

ABSTRACT
Certain major histocompatibility complex class I (MHC-I) alleles (e.g., HLA-B*27) are enriched among human immunodeficiency virus type 1 (HIV-1)-infected individuals who suppress viremia without treatment (termed “elite controllers” [ECs]). Likewise, Mamu-B*08 expression also predisposes rhesus macaques to control simian immunodeficiency virus (SIV) replication. Given the similarities between Mamu-B*08 and HLA-B*27, SIV-infected Mamu-B*08+ animals provide a model to investigate HLA-B*27-mediated elite control. We have recently shown that vaccination with three immunodominant Mamu-B*08-restricted epitopes (Vif RL8, Vif RL9, and Nef RL10) increased the incidence of elite control in Mamu-B*08+ macaques after challenge with the pathogenic SIVmac239 clone. Furthermore, a correlate analysis revealed that CD8+ T cells targeting Nef RL10 was correlated with improved outcome. Interestingly, this epitope is conserved between SIV and HIV-1 and exhibits a delayed and atypical escape pattern. These features led us to postulate that a monotypic vaccine-induced Nef RL10-specific CD8+ T-cell response would facilitate the development of elite control in Mamu-B*08+ animals following repeated intrarectal challenges with SIVmac239. To test this, we vaccinated Mamu-B*08+ animals with nef inserts in which Nef RL10 was either left intact (group 1) or disrupted by mutations (group 2). Although monkeys in both groups mounted Nef-specific cellular responses, only those in group 1 developed Nef RL10-specific CD8+ T cells. These vaccine-induced effector memory CD8+ T cells did not prevent infection. Escape variants emerged rapidly in the group 1 vaccinees, and ultimately, the numbers of ECs were similar in groups 1 and 2. High-frequency vaccine-induced CD8+ T cells focused on a single conserved epitope and therefore did not prevent infection or increase the incidence of elite control in Mamu-B*08+ macaques.
IMPORTANCE Since elite control of chronic-phase viremia is a classic example of an effective immune response against HIV/SIV, elucidating the basis of this phenomenon may provide useful insights into how to elicit such responses by vaccination. We have previously established that vaccine-induced CD8+ T-cell responses against three immunodominant epitopes can increase the incidence of elite control in SIV-infected Mamu-B*08+ rhesus macaques—a model of HLA-B*27-mediated elite control. Here, we investigated whether a monotypic vaccine-induced CD8+ T-cell response targeting the conserved “late-escaping” Nef RL10 epitope can increase the incidence of elite control in Mamu-B*08+ monkeys. Surprisingly, vaccine-induced Nef RL10-specific CD8+ T cells selected for variants within days after infection and, ultimately, did not facilitate the development of elite control. Elite control is, therefore, likely to involve CD8+ T-cell responses against more than one epitope. Together, these results underscore the complexity and multidimensional nature of virologic control of lentivirus infection.
INTRODUCTION
Elite controllers (ECs) are a small subset of untreated human immunodeficiency virus type 1 (HIV-1)-infected patients who spontaneously control viral replication (1). Since they manifest durable control of HIV-1 infection in the absence of antiretroviral therapy (ART), considerable effort has been devoted to elucidating the basis for their successful outcome. Despite great heterogeneity within the group, several major histocompatibility complex class I (MHC-I) alleles, including HLA-B*57, HLA-B*27, and HLA-B*5801, remain the major determinants of elite control (1, 2). These associations, together with extensive characterization of HIV-1-specific cellular immune responses in ECs and normal progressors (3–8), implicate immunosurveillance by CD8+ T cells as a key mediator of elite control. A better understanding of these efficacious CD8+ T-cell responses could therefore provide useful clues for the development of HIV-1 vaccines.
Expression of the MHC-I alleles Mamu-B*08 and Mamu-B*17 also predisposes simian immunodeficiency virus (SIV)-infected Indian rhesus macaques to control viral replication (9, 10). In the case of Mamu-B*08, approximately 50% of monkeys expressing the allele become ECs, defined here as having a set point viral load (VL) of <103 viral RNA (vRNA) copies/ml of plasma. It should be noted, however, that the penetrance of this effect can vary across experiments and may depend on the route of challenge (9, 11–13). In comparison, SIVmac239-infected monkeys lacking protective MHC-I alleles often experience set point viral loads that exceed 106 vRNA copies/ml (14). Consistent with a vital role of CD8+ T cells in establishing this phenotype, control of viral replication is impaired in Mamu-B*08+ monkeys infected with a SIVmac239 variant harboring escape mutations in multiple epitopes restricted by the allele (13). Furthermore, early CD8+ T-lymphocyte escape is a hallmark of Mamu-B*08+ animals that fail to contain SIVmac239 replication (11). Interestingly, Mamu-B*08-restricted epitopes conform to a peptide binding motif that greatly resembles that of HLA-B*27, characterized by a requirement for an Arg at position 2 and a predilection for hydrophobic or basic amino acids at the carboxyl termini of their epitopes (15). Given these similarities, SIV-infected Mamu-B*08+ macaques may provide a useful model to study HLA-B*27-mediated elite control in humans.
The dominance of HLA-B*27-restricted CD8+ T-cell responses observed during primary HIV-1 infection is also recapitulated in SIV-infected Mamu-B*08+ macaques. However, instead of focusing on Gag (16), three epitopes (Vif RL9 [amino acids {aa} 123 to 131], Vif RL8 [aa 172 to 179], and Nef RL10 [aa 137 to 146]) account for more than half of the antiviral CD8+ T-cell response in Mamu-B*08+ ECs (17). Importantly, we have recently shown that these three immunodominant CD8+ T-cell responses are responsible for elite control in this animal model. That is, after a high-dose intrarectal (i.r.) challenge with SIVmac239, Mamu-B*08+ macaques vaccinated with Vif RL9, Vif RL8, and Nef RL10 manifested better control of viral replication than MHC-I-matched animals immunized with SIV inserts lacking any Mamu-B*08-restricted determinants (12). Interestingly, an inverse association between peak VLs and the magnitude of CD8+ T cells directed against Nef RL10 was the only correlate of protection identified, implying that these Nef-specific responses were decisive for the challenge outcome.
Consistent with these results, CD8+ T cells directed against Nef RL10, but not Vif RL9 or Vif RL8, have also been linked to lower set point viremia in primary SIVmac239 infection (D. Douek, personal communication). Moreover, the Nef RL10-specific CD8+ T cells mobilized in ECs contained significantly more public T-cell receptor clonotypes than the responses detected in normal progressors (D. Douek, personal communication). Although conventional immunological measurements have been unable to explain these discordant associations (18), unique features of Nef RL10 suggest that it is indeed a useful target for CD8+ T-cell responses in Mamu-B*08+ animals. Namely, this epitope lies within the central region of Nef, which is relatively conserved and contains many CD8+ T-cell targets that contribute to immune control of HIV-1 replication (19). In fact, Nef RL10 is homologous to the HLA-B*27:05-restricted Nef RI10 (aa 105 to 114) epitope in HIV-1 (19). Furthermore, a recent analysis of viral sequence evolution in SIV-infected Mamu-B*08+ macaques revealed that Nef RL10 escape mutations occur later in the infection than those detected in Vif RL9 and Vif RL8 (11). Additionally, the mode by which SIV escapes from Nef RL10-specific CD8+ T cells is unusual compared to the mutational pathways described for other SIV epitopes, since it requires an Ala-to-Pro substitution immediately upstream of the Nef RL10 sequence (A136P) (20). This change at position −1 likely interferes with NH2-terminal trimming of the optimal epitope by the endoplasmic reticulum aminopeptidase I, as shown previously for the HLA-B*57:03-restricted Gag IW9 (aa 147 to 155) epitope (21). This atypical and delayed escape pattern suggests that it is difficult for the virus to avoid immunosurveillance by Nef RL10-specific CD8+ T cells. These observations, together with the correlate analysis from our previous experiment, prompted us to hypothesize that a vigorous Nef RL10-directed CD8+ T-cell response would be sufficient to establish elite control or even to block acquisition of SIV infection in Mamu-B*08+ animals.
To test this hypothesis, we engendered high-frequency CD8+ T-cell responses against Nef RL10 by vaccinating Mamu-B*08+ macaques several times with a nef minigene. Our controls consisted of a group of Mamu-B*08+ animals that were immunized with the same nef insert harboring amino acid substitutions around and within Nef RL10 designed to inactivate the epitope. Although macaques in both groups developed Nef-specific cellular immune responses, only those vaccinated with the intact epitope mounted CD8+ T cells against Nef RL10. These narrowly targeted CD8+ T-cell responses reached high frequencies, displayed markers of effector memory T cells (TEM), and were present at mucosal surfaces and secondary lymphoid organs (SLO) at the time of challenge. Here, we report the efficacy of these vaccine-induced Nef RL10-specific CD8+ T-cell responses after repeated i.r. challenges with SIVmac239.
MATERIALS AND METHODS
Research animals.
Eighteen Mamu-B*08+ Indian rhesus macaques (Macaca mulatta) were used in this study, all of which were housed at the Wisconsin National Primate Research Center (WNPRC). They were cared for in accordance with the guidelines of the Weatherall Report under a protocol approved by the University of Wisconsin Graduate School Animal Care and Use Committee. Vaccinations, biopsies, and SIV challenges were performed under anesthesia, and all efforts were made to minimize suffering. Additional animal information, such as MHC-I genotype, age, and sex, is shown in Table 1.
TABLE 1.
Animal characteristics
| Exptl group | Animal ID | MHC-I allele(s) | Age (yr)a | Gender | Infecting challenge no.b |
|---|---|---|---|---|---|
| 1 | r01068 | Mamu-B*08 | 11.2 | Male | 1 |
| r04001 | Mamu-B*08 | 9 | Female | 1 | |
| r06008 | Mamu-B*08 | 7 | Female | 3 | |
| r08017 | Mamu-A*02, -B*08, -B*17 | 4.4 | Male | 3 | |
| r08018 | Mamu-B*08 | 4.4 | Female | 7 | |
| r08020 | Mamu-B*08 | 4.4 | Female | 8 | |
| r09029 | Mamu-B*08 | 3.6 | Male | 3 | |
| r09089 | Mamu-B*08 | 3.2 | Male | 6 | |
| 2 | rh2099 | Mamu-B*08 | 23.5 | Male | 1 |
| r97076 | Mamu-B*08 | 15.3 | Female | 4 | |
| r08016 | Mamu-B*08 | 4.5 | Male | 1 | |
| r08032 | Mamu-B*08 | 4.5 | Female | 3 | |
| r08054 | Mamu-B*08 | 4 | Male | 3 | |
| r09014 | Mamu-B*08 | 3.8 | Male | 3 | |
| r09037 | Mamu-B*08 | 3.6 | Female | 2 | |
| r09068 | Mamu-B*08 | 3.4 | Female | 13 | |
| 3 | r04087 | Mamu-B*08 | 8.3 | Female | 13 |
| r09092 | Mamu-A*02, -B*08 | 3 | Male | 6 |
Animal age at beginning of study.
Intrarectal challenge at which the animal became productively infected with SIVmac239.
Vaccinations.
