

ABSTRACT
Equine herpesvirus type 1 (EHV-1) is a main cause of respiratory disease, abortion, and encephalomyelopathy in horses. Monocytic cells (CD172a+) are the main carrier cells of EHV-1 during primary infection and are proposed to serve as a “Trojan horse” to facilitate the dissemination of EHV-1 to target organs. However, the mechanism by which EHV-1 is transferred from CD172a+ cells to endothelial cells (EC) remains unclear. The aim of this study was to investigate EHV-1 transmission between these two cell types. We hypothesized that EHV-1 employs specific strategies to promote the adhesion of infected CD172a+ cells to EC to facilitate EHV-1 spread. Here, we demonstrated that EHV-1 infection of CD172a+ cells resulted in a 3- to 5-fold increase in adhesion to EC. Antibody blocking experiments indicated that α4β1, αLβ2, and αVβ3 integrins mediated adhesion of infected CD172a+ cells to EC. We showed that integrin-mediated phosphatidylinositol 3-kinase (PI3K) and ERK/MAPK signaling pathways were involved in EHV-1-induced CD172a+ cell adhesion at early times of infection. EHV-1 replication was enhanced in adherent CD172a+ cells, which correlates with the production of tumor necrosis factor alpha (TNF-α). In the presence of neutralizing antibodies, approximately 20% of infected CD172a+ cells transferred cytoplasmic material to uninfected EC and 0.01% of infected CD172a+ cells transmitted infectious virus to neighboring cells. Our results demonstrated that EHV-1 infection induces adhesion of CD172a+ cells to EC, which enhances viral replication, but that transfer of viral material from CD172a+ cells to EC is a very specific and rare event. These findings give new insights into the complex pathogenesis of EHV-1.
IMPORTANCE Equine herpesvirus type 1 (EHV-1) is a highly prevalent pathogen worldwide, causing frequent outbreaks of abortion and myeloencephalopathy, even in vaccinated horses. After primary replication in the respiratory tract, EHV-1 disseminates via cell-associated viremia in peripheral blood mononuclear cells (PBMC) and subsequently infects the endothelial cells (EC) of the pregnant uterus or central nervous system, leading in some cases to abortion and/or neurological disorders. Recently, we demonstrated that CD172a+ monocytic carrier cells serve as a “Trojan horse” to facilitate EHV-1 spread from blood to target organs. Here, we investigated the mechanism underlying the transmission of EHV-1 from CD172a+ cells to EC. We demonstrated that EHV-1 infection induces cellular changes in CD172a+ cells, promoting their adhesion to EC. We found that both cell-to-cell contacts and the secretion of soluble factors by EC activate EHV-1 replication in CD172a+ cells. This facilitates transfer of cytoplasmic viral material to EC, resulting mainly in a nonproductive infection. Our findings give new insights into how EHV-1 may spread to EC of target organs in vaccinated horses.
INTRODUCTION
Equine herpesvirus type 1 (EHV-1), a member of the subfamily Alphaherpesvirinae, is a ubiquitous pathogen in horses, causing serious economic losses in the horse industry. Primary EHV-1 infection usually results in the establishment of a lifelong latent infection within the first months of life, with subsequent viral reactivation causing clinical disease and viral shedding during periods of stress (1, 2). EHV-1 infection is characterized by upper respiratory tract disease, neurological disorders, abortion, or neonatal death (3, 4). The virus first replicates in the epithelial cells of the upper respiratory tract and disseminates through the body via a cell-associated viremia in peripheral blood mononuclear cells (PBMC) to target organs, such as the pregnant uterus or central nervous system (CNS). Secondary replication in the endothelial cells (EC) lining the blood vessels of those organs can cause vasculitis and ischemic thrombosis and may lead to severe symptoms, such as abortion and/or neurological disorders (5–7).
Monocytic cells (CD172a+) have been shown to be the main carrier cell type of EHV-1 during primary infection (8, 9). We previously reported that CD172a+ cells serve as a “Trojan horse” to facilitate the spread of EHV-1 to target organs and to evade immunosurveillance (10). This may contribute to the observation that current vaccines do not provide full protection against severe symptoms, as EHV-1 can cause viremia despite the presence of a virus-specific immune response in the horse (11–13).
EC are actively involved in a wide variety of pathological processes, such as thrombosis and vasculitis, and endothelial cell-monocyte interactions are known to play a central role in the pathogenesis of herpesviruses infections. For instance, human cytomegalovirus (HCMV) infection of EC has been shown to promote naïve-monocyte adhesion to and migration through the endothelium, and virus-mediated cellular activation was found to be responsible for HCMV-induced monocyte migration (14, 15). The mechanism of HCMV dissemination to host tissue is thought to be associated with HCMV-induced vascular diseases.
So far, several in vitro systems have demonstrated the potential utility of cultured EC in the study of the pathogenesis of EHV-1 (16, 17). Studies have shown that the infection of EC located in the vasculature of the late-gravid uterus or CNS was mediated by cell-to-cell contacts between infected PBMC and EC and occurred even in the presence of virus-neutralizing antibodies (18, 19). In addition, Smith et al. (18) provided evidence that activation of EC adhesion molecules may be involved in the transfer of virus from infected PBMC to EC and may determine the restricted tissue specificity of EHV-1. However, the precise mechanism underlying the transmission of EHV-1 from monocytic cells to EC is still unclear.
Given the importance of the interactions between monocytic cells and EC in the pathogenesis of EHV-1 infections, we studied the ability of EHV-1-inoculated CD172a+ cells to adhere and subsequently transmit EHV-1 infection to equine venous EC. We examined the contributions of specific cell adhesion molecules and the cellular signal transduction pathways involved in the adhesion process in vitro. Furthermore, we studied the replication kinetics of EHV-1 in CD172a+ cells upon adhesion to EC.
MATERIALS AND METHODS
Virus.
Two Belgian EHV-1 strains were included in this study. The neurovirulent strain 03P37 was originally isolated in 2003 from the blood of a paralytic horse (20, 21), and the nonneurovirulent strain 97P70 was first isolated in 1997 from the lungs of an aborted fetus (8). The virus stocks used for inoculation were at the sixth passage, with five passages in equine embryonic lung cells (EEL) and one passage in RK-13 cells for strain 97P70 and four passages in EEL and one passage in RK-13 cells for strain 03P37. For virus inactivation, a thin layer of viral suspension was exposed to short-wave UV light for 10 min. Absence of viral infectivity was checked by virus titration on RK-13 cells.
Cells. (i) Isolation of equine blood CD172a+ cells.
Healthy horses between 8 and 10 years old were used as blood donors. The horses were seropositive for EHV-1. The collection of blood was approved by the ethical committee of Ghent University (EC2013/17). Blood was collected by jugular venipuncture on heparin (15 U ml−1; Leo) and diluted in an equal volume of Dulbecco's phosphate-buffered saline (DPBS) without calcium and magnesium (Gibco). PBMC were isolated by density centrifugation on Ficoll-Paque (density = 1.077 g ml−1; GE Healthcare, Life Sciences) at 800 × g for 30 min at 18°C. The interphase cells containing the PBMC were collected and washed three times with DPBS. The cells were resuspended in leukocyte medium (LM) based on RPMI (Gibco) supplemented with 5% fetal calf serum (FCS) (Grainer), 1% penicillin, 1% streptomycin, 0.5% gentamicin (Gibco). Afterward, cells were seeded in 6-well plates (Nunc A/S) at a concentration of 106 cells per ml and cultivated at 37°C with 5% CO2. After 12 h, nonadhering lymphocytes were removed by washing the cells three times with RPMI. The adherent cells consisted of >90% monocytic cells, as assessed by flow cytometry after indirect immunofluorescence staining with a mouse anti-CD172a monoclonal antibody (MAb) (VMRD; clone DH59B; 1:100; IgG1) directed against cells from a myeloid lineage, followed by goat anti-mouse IgG fluorescein isothiocyanate (FITC) (Molecular Probes; 1:200).
(ii) Isolation of equine venous endothelial cells.
