

ABSTRACT
The A7(74) strain of Semliki Forest virus (SFV; genus Alphavirus) is avirulent in adult mice, while the L10 strain is virulent in mice of all ages. It has been previously demonstrated that this phenotypic difference is associated with nonstructural protein 3 (nsP3). Consensus clones of L10 (designated SFV6) and A7(74) (designated A774wt) were used to construct a panel of recombinant viruses. The insertion of nsP3 from A774wt into the SFV6 backbone had a minor effect on the virulence of the resulting recombinant virus. Conversely, insertion of nsP3 from SFV6 into the A774wt backbone or replacement of A774wt nsP3 with two copies of nsP3 from SFV6 resulted in virulent viruses. Unexpectedly, duplication of nsP3-encoding sequences also resulted in elevated levels of nsP4, revealing that nsP3 is involved in the stabilization of nsP4. Interestingly, replacement of nsP3 of SFV6 with that of A774wt resulted in a virulent virus; the virulence of this recombinant was strongly reduced by functionally coupled substitutions for amino acid residues 534 (P4 position of the cleavage site between nsP1 and nsP2) and 1052 (S4 subsite residue of nsP2 protease) in the nonstructural polyprotein. Pulse-chase experiments revealed that A774wt and avirulent recombinant virus were characterized by increased processing speed of the cleavage site between nsP1 and nsP2. A His534-to-Arg substitution specifically activated this cleavage, while a Val1052-to-Glu substitution compensated for this effect by reducing the basal protease activity of nsP2. These findings provide a link between nonstructural polyprotein processing and the virulence of SFV.
IMPORTANCE SFV infection of mice provides a well-characterized model to study viral encephalitis. SFV also serves as a model for studies of alphavirus molecular biology and host-pathogen interactions. Thus far, the genetic basis of different properties of SFV strains has been studied using molecular clones, which often contain mistakes originating from standard cDNA synthesis and cloning procedures. Here, for the first time, consensus clones of SFV strains were used to map virulence determinants. Existing data on the importance of nsP3 for virulent phenotypes were confirmed, another determinant of neurovirulence and its molecular basis was characterized, and a novel function of nsP3 was identified. These findings provide links between the molecular biology of SFV and its biological properties and significantly increase our understanding of the basis of alphavirus-induced pathology. In addition, the usefulness of consensus clones as tools for studies of alphaviruses was demonstrated.
INTRODUCTION
Semliki Forest virus (SFV) is an Alphavirus of the family Togaviridae. SFV is transmitted to vertebrate hosts by mosquitoes. In rodents, SFV infection can lead to lethal encephalitis. Various strains of SFV provide an experimental system to study the pathogenesis of viral encephalitis. The most commonly used SFV strains include A7(74), L10, and SFV4. Age-related virulence is a well-documented phenomenon; all strains of SFV can replicate in immature cells of the developing brain (1) and are able to infect the central nervous system (CNS) (2, 3). The outcome of infection in adult mice, however, is SFV strain dependent (4, 5).
SFV is an enveloped virus with a positive-stranded RNA genome that is approximately 11.5 kb in length. The genome contains two open reading frames encoding nonstructural (ns) or structural polyproteins (5, 6). The ns proteins nsP1, nsP2, nsP3, and nsP4 are formed by autoproteolytic cleavages of ns polyprotein P1234 (6, 7). P123, an intermediate product of this process, and nsP4 are responsible for the synthesis of negative-strand RNA during the early stages of infection, which last for approximately 3 to 4 h (8). The mature nsPs form a stable replication complex that is responsible for the synthesis of positive-strand genomic RNA and subgenomic RNA (sgRNA) from the negative-strand template (9).