The macaques in groups 1 and 2 were vaccinated with two minigenes that encoded aa 45 to 210 of SIVmac239 Nef. The minigene delivered to group 1 corresponded to the wild-type (WT) SIVmac239 nef sequence, while the one given to group 2 contained several amino acid substitutions designed to inactivate the Nef RL10 epitope (Fig. 1A). These nef minigenes were inserted into three vector platforms: recombinant DNA (rDNA), yellow fever vaccine virus 17D (rYF17D), and adenovirus type 5 (rAd5). The rDNA constructs consisted of two pCMVkan plasmids (22), each carrying either the WT nef or the mutated nef minigene mentioned above. Expression of these gene fragments was under the control of the human cytomegalovirus (CMV) promoter and the bovine growth hormone polyadenylation signal. The rDNA constructs were codelivered with the AG157 plasmid (23), which encodes the two subunits of rhesus interleukin 12 (IL-12) expressed from two separate transcription units. We refer to this plasmid as pIL-12 below. The animals were vaccinated intramuscularly with a mixture of 1.0 mg of rDNA plasmid containing the WT nef minigene or its mutated counterpart and 0.1 mg of pIL-12 using the TriGrid in vivo electroporation system (Ichor Medical Systems, Inc., San Diego, CA). We primed the animals in groups 1 and 2 three times at 4-week intervals with electroporated (EP) rDNA plus pIL-12.
FIG 1.

Experimental layout. (A) Amino acid alignment of the WT and mutated Nef immunogens delivered to animals in groups 1 and 2, respectively. Both constructs spanned aa 45 to 210 of SIVmac239 Nef. The box shows the position of Nef RL10 in the WT insert and the amino acid substitutions used to disrupt this epitope in the mutated immunogen. The asterisks below the sequence alignment indicate identical amino acid residues. (B) The Mamu-B*08+ macaques in group 1 (n = 8) and group 2 (n = 8) were primed three times with EP rDNA plus pIL-12, followed by the administration of rYF17D and then a final boost with rAd5. The intervals between vaccinations are shown. The Mamu-B*08+ monkeys in group 3 (n = 2) did not receive any vaccine regimen and served as additional controls for the experiment. Eight weeks after the rAd5 boost, we began challenging all the animals with 200 TCID50 of SIVmac239 delivered via the i.r. route.
Six weeks after the 3rd EP rDNA plus pIL-12 vaccination, we boosted immune responses with the subcutaneous administration of 2.0 × 105 PFU of rYF17D vectors carrying the above-mentioned nef minigenes. The codon usage of these SIV sequences matched that of the YF17D virus. These live attenuated viruses were generated as described previously (24).
The final rAd5 boost occurred 4 weeks after the rYF17D vaccination. The WT and mutated nef inserts were delivered by two rAd5 vectors that were made by Viraquest, Inc., according to the RAPAd method (25). Each rAd5 vector was administered intramuscularly at a dose of 1011 viral particles in a final volume of 0.4 ml of PBS. Of note, both the rDNA and rAd5 vectors expressed nef minigenes that were codon optimized for high expression in mammalian cells.
SIVmac239 challenges.
Eight weeks following the rAd5 boost, we began challenging all the animals with repeated intrarectal inoculations of 200 50% tissue culture infective doses (TCID50) of SIVmac239. This corresponded to 4.8 × 105 vRNA copies. The animals were exposed to SIV on a weekly basis, and their plasma VLs were assessed 7 days later. If a macaque had a positive VL, it was no longer challenged. The number of challenges required to infect each monkey in this study is shown in Table 1.
Sample processing.
We isolated PBMC from EDTA-treated blood by using Ficoll-Paque Plus (GE Health Sciences) density centrifugation. Red blood cells were lysed by treating PBMC with ACK buffer (H2O containing 8.3 g/liter NH4Cl, 1.0 g/liter KHCO3, and 0.42 g/liter Na2EDTA) for 5 min. The cells were subsequently washed in R10 medium (RPMI 1640 medium supplemented with GlutaMax [Life Technologies], 10% fetal bovine serum [FBS], and 1% antibiotic/antimycotic) and then resuspended at various concentrations depending on the application. Bronchoalveolar lavage (BAL) fluid was collected by the WNPRC staff by flushing saline solution into the bronchus and aspirating until fluid was no longer recovered. These cell suspensions were strained through a 100-μm cell strainer (BD Falcon), resuspended in R10, and then counted before flow cytometric analysis. Lymph node (LN) and rectal biopsy specimens were obtained as described previously (16). To process the LN biopsy specimens, we placed the tissue specimen in 100-μm cell strainers and gently pushed it against the nylon mesh using a 3-ml syringe plunger until the tissue was disrupted. We counted the cells in the ensuing cell suspension and then used it in immunological assays. Rectal biopsy specimens were washed once in R10 and then resuspended in collagenase medium (R10 with 0.5 mg/ml of type II collagenase from Clostridium histolyticum [Sigma-Aldrich]). After rocking the cells at 80 rpm for 30 min at 37°C on a tabletop shaker, we collected the supernatant and washed it twice with R10 medium. We repeated this digestion cycle two additional times. We then pooled the cells collected in each digestion cycle, layered them on top of a 40 to 90% Percoll gradient, and then centrifuged them at 450 × g for 30 min. Purified gut lymphocytes were collected from the interface between the 40% and 90% Percoll layers. We then washed the cells once with R10 medium, counted them, and finally used them in immunological assays. Of note, all the tissues described above were harvested at the WNPRC and subsequently refrigerated at 4°C until they were shipped overnight to the University of Miami in a temperature-controlled package. Blood draws intended for viral load measurements were an exception. In those cases, the blood was processed shortly after venipuncture, and plasma aliquots were immediately frozen at −80°C.
Memory phenotyping of MHC-I tetramer+ CD8+ T cells.
We used MHC-I tetramers produced at the NIH Tetramer Core Facility conjugated to either allophycocyanin (APC) or phycoerythrin (PE) to label PBMC, BAL fluid, and cells isolated from LN or rectal biopsy specimens. Up to 8.0 × 105 cells were incubated in the presence of the appropriate fluorochrome-labeled tetramer at 37°C for 1 h and then stained with monoclonal antibodies (MAbs) to the surface molecules CD3 (peridinin chlorophyll protein [PerCP] Cy5.5; clone SP34-2; BD Biosciences), CD8 (brilliant violet 421 [BV421]; clone RPA-T8; Biolegend, Inc.), CD28 (PE Cy7; clone CD28.2; Biolegend, Inc.), and CCR7 (fluorescein isothiocyanate [FITC]; clone 150503; BD Biosciences). To create a “dump channel,” we included LIVE/DEAD Fixable Aqua Dead Cell Stain (Life Technologies) and BV510-conjugated MAbs to CD14 (clone M5E2), CD16 (clone 3G8), and CD20 (clone 2H7) (all from Biolegend, Inc.) to the surface-staining mixture. After a 25-min incubation at room temperature, we treated the cells with BD FACS (fluorescence-activated cell sorter) lysing solution (BD Biosciences) for 10 min and subsequently washed them with wash buffer (Dulbecco's phosphate-buffered saline [PBS] with 0.1% bovine serum albumin [BSA] and 0.45 g/liter NaN3). We permeabilized the cells by resuspending them in “Perm buffer” (1× BD FACS lysing solution 2 [Becton Dickinson] and 0.05% Tween 20 [Sigma-Aldrich]) for 10 min. We then washed the cells once and stained them with a granzyme B (Gzm B)-specific MAb (PE; clone GB12; Life Technologies). After a 30-min incubation in the dark at room temperature, we washed the cells and stored them at 4°C until acquisition. Samples were acquired using FACS Diva version 6 on a Special Order Research Product BD LSR II apparatus equipped with a 50-mW 405-nm violet laser, a 100-mW 488-nm blue laser, and a 30-mW 635-nm red laser.
We used FlowJo 9.6 (Treestar, Inc.) to analyze data. First, we gated on diagonally clustered singlets by plotting forward scatter height (FSC-H) versus FSC area (FSC-A) and then side scatter height (SSC-H) versus SSC area (SSC-A). Next, we created a time gate that included only those events that were recorded within the 5th and 90th percentiles and then gated on dump channel-negative, CD3+ cells. At this stage, we delineated the lymphocyte population based on its FSC-A and SSC-A properties and subsequently gated on CD8+ cells. After outlining tetramer+ cells, we conducted our memory phenotyping analysis within this gate. Cells stained with fluorochrome-labeled MAbs of the same isotypes as the anti-granzyme B, anti-CD28, and anti-CCR7 MAbs guided the identification of the memory subsets within the tetramer+ population. Based on this gating strategy, all tetramer frequencies mentioned in this paper correspond to percentages of live CD3+ CD8+ tetramer+ lymphocytes.
ICS assay.
For T-cell stimulation, we used two pools of 15-mers overlapping by 11 aa. The “Nef ORF” pool contained 64 15-mers spanning the entire SIV Nef open reading frame (ORF), while the “Nef ORFΔRL10” pool lacked the four 15-mers that contained the Nef RL10 epitope. The final assay concentration of each 15-mer was 1.0 μM. These peptide mixtures were used in the presence of costimulatory MAbs directed against CD28 (clone L293; BD Biosciences) and CD49d (clone 9F10; BD Pharmingen). We used leukocyte activation cocktail (LAC) (BD Pharmingen) as the positive control and tissue culture medium devoid of stimulatory peptides as the negative control. To measure CD107a externalization, we included a MAb specific for CD107a (PE; clone H4A3; Biolegend, Inc.) to the stimulation cocktail. We set up the intracellular cytokine staining (ICS) assay in 12- by 75-mm polypropylene tubes (Corning, Inc.) containing 1.5 × 106 PBMC in a final volume of 1.0 ml of R10. After adding the appropriate stimulation cocktails to each tube, the cells were placed in a 5.0% CO2 incubator at 37°C for 9 h and then refrigerated to 4°C until staining with fluorochrome-labeled MAbs. To inhibit protein transport, we added brefeldin A (Biolegend, Inc.) and GolgiStop (BD Biosciences) to all tubes 1 h into the incubation period at concentrations of 5.0 μg/ml and 0.7 μg/ml, respectively.
We employed the same steps outlined above to stain molecules on the surfaces of cells and subsequently permeabilize them for the ICS step. The surface-staining master mixture included the following MAbs: CD3 (PerCP Cy5.5; clone SP34-2; BD Biosciences), CD4 (Alexa Fluor 700; clone L200; BD Biosciences), and CD8 (FITC; clone RPA-T8; Biolegend, Inc.). The dump channel reagents were the same as described above. For the ICS step, we stained the cells with MAbs to gamma interferon (IFN-γ) (BV421; clone 4S.B3; Biolegend, Inc.), tumor necrosis factor alpha (TNF-α) (APC; clone Mab11; BD Biosciences), and IL-2 (PE Cy7; clone MQ1-17H12; Biolegend, Inc.). After a 1-h incubation in the dark at room temperature, we washed the cells and stored them at 4°C until acquisition. Samples were acquired in the same flow cytometer mentioned above and analyzed with FlowJo 9.6 (Treestar, Inc.).
After gating on the lymphocyte population according the strategy described above, we analyzed subsets based on their expression of either CD4 or CD8, but not both markers. To conduct functional analyses within these two compartments, we made gates for each function (IFN-γ, TNF-α, IL-2, and CD107a) and then employed the Boolean gate platform to generate a full array of possible combinations, equating to 16 response patterns when testing four functions (24 = 16). LAC-stimulated cells stained with fluorochrome-labeled MAbs of the same isotypes as the anti-IFN-γ, anti-TNF-α, anti-IL-2, and anti-CD107a MAbs guided the identification of populations that were positive for these effector markers. We used two criteria to determine the positivity of responses. First, the frequency of events in each Boolean gate had to be at least 2-fold higher than their corresponding values in negative-control tests (background). Second, the Boolean gates for each response had to contain ≥10 events. The percentages of responding CD4+ or CD8+ T cells (see Fig. 3; see Fig. S1 in the supplemental material) were calculated by adding the frequencies of positive responses producing any combination of the four immunological functions described above. Background subtraction and calculation of the frequencies of responding cells were performed with Microsoft Excel.
FIG 3.