Equine endothelial cells were obtained from the vena cava of a healthy horse at the slaughterhouse. The vena cava was harvested in a bottle containing Dulbecco's modified Eagle medium (DMEM) (Gibco) supplemented with 1% penicillin, 1% streptomycin, 0.5% gentamicin, and 0.1% Fungizone. One end of the vessel was closed using a hemostatic clamp. A prewarmed enzyme mixture of 0.1% type I collagenase (Invitrogen) and 0.12% dispase (Sigma-Aldrich) in DMEM was infused into the segment until there was moderate distention of the vessel. After closing the segment with a second hemostatic clamp, the vessel was incubated for 30 to 40 min at 37°C. Then, one of the hemostatic clamps was opened. The loose endothelial cells were collected by flushing the vessel with warm DMEM. The effluent was collected with sterile syringes and transferred into chilled centrifuge tubes with FCS. The cells were pelleted by centrifugation at 200 × g at 4°C for 10 min. The supernatant was discarded, and the cell pellet was resuspended in endothelial growth medium based on DMEM supplemented with 5% FCS, 1% penicillin, 1% streptomycin, 0.5% gentamicin, 1% sodium pyruvate, 1% nonessential amino acids (Gibco), 50 μg/ml endothelial cell growth supplement (ECGS) (Biomedical Technologies Inc.). Cells were plated on 0.5% gelatin-coated plasticware (Nunc A/S) and incubated at 37°C with 5% CO2. After overnight incubation, nonadherent cells were removed by washing with prewarmed DMEM, and fresh endothelial growth medium was added. Thereafter, the culture medium was refreshed every 2 to 3 days.
(iii) Immortalization of endothelial cells.
Endothelial cell lines were immortalized by transduction of the genes encoding the simian virus 40 large T-antigen (SV40LT) and human telomerase reverse transcriptase (hTERT). Primary endothelial cells (50% confluent) were subsequently exposed to either a recombinant lentiviral vector containing the gene encoding SV40LT-transforming protein, a lentiviral vector carrying the gene encoding hTERT, or a combination of the two (Applied Biological Materials Inc.). All primary EC were incubated in the presence of 8 μg/ml Polybrene (Applied Biological Materials Inc.). To avoid cytotoxicity, the viral supernatant was diluted after 30 min with heparin-free EC growth medium (1:1) and further incubated overnight. The medium was refreshed the next day and then every other day until the cells reached confluence. At 72 h postransduction, the cells were incubated with EC growth medium supplemented with 10 μg/ml puromycin (Applied Biological Materials Inc.) to select for stable transduced cells. Selection continued until the cultures consisted of resistant surviving cells only. They were further expanded in standard medium and routinely passaged at a 1:3 split ratio using 10% trypsin (Sigma-Aldrich), 1% EDTA (Vel Chemicals) in phosphate-buffered saline (PBS). Immortalized EC (IEC) were analyzed by indirect immunofluorescence for the expression of hTERT and SV40LT antigen.
(iv) Purity of primary and immortalized EC.
The purity of primary and immortalized EC was analyzed by fluorescence staining with 1,1-diocatadecyl-1-3,3,3′,3′-tetramethylindocarbocyanine-perchlorate-acetylated low-density lipoprotein (DiI-Ac-LDL) (Biomedical Technologies Inc.) and was always >90%.
(v) Cell viability.
Cell viability was determined by flow cytometry prior to virus inoculation, using 1 μg ml−1 propidium iodide (Sigma-Aldrich), and was >90% in all cell populations.
EHV-1 inoculation.
Cells were inoculated in vitro with both replication-competent and UV-inactivated EHV-1 (UV-EHV-1) strains 03P37 and 97P70 at a multiplicity of infection (MOI) of 1 in 1 ml of LM for 1 h at 37°C with 5% CO2. The cells were gently washed twice with RPMI to remove the inoculum and further incubated with fresh medium. Mock inoculations were carried out in parallel.
Inhibition of the MEK/ERK signaling pathway was performed by using the MEK/ERK signaling inhibitor U0126 (1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene) (Cell Signaling). The stock solution of U0126 was prepared in dimethyl sulfoxide (DMSO) at a concentration of 10 mM. U0126 interferes with MEK1 and MEK2 directly by inhibiting the catalytic activity of the active enzyme and consequently blocks the phosphorylation and activation of ERK1 and ERK2. Inhibition of the phosphatidylinositol 3-kinase (PI3K) signaling pathway was performed by using the PI3K inhibitor LY294002 (Invivogen). The stock solution of LY294002 was prepared in DMSO at a concentration of 50 mM. Cells were preincubated with U0126 (1 and 10 μM), LY294002 (25 and 50 μM), or DMSO-based diluent, used as a solvent control, for 30 min at 37°C before EHV-1 inoculation. MEK/ERK and PI3K inhibitors were maintained in the culture media throughout the course of the experiment. The concentration of inhibitors used in this study and the DMSO-based diluent did not decrease cell viability.
Where indicated, 10 ng/ml of equine recombinant tumor necrosis factor alpha (TNF-α) or 10 ng/ml of TNF-α-neutralizing polyclonal antibody (R&D Systems) was added at the time of inoculation and maintained in the medium throughout the course of inoculation. TNF-α was reconstituted in PBS and diluted in LM.
Adhesion assay.
After virus inoculation, cells were incubated with EHV-1-neutralizing IgG antibodies for 1 h at 37°C, 5% CO2. Mock-, EHV-1-, or UV-EHV-1-inoculated CD172a+ cells were then detached using accutase solution (Sigma-Aldrich), counted, and resuspended in LM supplemented with 10 μg ml−1 polymyxin B (LMPB) (Sigma-Aldrich). Polymyxin B is an antibiotic used to neutralize circulating lipopolysaccharide (LPS) in the medium and thus was used in this study to prevent LPS-mediated adhesion of CD172a+ cells to endothelial cell monolayers. One hundred thousand CD172a+ cells were added to each well of EC, which were grown on Lab-Tek chamber slides with eight compartments (VWR). The monocyte/endothelial cell ratio in cocultures was 1:3. CD172a+ cells adherent to the plastic plate were included as a control. At different time points of cocultivation (2, 4, 6, and 12 h), adherent cells were fixed with 1 ml of methanol at −20°C for 20 min. Nonadherent cells were harvested, fixed with 1 ml of 1% (paraformaldehyde [PFA]) for 10 min at room temperature (RT), permeabilized with 1 ml of 0.1% Triton X-100, and cytospun on a slide. After 2, 4, 6, and 12 h of cocultivation, both adherent and nonadherent cells were incubated for 1 h at 37°C with a rabbit polyclonal anti-immediate-early protein (IEP) antibody (Ab) (1:1,000) to detect IEP expression. At 12 h of cocultivation, cells were also double stained with a rabbit polyclonal anti-IEP Ab (1:1,000) and a mouse monoclonal anti-glycoprotein B (gB) (4B6; 1:100) against late gB protein expression. The IEP and 4B6 antibodies were kindly provided by D. O'Callaghan (Louisiana State University) and N. Osterrieder (Freie Universität Berlin, Berlin, Germany), respectively. Where indicated, cells were fixed with 1 ml of 1% PFA for 10 min at RT, washed twice with PBS, and stained with a horse polyclonal antibody against EHV-1 late proteins (1:100). The horse polyclonal anti-EHV-1 antibody was obtained by hyperimmunization of a horse (22). The polyclonal antibody was purified on a protein G column and subsequently biotinylated (Amersham International, Buckinghamshire, United Kingdom). This antibody could recognize the late proteins of EHV-1, as described previously (9). Subsequently, samples were incubated for 50 min at 37°C with goat anti-rabbit IgG FITC (1:200) and goat anti-mouse IgG Texas Red (1:200) or Streptavidin-FITC (1:200) antibodies (Molecular Probes). All antibodies were diluted in DPBS. The nuclei were counterstained with Hoechst 33342 (10 μg ml−1; Molecular Probes) for 10 min at 37°C. As a negative control, mock-inoculated cells were stained following the above-mentioned protocols. In addition, appropriate isotype-matched controls were included.
For each time point, the IEP- and gB-positive adherent CD172a+ cells were counted based on the total number of adherent CD172a+ cells counted in 5 randomly selected microscopic fields of 0.2 mm2 using a fluorescence microscope (40× objective; Leica Microsystems DMRBE, Wetzlar, Germany). The results were shown as the number of cells per square millimeter and were also expressed as a percentage. In the nonadherent cell fraction, the number of IEP-positive CD172a+ cells was calculated based on 300 cells counted in distinct fields, and the results were expressed as a percentage. Samples were analyzed by confocal microscopy (Leica TCS SP2 laser scanning spectral confocal system; Leica Microsystems GmbH, Germany).
Adhesion-blocking assay.
To determine the role of α4β1 (CD29/CD49d), αLβ2 (CD11a/CD18), and αVβ3 (CD51/61) integrins in EHV-1-induced adhesion to endothelial cells, adhesion assays were performed as described above with some modifications. Prior to addition of CD172a+ cells to endothelial cells, EHV-inoculated CD172a+ cells were incubated with LMPB supplemented with 10 μg/ml of neutralizing monoclonal antibodies directed against CD29 (clone TDM29; IgG1; Merck Millipore); CD49d (clone 9F10; IgG2b; BD Pharmingen); CD11a/CD18 (clone CVS9; IgG1; AbD Serotec); CD51/61 (clone 23C6; IgG1; BioLegend); or a mixture of CD29, CD49d, CD11a/18, and CD51/61 or with isotype-matched control antibodies for 30 min at 4°C. After washing, EHV-1-inoculated CD172a+ cells were added to wells of IEC. The cells were fixed after 2 h of cocultivation and stained for IEP expression, as previously described.