nsP1 serves as a membrane anchor for the replicase complex (10) and is responsible for capping the genomic and sgRNAs (11). It is also required for the initiation of synthesis and elongation of negative-strand RNA (12). Additionally, nsP1 is involved in the induction of type I interferon (6, 13, 14). nsP2 is a multidomain protein. The N-terminal half of nsP2 contains domains required for NTPase and RNA triphosphatase activities (15, 16), while its C-terminal half represents the core of the protease responsible for the processing of the ns polyprotein. Different domains of nsP2 interact with each other; as a consequence, only full-length nsP2 has RNA helicase activity and is capable of performing all cleavages required for the processing of P1234 (17, 18). nsP2 is the only ns protein shown to translocate to the nucleus. The nuclear fraction of nsP2 is responsible for cytotoxic effects and suppression of the interferon response; such effects are important for the replication of SFV in the brain (6, 19–21). The functions of nsP3 remain more elusive. It has an N-terminal macrodomain, a central zinc-binding domain (22), and a C-terminal nonconserved region which is intrinsically disordered. This protein is important for the correct localization of the replication complexes in intracellular vesicles (10) and for alphavirus-host interactions and in SFV replication (23). nsP3 is the only phosphorylated ns protein of SFV, and its phosphorylation is important for neurovirulence (24). nsP4 is the catalytic subunit of viral RNA-dependent RNA polymerase (25). Compared to the other ns proteins, it is less abundant in infected cells, as free nsP4 is rapidly degraded (26). In most alphaviruses and individual SFV strains, the translation of nsP4 requires read-through of an opal termination codon located in the nsP3-encoding region (6). In replication complexes, nsP4 appears to be stable, but the mechanism responsible for this stabilization is not known.
SFV strains fall into two groups according to their neurovirulence in adult mice: virulent and avirulent (7). The A7(74) strain derived from the AR20266 strain isolated in Mozambique in 1959 (27) exhibits an age-related virulence phenotype. In neonatal and suckling mice, the infection is lethal, while in adult mice A7(74) is avirulent. In adult animals, intraperitoneal (i.p.) inoculation of the A7(74) strain causes high levels of viremia, but infection in mature neurons is restricted and the virus causes subclinical encephalitis (4, 28, 29). The L10 strain was isolated from a pool of mosquitoes collected in Uganda in 1942 (30). L10 is virulent in mice of all ages independent of the route of inoculation (4, 7, 31). Intraperitoneal inoculation of L10 results in high-titer viremia that develops into fatal panencephalitis. The SFV4 strain has the same origin as L10, but with a different phenotype in vivo. Adult mice inoculated intracerebrally with SFV4 develop panencephalitis rapidly, whereas when inoculated i.p. or intranasally, SFV4 is virulent only at high doses (32).
It has been reported that a chimera containing the structural region from the molecular clone (rA774) of avirulent A7(74) and the ns region of SFV4 is virulent. Replacement of the nsP3 region of rA774 with that of SFV4 also resulted in virulent virus, showing that nsP3 of SFV4 alone is sufficient to restore neurovirulence in an avirulent strain (33). Subsequent analysis, however, failed to pinpoint the differences in the virulence of SFV4 and rA774 to any specific position of the nsP3 (32). It was also suggested that other determinants of virulence must be present in the ns region (33).
Prior studies using molecular clones such as rA774 and SFV4 are difficult to interpret because these clones were not based on consensus sequences. Recently, we demonstrated that the sequence of SFV4 differs from the consensus sequence of the L10 strain; furthermore, one of the amino acid differences, located in the structural region, attenuates the replication of SFV4 in the periphery. A fully virulent consensus clone of the SFV L10 strain, designated SFV6, was constructed (34). In vitro transcripts of rA774 demonstrated abnormally low infectivity, suggesting that the rA774 clone carries an uncharacterized functional defect(s). Consequently, deep sequencing was undertaken to identify a consensus sequence and generate new clones to confirm prior findings.
