 countries had submitted fewer than 10 sequences to public databases and 95 countries had submitted fewer than 100 sequences. Only 22 countries had submitted more than 500 sequences (see Table S2 in the supplemental material). In addition to reporting effort, we found that health care spending per capita in 2010 was strongly predictive of observed HA subtype diversity, indicating that nations spending more on health care may be able to allocate more resources to influenza detection, including the application of broad testing followed by subtyping, thus enabling the detection of a greater variety of IAV subtypes and the availability of a larger number of samples to test. While the number of strains reported and the level of health care spending were correlated (r = 0.40), this correlation is weak enough for the data to have distinct predictive power. Interestingly, among biological predictors, pig population density was mildly predictive of observed HA subtype diversity whereas neither poultry population density nor avian population density was associated with such diversity. There may be other relevant biological predictors of subtype diversity, but the inconsistent quality of the data at the country level meant that we did not have the power to detect these relationships. Thus, our models could be largely improved if there were greater uniformity in IAV sampling, testing, and reporting methods worldwide. Simple efforts to improve publically available data, such as georeferencing of submitted strains and reporting the total number of samples tested in a given study, would greatly improve future surveillance and modeling efforts.
It is clear from our search of sequence databases that there is a paucity of influenza virus sequence data from many regions of the world. In particular, there is very little sequence data available from the majority of African countries. Data from many parts of Latin America and Eastern Europe were also limited. In Asia, most of the sequence data for strains other than H9N2, H5N1, H3N2, and H1N1 were limited to just a few East Asian countries, including China, Japan, South Korea, and Taiwan. While not all subtypes likely exist in all countries or regions, current strategies of targeted testing for specific influenza virus subtypes such as H5N1 severely limit our understanding of the total diversity of subtypes present and circulating in many countries. These strategies, in turn, limit our ability to monitor the evolution and diversity of influenza virus subtypes circulating globally. As such, there is a great need to encourage all countries currently conducting only targeted IAV testing to perform broader testing that includes protocols to detect all subtypes, followed by sequencing and subtyping procedures, in at least a subset of surveillance samples.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kate Thomas for producing the maps presented in the manuscript. We also thank everyone who submitted IAV sequence data to public databases for making our analyses possible.
This study was made possible by the generous support of the American people through the United States Agency for International Development (USAID) Emerging Pandemic Threats PREDICT project.
The contents are our responsibility and do not necessarily reflect the views of USAID or the United States government.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01573-15.
REFERENCES