Flow cytometry.
To evaluate integrin expression on CD172a+ cells, the cells were incubated with 10 μg/ml of the anti-integrin monoclonal antibodies CD29, CD49d, CD11a/CD18, and CD51/61 or with isotype-matched control antibodies for 30 min at 4°C. The cells were washed twice in PBS and incubated with Alexa Fluor 647-labeled goat anti-mouse IgG (1:200 dilution; Molecular Probes) for 30 min at 4°C. After a final wash in PBS, 10,000 cells were analyzed with a FACSCanto flow cytometer equipped with FACSDiva software (BD Biosciences, Mountain View, CA, USA).
Western blot analysis.
One million CD172a+ cells were mock inoculated or inoculated with EHV-1 strain 03P37 or 97P70 (MOI = 1) and harvested at 0, 5, and 15 min postinoculation (p.i.). The cells were resuspended in ice-cold lysis buffer (TNE buffer [10% NP-40, 1 mM NA3VO4, 10 mM NaF, protease inhibitor cocktail]) and incubated for 20 min at 4°C. Then, samples were boiled for 10 min. SDS-PAGE and Western blotting were performed as described previously (23). Protein concentrations of 20 to 30 μg/ml were used for all experiments. The protein concentration was determined using the bicinchoninic acid (BCA) protein assay reagent (Thermo Scientific) according to the manufacturer's instructions. Blots were blocked in 5% nonfat dry milk in PBS-Tween 20 for 1 h at RT and incubated with primary antibodies for 1 h or overnight (according to the manufacturer's instructions). After several washes in 0.1% Tris-buffered saline (TBS)-Tween 20, the blots were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at RT and developed using enhanced chemiluminescence (ECL). Phosphospecific ERK1/2 antibody (1:1,000; Cell Signaling) and phospho-Akt antibody (1:1,000; Cell Signaling) signals were detected with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific). Total ERK1/2 antibody (1:1,000; Cell Signaling) and total Akt (1:1,000; LSBio) signals were detected with Pierce ECL (Thermo Scientific). As a control for loading, total β-actin (Abcam) levels were assessed.
ELISA.
The equine TNF-α contents in culture supernatants of mock- and EHV-1-inoculated CD172a+ cells cultured on plastic or on top of IEC were measured with an enzyme-linked immunosorbent assay (ELISA) kit obtained from Thermo Scientific. The assay was performed according to the manufacturer's instructions. Recombinant equine TNF-α was used as a standard, and the assay sensitivity was 16 pg/ml.
Cocultivation assay.
A cocultivation assay was used to detect and quantify EHV-1-producing CD172a+ cells by cocultivation of these EHV-1-inoculated CD172a+ cells with EC, where a semisolid overlay technique was applied. Briefly, EHV-1-inoculated and mock-inoculated cells were harvested at 12 h p.i. One hundred thousand cells per milliliter were 10-fold diluted in LM, and 0.5 ml of each dilution was added onto EC monolayers in a 6-well plate and overlaid with a 0.94% carboxymethylcellulose medium (Sigma-Aldrich) prepared in 2× RPMI and centrifuged at 800 × g for 30 min at 18°C, as previously described (8). The cells were further incubated for 5 days at 37°C, 5% CO2. The cell monolayers were then stained with 5% crystal violet, and the plaques were counted. The percentage of infected cells producing infectious EHV-1 was calculated based on the number of plaques counted and the number of cells seeded per milliliter according to the volume plated. This experiment was performed three times.
Statistical analysis.
Data were analyzed with GraphPad Prism 6 software (GraphPad Software Inc.). The data analyzed for statistical significance were subjected to a two-way analysis of variance (ANOVA). All results shown represent means and standard deviations (SD) of three independent experiments. Results with P values of ≤0.05 were considered statistically significant.
RESULTS
EHV-1 infection induces adhesion of CD172a+ cells to EC.
To determine whether EHV-1 infection may enhance adhesion of CD172a+ cells to EC, we compared the adhesion kinetics of mock-, UV-inactivated EHV-1-, and EHV-1-inoculated CD172a+ cells to EC monolayers (Fig. 1).
FIG 1.

Kinetics of adhesion of mock-inoculated or EHV-1-inoculated blood CD172a+ cells to equine EC. Cells were inoculated with replication-competent EHV-1 or UV-EHV-1 strain 03P37 or 97P70 (MOI = 1) and directly cocultured with primary (A) or immortalized (B) monolayers for 2, 4, and 6 h. The number of adherent CD172a+ cells per square millimeter was calculated. Experiments were performed three times. The error bars show the SD, and a two-way ANOVA test was performed to evaluate significant differences between EHV-1- and mock-inoculated and UV-inactivated EHV-1 samples (ns, not significant; *, P < 0.001). (C) Low-magnification confocal images of blood CD172a+ cell adhesion to EC after 2 h coculture (merged z-stacks). Nuclei were counterstained with Hoechst (blue). The arrows point to adhering monocytic cells; the arrowheads indicate EC. Scale bar, 100 μm.
EHV-1 infection resulted in a significant increase (P < 0.001) in adhesion of CD172a+ cells to primary and immortalized venous EC monolayers that was independent of the EHV-1 strain used (03P37 or 97P70) (Fig. 1A, B, and C). A maximum of 200 to 230 adherent cells per mm2 was reached with EHV-1-inoculated CD172a+ cells after 2 h of coculture compared to approximately 50 adherent cells per mm2 with mock-inoculated CD172a+ cells. In addition, no significant difference in the kinetics of adhesion of CD172a+ cells to EC monolayers was observed between UV-EHV-1- and EHV-1-treated cells. These data indicate that active EHV-1 gene expression was not required to induce adhesion of CD172a+ cells to EC monolayers, and thus, viral entry was sufficient to activate CD172a+ cell adhesion to EC. As the kinetics of CD172a+ cell adhesion were similar in primary EC cultures and immortalized EC lines, the subsequent experiments were performed using immortalized EC only.
EHV-1-mediated CD172a+ cell adhesion to EC depends on cellular adhesion molecules.
To determine the contribution of specific cellular adhesion molecules in EHV-1-induced adhesion of activated CD172a+ cells to EC, we performed adhesion-blocking experiments using blocking antibodies against α4β1, αLβ2, and αVβ3 integrins. These integrins are known to play a role in mediating firm adhesion of monocytes to the endothelium (24–26). The presence of α4β1 (CD49d/CD29), αLβ2 (CD11a/CD18), and αVβ3 (CD51/CD61) integrins on CD172a+ cells was confirmed by flow cytometry (Fig. 2A), but it was not possible to compare the expression levels of mock- and EHV-1-infected CD172a+ cells due to the low percentage of infected cells detected at early stages of infection (data not shown).
FIG 2.

Adhesion of mock-inoculated or EHV-1 (03P37 or 97P70)-inoculated blood CD172a+ cells to EC monolayers after preincubation of monoclonal antibodies against α4β1 (CD49d/CD29), αLβ2 (CD11a/CD18), and αVβ3 (CD51/CD61) integrins. (A) Flow cytometric analyses of the expression of CD49d, CD29, CD11a/CD18, and CD51/CD61 integrins on CD172a+ cells. (B) Cells were mock inoculated or inoculated with EHV-1 strain 03P37 or 97P70 (MOI = 1), preincubated with 10 μg/ml of specific blocking antibodies for 30 min at 4°C, and cocultured with immortalized EC for 2 h at 37°C. As a control, cells were preincubated with isotype-matched antibodies. The number of adherent CD172a+ cells per square millimeter was calculated. Experiments were performed three times. The error bars show the SD, and a two-way ANOVA test was performed to evaluate significant differences from controls (*, P < 0.001). (C) Low-magnification confocal images of mock-inoculated or EHV-1 (03P37 or 97P70)-inoculated blood CD172a+ cells adherent to EC after preincubation with αLβ2 (CD11a/18) blocking antibody. Nuclei were counterstained with Hoechst (blue). The arrow points to an adhering monocytic cell. All the confocal images shown represent merged z-stacks. Scale bars, 100 μm.