ICS analysis of vaccine-induced T-cell responses in groups 1 and 2 following the rAd5 boost. We carried out ICS assays in PBMC from the group 1 and group 2 macaques at weeks 0, 1, and 2 post-rAd5 boost. Our stimuli consisted of pools of peptides (15-mers overlapping by 11 aa) spanning the entire SIVmac239 Nef ORF (Nef ORF peptide pool) or lacking the four 15-mers containing the Nef RL10 sequence (Nef ORFΔRL10 peptide pool). The percentages of responding CD4+ or CD8+ T cells displayed in panels A to C were calculated by adding the frequencies of positive responses producing any combination of four immunological functions (IFN-γ, TNF-α, IL-2, and CD107a). (A) Magnitudes of vaccine-induced CD8+ T-cell responses directed against the Nef ORF peptide pool in groups 1 and 2. (B) Magnitudes of vaccine-induced CD8+ T-cell responses directed against the Nef ORFΔRL10 peptide pool in groups 1 and 2. (C) Magnitudes of vaccine-induced CD4+ T-cell responses directed against the Nef ORF peptide pool in groups 1 and 2. The gating strategy to analyze these data is described in Materials and Methods. The lines represent medians, and each symbol denotes one monkey.
SIV and rYF17D viral load measurements.
SIV VLs were measured from EDTA-anticoagulated plasma. VL quantification was performed according to a previously published protocol (26). The limit of reliable quantification as performed on an input volume of 0.5 ml of plasma was 30 vRNA copies per ml. To quantify rYF17D replication, we isolated RNA in duplicate from 250 μl of EDTA-anticoagulated plasma using the MasterPure RNA purification kit (Epicentre) according to the manufacturer's instructions. Viral RNA was reverse transcribed and amplified using the TaqMan Fast Virus 1-Step master mix (Applied Biosystems) in an Applied Biosystems 7500 Fast instrument. Each reaction mixture contained 7.5 μl of 4× TaqMan Fast Virus 1-Step master mix, 400 nM forward primer, 900 nM reverse primer, 250 nM probe (5′ FAM [6-carboxyfluorescein]/ZEN/3′ IBFQ [Iowa Black FQ quencher]), and 20 μl of RNA sample in a final volume of 30 μl. The sequences of the primers and probe have been described previously (27). The reverse transcription was performed at 50°C for 5 min. The quantitative-PCR (qPCR) conditions were 95°C for 20 s, followed by 40 amplification cycles of 95°C for 15 s and 60°C for 1 min. Samples were run in triplicate. The copy numbers of YF17D viral RNA were calculated by absolute quantitation using a standard curve for each run. The standard curve was generated by 10-fold dilutions of an in vitro transcript made with a Megascript ultra-high-yield kit (Invitrogen) and purified using a Megaclear kit (Ambion) according to the manufacturer's instructions. The plasmid pCRII-TOPO vector (Invitrogen) containing a PCR amplicon with a YF17D sequence identical to nucleotides 9833 to 10333 of GenBank accession number U17066.1 served as the template for transcription. The limit of detection under standard assay conditions was approximately 14 vRNA copies/ml, and the accurate quantification range was between 140 and 1.4 × 107 vRNA copies/ml of plasma.
Amplicon-based 454 sequencing of Nef RL10.
One to 3 ml of plasma was thawed on ice and centrifuged at 20,817 × g for 1.5 h at 4°C, after which the pellet was resuspended in 140 μl of the supernatant. The QIAamp viral RNA minikit (Qiagen) was used to isolate viral RNA according to the manufacturer's protocol. The viral RNA was eluted in 60 μl of AVE buffer, aliquoted, and stored at −80°C for future reverse transcription (RT)-PCR. An amplicon-based 454 sequencing approach was used to analyze the Nef RL10 epitope, as previously described (11). In brief, primers were synthesized with Roche 454 amplicon (Lib-A) adaptor sequences, multiplex identifier tags (MID) 1 to 18, and sequence-specific regions (Nef; forward, 5′-ACTGGAAGGGATTTATTAC-3′, and reverse, 5′-GAGTTTCCTTCTTGTCAGCC-3′), which allowed multiplexing of up to 16 animals per amplicon combination per sequencing run. A single-step RT-PCR was carried out for each of the unique amplicon-animal-MID sequence combinations using a SuperScript III one-step RT-PCR system with platinum Taq high fidelity (Invitrogen). Each 25-μl reaction mixture contained 12.5 μl of 2× reaction mixture; a further 0.3 mM MgSO4; 1.0 μl of enzyme mixture; 0.2 μM (each) the sequence specific, adaptor/MID-tagged forward and reverse primers; and up to 10 μl of template RNA. Cycling parameters for the RT-PCR were as follows: 54°C for 30 min and 94°C for 2 min, followed by 40 cycles of 94°C for 15 s, 54°C for 30 s, and 68°C for 30 s. The last two steps were 68°C for 5 min and then holding at 10°C. Amplicons were visualized on a 1.0% agarose gel and purified using the Purelink quick gel extraction kit (Invitrogen). RT-PCR products were quantified using a Promega quantiflor-ST fluorometer (Promega) and analyzed for quality using an Agilent 2100 bioanalyzer with high-sensitivity DNA chips. For each sequencing run, up to 16 animal/amplicon samples were pooled in equimolar ratios, and 2.0 × 107 molecules of pooled sample were added to 107 DNA capture beads for a final ratio of 2 DNA molecules per bead. Emulsion PCR, enrichment, breaking, and DNA sequencing were all performed according to the GS Junior FLX titanium series manuals for Lib-A (Roche). Sequencing and run processing were performed on a GS Junior 454 sequencing instrument (Roche). We used the ReadClean 454 (RC454) and V-Phaser algorithms as previously described to call variants from the 454 data sets (28). In brief, RC454 was used to align reads to SIVmac239. They were subsequently corrected for sequencing-related artifacts, such as indels resulting from overcalls and undercalls in homopolymeric regions and carry forward and incomplete extension (CAFIE) errors. Furthermore, RC454 optimizes read alignments using coding-frame information. The V-Phaser algorithm was then used to distinguish an observed variant as a true variant from an amplification or sequencing artifact (29). All raw read files generated as part of this study are available at the NCBI sequence read archive under experiment accession number SRA278133.
Statistics.
We defined each animal's peak VL as the highest VL measurement obtained during the first 4 weeks after infection. The set point viremia was determined as the geometric mean of VLs measured between week 10 postinfection (p.i.) and the last time point available for each animal. We compared VLs among groups by using the Mann-Whitney test. To determine if vaccinees in groups 1 and 2 differed in the rate at which they acquired SIV infection, we used the log rank test. For the correlation analysis, we compared the magnitudes of vaccine-induced CD8+ T-cell responses and the percentages of WT epitope reads by using the Spearman rank correlation test.
RESULTS
Here, we explored the hypothesis that a monotypic CD8+ T-cell response targeting Nef RL10 might be sufficient to control replication of the pathogenic SIVmac239 clone. To do this, we vaccinated 16 Mamu-B*08+ macaques with two different minigenes encoding the central region of Nef (aa 45 to 210) (Fig. 1A). These inserts were identical, except that one of them harbored several mutations (including the A136P substitution) designed to inactivate the Nef RL10 epitope (Fig. 1A). We divided the animals into two groups based on which minigene they received; group 1 (n = 8) was vaccinated with the WT nef sequence, whereas group 2 (n = 8) received the mutated construct (Fig. 1B). Thus, animals in both groups would develop Nef-specific T-cell responses, but only those in group 1 would mount CD8+ T-cell responses against Nef RL10.
In our previous Mamu-B*08 efficacy trial, we vaccinated animals with an rYF17D prime followed by an rAd5 boost encoding Vif RL9, Vif RL8, and Nef RL10 (12). Although this regimen elicited sizeable SIV-specific responses shortly after the rAd5 boost, less than 0.3% of vaccine-induced CD8+ T cells targeted these Mamu-B*08-restricted epitopes on the day of challenge (DOC) (Fig. 2A). To improve immunogenicity in the present experiment, we primed the macaques three times with EP rDNA plus pIL-12 before delivering the rYF17D and rAd5 vectors (Fig. 1B). Consistent with the efficiency of electroporated rDNA to prime cellular immunity (30, 31), the new EP rDNA plus pIL-12/rYF17D/rAd5 regimen was significantly more immunogenic than the previously reported rYF17D/rAd5 protocol. Indeed, we could already detect Nef RL10-specific CD8+ T cells by fluorochrome-labeled MHC-I tetramer staining in PBMC from the group 1 vaccinees as early as 2 weeks after the 3rd EP rDNA plus pIL-12 immunization (data not shown). The rYF17D boost further expanded these responses in nearly all the animals, with one vaccinee (r04001) having 7.2% of its CD8+ T cells targeting Nef RL10 at week 2 postvaccination (Fig. 2B). Remarkably, these responses underwent massive proliferation following the rAd5 boost, peaking at 46%, 14%, and 13% of all peripheral CD8+ T cells measured in r04001, r09029, and r08018, respectively (Fig. 2B). Despite considerable contraction in the ensuing weeks, the median frequency of vaccine-elicited Nef RL10-specific CD8+ T cells in PBMC among the group 1 animals was 1.1% on the DOC, with two macaques at 6% and 17% (Fig. 2A and B). This corresponded to a 3.8-fold increase compared to the combined magnitude of DOC responses targeting Vif RL9, Vif RL8, and Nef RL10 measured in the rYF17D/rAd5 vaccinees from our previous study (Fig. 2A) (12).
FIG 2.

Kinetics of vaccine-induced, Nef RL10-specific CD8+ T-cell responses in the group 1 animals. We employed fluorochrome-labeled MHC-I tetramers to monitor the expansion of Nef RL10-specific CD8+ T cells in the group 1 vaccinees at several time points after the rYF17D vaccination. (A) Comparison of the total magnitudes of vaccine-induced CD8+ T-cell responses detected in PBMC on the DOC between animals in group 1 and the rYF17D/rAd5 vaccinees (n = 8) from our latest Mamu-B*08 trial (12). The group 1 frequencies correspond only to Nef RL10-specific CD8+ T cells, whereas those computed for the rYF17D/rAd5 vaccinees include responses to Vif RL9, Vif RL8, and Nef RL10. The lines represent medians, and each symbol denotes one monkey. (B to E) We also performed tetramer staining in several tissues obtained from the group 1 vaccinees, including PBMC (B) and BAL fluid (C), and on single-cell suspensions obtained from biopsy specimens from LN (D) and rectum (E). The gating strategy to determine the percentages of tetramer+ CD8+ T cells is described in Materials and Methods. The vertical dashed lines in panels B to E indicate the time of the rAd5 boost. The arrows below the x axes of the graphs correspond to the day of the first i.r. SIV challenge (DOC).
To determine if these vaccine-induced CD8+ T cells accessed mucosal surfaces and SLO, we also performed tetramer staining on single-cell suspensions obtained from BAL fluid and biopsy specimens of rectum and LN. We could easily detect tetramer+ CD8+ T cells at these anatomical sites shortly after the rAd5 boost, although at variable frequencies (Fig. 2C to E). While the lungs harbored the greatest percentages of Nef RL10-specific CD8+ T cells at all time points analyzed (Fig. 2E), these responses peaked at less than 3.0% CD8+ T cells present in the LN and gut in the majority of the animals (Fig. 2D and E). Of note, we did not sample the gut on the DOC to avoid compromising the rectal mucosa immediately before the i.r. SIV challenge. Similar to the pattern observed in PBMC, r04001 and r08018 had the highest frequencies of Nef RL10-specific CD8+ T cells in the lungs, LN, and gut (Fig. 2C to E). Importantly, we did not detect vaccine-induced CD8+ T-cell responses against Nef RL10 in any of the group 2 macaques (data not shown).