In this study, infectious cDNA (icDNA) clone A774wt, corresponding to the consensus sequence of the A7(74) strain, was constructed. The efficiency of rescue of infectious virus from the pCMV-A774wt plasmid was more than 100-fold higher than that with a similar plasmid containing original rA774 cDNA. Recombinants of A774wt and SFV6 carrying two copies of nsP3 as well as viruses in which the sequence of nsP3 in A774wt was replaced with that of SFV6 (A774wt-6) or vice versa (SFV6-74) were constructed. It was found that the presence of an extra copy of nsP3 remarkably increased intracellular amounts of nsP4, indicating that nsP3 is involved in stabilization of this protein. In vivo studies revealed that all the viruses with two copies of nsP3 (at least one of them was always from SFV6) were neurovirulent. Furthermore, both A774wt-6 and SFV6-74 were also neurovirulent. Amino acid residues affecting the speed of P123 polyprotein processing were found to represent a second crucial determinant of SFV neurovirulence. These results show that both nsP3 and ns polyprotein processing contribute to the neurovirulence of SFV.
MATERIALS AND METHODS
Cell culture.
BHK-21 cells (ATCC) were grown in Glasgow's minimal essential medium (GMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; GE Healthcare), 2% tryptose phosphate broth (TPB), and 20 mM HEPES (pH 7.2). NIH 3T3 cells (ATCC) were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% FBS. All growth media contained 100 U/ml penicillin and 100 μg/ml streptomycin (PAA). The cells were grown in a humidified incubator at 37°C with 5% CO2.
Virus strains.
SFV A7(74) was obtained from H. E. Webb (St. Thomas Hospital, London, England), who received it from Claude Bradish, Porton Down, England. The virus was stored at −80°C with minimum passage. An icDNA clone, rA774 (33), was used to construct an infectious plasmid designated pCMV-rA774 by changing the plasmid backbone of the rA774 clone to that of pCMV-SFV4 (35). Subsequently, pCMV-rA774 was used to construct pCMV-A774wt by replacing the codon for Val1347 with that of Ile. The virus rescued from this plasmid was designated A774wt. Construction of a consensus clone of the L10 strain (designated pCMV-SFV6) has been described elsewhere (34).
High-throughput sequencing of SFV A7(74).
Approximately 6 × 105 NIH 3T3 cells were grown in 6-well plates and were infected with A7(74) at a multiplicity of infection (MOI) of 10. After 8 h, total RNA was extracted from the cells by using the RNeasy kit (Qiagen) according to the manufacturer's instructions. RNA integrity and concentration were measured with the RNA 6000 Nano assay (Agilent Technologies). Subsequent RNA processing and sequencing were carried out by ARK Genomics (The Roslin Institute, University of Edinburgh). To generate the consensus sequence of A7(74), obtained reads were mapped to the reference sequence of rA774 (kindly provided by Ari Hinkkanen) by using the Novoalign software tool (Novocraft). Differences between the sequences of rA774, L10 (GenBank accession number KP271965), and the obtained consensus sequence were detected using Jalview 2.7 software (36).
Construction of virus chimeras and mutants.
pCMV-SFV4 and pCMV-rA774 plasmids were used to obtain the initial panel of recombinant constructs. Recombinant clones used in subsequent experiments were engineered using pCMV-A774wt and pCMV-SFV6. To increase the stability of the duplicated nsP3 region, a fragment of the SFV6 genome encoding nsP3 was redesigned using synonymous changes and obtained as synthetic DNA (Genewiz). Native fragments of A774wt or SFV6, encoding nsP3, were inserted downstream of the codon-altered region. Obtained cassettes were used to replace nsP3-encoding regions in pCMV-SFV6 or pCMV-A774wt, respectively. The resulting clones were designated pCMV-A774wt-6/74, pCMV-A774wt-6/6, pCMV-SFV6-6/74, and pCMV-SFV6-6/6; A774wt-6/74, A774wt-6/6, SFV6-6/74, and SFV6-6/6 were used as the rescued viruses (Fig. 1B).
FIG 1.

Recombinant constructs and viruses carrying two copies of nsP3-encoding sequences. (A and B) Schematic representations show the recombinant SFV constructs based on original rA774 and SFV4 sequences (A) and constructs based on corrected A774wt and SFV6 sequences containing codon-altered copy of nsP3 encoding region (B). (C and D) Results of ICA and titers of collected P0 stocks. Multistep (C) and single-step (D) growth curves of recombinant viruses were determined. BHK-21 cells were infected at the indicated MOI, and samples were collected at the indicated time points. Data from one of two reproducible experiments are shown in each panel.