Pretreatment of EHV-1-inoculated CD172a+ cells with blocking antibodies resulted in a 3-fold decrease in adhesion to EC after 2 h of coculture compared to isotype controls (P < 0.001) (Fig. 2B and C). After preincubation with blocking antibodies, the number of adherent CD172a+ cells per square millimeter in EHV-1-inoculated cells decreased to levels at or slightly below those of mock-inoculated cells (±50 adherent cells per mm2). Pretreating cells with a mixture of blocking antibodies had no cumulative effect on decreasing CD172a+ cell adhesion to EC compared to treatment of cells with a single antibody. Interestingly, no significant decrease in cell adhesion was observed with mock-inoculated CD172a+ cells. Taken together, these results show that interactions between EHV-1-inoculated CD172a+ cells and EC are in part mediated by integrins.
EHV-1-induced adhesion of CD172a+ cells to EC is dependent on integrin-mediated PI3K and ERK/MAPK signaling pathways.
Next, we wanted to identify the mechanism by which EHV-1 induces adhesion of CD172a+ cells to EC. Binding of herpesviruses to integrins is known to activate cellular pathways, including PI3K/Akt (PI3K stands for phosphatidylinositol 3-kinase and Akt for protein kinase B), one of the early events of integrin-ligand interaction, and the downstream ERK/MAPK (MAPK stands for mitogen-activated protein kinase and ERK for extracellular signal-regulated kinases). Both these pathways have been implicated in the manipulation of cellular functions, such as adhesion, and in the control of herpesvirus infection and replication in target cells (15, 27, 28). Hence, we hypothesized that EHV-1 would also bind to cellular integrins present on CD172a+ cells and induce PI3K/Akt and ERK/MAPK activation, leading to increased adhesion of CD172a+ cells to EC.
We first tested whether EHV-1 infection upregulates ERK/MAPK and PI3K activities in CD172a+ cells. We compared by Western blotting analyses the levels of phosphospecific ERK1/2 and total ERK1/2, as well as the levels of phosphospecific Akt and total Akt, between mock- and virus-inoculated CD172a+ cells in the presence or absence of the specific MEK/ERK noncompetitive inhibitor U0126 and the PI3K reversible inhibitor LY294002. We found that mock- and EHV-1-inoculated CD172a+ cells showed similar levels of total ERK1/2 and total Akt expression at 0, 5, and 15 min postinoculation. However, we failed to detect any levels of ERK1/2 and Akt phosphorylation, even by using a highly sensitive detection kit (Fig. 3A). It is possible that the concentration of cells used in this assay (106 cells per ml) was not sufficient to detect any signal. Due to technical limitations, it was not possible to increase the cell concentration. Thus, we could not test whether EHV-1 infection upregulates ERK/MAPK and PI3K activities in CD172a+ cells.
FIG 3.

Effects of inhibition of the ERK/MAPK and PI3K signaling pathways on EHV-1-induced CD172a+ cell adhesion to EC monolayers. (A) Western blot analyses of the levels of phosphospecific ERK1/2 and total ERK1/2, as well as the levels of phosphospecific Akt and total Akt, between mock- and EHV-1-inoculated CD172a+ cells in the presence or absence of ERK/MAPK and PI3K inhibitors. One million CD172a+ cells were preincubated with the MEK/ERK inhibitor U0126 (10 μM), the PI3K inhibitor LY294002 (50 μM), or DMSO-based diluent for 30 min at 37°C. Cells were (mock) inoculated with EHV-1 strain 03P37 or 97P70 (MOI = 1) and lysed at 0, 5, and 15 min postinoculation. As a control for loading, total β-actin levels were assessed. (B and C) CD172a+ cells were preincubated with U0126 (1 or 10 μM) (B) or LY294002 (25 or 50 μM) (C) for 30 min at 37°C. The cells were (mock) inoculated with EHV-1 strain 03P37 or 97P70 (MOI = 1) and cocultured for 2 h with immortalized EC. The number of adherent CD172a+ cells per square millimeter was calculated. (D) Low-magnification confocal images of the reduced adhesion of blood CD172a+ cells to EC upon inhibition of the ERK/MAPK signaling pathway. Nuclei were counterstained with Hoechst (blue). The arrow points to an adhering monocytic cell. All the confocal images shown represent merged z-stacks. Scale bars, 100 μm. (E) U0126 or LY294002 pretreatment did not decrease EHV-1 infection of CD172a+ cells. CD172a+ cells adherent to plastic were pretreated with U0126 (10 μM), LY294002 (25 μM), or DMSO-based diluent for 30 min at 37°C and inoculated with EHV-1 strain 03P37 or 97P70 (MOI = 1) for 1 h at 37°C in LM supplemented with inhibitors. The cells were further incubated in LM for 12 h at 37°C. The percentage of IEP-positive cells was calculated based on 300 cells counted in distinct fields. Experiments were performed three times. The error bars show the SD, and a two-way ANOVA test was performed to evaluate significant differences from controls (ns, not significant; *, P ≤ 0.001; **, P < 0.0001).
However, we could analyze the potential role of ERK/MAPK and PI3K activation in EHV-1-induced CD172a+ cell adhesion to EC by pretreating CD172a+ cells with U0126 (1 and 10 μM) or LY294002 (25 and 50 μM), or with DMSO-based diluent as a control, for 30 min before EHV-1 inoculation. The treated CD172a+ cells were then added to immortalized EC monolayers and cocultured for 2 h. Inhibition of the ERK/MAPK signaling pathway prior to EHV-1 inoculation resulted in a dose-dependent significant decrease (P < 0.001) in CD172a+ cell adhesion to mock levels (Fig. 3B and D). When CD172a+ cells were pretreated with 10 μM U0126, EHV-1 (03P37 and 97P70)-induced CD172a+ cell adhesion to EC monolayers was reduced by 55 and 61%, respectively. Confocal images of the reduced adhesion of blood CD172a+ cells to EC upon inhibition of the ERK/MAPK signaling pathway are shown in Fig. 3. Inhibition of PI3K activity prior to EHV-1 inoculation resulted in a similar decrease of CD172a+ cell adhesion (Fig. 3C). Pretreating mock-inoculated cells with the inhibitor U0126 or LY294002 had no significant effect on CD172a+ cell adhesion. Furthermore, we found that inhibition of the ERK/MAPK and PI3K pathways did not significantly decrease EHV-1 infection of CD172a+ cells compared to controls (Fig. 3E).Taken together, these data suggest that EHV-1 infection induces CD172a+ cell adhesion in a PI3K- and ERK/MAPK-dependent manner at a very early time of infection.
EHV-1 replication is enhanced in CD172a+ cells upon adhesion to EC.
To determine whether EHV-1 replication was enhanced in CD172a+ cells upon adhesion to EC, we compared the expression kinetics of IEP and gB protein in CD172a+ cells cultured on plastic (without EC) or on top of immortalized EC monolayers for 2, 4, 6, and 12 h.
From 2 to 12 h postadhesion (p.a.) on EC monolayers, the percentage of IEP-positive CD172a+ cells significantly increased from 4% to 14% for EHV-1 strain 03P37 (P < 0.01). At 12 h p.a., the percentage of IEP-positive CD172a+ cells was significantly higher in cells adherent to EC than in cells adherent to plastic (P < 0.01) (Fig. 4A, left). In contrast, the percentage of IEP-positive CD172a+ cells remained stable from 2 to 6 h p.a. (4%) and significantly increased to 10% at 12 h p.a. for EHV-1 strain 97P70 (P < 0.01) (Fig. 4A, right). No significant difference in the percentage of IEP-positive cells was observed in CD172a+ cells inoculated with EHV-1 strain 03P37 or 97P70 and cultured on plastic. The percentage of IEP-positive cells decreased in the nonadherent CD172a+ cell fraction over time postadhesion for both EHV-1 strains (Fig. 4B). No gB protein was detected in cells from 2 to 6 h p.a. An upward trend and a significant increase in gB expression (P < 0.05) were observed in IEP-positive cells inoculated with EHV-1 strains 03P37 and 97P70, respectively, and cocultured for 12 h with EC monolayers compared to cells cultured on plastic (Fig. 4C). Confocal images showed the expression of IEP and gB protein in EHV-1-infected CD172a+ cells after 12 h coculture with immortalized EC (Fig. 4D). The expression levels of gB in IEP-positive CD172a+ cells upon adhesion to EC were comparable between the two strains (60%).
FIG 4.

Expression kinetics of EHV-1 proteins in blood CD172a+ cells upon adhesion to EC. (A) Expression kinetics of EHV-1 IEP in adherent blood CD172a+ cells. Cells were inoculated with EHV-1 strain 03P37 or 97P70 (MOI = 1) and cocultured with immortalized EC monolayers for 2, 4, 6, and 12 h. As a control, EHV-1-inoculated CD172a+ cells were incubated on plastic (without EC). (B) Expression kinetics of EHV-1 IEP in the nonadherent CD172a+ cell fraction after 2, 4, 6, and 12 h coculture with immortalized EC. (C) Expression of EHV-1 gB protein in infected CD172a+ cells after 12 h coculture with immortalized EC in the presence of neutralizing EHV-1 antibodies. Three independent experiments were performed, and the data are represented as means plus SD. A two-way ANOVA test was performed to evaluate significant differences from the control (*, P < 0.05; **, P < 0.01). (D) Double immunofluorescence of EHV-1 IEP (green) and gB (red) proteins in blood CD172a+ cells adherent to immortalized EC for 12 h in the presence of neutralizing EHV-1 antibodies. Nuclei were counterstained with Hoechst (blue). All the confocal images represent merged z-stacks. Scale bars = 50 μm.