We also evaluated whether vaccinees in groups 1 and 2 mounted CD4+ and CD8+ T-cell responses to other epitopes in the WT and mutated Nef immunogens after the rAd5 boost (Fig. 3). To do this, we stimulated PBMC with two pools of Nef peptides (15-mers overlapping by 11 aa); the first spanned the entire Nef ORF, whereas the second pool included the same peptides, except for the four 15-mers that contained the Nef RL10 epitope (Nef ORFΔRL10). This approach revealed that vaccine-induced CD8+ T-cell responses were equivalent in groups 1 and 2 at the time of the rAd5 boost but trended higher in group 1 in the next 2 weeks (Fig. 3A). Surprisingly, the group 2 macaque rh2099 had extremely high frequencies of CD8+ T-cell responses on the day of the rAd5 boost; 60% of its CD8+ T cells recognized both the Nef ORF and Nef ORFΔRL10 pools (Fig. 3A and B). These extraordinary CD8+ T-cell responses likely stemmed from the high levels of rYF17D replication experienced by the animal shortly after vaccination with this live-attenuated vector (see Fig. S1A in the supplemental material). Indeed, rh2099's rYF17D viremia reached 3.4 × 105 vRNA copies/ml of plasma on day 6 post-rYF17D vaccination, the largest plasma concentration of rYF17D detected in >25 rYF17D-immunized animals examined in previous studies conducted by our laboratory (see Fig. S1A in the supplemental material) (27, 32, 33). Subsequent ICS assays demonstrated that these high-frequency CD8+ T-cell responses were directed against a single epitope within aa 170 to 179 (TFGWLWKLVP) (see Fig. S1B in the supplemental material). Except for r06008 and r08017, vaccinees in group 1 did not recognize the Nef ORFΔRL10 pool, confirming that their CD8+ T-cell responses were focused on Nef RL10 (Fig. 3B). The reactivity observed in r06008 and r08017 was likely due to vaccine-induced CD8+ T cells targeting epitopes restricted by other MHC class I alleles. The latter animal, for example, was also positive for Mamu-B*17 and developed CD8+ T-cell responses directed against two epitopes restricted by this allele, Nef IW9 (aa 163 to 173) and Nef MW9 (aa 195 to 203) (see Fig. S1C in the supplemental material) (34). Furthermore, macaques in both groups developed similar levels of CD4+ T-cell responses following the rAd5 boost, with r04001 in group 1 and rh2099 in group 2 mounting the highest frequencies of Nef-specific CD4+ T cells (Fig. 3C).
Next, we assessed the memory phenotype of vaccine-induced CD8+ T cells on the DOC by costaining PBMC with the Mamu-B*08/Nef RL10 tetramer and antibodies directed against CD28, CCR7, and granzyme B (Gzm B) (Fig. 4A). Despite some intragroup variability, the majority of tetramer+ cells expressed markers of either fully differentiated effector memory (TEM2; median, 57%) or transitional memory (TEM1; median, 27%) T cells (Fig. 4B). Gzm B was also expressed by these antigen (Ag)-specific CD8+ T cells (median, 64%), indicating their cytotoxic potential (Fig. 4C). Of note, these tetramer+ cells were phenotypically similar to those elicited in our previous trial (Fig. 4D and E), suggesting that the three EP rDNA plus pIL-12 priming immunizations delivered to the group 1 animals did not significantly alter the memory phenotype of their vaccine-induced CD8+ T cells compared to the responses engendered by the rYF17D/rAd5 regimen.
FIG 4.

Memory phenotype of vaccine-induced, Nef RL10-specific CD8+ T cells in PBMC on the DOC. (A) Gating strategy used to delineate fully differentiated effector memory (TEM2), transitional memory (TEM1), or central memory (TCM) subsets within tetramer+ CD8+ T cells in peripheral blood. We also evaluated Gzm B (black lines) expression within tetramer+ CD8+ T cells, as shown in the histogram. The shaded area corresponds to tetramer+ CD8+ T cells stained with an isotype-matched control MAb. (B to E) We evaluated the memory phenotype (B and D) and Gzm B expression (C and E) of Nef RL10-specific CD8+ T cells measured in PBMC from the group 1 animals (B and C) or the rYF17D/rAd5 vaccinees (D and E) from our latest Mamu-B*08 trial (12). The group 1 and rYF17D/rAd5 tetramer staining assays were performed on freshly isolated and cryopreserved PBMC, respectively, using the same experimental conditions. Samples from only six of the latter vaccinees were available for this analysis. The lines represent medians, and each symbol denotes one monkey.
Given the high frequencies of vaccine-induced CD8+ T-cell responses in our vaccinees, we set out to determine whether these Nef-specific responses could either affect acquisition of SIV infection or facilitate the establishment of elite control. To do this, we began challenging all the monkeys intrarectally with 200 TCID50 of SIVmac239 at week 8 following the rAd5 boost (Fig. 1B). These challenges occurred on a weekly basis and were discontinued once an animal became infected. As additional controls, we also subjected two unvaccinated Mamu-B*08+ rhesus macaques to this challenge regimen (group 3) (Fig. 1B). Three vaccinees were noteworthy on the DOC because of their high-frequency T-cell responses. The group 1 animals r04001 and r08018 had 17% and 6% of their peripheral CD8+ T cells targeting Nef RL10 (Fig. 2A and B), respectively, and most of these cells displayed TEM markers (Fig. 4B and C). Moreover, 11% of rh2099's CD8+ T cells recognized the Nef 170TFGWLWKLVP179 peptide on the DOC (see Fig. S1B in the supplemental material). In spite of these high-magnitude vaccine-induced CD8+ T-cell responses, macaques in groups 1 and 2 acquired SIV infection at similar rates (Fig. 5). Strikingly, r04001 and rh2099 became infected after the first i.r. challenge, while r08018 became viremic after the 7th virus exposure (Table 1). These results suggest that vaccine-induced CD8+ T cells directed against a single epitope cannot prevent lentivirus infection following i.r. challenge.
FIG 5.
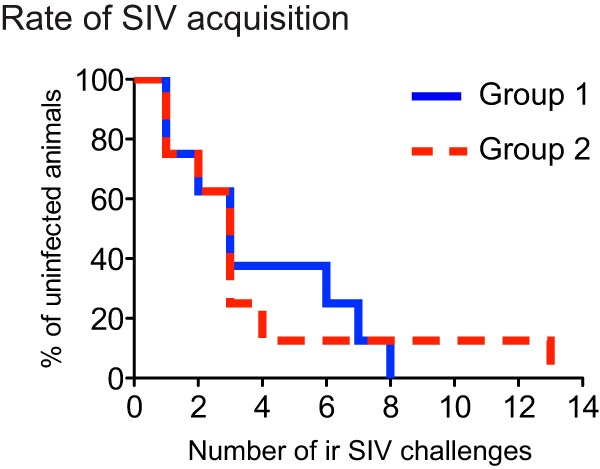
Kaplan-Meier rate of infection after repeated intrarectal challenges with SIVmac239. Macaques in groups 1 and 2 were inoculated with 200 TCID50 of SIVmac239 via the i.r. route on a weekly basis. The infecting exposure for each animal is shown in Table 1. The rate at which vaccinees in group 1 acquired SIV infection was not significantly different from that exhibited by their group 2 counterparts (P = 0.992, according to the log rank test).
Unexpectedly, it took 13 i.r. challenges to infect r09068 in group 2 and the unvaccinated Mamu-B*08+ monkey r04087 (Table 1). While r09068 remained aviremic until 1 week after its 13th virus exposure, r04087 experienced a VL “blip” of 40 vRNA copies/ml of plasma on the day of the 5th SIV inoculation. Since we could not confirm infection on a plasma sample obtained 3 days later, we did not interrupt the challenge schedule. Shortly after the 7th exposure, we detected a low-frequency Nef-specific CD8+ T-cell response in r04087 and decided to halt the challenges in order to determine whether the animal had acquired SIV infection. Attempts to recover SIV by coculturing PBMC or LN cells with CEMx174 cells yielded no replication-competent virus (data not shown). Although we mapped r04087's Nef-specific CD8+ T-cell response to aa 65 to 83—a region lacking any previously identified epitopes—we could not detect T-cell responses to Gag, Env, or Vif in the animal. Once we resumed challenging r04087 22 weeks after its last SIV exposure, it took 6 additional i.r. inoculations (13 in total) to productively infect the animal. Neither r04087 nor r09068 controlled viral replication after they became infected (Fig. 6B and C). It is unclear why these animals resisted more i.r. challenges than the rest of the monkeys in this experiment.
FIG 6.

Plasma virus concentrations after SIVmac239 infection. (A to C) Macaques in group 1 (A), group 2 (B), and group 3 (C) acquired SIV infection after various i.r. exposures (Table 1). The VLs were log transformed and correspond to the number of vRNA copies/ml of plasma. The dashed lines in all the graphs are for reference only and indicate a VL of 103 vRNA copies/ml. The solid lines in panels A to C are also for reference only and denote a VL of 106 vRNA copies/ml. (D to F) We compared the median peak (D), set point (E), and week 4 (F) VLs among animals in group 1 and group 2 and unvaccinated Mamu-B*08+ controls using the Mann-Whitney test. The last group includes the two unimmunized monkeys in group 3 and four historical controls from a recent SIV challenge experiment (12). We determined the set point viremia for each animal by calculating the geometric mean of viral loads measured at week 10 p.i. onward. (G) We compared the cumulative levels of SIV replication experienced by macaques in groups 1 and 2 until week 6 p.i. by an AUC analysis.
Our next step was to determine if the robust Nef RL10-specific CD8+ T cells engendered in the group 1 vaccinees improved their ability to contain SIV infection compared to their group 2 counterparts and unvaccinated Mamu-B*08+ macaques. We found that infection outcomes varied considerably from animal to animal (Fig. 6A and B). For example, the two macaques mounting the highest frequencies of SIV-specific CD8+ T-cell responses on the DOC—r04001 in group 1 and rh2099 in group 2—did not control acute-phase viremia and subsequently had escalating VLs until week 20 p.i. (Fig. 6A and B). The group 2 macaques r97076, r09014, and r09068 also failed to establish durable control of SIV replication (Fig. 6B). Furthermore, while several group 1 vaccinees (r01068, r08017, r08020, and r09029) experienced episodes of controlled VLs (<1,000 vRNA copies/ml), SIV viremia eventually rebounded in those animals at later time points (Fig. 6A and B). Notably, however, three animals in group 1 (r06008, r08018, and r09089) and four in group 2 (r08016, r08032, r08054, and r09037) became ECs, as defined by their chronic-phase VLs of less than 1,000 vRNA copies/ml (Fig. 6A and B). Additional follow-up was available for most of these ECs and revealed that they had undetectable VLs at weeks 69 to 73 p.i. (Fig. 6A and B). Together, these results demonstrated that vaccine-induced Nef RL10-specific CD8+ T cells did not affect the rate of elite control in the Mamu-B*08+ macaques in group 1 compared to their counterparts in group 2. This conclusion was corroborated by the similar peak and set point VLs measured in groups 1 and 2 (Fig. 6D and E). Although macaques in group 1 manifested a modicum of control at week 4 p.i. (Fig. 6F), this effect was transient, since groups 1 and 2 did not differ in their cumulative levels of viremia, as determined by a time-weighted area under the curve (AUC) analysis (Fig. 6G). The similarity of these virologic parameters was unexpected, considering recent data suggesting that Nef RL10 is a useful target for CD8+ T-cell responses in Mamu-B*08+ macaques (D. Douek, personal communication) (12, 17, 20). Nevertheless, compared to SIV infection of unvaccinated Mamu-B*08+ monkeys (two animals in group 3 and four historical controls), vaccinees in group 1 still achieved significant reductions in plasma viremia (Fig. 6D to F). This analysis also revealed trends toward lower week 4 and set point VLs in the group 2 animals (Fig. 6E and F). Thus, vaccine-induced Nef RL10-specific CD8+ T cells decreased viral replication but failed to influence either the rate of SIV acquisition or the development of EC status.