To replace the nsP3 in A774wt with that of SFV6 or to perform a reciprocal change, the nsP3-encoding fragments of A774wt and SFV6 were PCR amplified. Amplified fragments were then cloned into a heterologous context. Resulting clones were designated pCMV-A774wt-6 and pCMV-SFV6-74, and the designations A774wt-6 and SFV6-74 were used for rescued viruses (see Fig. 5A, below). In addition to the swapping of nsP3 coding sequences, these cloning procedures also resulted in a few synonymous changes in the sequences encoding the nsP2/nsP3 and nsP3/nsP4 junctions. PCR-based site-directed mutagenesis and subcloning were used to obtain the recombinant viruses with substitutions in position P4 of the processing site between nsP1 and nsP2 (the 1/2 site) and amino acid residue 515 of nsP2 (residues 534 and 1052 of P1234, respectively). The fragment containing substitutions of codons for Arg534 (to His) and Glu1052 (to Val) was designated HV and used to replace a similar fragment in pCMV-A774wt; the resulting clone and virus were designated pCMV-A774wt-HV and A774wt-HV, respectively. Similarly, two amino acid substitutions (His534 to Arg and Val1052 to Glu [RE]) were introduced into the ns polyprotein encoded by pCMV-SFV6-74; the obtained clone and virus were designated pCMV-SFV6-74-RE and SFV6-74-RE, respectively (see Fig. 6A, below). All constructs were verified by sequencing; sequences of all recombinants are available upon request from the authors.
FIG 5.

Swapping of nsP3 regions between SFV6 and A774wt results in virulent chimeras. (A) Schematic representations of the SFV constructs, the results of ICA, and titers of P0 virus stocks. (B) Groups (n = 7) of 4- to 5-week-old mice were inoculated i.p. with 5,000 PFU of the indicated virus (mice infected with SFV6 are the same as in Fig. 4B) and monitored for survival for 10 days. (C and D) Virus titers in the blood on PID 1 (C) and on PID 3 (D). (E) Virus titers in brain tissue on PID 3. Each symbol represents the result for an individual mouse. The line depicts the mean titer of each group. The dotted line depicts the limit of detection. Significance was determined using the Mann-Whitney test. Only statistically significant differences are indicated: *, P < 0.05; **, P < 0.01.
FIG 6.

Mutations at positions associated with ns polyprotein processing affect SFV neurovirulence. (A) Schematic representations of the recombinant SFV constructs and the results of ICA and titers of P0 virus stocks. (B) Groups (n = 7) of 4- to 5-week-old mice were inoculated i.p. with 5,000 PFU of the indicted virus (note that the mice infected with SFV6 are different from those shown in Fig. 4 and 5) and monitored for survival for 10 days. (C and D) Virus titers in the blood on PID 1 (C) and PID 3 (D). (E and F) Virus titers in brain tissue on PID 1 (E) and PID 3 (F). Each symbol represents an individual mouse. The line depicts the mean titer of each group. The dotted line depicts the limit of detection. Differences between brain titers of A774wt-HV at PID 1 (E) and at PID 3 (F) as well as differences between brain titers of SFV6-74-RE at PID 1 (E) and PID 3 (F) are statistically significant (P = 0.008 and P = 0.02, respectively).
Infectious center assay, virus rescue, propagation, and growth curve analysis.
Plasmids containing icDNA of rA774, A774wt, SFV6, or obtained recombinant viruses were used for the transfection of BHK-21 cells via electroporation (220 V, 975 μF, one pulse in a cuvette with a 4-mm electrode gap). An infectious center assay (ICA) was performed as previously described (37). Briefly, 10-fold dilutions of electroporated BHK-21 cells were transferred onto confluent monolayers of BHK-21 cells. After incubation for 2 to 3 h at 37°C, the cells were overlaid with 2 ml of GMEM supplemented with 2% FBS and containing 0.8% carboxymethylcellulose (CMC; Sigma Life Science). After 2 days of incubation, the cells were fixed and stained with crystal violet. The rest of the electroporation mixture was transferred onto 100-mm cell culture dishes. After incubation for 24 h at 37°C, the stocks of rescued viruses (P0 stocks) were collected and titrated in a plaque assay on BHK-21 cells as previously described (4, 5). To obtain P1 stocks, confluent BHK-21 cells grown on 100-mm dishes were infected with P0 stocks at an MOI of 5. At 24 h postinfection (p.i.), the supernatants (P1 stocks) were collected and virus titers were determined. Passages up to P5 were obtained by repeating this procedure four more times.