Taken together, these results show that the adhesion of EHV-1-inoculated CD172a+ cells to EC speeds up the viral replication process in CD172a+ cells inoculated with EHV-1 strain 97P70, but to a lesser extent in CD172a+ cells inoculated with EHV-1 strain 03P37, compared to EHV-1-inoculated CD172a+ cells adherent to plastic. Moreover, the enhancement of EHV-1 replication in CD172a+ cells upon adhesion to EC suggests that direct cell-to-cell contacts provide a favorable environment for EHV-1 replication in CD172a+ cells.
Increased EHV-1 replication in CD172a+ cells upon adhesion to EC correlates with the production of TNF-α.
Since TNF-α has been described as a potent inducer of viral gene expression in monocytes/macrophages, we investigated whether TNF-α is produced during interaction of EHV-1-inoculated CD172a+ cells and EC and whether TNF-α enhances EHV-1 replication in CD172a+ cells.
The concentration of TNF-α in supernatants of mock-inoculated and EHV-1-inoculated CD172a+ cells cultured on plastic or on top of immortalized EC monolayers was determined by ELISA. At 12 h p.a., the concentration of TNF-α in supernatants of CD172a+ cells inoculated with EHV-1 strain 03P37 or 97P70 and cocultured with EC was significantly higher (P < 0.05 and P < 0.001, respectively) than the TNF-α concentration in supernatants of EHV-1-inoculated cells cultured on plastic (Fig. 5A). No significant difference was detected in TNF-α concentrations between supernatants of mock-inoculated CD172a+ cells cultured on plastic or on top of EC.
FIG 5.

Effect of TNF-α on the enhancement of EHV-1 replication in CD172a+ cells upon adhesion to EC. (A) Concentrations of TNF-α in supernatants of CD172a+ cells mock inoculated or inoculated with EHV-1 strain 03P37 or 97P70 (MOI = 1) and cultured on plastic or on top of immortalized EC for 12 h. (B) Expression of EHV-1 gB protein in CD172a+ cells adherent to plastic and cultured in LM supplemented or not with 10 ng/ml of equine recombinant TNF-α (+ TNF-α) for 12 h. (C) Expression of EHV-1 gB protein in CD172a+ cells adherent to EC and cultured for 12 h in LM supplemented or not with 10 ng/ml of TNF-α-neutralizing antibody (+ Anti-TNF-α). The data represent means plus SD of three independent experiments; ns, not significant; *, P < 0.05; **, P < 0.001; ***, P < 0.0001 (by two-way ANOVA test).
We also tested if the addition of exogenous TNF-α to CD172a+ cells at the time of virus inoculation affected EHV-1 replication. The expression levels of gB in CD172a+ cells cultured on plastic in the absence or presence of 10 ng/ml recombinant equine TNF-α were compared at 12 h p.i. We found that the number of cells positive for gB among IEP-positive cells was significantly higher (P < 0.001) when stimulated with equine recombinant TNF-α (Fig. 5B). Moreover, we demonstrated that the addition of neutralizing TNF-α antibody to cocultures of CD172a+ cells adherent to EC generated a trend, although not statistically significant, toward a decrease in the percentage of gB-positive cells (Fig. 5C). Taken together, these results indicate that TNF-α is an important soluble mediator enhancing EHV-1 replication in CD172a+ cells upon adhesion to EC.
Transfer of cytoplasmic EHV-1 material from infected CD172a+ cells to uninfected adjacent EC.
To investigate whether CD172a+ cells transmit EHV-1 to EC, we analyzed the intracellular distribution dynamics of IEP and gB proteins in EHV-1-inoculated CD172a+ cells after 12 h coculture with immortalized EC monolayers in the presence of neutralizing antibodies (Fig. 6). We found that IEP was expressed in nuclear replicative compartments and in the cytoplasm of infected CD172a+ cells adherent to EC. The gB protein was expressed only in the cytoplasm of these cells, and no gB expression was detected at the plasma membrane in CD172a+ cells after 12 h coculture with EC. However, when immunostaining was performed with a biotinylated horse polyclonal antibody that recognizes EHV-1 late proteins, some signal could be detected at the plasma membranes of 2 and 0.6% of CD172a+ cells adherent to EC monolayers and inoculated with EHV-1 03P37 and 97P70, respectively (Fig. 6A). With both EHV-1 strains, a cytoplasmic IEP-positive “bridge” was formed from an infected CD172a+ cell to an adjacent EC (Fig. 6B). Some cytoplasmic IEP staining was also observed in the adjacent EC. The late protein gB expressed a weak signal colocalized with IEP in some regions of both the cytoplasmic bridge and the cytoplasm of the EC. Eighteen and 27% of IEP/gB (03P37 and 97P70)-positive CD172a+ cells showed cytoplasmic transfer of EHV-1 material to adjacent EC. However, IEP/gB-positive plaques also could not be observed in EC at later time points postinoculation, indicating that spread of infection from CD172a+ cells to EC did not automatically lead to productive infection in EC.
FIG 6.
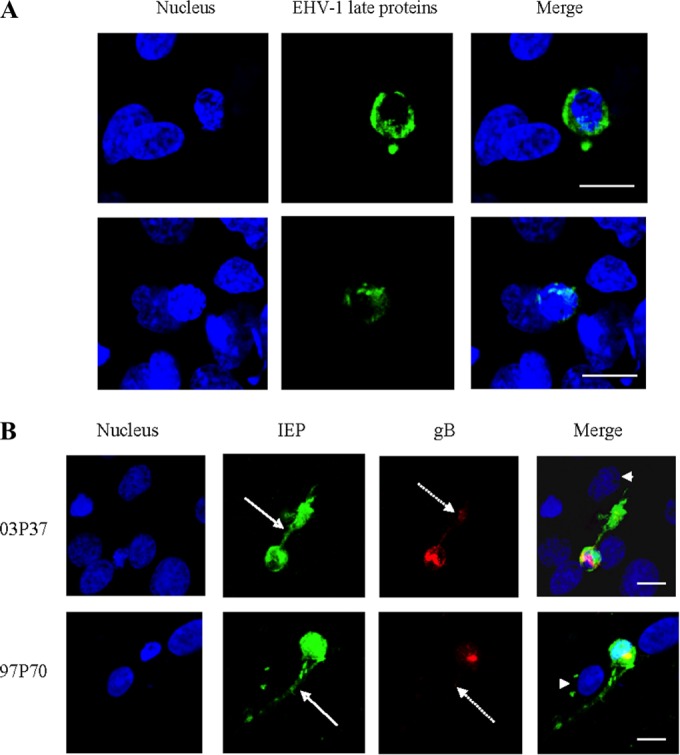
Transfer of cytoplasmic EHV-1 material from EHV-1-infected CD172a+ cells to uninfected EC. Cells were inoculated with EHV-1 strains 03P37 and 97P70 (MOI = 1) and cocultured with immortalized EC for 12 h in the presence of neutralizing EHV-1 antibodies. (A) Expression of EHV-1 late proteins at the plasma membranes of infected CD172a+ cells adherent to immortalized EC monolayers. Cells were fixed with 1% PFA without permeabilization and stained with a biotinylated horse polyclonal antibody directed against EHV-1 late proteins (green). (B) EHV-1 IEP (green) and gB protein (red) were detected by indirect immunofluorescence. The arrows indicate cytoplasmic transfer of IEP (solid arrows) and gB protein (dotted arrows) from a late-stage infected CD172a+ cell to one adjacent EC (arrowheads). Nuclei were counterstained with Hoechst (blue). All the confocal images represent merged z-stacks. Scale bars, 10 μm.
To address whether the enhanced EHV-1 replication in CD172a+ cells upon adhesion to EC led to a productive infection, we determined the percentage of CD172a+ cells that produced and transmitted infectious EHV-1 to neighboring endothelial cells by cocultivation assay. After 5 days of coculture of inoculated CD172a+ cells with EC monolayers, we showed that 0.1% and 0.05% of CD172a+ cells inoculated with EHV-1 strains 03P37 and 97P70, respectively, transmitted the infection to EC. Taken together, these results provide evidence for direct cytoplasmic transfer of EHV-1 material from late infected CD172a+ cells to adjacent EC, which typically leads to a nonproductive infection in these cells.