We then investigated whether a rapid emergence of escape variants within the Nef RL10 epitope could be one reason for the largely equivalent postchallenge performances observed in groups 1 and 2 and the unvaccinated Mamu-B*08+ monkeys. It would explain, for example, why r04001 failed to suppress viral replication despite mounting high-frequency TEM-biased, Nef RL10-directed responses on the DOC. Indeed, two rYF17D/rAd5 vaccinees in our previous study lost control of chronic-phase viremia due to escape in Mamu-B*08-restricted epitopes (12). To test this possibility, we characterized the viral quasispecies that encode the Nef RL10 epitope in plasma from animals in groups 1 and 2 using an amplicon-based 454 pyrosequencing approach (12). Remarkably, we could not detect WT Nef RL10 sequences in r04001 at week 6 p.i.; nearly all of its circulating viral quasispecies contained the canonical A136P escape mutation (Fig. 7A). While viral variants were also dominant in r08018, r06008, and r08020 (<25% WT reads), the majority of the virus replicating in the remaining group 1 animals had an intact epitope (>80% WT sequences) (Fig. 7A and C). Sequence variation within Nef RL10 was less frequent in group 2 but not significantly different than the rate observed in group 1 (Fig. 7B and C). The group 2 macaque r08032 was an outlier in that its virus had completely escaped using the A136P mutation by this time point (Fig. 7B). Of note, a characterization of the viral populations replicating in the chronic phase revealed that the A136P mutation had reached fixation in nearly all animals in groups 1 and 2 (see Fig. S2A in the supplemental material).
FIG 7.

Frequencies of WT and mutated Nef RL10 epitope reads at week 6 p.i. (A and B) We used an amplicon-based 454 pyrosequencing approach to characterize the viral quasispecies that constitute the Nef RL10 epitope in virus replicating in the group 1 (A) and group 2 (B) animals at week 6 p.i. All variants detected in >1% of sequenced reads are listed. Individual mutants that comprised <1% of sequenced reads were grouped in the same category (Variants <1%), and their combined frequencies are shown. The frequency of the predominant viral quasispecies in each animal is in boldface. Elite-controller macaques are marked with “EC” above their identifiers (IDs). (C) Comparison of the percentages of WT epitope reads detected in plasma from the group 1 and group 2 macaques at week 6 p.i. using the Mann-Whitney test. The lines represent medians, and each symbol denotes one monkey. The average numbers of sequencing reads obtained from animals in groups 1 and 2 at this time point were 2,747 and 6,971, respectively.
Since these data suggested that several animals mounted postchallenge Nef RL10-specific CD8+ T-cell responses that selected for viral variants even before week 6 p.i., we analyzed virus in plasma collected in the acute phase, specifically at week 2.4 p.i. (Fig. 8A and B). We found that Nef RL10 sequences were unaltered in all group 2 animals, including r08032 (Fig. 8B). However, the median frequency of WT epitope reads was slightly reduced in group 1 and significantly lower than that in group 2 (Fig. 8A and C), indicating that escape was already under way in the group 1 monkeys. This was especially evident in r04001, where Nef RL10 mutants had practically replaced WT virus at this early stage after infection (Fig. 8A). Interestingly, the A136P substitution was absent from the viral populations replicating in the animals at this time point (Fig. 8A). Together, these results demonstrate that selective pressure imposed by vaccine-induced CD8+ T cells can precipitate sequence evolution in the AIDS virus within days of mucosal transmission, even if these responses are directed against a “late-escaping” epitope, such as Nef RL10 (Table 2).
FIG 8.

Frequencies of WT and mutated Nef RL10 epitope reads at week 2.4 p.i. (A and B) Similar to the description in the legend to Fig. 7, we characterized the viral quasispecies that constitute the Nef RL10 epitope in virus replicating in the group 1 (A) and group 2 (B) animals at week 2.4 p.i. All variants detected in >1% of sequenced reads are listed. Individual mutants that comprised <1% of sequenced reads were grouped in the same category (Variants <1%), and their combined frequencies are shown. The frequency of the predominant viral quasispecies in each animal is in boldface. Elite-controller macaques are marked with “EC” above their IDs. (C) Comparison of the percentages of WT epitope reads detected in plasma from the group 1 and group 2 macaques at week 2.4 p.i. using the Mann-Whitney test. The lines represent medians, and each symbol denotes one monkey. The average numbers of sequencing reads obtained from animals in groups 1 and 2 at this time point were 3,194 and 2,333, respectively.
TABLE 2.
Summary of Nef-specific CD8+ T-cell responses, infection outcome, and extent of sequence variation in the Nef RL10 epitope
| Group | Animal ID | % tetramer+ CD8+ T cells on DOCa | % Nef ORF-reactive CD8+ T cells at wk 2 post-rAd5b | VL |
% of WT Nef RL10 reads |
||
|---|---|---|---|---|---|---|---|
| Peakc | Set pointd | Wk 2.4 p.i.e | Wk 6 p.i.f | ||||
| 1 | r01068 | 1.31 | 0.09 | 1.4 × 106 | 3.7 × 103 | 96.3 | 95 |
| r04001 | 17 | 1.75 | 1.2 × 106 | 2.3 × 105 | 11.7 | 0 | |
| r06008 | 0.87 | 2.82 | 4.5 × 105 | 4.0 × 101 | 100 | 16 | |
| r08017 | 0.63 | 2.15 | 4.4 × 106 | 7.0 × 103 | 100 | 100 | |
| r08018 | 6.33 | 1.53 | 9.4 × 105 | 1.6 × 102 | 99.7 | 7.9 | |
| r08020 | 1.3 | 0.81 | 2.4 × 107 | 9.7 × 102 | 96.3 | 24.3 | |
| r09029 | 0.63 | 2.19 | 9.0 × 104 | 1.7 × 103 | 96.6 | 82.8 | |
| r09089 | 0.58 | 0.4 | 4.5 × 106 | 4.4 × 101 | 100 | 98 | |
| 2 | rh2099 | NA | 48.4 | 3.0 × 107 | 1.6 × 106 | 100 | 80 |
| r97076 | NA | 0.01 | 7.4 × 105 | 9.1 × 103 | 99.8 | 96.8 | |
| r08016 | NA | 0.54 | 4.2 × 107 | 3.2 × 103 | 100 | 69.3 | |
| r08032 | NA | 0.07 | 3.0 × 106 | 7.7 × 102 | 100 | 1.7 | |
| r08054 | NA | 1.25 | 8.6 × 105 | 7.0 × 101 | 100 | 100 | |
| r09014 | NA | 8.41 | 1.2 × 106 | 7.0 × 103 | 100 | 97.1 | |
| r09037 | NA | 0.03 | 1.1 × 107 | 1.1 × 102 | 100 | 61.2 | |
| r09068 | NA | 0.32 | 1.0 × 107 | 4.1 × 104 | 100 | 47.8 | |
| 3 | r04087 | NA | NA | 7.8 × 106 | 4.6 × 105 | 100 | ND |
| r09092 | NA | NA | 9.6 × 106 | 3.9 × 103 | 100 | ND | |
Tetramer staining performed in PBMC on the day of the 1st SIV challenge (DOC). The graphical representation of these data is shown in Fig. 2A. NA, not applicable.
The percentages refer to the frequency of vaccine-induced CD8+ T cells responding to stimulation by the Nef ORF pool in an ICS assay performed at week 2 post-rAd5 boost. The graphical representation of these data is shown in Fig. 3A.
Highest VL measured within the first 4 weeks of infection. The values correspond to vRNA copies/ml of plasma. The graphical representation of these data is shown in Fig. 6D.
Set point VLs were determined as the geometric mean of VLs measured between week 10 p.i. and the last time point available for each animal. The values correspond to vRNA copies/ml of plasma. The graphical representation of these data is shown in Fig. 6E.
The graphical representation of these data is shown in Fig. 8.
The graphical representation of these data is shown in Fig. 7.
Lastly, we set out to identify biological variables that dictated the patterns of immune escape observed in groups 1 and 2. We began by comparing sequencing results obtained at week 6 p.i. between the present study and our latest Mamu-B*08+ trial (12). Unfortunately, data from only four of the latter animals were available for this analysis, which limited our statistical power (Fig. 9A). In spite of this caveat, the percentage of WT epitope reads in the group 1 vaccinees trended lower than that measured in the four rYF17D/rAd5-immunized animals from our earlier study (Fig. 9A). Since viral replication is needed to spawn epitope mutants, we examined whether the group 1 macaques selected for faster sequence variation because they experienced greater levels of acute-phase viremia than their rYF17D/rAd5 counterparts. In contrast to this prediction, a reevaluation of plasma virus concentrations on freshly thawed samples from animals in both groups using the same quantitative-PCR conditions showed that they had equivalent peak VLs (Fig. 9B). Our next step was to test if the total size of vaccine-induced Nef RL10-specific CD8+ T-cell responses dictated the escape rates observed after infection. Unfortunately, we could not perform this analysis on the rYF17D/rAd5-immunized macaques because of the limited size of the sequencing data set. Tellingly, however, we observed an inverse trend between the magnitude of Nef RL10-directed CD8+ T cells measured in the group 1 animals on the DOC and the frequency of WT epitope reads at week 2.4 (Fig. 9C). To further explore this issue, we examined the levels of tetramer+ CD8+ T cells present in PBMC, LN, and rectum samples from animals in groups 1 and 2 at week 2.4 p.i. As expected, Nef RL10-specific CD8+ T cells in group 1 greatly outnumbered those measured in the group 2 macaques, whereas the frequencies of primary CD8+ T-cell responses targeting Vif RL8 and Vif RL9 were similar in the two groups (Fig. 9D to F). Notably, for all three tissues assessed, the frequency of WT epitope reads was inversely associated with the number of Nef RL10-specific CD8+ T cells detected at this time point (Fig. 9G). Since r04001 was an outlier in these analyses, we repeated the above-mentioned comparisons in the absence of its data and found that all associations remained significant (PBMC, P = 0.01, r = −0.63; LN, P = 0.0009, r = −0.78; rectum, P = 0.02, r = −0.6). Curiously, however, the magnitude of acute-phase Nef RL10-specific CD8+ T-cell responses did not predict the frequency of epitope mutants observed at week 6 p.i. (Fig. 9H). Collectively, these findings indicate that during acute SIV infection, the magnitude of contemporaneous CD8+ T-cell responses directed against Nef RL10 dictated the amount of sequence evolution within the epitope.
FIG 9.

Biological variables affecting the kinetics of CD8+ T-cell escape within Nef RL10. (A) Comparison of the frequencies of WT Nef RL10 sequence reads detected at week 6 p.i. between group 1 and the rYF17D/rAd5-immunized animals in our latest Mamu-B*08 trial (12). Data from only four rYF17D/rAd5 vaccinees were available for this comparison. (B) Reevaluation of peak viral loads in macaques in group 1 and the rYF17D/rAd5 vaccinees. To avoid interassay variability, we determined plasma virus concentrations on freshly thawed samples using the same quantitative-PCR conditions. We used the Mann-Whitney test to calculate P values for the analyses depicted in panels A and B. The lines represent medians. (C) Magnitudes of Nef RL10-specific CD8+ T cells in PBMC of the group 1 animals on the DOC compared to their corresponding frequencies of viral quasispecies encoding WT Nef RL10 (WT epitope reads) measured at week 2.4 p.i. (D to F) Magnitudes of CD8+ T cells specific for the Mamu-B*08-restricted Nef RL10, Vif RL8, and Vif RL9 epitopes present in PBMC (D), LN (E), and rectum (F) from animals in groups 1 and 2 at week 2.4 p.i. (G and H) Magnitudes of acute-phase Nef RL10-specific CD8+ T cells in PBMC, LN, and rectum from animals in groups 1 and 2 compared to their corresponding frequencies of WT epitope reads at week 2.4 (G) and 6 (H) p.i. We determined the correlation coefficients in panels C, G, and H by using the Spearman rank correlation test. Each symbol denotes one monkey. ND, not done.