- 1.Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. 1992. Evolution and ecology of influenza A viruses. Microbiol Rev 56:152–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu S, Ji K, Chen J, Tai D, Jiang W, Hou G, Chen J, Li J, Huang B. 2009. Panorama phylogenetic diversity and distribution of type A influenza virus. PLoS One 4:e5022. doi: 10.1371/journal.pone.0005022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, Yang H, Chen X, Recuenco S, Gomez J, Chen LM, Johnson A, Tao Y, Dreyfus C, Yu W, McBride R, Carney PJ, Gilbert AT, Chang J, Guo Z, Davis CT, Paulson JC, Stevens J, Rupprecht CE, Holmes EC, Wilson IA, Donis RO. 2013. New world bats harbor diverse influenza A viruses. PLoS Pathog 9:e1003657. doi: 10.1371/journal.ppat.1003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bahl J, Vijaykrishna D, Holmes EC, Smith GJD, Guan Y. 2009. Gene flow and competitive exclusion of avian influenza A virus in natural reservoir hosts. Virology 390:289–297. doi: 10.1016/j.virol.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Webster RG, Krauss S, Hulse-Post D, Sturm-Ramirez K. 2007. Evolution and ecology of influenza A viruses. J Wildl Dis 43(Suppl):S1–S6. [Google Scholar]
- 6.Arzey GG, Kirkland PD, Arzey KE, Frost M, Maywood P, Conaty S, Hurt AC, Deng Y-M, Iannello P, Barr I, Dwyer DE, Ratnamohan M, McPhie K, Selleck P. 2012. Influenza virus A (H10N7) in chickens and poultry abattoir workers, Australia. Emerg Infect Dis 18:814–816. doi: 10.3201/eid1805.111852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan J, Zhang L, Kan X, Jiang L, Yang J, Guo Z, Ren Q. 2013. Origin and molecular characteristics of a novel 2013 avian influenza A(H6N1) virus causing human infection in Taiwan. Clin Infect Dis 57:1367–1368. doi: 10.1093/cid/cit479. [DOI] [PubMed] [Google Scholar]
- 8.Lopez-Martinez I, Balish A, Barrera-Badillo G, Jones J, Nuñez-García TE, Jang Y, Aparicio-Antonio R, Azziz-Baumgartner E, Belser JA, Ramirez-Gonzalez JE, Pedersen JC, Ortiz-Alcantara J, Gonzalez-Duran E, Shu B, Emery SL, Poh MK, Reyes-Teran G, Vazquez-Perez JA, Avila-Rios S, Uyeki T, Lindstrom S, Villanueva J, Tokars J, Ruiz-Matus C, Gonzalez-Roldan JF, Schmitt B, Klimov A, Cox N, Kuri-Morales P, Davis CT, Diaz-Quiñonez JA. 2013. Avian influenza A (H7N3) virus in poultry workers, Mexico, 2012. Emerg Infect Dis 19:1531–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peiris M, Yuen KY, Leung CW, Chan KH, Lai RW, Orr WK, Shortridge KF. 1999. Human infection with influenza H9N2. Lancet 354:916–917. doi: 10.1016/S0140-6736(99)03311-5. [DOI] [PubMed] [Google Scholar]
- 10.Fouchier RA, Schneeberger PM, Rozendaal FW, Broekman JM, Kemink SA, Munster V, Kuiken T, Rimmelzwaan GF, Schutten M, Van Doornum GJ, Koch G, Bosman A, Koopmans M, Osterhaus AD. 2004. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proc Natl Acad Sci U S A 101:1356–1361. doi: 10.1073/pnas.0308352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.To KKW, Tsang AKL, Chan JFW, Cheng VCC, Chen H, Yuen K-Y. 2014. Emergence in China of human disease due to avian influenza A(H10N8)—cause for concern? J Infect 68:205–215. doi: 10.1016/j.jinf.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 12.Rambaut A, Pybus OG, Nelson MI, Viboud C, Taubenberger JK, Holmes EC. 2008. The genomic and epidemiological dynamics of human influenza A virus. Nature 453:615–619. doi: 10.1038/nature06945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murcia PR, Wood JLN, Holmes EC. 2011. Genome-scale evolution and phylodynamics of equine H3N8 influenza A virus. J Virol 85:5312–5322. doi: 10.1128/JVI.02619-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bedford T, Cobey S, Beerli P, Pascual M. 2010. Global migration dynamics underlie evolution and persistence of human influenza A (H3N2). PLoS Pathog 6:e1000918. doi: 10.1371/journal.ppat.1000918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vijaykrishna D, Bahl J, Riley S, Duan L, Zhang JX, Chen H, Peiris JSM, Smith GJD, Guan Y. 2008. Evolutionary dynamics and emergence of panzootic H5N1 influenza viruses. PLoS Pathog 4:e1000161. doi: 10.1371/journal.ppat.1000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen R, Holmes EC. 2006. Avian influenza virus exhibits rapid evolutionary dynamics. Mol Biol Evol 23:2336–2341. doi: 10.1093/molbev/msl102. [DOI] [PubMed] [Google Scholar]
- 17.Lebarbenchon C, Stallknecht DE. 2011. Host shifts and molecular evolution of H7 avian influenza virus hemagglutinin. Virol J 8:328. doi: 10.1186/1743-422X-8-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawaoka Y, Bean WJ, Webster RG. 1989. Evolution of the hemagglutinin of equine H3 influenza viruses. Virology 169:283–292. doi: 10.1016/0042-6822(89)90153-0. [DOI] [PubMed] [Google Scholar]