DISCUSSION
Monocytic cells are assumed to play a key role in EHV-1 spread from the blood to target organs, but the precise interactions between monocytic cells and EC have not been unraveled. In the present study, we demonstrated that EHV-1 infection increased adhesion of CD172a+ monocytic cells to EC in vitro. Significantly more EHV-1-inoculated CD172a+ cells adhered to EC than to mock-inoculated cells. UV treatment of EHV-1 did not affect the EHV-1-triggered adhesion process, indicating that the binding of EHV-1 to CD172a+ cells was sufficient to induce their adhesion to EC. We hypothesized that viral binding induces cellular signaling pathways that trigger functional changes in CD172a+ cells, which subsequently promote their adhesion to EC. By the use of an in vitro flow system, Smith et al. (18) suggested that activation of adhesion molecules present on both leukocyte and EC surfaces is a prerequisite for the transfer of EHV-1 from leukocytes to the endothelium. However, the authors did not characterize in detail which adhesion molecules were activated on the endothelium or examine the exact nature of the adhesion molecules present on the surfaces of the carrier cells. Monocytes constitutively express a selection of adhesion molecules, including selectins and integrins (29, 30). Here, we showed that blocking α4β1 (VLA-4), αLβ2 (LFA-1), and αVβ3 integrins on EHV-1-inoculated CD172a+ cells significantly reduced adhesion to levels comparable to those of mock-inoculated cells. These results indicated that integrins mediated the adhesion of CD172a+ cells to EC. However, adhesion was not completely inhibited by the addition of blocking antibodies, suggesting that other adhesion molecules may be involved in this process. In addition to integrins, l-selectins are expressed on myeloid cells and have been shown to play an important role in the initial interaction with or tethering of monocytes to the endothelium (31, 32). Unfortunately, there is no antibody available against l-selectin that cross-reacts specifically with equine monocytes to allow further experiments. Moreover, although we demonstrated that EHV-1 infection induces integrin-dependent adhesion of CD172a+ cells to endothelial cells, at this stage, it is not clear whether EHV-1 infection induces activation or increased expression of specific integrins. Indeed, the antibodies used in this study do not discriminate between inactive and activated forms of the integrins, and it was technically impossible to assess any potential upregulation of integrin expression on EHV-1-inoculated cells compared to mock-inoculated cells, due to a low percentage of infected CD172a+ cells detected.
Next, we addressed the mechanism by which EHV-1 infection induces adhesion of CD172a+ cells to EC in the absence of viral gene expression. We demonstrated that EHV-1 infection induces CD172a+ cell adhesion in PI3K- and ERK/MAPK-dependent processes at a very early stage of infection. The specific MEK/ERK inhibitor U0126 and the PI3K inhibitor LY294002 were able to efficiently decrease CD172a+ cell adhesion to EC in a dose-dependent manner. Together, these data show that integrin engagement and the activation of integrin-mediated signaling pathways are essential in the induction of EHV-1-infected CD172a+ adhesion to EC. Several herpesviruses, such as pseudorabies virus (PRV), herpes simplex virus 1 (HSV-1), human herpesvirus 8 (HHV-8), and HCMV, have been reported to activate integrin-mediated ERK-MAPK and PI3K signaling pathways through the interactions of viral glycoproteins with putative receptors present at the plasma membranes of host cells (23, 28, 33–35). To date, the EHV-1 envelope glycoprotein D (gD) has been shown to interact with αV integrins present on PBMC and to be critical for viral entry via endocytosis (36). Here, we hypothesized that EHV-1 gD interacts with αV integrins on CD172a+ cells and subsequently activates integrin-mediated signaling pathways (PI3K and ERK/MAPK) (Fig. 7A). We believe that this activation may induce specific functional changes in CD172a+ cells, including the activation of integrins, which in turn may facilitate their adhesion to specific endothelial adhesion molecules (37) (Fig. 7B). The latter hypothesis is supported by the study of Chang et al. (27), which demonstrated that PKC-dependent ERK activation contributes to monocyte adhesion, and by studies of Smith et al. (15, 38), demonstrating that PI3K activity is essential for HCMV-mediated induction of monocyte motility and firm adhesion to EC. However, further studies are still needed to decipher the precise mechanism of regulation of integrins by EHV-1 in CD172a+ cells and to directly address the potential relationship between integrin activation and cellular signal transduction pathways in cell adhesion.
FIG 7.

Hypothetical model of the transmission of EHV-1 from a CD172a+ cell to EC. (A) Interaction of EHV-1 via gD with αVβ3 integrin on the surface of a CD172a+ cell activates the intracellular integrin-mediated PI3K and ERK/MAPK signaling pathways. (B) This activation leads to the upregulation of LFA-1 (αLβ2), VLA-4 (α4β1), and αVβ3 integrin expression on the surface of the infected CD172a+ cell. Thus, activated LFA-1, VLA-4, and αVβ3 integrins can interact with some of their respective endothelial ligands and mediate CD172a+ cell adhesion to EC. (C) Both cell-to-cell contacts and secretion of soluble factors, such as TNF-α, activate the replication of EHV-1 in a CD172a+ cell. (D) These processes facilitate direct transfer of cytoplasmic EHV-1 material from infected CD172a+ cell to adjacent EC.
In addition, we showed that EHV-1 replication was enhanced in CD172a+ cells upon adhesion to EC. The percentages of IEP- and gB-positive CD172a+ cells were higher in cells adherent to EC than in cells adherent to plastic. These findings corroborate in vitro studies on HIV, demonstrating that contacts with EC enhanced HIV-1 replication in infected monocytes and macrophages (39, 40). Interestingly, we demonstrated that the percentage of IEP-positive CD172a+ cells significantly increased from 2 to 12 h p.a. for EHV-1 strain 03P37 while it increased only at 12 h p.a. for EHV-1 (97P70). Moreover, we observed a greater increase in gB protein expression in IEP-positive cells inoculated with EHV-1 strain 97P70 and cocultured for 12 h with EC monolayers than in cells inoculated with EHV-1 03P37. Taken together, we believe that these results are due to differences in the replication kinetics of EHV-1 in target monocytic cells between the two strains. This hypothesis is based on a previous study by Laval et al. (10) demonstrating that EHV-1 (97P70) replication was restricted and delayed in CD172a+ cells adherent to plastic and ongoing in vitro studies on the replication kinetics of EHV-1 (03P37) in monocytic cells indicating strikingly different outcomes (unpublished data). We think that the adhesion of EHV-1-inoculated CD172a+ cells to EC speeds up the viral replication machinery in CD172a+ cells inoculated with EHV-1 strain 97P70, but to a lesser extent in CD172a+ cells inoculated with EHV-1 strain 03P37.
In addition, we showed that TNF-α was produced via direct interaction between CD172a+ cells and EC and that TNF-α triggered the enhanced EHV-1 replication observed in CD172a+ cells. This is in agreement with previous studies on HCMV and HIV infections, which demonstrated that the production of TNF-α induces cellular changes in monocytic cells, resulting in increased viral gene expression (41, 42). TNF-α secreted by EC was also found to be associated with increased HIV expression in monocytic cells (43). From our findings, we hypothesize that TNF-α indirectly influences IEP expression in CD172a+ cells by inducing differentiation of CD172a+ monocytic cells into EHV-1-permissive macrophages. TNF-α may induce the production of cellular factors important for EHV-1 replication in CD172a+ cells. However, further studies are still needed to determine the specific role played by TNF-α in the pathogenesis of EHV-1 infection.
We also demonstrated that the percentage of gB-positive CD172a+ cells adherent to EC was reduced following treatment with TNF-α-neutralizing antibody. We considered that other cytokines besides TNF-α might also play a role in the enhanced EHV-1 expression observed in CD172a+-EC cocultures, as both monocytic cells and EC are known to secrete a variety of inflammatory cytokines, which are important in the regulation of viral replication. However, we failed to detect substantial amounts of interleukin 1β (IL-1β) and alpha interferon (IFN-α) produced upon coculture of mock- or EHV-1-inoculated CD172a+ cells with ECs (data not shown). Taken together, our results indicate that the enhanced EHV-1 replication observed in CD172a+ cells is a complex process involving both cell contacts and the production of soluble factors, with major implications for EHV-1 spread to EC (Fig. 7C). Interestingly, EHV-1 neurovirulent and nonneurovirulent strains both enhanced EHV-1 replication in CD172a+ cells upon adhesion to EC. These findings suggest that additional factors account for the difference in pathogenesis of EHV-1 in vivo.