DISCUSSION
In HIV-1-infected HLA-B*27+ ECs, virologic control may be critically dependent on CD8+ T-cell responses targeting a single epitope, Gag KK10 (aa 263 to 272) (5, 16, 35). Given the similarities between HLA-B*27-mediated elite control and this phenomenon in SIV-infected Mamu-B*08+ rhesus macaques (15, 36), we investigated whether narrowly focused CD8+ T-cell responses against a single Nef epitope would also be sufficient to suppress SIV replication. We chose to investigate the protective efficacy of immunization with the Mamu-B*08-restricted Nef RL10 epitope due to its delayed and unusual escape pattern (17) and sequence conservation (19) and recent immunological data linking Nef RL10-specific CD8+ T cells with improved challenge outcome (12). In spite of these presumably desirable characteristics for a vaccine target, the high-frequency Nef RL10-directed CD8+ T cells elicited in the group 1 animals failed to affect SIV acquisition and did not increase the incidence of EC status. Furthermore, these high-frequency TEM responses afforded only a transient reduction in plasma viremia compared to the Nef-specific cellular responses engendered in the group 2 macaques that targeted epitopes other than Nef RL10. The most striking finding of this study was the rapidity with which Nef RL10-specific CD8+ T cells selected for escape variants after infection; indeed, viral mutants comprised almost 90% of the virus replicating in the group 1 vaccinee r04001 at week 2.4 p.i. and became widespread in half of the animals in this group by week 6 p.i. This rate was considerably higher than those reported in primary infection and in the rYF17D/rAd5 vaccinees from our latest Mamu-B*08 trial (12, 17).
These results are best interpreted in light of past reports of SIV sequence evolution in the face of strong selective pressure mediated by the host's CD8+ T-cell response. For example, a similar outcome has been achieved in Tat-vaccinated Mamu-A*01+ macaques, where monotypic vaccine-induced CD8+ T-cell responses against Tat SL8 (aa 28 to 35) were swiftly evaded by mutational escape shortly after challenge with SIVmac239 (37). However, an important distinction between this past study and the present one is the fact that even in unvaccinated monkeys, Tat SL8 mutants can largely replace WT virus within 4 weeks of infection without incurring any fitness cost to the virus (38, 39). Although Nef RL10 exhibits a lower escape rate, with most viral variants beginning to appear at week 8 p.i. (11, 17), it is not clear if this epitope is functionally constrained. Notably, a 12-bp deletion corresponding to the amino acids 143DMYL146 in SIVmacC8 Nef has been shown to decrease protein stability and function in vitro and likely contributes to the attenuated phenotype of this strain in vivo (40). The overlap between this deletion and Nef RL10 indicates that the region is indeed functionally constrained. Longitudinal sequencing analyses, such as the one conducted here, also support this conclusion. For instance, during primary infection of Mamu-B*08+ macaques, the earliest Nef RL10 mutants to emerge tend to harbor amino acid changes at various positions within the epitope, but not in the A136 residue that immediately precedes the minimal optimal peptide (11, 12). Interestingly, once the putative A136P processing mutation becomes fixed in the viral population, which typically occurs later in the chronic phase, the internal sequence of the Nef RL10 epitope almost invariably reverts to WT (11, 12, 17). Indeed, that was the case with nearly all the monkeys in the present study, although the intense selective pressure imposed by the anamnestic CD8+ T cells mounted by the group 1 vaccinees accelerated this process. We also recently inoculated Mamu-B*08-negative animals with a SIVmac239 variant encoding several Mamu-B*08-associated escape mutations, including A136P (41). Of note, this “escaped” virus did not replicate as well as WT SIVmac239 in vivo, but it is not clear if this impaired replicative capacity was due to A136P or the other changes introduced in its backbone. Notably, a characterization of the viral quasispecies circulating in those animals in the chronic phase revealed that the A136P substitution did not revert to WT, indicating that it can be stably maintained in vivo in the absence of CD8+ T-cell-mediated pressure (41). Based on these data, one might envision a scenario in which SIV initially tries to evade the host CD8+ T-cell response by mutating the internal residues of Nef RL10, even though sequence variation within this highly conserved epitope is probably disadvantageous for the virus. Once the A136P mutation arises, purifying selection would act to restore the internal epitope sequence to its WT status. Additional studies are obviously needed to confirm these predictions.
The rapid escape kinetics observed in r04001 preceded its loss of virologic control, but this was not a general pattern in the remaining animals. In fact, several macaques became ECs despite exhibiting fast selection of viral mutants. The group 1 vaccinee r06008, for instance, had 76% of its viral population expressing the I144T substitution at position 8 of the Nef RL10 epitope at week 6 p.i. and still suppressed viral replication to undetectable levels in the ensuing weeks. A notable feature of the I144T mutation is that it does not completely abrogate recognition of SIV-infected cells by Nef RL10-specific CD8+ T cells (13), which likely contributed to the impressive virologic control mediated by this animal. The infection outcomes for monkeys r08018 (group 1) and r08032 (group 2) provided further evidence of the poor prognostic value of Nef RL10 escape, since WT epitope reads were nearly absent from their viral quasispecies at week 6 p.i. and yet they ended up controlling chronic-phase viremia. On the other hand, the fact that Nef RL10 was intact in the virus replicating in the group 2 macaques rh2099, r97076, and r09014 at week 6 p.i. also did not predispose them to control viral replication. These discordant associations between the extents of sequence variation within Nef RL10 and disease progression are not surprising considering that multiple genetic, virologic, and immune factors have been shown to affect control of lentivirus infection (1, 42).
Of note, rapid escape driven by vaccine-induced CD8+ T cells has been linked to virologic control in SIV-infected Burmese rhesus macaques (43). In that study, Matano et al. vaccinated eight animals with an rDNA prime followed by a recombinant Sendai virus (rSeV) vector boost encoding Gag and subsequently challenged them with SIVmac239 intravenously. Notably, 5/8 vaccinees controlled viral replication to undetectable levels after week 5 p.i. Subsequent analyses of SIV gag sequences in all the immunized animals revealed that virologic control was linked to escape in a few Gag epitopes, which resulted in less fit viruses (43, 44). These results are in contrast to those reported here, where vaccine-elicited CD8+ T cells directed against Nef RL10 precipitated escape in this epitope without any overt effect on viral replication. The reasons for these discrepant outcomes are difficult to assess given the many biological variables that distinguish our experiment from that conducted by Matano and colleagues. However, the fact that the rDNA/rSeV vaccinees targeted multiple Gag epitopes (44), some of them under structural constraints, suggests that the fitness cost for escaping from these Gag-directed CD8+ T-cell responses would be higher than that required to evade the Nef RL10-specific CD8+ T cells elicited in the present study.
Although the magnitude of Nef RL10-specific CD8+ T cells measured in the group 1 and group 2 animals at week 2.4 p.i. was correlated with their contemporaneous rates of immune escape, this association did not hold for the sequence variation detected at week 6 p.i. The fact that these acute-phase CD8+ T-cell responses lost their predictive power during the 4-week interval that separated the above-mentioned time points underscores the complexity of virus-host interactions during lentivirus infection. The high antigen loads that result from HIV/SIV infection, for instance, are well known to be detrimental to T-cell immunity (45). Along these lines, Fujita et al. have recently reported the expansion of dysfunctional CD8+ T cells expressing programmed death 1 (PD-1) and T-cell Ig- and mucin domain-containing molecule 3 (Tim-3) during acute SIV infection (46). Differential upregulation of these inhibitory receptors could therefore help explain the discordance between the postchallenge responses developed by r01068 and r04001 in group 1 and their respective rates of immune escape at week 6 p.i. Despite mounting equally abundant anamnestic Nef RL10-specific CD8+ T cells, the virus replicating in the former animal encoded an intact epitope whereas only escape mutants were detected in r04001. It was also unclear why viral variants had replaced WT sequences in the group 2 macaque r08032 at week 6 p.i., given its scant CD8+ T-cell response to Nef RL10 after infection. Nevertheless, it is important to bear in mind that interhost heterogeneity can exist in CD8+ T cells restricted by the same MHC-I molecule and specific for the same epitope, even among genetically identical mice (47). Indeed, continuous priming of naive CD8+ T cells that are phenotypically distinct from their Ag-specific memory counterparts can affect the outcome of chronic viral infections (48). T-cell receptor (TCR) clonotype usage has also been shown to govern patterns of viral sequence evolution and the ability of CD8+ T cells to recognize epitopes bearing common escape mutations (49, 50). Given these intricate biological variables, CD8+ T-cell frequency alone may no longer be able to dictate the kinetics of immune escape once productive HIV/SIV infection is established.
Recent studies have demonstrated that TEM responses induced by persistent herpesviral vectors, such as rhesus cytomegalovirus (RhCMV), can afford superior virologic control in SIV-infected monkeys. Notably, Hansen et al. have reported that approximately 50% of RhCMV-vaccinated macaques manifest profound and early control of SIVmac239 replication, regardless of the challenge route, and subsequently clear the virus (51, 52). In the present study, we delivered a total of five immunizations as part of the EP rDNA plus pIL-12/rYF17D/rAd5 protocol in an attempt to elicit TEM responses, since repetitive boosting has also been shown to drive TEM differentiation (53). Consistent with this expectation, vaccine-induced Nef RL10-specific CD8+ T cells in the majority of group 1 animals displayed a TEM signature on the DOC and accessed relevant sites of virus transmission and replication. A comparison of the protective efficacy of the TEM responses engendered by our EP rDNA plus pIL-12/rYF17D/rAd5 regimen versus that achieved by RhCMV vaccination is complicated, not only by the fast selection of escape variants observed here, but also because of the unconventional nature of the CD8+ T-cell responses elicited by the RhCMV strain utilized by Hansen and colleagues. Indeed, RhCMV vaccinees do not recognize classically immunodominant CD8+ T-cell determinants associated with SIV infection; instead, these animals develop CD8+ T-cell responses against SIV epitopes restricted by MHC-II and nonclassical MHC-I molecules (54, 55). Furthermore, in contrast to the narrowly focused CD8+ T-cell responses elicited by our EP rDNA plus pIL-12/rYF17D/rAd5 modality, RhCMV vaccination results in T-cell repertoires of greater breadth than conventional vaccine platforms. The latter feature may be particularly important for the impressive protection conferred by this herpesviral vector, considering the apparent ease with which SIV evaded Nef RL10-directed CD8+ T-cell responses in the present experiment. A similar outcome has actually been reported in a vaccinated HLA-B*27+ person who subsequently acquired HIV-1 infection (56). Despite mounting CD8+ T-cell responses to Gag KK10, a structurally constrained epitope whose canonical escape mutation (R264K) imparts a severe fitness cost to the virus, this individual did not exhibit clinical signs of controlled infection. Alarmingly, Gag KK10 escape variants emerged at a higher rate than previously reported during primary infection (5, 56). Thus, monotypic CD8+ T-cell responses can accelerate the selection of viral mutants even if the targeted epitope lies within conserved, functionally constrained regions of HIV/SIV.