- 19.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nielsen R, Yang Z. 2003. Estimating the distribution of selection coefficients from phylogenetic data with applications to mitochondrial and viral DNA. Mol Biol Evol 20:1231–1239. doi: 10.1093/molbev/msg147. [DOI] [PubMed] [Google Scholar]
- 21.Burnham K, Anderson D. 2002. Model selection and multi-model inference: a practical information-theoretic approach, 2nd ed Springer-Verlag, New York, NY. [Google Scholar]
- 22.Cattoli G, Fusaro A, Monne I, Coven F, Joannis T, El-Hamid HSA, Hussein AA, Cornelius C, Amarin NM, Mancin M, Holmes EC, Capua I. 2011. Evidence for differing evolutionary dynamics of A/H5N1 viruses among countries applying or not applying avian influenza vaccination in poultry. Vaccine 29:9368–9375. doi: 10.1016/j.vaccine.2011.09.127. [DOI] [PubMed] [Google Scholar]
- 23.Poovorawan Y, Pyungporn S, Prachayangprecha S, Makkoch J. 2013. Global alert to avian influenza virus infection: from H5N1 to H7N9. Pathog Glob Health 107:217–223. doi: 10.1179/2047773213Y.0000000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.García M, Suarez DL, Crawford JM, Latimer JW, Slemons RD, Swayne DE, Perdue ML. 1997. Evolution of H5 subtype avian influenza A viruses in North America. Virus Res 51:115–124. doi: 10.1016/S0168-1702(97)00087-7. [DOI] [PubMed] [Google Scholar]
- 25.Capua I, Marangon S. 2007. Control and prevention of avian influenza in an evolving scenario. Vaccine 25:5645–5652. doi: 10.1016/j.vaccine.2006.10.053. [DOI] [PubMed] [Google Scholar]
- 26.Ellis TM, Sims LD, Wong HK, Dyrting KC, Chow KW, Leung C, Peiris JS. 2006. Use of avian influenza vaccination in Hong Kong. Dev Biol (Basel) 124:133–143. [PubMed] [Google Scholar]
- 27.Lee C, Senne DA, Suarez DL. 2004. Effect of vaccine use in the evolution of Mexican lineage H5N2 avian influenza virus. J Virol doi: 10.1128/JVI.78.15.8372-8381.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naeem K, Siddique N. 2006. Use of strategic vaccination for the control of avian influenza in Pakistan. Dev Biol (Basel) 124:145–150. [PubMed] [Google Scholar]
- 29.Suarez DL. 2000. Evolution of avian influenza viruses. Vet Microbiol 74:15–27. doi: 10.1016/S0378-1135(00)00161-9. [DOI] [PubMed] [Google Scholar]
- 30.Schäfer JR, Kawaoka Y, Bean WJ, Suss J, Senne D, Webster RG. 1993. Origin of the pandemic 1957 H2 influenza A virus and the persistence of its possible progenitors in the avian reservoir. Virology 194:781–788. doi: 10.1006/viro.1993.1319. [DOI] [PubMed] [Google Scholar]
- 31.Zhou NN, Shortridge KF, Claas EC, Krauss SL, Webster RG. 1999. Rapid evolution of H5N1 influenza viruses in chickens in Hong Kong. J Virol 73:3366–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dugan VG, Chen R, Spiro DJ, Sengamalay N, Zaborsky J, Ghedin E, Nolting J, Swayne DE, Runstadler JA, Happ GM, Senne DA, Wang R, Slemons RD, Holmes EC, Taubenberger JK. 2008. The evolutionary genetics and emergence of avian influenza viruses in wild birds. PLoS Pathog 4:e1000076. doi: 10.1371/journal.ppat.1000076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill NJ, Takekawa JY, Cardona CJ, Meixell BW, Ackerman JT, Runstadler JA, Boyce WM. 2012. Cross-seasonal patterns of avian influenza virus in breeding and wintering migratory birds: a flyway perspective. Vector Borne Zoonotic Dis 12:243–253. doi: 10.1089/vbz.2010.0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gunnarsson G, Latorre-Margalef N, Hobson KA, Van Wilgenburg SL, Elmberg J, Olsen B, Fouchier RA, Waldenström MJ. 2012. Disease dynamics and bird migration–linking mallards Anas platyrhynchos and subtype diversity of the influenza A virus in time and space. PLoS One 7:e35679. doi: 10.1371/journal.pone.0035679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pond SL, Frost SD, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.