Finally, we demonstrated that approximately 20% of late infected CD172a+ cells were able to transfer cytoplasmic EHV-1 material to uninfected, adjacent EC in the presence of neutralizing antibodies (Fig. 7D). Our data are in line with the findings of Digel et al. (44), showing direct transfer of HCMV cytoplasmic material from infected to neighboring uninfected cells. Although it is speculative at this point, we hypothesize that EHV-1 spreads from CD172a+ cells to EC through “microfusion” events, as described for other herpesviruses (45, 46). This strategy would allow the virus to spread from CD172a+ cells to the endothelium of target organs in the presence of neutralizing antibodies. In our experiments, transfer of EHV-1 from late infected CD172a+ cells to EC predominantly resulted in a nonproductive infection in EC. These data suggest that (i) only a minority of infected CD172a+ cells can efficiently transfer EHV-1 to EC and initiate an infection and (ii) EHV-1 infection of EC does not automatically result in viral spread to the surrounding tissues.
Previous in vivo studies showed that EHV-1 infection of endothelial cells of the pregnant uterus or CNS resulted in damage to the microvasculature of target organs due to the initiation of an inflammatory cascade, vasculitis, and microthrombosis (47–49). EHV-1 infection of endothelial cells also induced extravasation of mononuclear cells, resulting in perivascular cuffing and local hemorrhage (50). Based on this, we believe that the adhesion and transfer of EHV-1 from CD172a+ monocytic cells to endothelial cells may contribute to clinical thrombosis in horses with EHV-1 infection. The use of specific antiadhesive molecules may represent a valuable therapeutic tool to reduce adhesion of infected CD172a+ cells and subsequent transfer of EHV-1 to EC of target organs.
In conclusion, our data suggest that EHV-1 uses specific cellular signaling pathways to trigger cellular changes in CD172a+ cells, which result in increased adhesion to EC. The interaction between EHV-1-infected CD172a+ cells and EC enhances EHV-1 replication in CD172a+ cells through cell-to-cell contacts and secretion of soluble factors, which subsequently facilitates viral transfer to EC.
ACKNOWLEDGMENTS
This research was supported by the Concerted Research Action 01G01311 of the Research Council of Ghent University, Merelbeke, Belgium. Kathlyn Laval and Hans J. Nauwynck are members of the BELVIR consortium (IAP, phase VII) sponsored by Belgian Science Policy (BELSPO).
We are grateful to D. O'Callaghan and N. Osterrieder for supplying the antibodies against EHV-1 proteins. We also acknowledge Carine Boone and Lieve Sys for their excellent technical support and Maria Pontes and Korneel Grauwet for their technical advice.
We declare that we have no competing interests.
REFERENCES
- 1.Allen GP, Kydd JH, Slater JD, Smith KC. 2004. Equid herpesvirus-1 and equid herpesvirus-4 infections, p 829–859. In Coetzer JAW, Tustin RC (ed), Infectious diseases of livestock, 2nd ed Oxford University Press, London, United Kingdom. [Google Scholar]
- 2.Foote CE, Love DN, Gilkerson JR, Whalley JM. 2004. Detection of EHV-1 and EHV-4 DNA in unweaned thoroughbred foals from vaccinated mares on a large stud farm. Equine Vet J 36:341–345. [DOI] [PubMed] [Google Scholar]
- 3.Allen GP, Bryans JT. 1986. Molecular epizootiology, pathogenesis, and prophylaxis of equine herpesvirus-1 infections. Prog Vet Microbiol Immunol 2:78–144. [PubMed] [Google Scholar]
- 4.Patel JR, Heldens J. 2005. Equine herpesviruses 1 (EHV-1) and 4 (EHV-4) epidemiology, disease and immunoprophylaxis: a brief review. Vet J 170:14–23. doi: 10.1016/j.tvjl.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 5.Edington N, Smith B, Griffiths L. 1991. The role of endothelial cell infection in the endometrium, placenta and fetus of equid herpesvirus 1 (EHV-1) abortions. J Comp Pathol 104:379–387. doi: 10.1016/S0021-9975(08)80148-X. [DOI] [PubMed] [Google Scholar]
- 6.Smith KC, Mumford JA, Lakhani K. 1996. A comparison of equid herpesvirus-1 (EHV-1) vascular lesions in the early versus late pregnant equine uterus. J Comp Pathol 114:231–247. doi: 10.1016/S0021-9975(96)80045-4. [DOI] [PubMed] [Google Scholar]
- 7.Wilson WD. 1997. Equine herpesvirus 1 myeloencephalopathy. Vet Clin North Am Equine Pract 13:53–72. [DOI] [PubMed] [Google Scholar]
- 8.Van der Meulen KM, Nauwynck HJ, Buddaert W, Pensaert MB. 2000. Replication of equine herpesvirus type 1 in freshly isolated equine peripheral blood mononuclear cells and changes in susceptibility following mitogen stimulation. J Gen Virol 81:21–25. doi: 10.1099/0022-1317-81-1-21. [DOI] [PubMed] [Google Scholar]
- 9.Gryspeerdt AC, Vandekerckhove AP, Baghi HB, Van de Walle GR, Nauwynck HJ. 2012. Expression of late viral proteins is restricted in nasal mucosal leucocytes but not in epithelial cells during early-stage equine herpes virus-1 infection. Vet J 193:576–578. doi: 10.1016/j.tvjl.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 10.Laval K, Favoreel HW, Nauwynck HJ. 2015. Equine herpesvirus type 1 (EHV-1) replication is delayed in CD172a+ monocytic cells and controlled by histone deacetylases. J Gen Virol 96:118–130. doi: 10.1099/vir.0.067363-0. [DOI] [PubMed] [Google Scholar]
- 11.Mumford JA, Rossdale PD, Jessett DM, Gann S, Ousey J, Cook RF. 1987. Serological and virological investigations of an equid herpesvirus 1 (EHV-1) abortion storm on a stud farm in 1985. J Reprod Fertil 35:509–518. [PubMed] [Google Scholar]
- 12.O'Neill T, Kydd JH, Allen GP, Wattrang E, Mumford JA, Hannant D. 1999. Determination of equid herpesvirus 1-specific, CD8+ cytotoxic T lymphocyte precursor frequencies in ponies. Vet Immunol Immunopathol 70:43–54. doi: 10.1016/S0165-2427(99)00037-9. [DOI] [PubMed] [Google Scholar]
- 13.Kydd JH, Townsend HG, Hannant D. 2006. The equine immune response to equine herpesvirus-1: the virus and its vaccines. Vet Immunol Immunopathol 111:15–30. doi: 10.1016/j.vetimm.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 14.Bentz GL, Jarquin-Pardo M, Chan G, Smith MS, Sinzger C, Yurochko AD. 2006. Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J Virol 80:11539–11555. doi: 10.1128/JVI.01016-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith MS, Bivins-Smith ER, Tilley AM, Bentz GL, Chan G, Minard J, Yurochko AD. 2007. Roles of phosphatidylinositol 3-kinase and NF-kappaB in human cytomegalovirus-mediated monocyte diapedesis and adhesion: strategy for viral persistence. J Virol 81:7683–7694. doi: 10.1128/JVI.02839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tearle JP, Smith KC, Platt AJ, Hannant D, Davis-Poynter NJ, Mumford JA. 2003. In vitro characterisation of high and low virulence isolates of equine herpesvirus-1 and -4. Res Vet Sci 75:83–86. doi: 10.1016/S0034-5288(03)00031-6. [DOI] [PubMed] [Google Scholar]
- 17.Chiam R, Smid L, Kydd JH, Smith KC, Platt A, Davis-Poynter NJ. 2006. Use of polarised equine endothelial cell cultures and an in vitro thrombosis model for potential characterisation of EHV-1 strain variation. Vet Microbiol 113:243–249. doi: 10.1016/j.vetmic.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Smith DJ, Hamblin AS, Edington N. 2001. Infection of endothelial cells with equine herpesvirus-1 (EHV-1) occurs where there is activation of putative adhesion molecules: a mechanism for transfer of virus. Equine Vet J 33:138–142. [DOI] [PubMed] [Google Scholar]
- 19.Goehring LS, Hussey GS, Ashton LV, Schenkel AR, Lunn DP. 2011. Infection of central nervous system endothelial cells by cell-associated EHV-1. Vet Microbiol 148:389–395. doi: 10.1016/j.vetmic.2010.08.030. [DOI] [PubMed] [Google Scholar]
- 20.Garré B, Gryspeerdt A, De Backer P, Nauwynck HJ. 2009. Evaluation of orally administered valacyclovir in experimentally EHV1-infected ponies. Vet Microbiol 135:214–221. doi: 10.1016/j.vetmic.2008.09.062. [DOI] [PubMed] [Google Scholar]
- 21.Van der Meulen KM, Vercauteren G, Nauwynck HJ, Pensaert M. 2003. A local epidemic of equine herpesvirus 1-induced neurological disorders in Belgium. Vlaams Diergen Tijds 72:186–194. [Google Scholar]