In conclusion, here, we show that high-frequency effector memory Nef RL10-specific CD8+ T-cell responses elicited by vaccination do not affect either the rate of SIV acquisition following repeated i.r. challenges or the incidence of elite control in Mamu-B*08+ macaques. Despite the conserved nature of Nef RL10 and its delayed escape pattern, the intense selective pressure exerted by these vaccine-induced CD8+ T cells accelerated the emergence of epitope variants after infection. Furthermore, we observed instances of mutational escape preceding virologic control or leading up to breakthrough viremia, suggesting that the protective efficacy of Nef RL10-specific CD8+ T cells can vary substantially among Mamu-B*08+ macaques. Our results also have implications for vaccine strategies aimed at eliciting cellular immunity against HIV-1, since they demonstrate that lentiviruses can exploit their genetic malleability to evade narrowly focused CD8+ T cells within days of infection, even if these responses are directed against a conserved epitope. This may be analogous to treating HIV-1-infected individuals with a single drug (leading to rapid escape) compared to several drugs that ultimately delay the emergence of escape mutations (57). Alternatively, considering that an rYF17D/rAd5 regimen encoding Vif RL8, Vif RL9, and Nef RL10 resulted in the establishment of elite control in 6/8 Mamu-B*08+ macaques following challenge with SIVmac239 in our previous experiment (12), it is possible that Nef-specific CD8+ T cells may not be as effective as those directed against Vif. Ongoing studies in our laboratory will hopefully address this hypothesis.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by Public Health Service grant R37AI052056 to D.I.W. and was supported in part by federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E and from the National Institute of Allergy and Infectious Diseases through P01 AI074415 (T.M.A.). This work was also partially funded by the International AIDS Vaccine Initiative (IAVI) with the generous support of the U.S. Agency for International Development (USAID) and the Bill and Melinda Gates Foundation. A full list of IAVI donors is available at http://www.iavi.org.
The contents of the manuscript are our responsibility and do not necessarily reflect the views of USAID or the U.S. government.
We thank Teresa Maidana Giret for confirming the MHC-I genotype of the monkeys in this study; Iris Castro for administrative assistance; Michael Ricciardi, Rebeca Alvarez, Kim Weisgrau, Jessica Furlott, Randy Fast, Kelli Oswald, and Rebecca Shoemaker for technical support; Peng Li for assistance with statistical analyses; and Eric Peterson and Kristin Crosno for providing excellent care of the rhesus macaques used in the experiment. We also thank Louis Picker, Scott Hansen, and Andrew Sylwester for sharing their expertise on intracellular-cytokine-staining measurements.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01440-15.
REFERENCES
- 1.Walker BD, Yu XG. 2013. Unravelling the mechanisms of durable control of HIV-1. Nat Rev Immunol 13:487–498. doi: 10.1038/nri3478. [DOI] [PubMed] [Google Scholar]
- 2.Goulder PJ, Walker BD. 2012. HIV and HLA class I: an evolving relationship. Immunity 37:426–440. doi: 10.1016/j.immuni.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, Walker BD. 2006. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 5.Goulder PJ, Phillips RE, Colbert RA, McAdam S, Ogg G, Nowak MA, Giangrande P, Luzzi G, Morgan B, Edwards A, McMichael AJ, Rowland-Jones S. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med 3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 6.Hersperger AR, Pereyra F, Nason M, Demers K, Sheth P, Shin LY, Kovacs CM, Rodriguez B, Sieg SF, Teixeira-Johnson L, Gudonis D, Goepfert PA, Lederman MM, Frank I, Makedonas G, Kaul R, Walker BD, Betts MR. 2010. Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS Pathog 6:e1000917. doi: 10.1371/journal.ppat.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW, Van Baarle D, Kostense S, Miedema F, McLaughlin M, Ehler L, Metcalf J, Liu S, Connors M. 2002. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat Immunol 3:1061–1068. doi: 10.1038/ni845. [DOI] [PubMed] [Google Scholar]
- 8.Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA, Rood JE, Berkley AM, Sacha JB, Cogliano-Shutta NA, Lloyd M, Roby G, Kwan R, McLaughlin M, Stallings S, Rehm C, O'Shea MA, Mican J, Packard BZ, Komoriya A, Palmer S, Wiegand AP, Maldarelli F, Coffin JM, Mellors JW, Hallahan CW, Follman DA, Connors M. 2008. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity 29:1009–1021. doi: 10.1016/j.immuni.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loffredo JT, Maxwell J, Qi Y, Glidden CE, Borchardt GJ, Soma T, Bean AT, Beal DR, Wilson NA, Rehrauer WM, Lifson JD, Carrington M, Watkins DI. 2007. Mamu-B*08-positive macaques control simian immunodeficiency virus replication. J Virol 81:8827–8832. doi: 10.1128/JVI.00895-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yant LJ, Friedrich TC, Johnson RC, May GE, Maness NJ, Enz AM, Lifson JD, O'Connor DH, Carrington M, Watkins DI. 2006. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J Virol 80:5074–5077. doi: 10.1128/JVI.80.10.5074-5077.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mudd PA, Ericsen AJ, Burwitz BJ, Wilson NA, O'Connor DH, Hughes AL, Watkins DI. 2012. Escape from CD8(+) T cell responses in Mamu-B*00801(+) macaques differentiates progressors from elite controllers. J Immunol 188:3364–3370. doi: 10.4049/jimmunol.1102470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mudd PA, Martins MA, Ericsen AJ, Tully DC, Power KA, Bean AT, Piaskowski SM, Duan L, Seese A, Gladden AD, Weisgrau KL, Furlott JR, Kim YI, Veloso de Santana MG, Rakasz E, Capuano S, Wilson NA, Bonaldo MC, Galler R, Allison DB, Piatak MJ, Haase AT, Lifson JD, Allen TM, Watkins DI. 2012. Vaccine-induced CD8+ T cells control AIDS virus replication. Nature 491:129–133. doi: 10.1038/nature11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valentine LE, Loffredo JT, Bean AT, Leon EJ, MacNair CE, Beal DR, Piaskowski SM, Klimentidis YC, Lank SM, Wiseman RW, Weinfurter JT, May GE, Rakasz EG, Wilson NA, Friedrich TC, O'Connor DH, Allison DB, Watkins DI. 2009. Infection with “escaped” virus variants impairs control of simian immunodeficiency virus SIVmac239 replication in Mamu-B*08-positive macaques. J Virol 83:11514–11527. doi: 10.1128/JVI.01298-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dang Q, Hirsch VM. 2008. Rapid disease progression to AIDS due to simian immunodeficiency virus infection of macaques: host and viral factors. Adv Pharmacol 56:369–398. doi: 10.1016/S1054-3589(07)56012-6. [DOI] [PubMed] [Google Scholar]
- 15.Loffredo JT, Sidney J, Bean AT, Beal DR, Bardet W, Wahl A, Hawkins OE, Piaskowski S, Wilson NA, Hildebrand WH, Watkins DI, Sette A. 2009. Two MHC class I molecules associated with elite control of immunodeficiency virus replication, Mamu-B*08 and HLA-B*2705, bind peptides with sequence similarity. J Immunol 182:7763–7775. doi: 10.4049/jimmunol.0900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altfeld M, Kalife ET, Qi Y, Streeck H, Lichterfeld M, Johnston MN, Burgett N, Swartz ME, Yang A, Alter G, Yu XG, Meier A, Rockstroh JK, Allen TM, Jessen H, Rosenberg ES, Carrington M, Walker BD. 2006. HLA alleles associated with delayed progression to AIDS contribute strongly to the initial CD8(+) T cell response against HIV-1. PLoS Med 3:e403. doi: 10.1371/journal.pmed.0030403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loffredo JT, Bean AT, Beal DR, Leon EJ, May GE, Piaskowski SM, Furlott JR, Reed J, Musani SK, Rakasz EG, Friedrich TC, Wilson NA, Allison DB, Watkins DI. 2008. Patterns of CD8+ immunodominance may influence the ability of Mamu-B*08-positive macaques to naturally control simian immunodeficiency virus SIVmac239 replication. J Virol 82:1723–1738. doi: 10.1128/JVI.02084-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vojnov L, Reed JS, Weisgrau KL, Rakasz EG, Loffredo JT, Piaskowski SM, Sacha JB, Kolar HL, Wilson NA, Johnson RP, Watkins DI. 2010. Effective simian immunodeficiency virus-specific CD8+ T cells lack an easily detectable, shared characteristic. J Virol 84:753–764. doi: 10.1128/JVI.01596-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adland E, Carlson JM, Paioni P, Kloverpris H, Shapiro R, Ogwu A, Riddell L, Luzzi G, Chen F, Balachandran T, Heckerman D, Stryhn A, Edwards A, Ndung'u T, Walker BD, Buus S, Goulder P, Matthews PC. 2013. Nef-specific CD8+ T cell responses contribute to HIV-1 immune control. PLoS One 8:e73117. doi: 10.1371/journal.pone.0073117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loffredo JT, Friedrich TC, Leon EJ, Stephany JJ, Rodrigues DS, Spencer SP, Bean AT, Beal DR, Burwitz BJ, Rudersdorf RA, Wallace LT, Piaskowski SM, May GE, Sidney J, Gostick E, Wilson NA, Price DA, Kallas EG, Piontkivska H, Hughes AL, Sette A, Watkins DI. 2007. CD8+ T cells from SIV elite controller macaques recognize Mamu-B*08-bound epitopes and select for widespread viral variation. PLoS One 2:e1152. doi: 10.1371/journal.pone.0001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Draenert R, Le Gall S, Pfafferott KJ, Leslie AJ, Chetty P, Brander C, Holmes EC, Chang SC, Feeney ME, Addo MM, Ruiz L, Ramduth D, Jeena P, Altfeld M, Thomas S, Tang Y, Verrill CL, Dixon C, Prado JG, Kiepiela P, Martinez-Picado J, Walker BD and Goulder PJ. 2004. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J Exp Med 199:905–915. doi: 10.1084/jem.20031982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosati M, von Gegerfelt A, Roth P, Alicea C, Valentin A, Robert-Guroff M, Venzon D, Montefiori DC, Markham P, Felber BK, Pavlakis GN. 2005. DNA vaccines expressing different forms of simian immunodeficiency virus antigens decrease viremia upon SIVmac251 challenge. J Virol 79:8480–8492. doi: 10.1128/JVI.79.13.8480-8492.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jalah R, Patel V, Kulkarni V, Rosati M, Alicea C, Ganneru B, von Gegerfelt A, Huang W, Guan Y, Broderick KE, Sardesai NY, LaBranche C, Montefiori DC, Pavlakis GN, Felber BK. 2012. IL-12 DNA as molecular vaccine adjuvant increases the cytotoxic T cell responses and breadth of humoral immune responses in SIV DNA vaccinated macaques. Hum Vaccin Immunother 8:1620–1629. doi: 10.4161/hv.21407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonaldo MC, Mello SM, Trindade GF, Rangel AA, Duarte AS, Oliveira PJ, Freire MS, Kubelka CF, Galler R. 2007. Construction and characterization of recombinant flaviviruses bearing insertions between E and NS1 genes. Virol J 4:115. doi: 10.1186/1743-422X-4-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson RD, Haskell RE, Xia H, Roessler BJ, Davidson BL. 2000. A simple method for the rapid generation of recombinant adenovirus vectors. Gene Ther 7:1034–1038. doi: 10.1038/sj.gt.3301197. [DOI] [PubMed] [Google Scholar]
- 26.Cline AN, Bess JW, Piatak MJ, Lifson JD. 2005. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J Med Primatol 34:303–312. doi: 10.1111/j.1600-0684.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- 27.Martins MA, Bonaldo MC, Rudersdorf RA, Piaskowski SM, Rakasz EG, Weisgrau KL, Furlott JR, Eernisse CM, Veloso de Santana MG, Hidalgo B, Friedrich TC, Chiuchiolo MJ, Parks CL, Wilson NA, Allison DB, Galler R, Watkins DI. 2013. Immunogenicity of seven new recombinant yellow fever viruses 17D expressing fragments of SIVmac239 Gag, Nef, and Vif in Indian rhesus macaques. PLoS One 8:e54434. doi: 10.1371/journal.pone.0054434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, Macalalad AR, Berlin AM, Malboeuf CM, Ryan EM, Gnerre S, Zody MC, Erlich RL, Green LM, Berical A, Wang Y, Casali M, Streeck H, Bloom AK, Dudek T, Tully D, Newman R, Axten KL, Gladden AD, Battis L, Kemper M, Zeng Q, Shea TP, Gujja S, Zedlack C, Gasser O, Brander C, Hess C, Gunthard HF, Brumme ZL, Brumme CJ, Bazner S, Rychert J, Tinsley JP, Mayer KH, Rosenberg E, Pereyra F, Levin JZ, Young SK, Jessen H, Altfeld M, Birren BW, Walker BD, Allen TM. 2012. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog 8:e1002529. doi: 10.1371/journal.ppat.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macalalad AR, Zody MC, Charlebois P, Lennon NJ, Newman RM, Malboeuf CM, Ryan EM, Boutwell CL, Power KA, Brackney DE, Pesko KN, Levin JZ, Ebel GD, Allen TM, Birren BW, Henn MR. 2012. Highly sensitive and specific detection of rare variants in mixed viral populations from massively parallel sequence data. PLoS Comput Biol 8:e1002417. doi: 10.1371/journal.pcbi.1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sardesai NY, Weiner DB. 2011. Electroporation delivery of DNA vaccines: prospects for success. Curr Opin Immunol 23:421–429. doi: 10.1016/j.coi.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vasan S, Hurley A, Schlesinger SJ, Hannaman D, Gardiner DF, Dugin DP, Boente-Carrera M, Vittorino R, Caskey M, Andersen J, Huang Y, Cox JH, Tarragona-Fiol T, Gill DK, Cheeseman H, Clark L, Dally L, Smith C, Schmidt C, Park HH, Kopycinski JT, Gilmour J, Fast P, Bernard R, Ho DD. 2011. In vivo electroporation enhances the immunogenicity of an HIV-1 DNA vaccine candidate in healthy volunteers. PLoS One 6:e19252. doi: 10.1371/journal.pone.0019252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonaldo MC, Martins MA, Rudersdorf R, Mudd PA, Sacha JB, Piaskowski SM, Costa Neves PC, Veloso de Santana MG, Vojnov L, Capuano S, Rakasz EG, Wilson NA, Fulkerson J, Sadoff JC, Watkins DI, Galler R. 2010. Recombinant yellow fever vaccine virus 17D expressing simian immunodeficiency virus SIVmac239 gag induces SIV-specific CD8+ T-cell responses in rhesus macaques. J Virol 84:3699–3706. doi: 10.1128/JVI.02255-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mudd PA, Piaskowski SM, Neves PC, Rudersdorf R, Kolar HL, Eernisse CM, Weisgrau KL, de Santana MG, Wilson NA, Bonaldo MC, Galler R, Rakasz EG, Watkins DI. 2010. The live-attenuated yellow fever vaccine 17D induces broad and potent T cell responses against several viral proteins in Indian rhesus macaques—implications for recombinant vaccine design. Immunogenetics 62:593–600. doi: 10.1007/s00251-010-0461-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mothe BR, Sidney J, Dzuris JL, Liebl ME, Fuenger S, Watkins DI, Sette A. 2002. Characterization of the peptide-binding specificity of Mamu-B*17 and identification of Mamu-B*17-restricted epitopes derived from simian immunodeficiency virus proteins. J Immunol 169:210–219. doi: 10.4049/jimmunol.169.1.210. [DOI] [PubMed] [Google Scholar]
- 35.Feeney ME, Tang Y, Roosevelt KA, Leslie AJ, McIntosh K, Karthas N, Walker BD, Goulder PJ. 2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J Virol 78:8927–8930. doi: 10.1128/JVI.78.16.8927-8930.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mudd PA, Watkins DI. 2011. Understanding animal models of elite control: windows on effective immune responses against immunodeficiency viruses. Curr Opin HIV AIDS 6:197–201. doi: 10.1097/COH.0b013e3283453e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allen TM, Mortara L, Mothe BR, Liebl M, Jing P, Calore B, Piekarczyk M, Ruddersdorf R, O'Connor DH, Wang X, Wang C, Allison DB, Altman JD, Sette A, Desrosiers RC, Sutter G, Watkins DI. 2002. Tat-vaccinated macaques do not control simian immunodeficiency virus SIVmac239 replication. J Virol 76:4108–4112. doi: 10.1128/JVI.76.8.4108-4112.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen TM, O'Connor DH, Jing P, Dzuris JL, Mothe BR, Vogel TU, Dunphy E, Liebl ME, Emerson C, Wilson N, Kunstman KJ, Wang X, Allison DB, Hughes AL, Desrosiers RC, Altman JD, Wolinsky SM, Sette A, Watkins DI. 2000. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407:386–390. doi: 10.1038/35030124. [DOI] [PubMed] [Google Scholar]
- 39.Friedrich TC, Dodds EJ, Yant LJ, Vojnov L, Rudersdorf R, Cullen C, Evans DT, Desrosiers RC, Mothe BR, Sidney J, Sette A, Kunstman K, Wolinsky S, Piatak M, Lifson J, Hughes AL, Wilson N, O'Connor DH, Watkins DI. 2004. Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat Med 10:275–281. doi: 10.1038/nm998. [DOI] [PubMed] [Google Scholar]
- 40.Carl S, Iafrate AJ, Skowronski J, Stahl-Hennig C, Kirchhoff F. 1999. Effect of the attenuating deletion and of sequence alterations evolving in vivo on simian immunodeficiency virus C8-Nef function. J Virol 73:2790–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mudd PA, Ericsen AJ, Walsh AD, Leon EJ, Wilson NA, Maness NJ, Friedrich TC, Watkins DI. 2011. CD8+ T cell escape mutations in simian immunodeficiency virus SIVmac239 cause fitness defects in vivo, and many revert after transmission. J Virol 85:12804–12810. doi: 10.1128/JVI.05841-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deeks SG, Walker BD. 2007. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27:406–416. doi: 10.1016/j.immuni.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 43.Matano T, Kobayashi M, Igarashi H, Takeda A, Nakamura H, Kano M, Sugimoto C, Mori K, Iida A, Hirata T, Hasegawa M, Yuasa T, Miyazawa M, Takahashi Y, Yasunami M, Kimura A, O'Connor DH, Watkins DI, Nagai Y. 2004. Cytotoxic T lymphocyte-based control of simian immunodeficiency virus replication in a preclinical AIDS vaccine trial. J Exp Med 199:1709–1718. doi: 10.1084/jem.20040432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawada M, Igarashi H, Takeda A, Tsukamoto T, Yamamoto H, Dohki S, Takiguchi M, Matano T. 2006. Involvement of multiple epitope-specific cytotoxic T-lymphocyte responses in vaccine-based control of simian immunodeficiency virus replication in rhesus macaques. J Virol 80:1949–1958. doi: 10.1128/JVI.80.4.1949-1958.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Virgin HW, Wherry EJ, Ahmed R. 2009. Redefining chronic viral infection. Cell 138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 46.Fujita T, Burwitz BJ, Chew GM, Reed JS, Pathak R, Seger E, Clayton KL, Rini JM, Ostrowski MA, Ishii N, Kuroda MJ, Hansen SG, Sacha JB, Ndhlovu LC. 2014. Expansion of dysfunctional Tim-3-expressing effector memory CD8+ T cells during simian immunodeficiency virus infection in rhesus macaques. J Immunol 193:5576–5583. doi: 10.4049/jimmunol.1400961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson S, Bergthaler A, Graw F, Flatz L, Bonilla WV, Siegrist CA, Lambert PH, Regoes RR, Pinschewer DD. 2015. Protective efficacy of individual CD8+ T cell specificities in chronic viral infection. J Immunol 194:1755–1762. doi: 10.4049/jimmunol.1401771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vezys V, Masopust D, Kemball CC, Barber DL, O'Mara LA, Larsen CP, Pearson TC, Ahmed R, Lukacher AE. 2006. Continuous recruitment of naive T cells contributes to heterogeneity of antiviral CD8 T cells during persistent infection. J Exp Med 203:2263–2269. doi: 10.1084/jem.20060995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen H, Ndhlovu ZM, Liu D, Porter LC, Fang JW, Darko S, Brockman MA, Miura T, Brumme ZL, Schneidewind A, Piechocka-Trocha A, Cesa KT, Sela J, Cung TD, Toth I, Pereyra F, Yu XG, Douek DC, Kaufmann DE, Allen TM, Walker BD. 2012. TCR clonotypes modulate the protective effect of HLA class I molecules in HIV-1 infection. Nat Immunol 13:691–700. doi: 10.1038/ni.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Price DA, West SM, Betts MR, Ruff LE, Brenchley JM, Ambrozak DR, Edghill-Smith Y, Kuroda MJ, Bogdan D, Kunstman K, Letvin NL, Franchini G, Wolinsky SM, Koup RA, Douek DC. 2004. T cell receptor recognition motifs govern immune escape patterns in acute SIV infection. Immunity 21:793–803. doi: 10.1016/j.immuni.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 51.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak MJ, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527. doi: 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hansen SG, Piatak MJ, Ventura AB, Hughes CM, Gilbride RM, Ford JC, Oswald K, Shoemaker R, Li Y, Lewis MS, Gilliam AN, Xu G, Whizin N, Burwitz BJ, Planer SL, Turner JM, Legasse AW, Axthelm MK, Nelson JA, Fruh K, Sacha JB, Estes JD, Keele BF, Edlefsen PT, Lifson JD, Picker LJ. 2013. Immune clearance of highly pathogenic SIV infection. Nature 502:100–104. doi: 10.1038/nature12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Masopust D, Ha SJ, Vezys V, Ahmed R. 2006. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol 177:831–839. doi: 10.4049/jimmunol.177.2.831. [DOI] [PubMed] [Google Scholar]
- 54.Hansen SG, Sacha JB, Hughes CM, Ford JC, Burwitz BJ, Scholz I, Gilbride RM, Lewis MS, Gilliam AN, Ventura AB, Malouli D, Xu G, Richards R, Whizin N, Reed JS, Hammond KB, Fischer M, Turner JM, Legasse AW, Axthelm MK, Edlefsen PT, Nelson JA, Lifson JD, Fruh K, Picker LJ. 2013. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 340:1237874. doi: 10.1126/science.1237874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu H, Hansen SG, Hammond KB, Hughes CM, Reed J, Ventura AB, Pathak R, Legasse AW, Burwitz BJ, Axthelm MK, Früh K, Picker LJ, Sacha JB. 2014. Universal, MHC-E restricted CD8+ T cells participate in RhCMV vaccine vector-induced protection against SIV, SY11.03, p 34 HIV R4P Conf Symp 11 Emerg Areas Immun. [Google Scholar]
- 56.Betts MR, Exley B, Price DA, Bansal A, Camacho ZT, Teaberry V, West SM, Ambrozak DR, Tomaras G, Roederer M, Kilby JM, Tartaglia J, Belshe R, Gao F, Douek DC, Weinhold KJ, Koup RA, Goepfert P, Ferrari G. 2005. Characterization of functional and phenotypic changes in anti-Gag vaccine-induced T cell responses and their role in protection after HIV-1 infection. Proc Natl Acad Sci U S A 102:4512–4517. doi: 10.1073/pnas.0408773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arts EJ and Hazuda DJ. 2012. HIV-1 antiretroviral drug therapy. Cold Spring Harb Perspect Med 2:a007161. doi: 10.1101/cshperspect.a007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.