- 22.Van der Meulen KM, Nauwynck HJ, Pensaert MB. 2003. Absence of viral antigens on the surface of equine herpesvirus-1-infected peripheral blood mononuclear cells: a strategy to avoid complement-mediated lysis. J Gen Virol 84:93–97. doi: 10.1099/vir.0.18864-0. [DOI] [PubMed] [Google Scholar]
- 23.Setas Pontes M, Devriendt B, Favoreel HW. 2015. Pseudorabies virus triggers glycoprotein gE-mediated ERK1/2 activation and ERK1/2-dependent migratory behavior in T cells. J Virol 89:2149–2156. doi: 10.1128/JVI.02549-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elangbam CS, Qualls CW, Dahlgren RR. 1997. Cell adhesion molecules-update. Vet Pathol 34:61–73. doi: 10.1177/030098589703400113. [DOI] [PubMed] [Google Scholar]
- 25.Mazzone A, Ricevuti G. 1995. Leukocyte CD11/CD18 integrins: biological and clinical relevance. Haematologica 80:161–175. [PubMed] [Google Scholar]
- 26.Weerasinghe D, McHugh KP, Ross FP, Brown EJ, Gisler RH, Imhof BA. 1998. A role for the alphavbeta3 integrin in the transmigration of monocytes. J Cell Biol 142:595–607. doi: 10.1083/jcb.142.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang MY, Huang DY, Ho FM, Huang KC, Lin WW. 2012. PKC-dependent human monocyte adhesion requires AMPK and Syk activation. PLoS One 7:e40999. doi: 10.1371/journal.pone.0040999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naranatt PP, Akula SM, Zien CA, Krishnan HH, Chandran B. 2003. Kaposi's sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: implications for infectivity. J Virol 77:1524–1539. doi: 10.1128/JVI.77.2.1524-1539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luscinskas FW, Kansas GS, Ding H, Pizcueta P, Schleiffenbaum BE, Tedder TF, Gimbrone MA. 1994. Monocyte rolling, arrest, and spreading on IL-4-activated vascular endothelium under flow is mediated via sequential action of L-selectin, beta1 integrins, and beta2 integrins. J Cell Biol 125:1417–1427. doi: 10.1083/jcb.125.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang S, Endo RI, Nemerow GR. 1995. Upregulation of integrins alphavbeta3 and alphavbeta5 on human monocytes and T lymphocytes facilitates adenovirus-mediated gene delivery. J Virol 69:2257–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vestweber D, Blanks JE. 1999. Mechanisms that regulate the function of the selectins and their ligands. Physiol Rev 79:181–213. [DOI] [PubMed] [Google Scholar]
- 32.Giuffrè L, Cordey A, Monai N, Tardy T, Schapira M, Spertini O. 1997. Monocyte adhesion to activated aortic endothelium: role of L-selectin and heparan sulfate proteoglycans. J Cell Biol 136:945–956. doi: 10.1083/jcb.136.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng K, Xiang Y, Wang X, Wang Q, Zhong M, Wang S, Wang X, Fan J, Kitazato K, Wanga Y. 2014. Epidermal growth factor receptor-PI3K signaling controls cofilin activity to facilitate herpes simplex virus 1 entry into neuronal cells. mBio 5:e00958–13. doi: 10.1128/mBio.00958-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reeves MB, Breidenstein A, Compton T. 2012. Human cytomegalovirus activation of ERK and myeloid cell leukemia-1 protein correlates with survival of latently infected cells. Proc Natl Acad Sci U S A 109:588–593. doi: 10.1073/pnas.1114966108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nogalski MT, Chan G, Stevenson EV, Gray S, Yurochko AD. 2011. Human cytomegalovirus-regulated paxillin in monocytes links cellular pathogenic motility to the process of viral entry. J Virol 85:1360–1369. doi: 10.1128/JVI.02090-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van de Walle G, Peters ST, VanderVen BC, O'Callaghan DJ, Osterrieder N. 2008. Equine herpesvirus 1 entry via endocytosis is facilitated by alphaV integrins and an RSD motif in glycoprotein D. J Virol 82:11859–11868. doi: 10.1128/JVI.00868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlos T, Kovack N, Schwartz B, Rosa M, Newman B, Wayner E, Benjamin C, Osborn L, Lobb R, Harlan J. 1991. Human monocytes bind to two cytokine-induced adhesive ligands on cultured human endothelial cells: endothelial-leukocyte adhesion molecule-1 and vascular cell adhesion molecule-1. Blood 77:2266–2271. [PubMed] [Google Scholar]
- 38.Smith MS, Bentz GL, Smith PM, Bivins ER, Yurochko AD. 2004. HCMV activates PI(3)K in monocytes and promotes monocyte motility and transendothelial migration in a PI(3)K-dependent manner. J Leukoc Biol 76:65–76. doi: 10.1189/jlb.1203621. [DOI] [PubMed] [Google Scholar]
- 39.Fan ST, Hsia K, Edington TS. 1994. Upregulation of human immunodeficiency virus-1 in chronically infected monocytic cell line by both contact with endothelial cells and cytokines. Blood 84:1565–1572. [PubMed] [Google Scholar]
- 40.Gilles PN, Lathey JL, Spector SA. 1995. Replication of macrophage-tropic and T-cell-tropic strains of human immunodeficiency virus type 1 is augmented by macrophage-endothelial cell contact. J Virol 69:2133–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Söderberg-Nauclér C, Kenneth NF, Nelson JA. 1997. Interferon-gamma and tumor necrosis factor-alpha specifically induce formation of cytomegalovirus-permissive monocyte-derived macrophages that are refractory to the antiviral activity of these cytokines. J Clin Invest 100:3154–3163. doi: 10.1172/JCI119871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poli G, Kinter A, Justement JS, Kehrl JH, Bresseler P, Stanley S, Fauci A. 1990. Tumor necrosis factor-alpha functions in an autocrine manner in the induction of human immunodeficiency virus expression. Proc Natl Acad Sci U S A 87:782–785. doi: 10.1073/pnas.87.2.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borghi MO, Panzeri P, Shattock R, Sozzani S, Dobrina A, Meroni PL. 2000. Interaction between chronically HIV-infected promonocytic cells and human umbilical vein endothelial cells: role of proinflammatory cytokines and chemokines in viral expression modulation. Clin Exp Immunol 120:93–100. doi: 10.1046/j.1365-2249.2000.01186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Digel M, Sampaio KL, Jahn G, Sinzger C. 2006. Evidence for direct transfer of cytoplasmic material from infected to uninfected cells during cell-associated spread of human cytomegalovirus. J Clin Virol 37:10–20. doi: 10.1016/j.jcv.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 45.Gerna G, Percivalle E, Baldanti F, Sozzani S, Lanzarini P, Genini E, Lilleri D, Revello MG. 2000. Human cytomegalovirus replicates abortively in polymorphonuclear leukocytes after transfer from infected endothelial cells via transient microfusion events. J Virol 74:5629–5638. doi: 10.1128/JVI.74.12.5629-5638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van de Walle G, Favoreel HW, Nauwynck HJ, Mettenleiter TC, Pensaert MB. 2003. Transmission of pseudorabies virus from immune-masked blood monocytes to endothelial cells. J Gen Virol 84:629–637. doi: 10.1099/vir.0.18796-0. [DOI] [PubMed] [Google Scholar]
- 47.Edington N, Bridges CG, Patel JR. 1986. Endothelial cell infection and thrombosis in paralysis caused by equid herpesvirus-1: equine stroke. Arch Virol 90:111–124. doi: 10.1007/BF01314149. [DOI] [PubMed] [Google Scholar]
- 48.Patel JR, Edington N, Mumford JA. 1982. Variation in cellular tropism between isolates of equine herpesvirus-1 in foals. Arch Virol 74:41–51. doi: 10.1007/BF01320781. [DOI] [PubMed] [Google Scholar]
- 49.Smith KC, Whitwell KE, Binns MM, Dolby CA, Hannant D, Mumford JA. 1992. Abortion of virologically negative foetuses following experimental challenge of pregnant pony mares with equid herpesvirus 1. Equine Vet J 24:256–259. doi: 10.1111/j.2042-3306.1992.tb02830.x. [DOI] [PubMed] [Google Scholar]
- 50.Stierstorfer B, Eichhorn W, Schmahl W, Brandmüller C, Kaaden OR, Neubauer A. 2002. Equine herpesvirus type 1 (EHV-1) myeloencephalopathy: a case report. J Vet Med B Infect Dis Vet Public Health 49:37–41. doi: 10.1046/j.1439-0450.2002.00537.x. [DOI] [PubMed] [Google Scholar]