For single-step and multistep growth curves, confluent BHK-21 cells grown on 60-mm dishes were infected with P0 stocks of A774wt, A774wt-6/74, A774wt-6/6, SFV6, SFV6-6/74, and SFV6-6/6 at an MOI of 5 or 0.01. The inoculum was removed, cells were washed with phosphate-buffered saline (PBS), overlaid with 4 ml of GMEM supplemented with 2% FBS, and incubated in a humidified incubator at 37°C with 5% CO2. Then, 200-μl aliquots were collected at 2, 4, 6, 8, 12, 16, and 24 h p.i. for single-step growth curves and at 2, 4, 8, 12, 24, 36, 48, and 60 h p.i for multistep growth curves. Virus titers were determined by plaque assay.
RNA extraction, reverse transcription, and PCR.
For sequence analysis, total RNA was extracted from infected cell monolayers by using the RNeasy minikit (Qiagen), and cDNA was synthesized using SuperScript III reverse transcriptase (RT; Invitrogen) and oligo(dT)15 (Promega) as the primer. Regions of interest were amplified by PCR using GoTaq DNA polymerase (Promega) and analyzed by Sanger sequencing.
To analyze the stability of genomes harboring two copies of nsP3-encoding sequences, RNA was purified from either 100 μl of P0, P3, and P5 stocks or from 100 μl of homogenate prepared from half the brain of an animal (euthanized at the clinical endpoint of the experiment) by using the RNeasy minikit (Qiagen) and reverse transcribed using the First-Strand cDNA synthesis kit (Thermo Scientific) with a random hexamer primer according to the manufacturer's instructions. PCRs were performed using either of two primer sets. The first set had a sense primer that matched the 3′ end of the nsP2-encoding sequence and a primer complementary to the sequence in the beginning of the native (but not to the codon-altered) nsP3 as the antisense primer. Obtained RT-PCR fragments corresponding to viruses with one copy of nsP3 were 292 bp long, while fragments corresponding to viruses containing two copies of nsP3 had a length of 1,738 bp (see Fig. 3A and 4F, below). The second set of primers amplified the nsP3/nsP3 junction region. It contained a primer corresponding to the 3′ end of the codon-altered (but not to the native) nsP3-encoding sequence as the sense primer and a primer complementary to the sequence in the beginning of the native (but not to the codon-altered) nsP3 as the antisense primer. Obtained RT-PCR fragments corresponding to viruses with two copies of nsP3 were 311 bp long, while viruses containing one copy of nsP3 gave no product (see Fig. 4F).
FIG 3.

Analysis of in vitro genetic stability, ns protein expression, and ns polyprotein processing of the viruses harboring two copies of nsP3. (A) Schematic representation of the assay and results of the RT-PCR analysis of the region containing duplicated nsP3-encoding sequences. RNA obtained from P0 (left panel), P3 (middle panel), and P5 (right panel) stocks was reverse transcribed, cDNA was PCR amplified using primers corresponding to the 3′ region of the nsP2-encoding sequence and complementary to the 5′ region of the native nsP3-encoding sequence. PCR products were analyzed on a 1% agarose gel. (B) BHK-21 cells infected at an MOI of 1 were collected 8 h p.i. and lysed, and SFV ns proteins were analyzed using SDS-PAGE. nsPs were detected using corresponding polyclonal antisera; their positions and that of P34 are shown on the right side of the panel. β-Actin was used as a loading control. (C) Processing of ns polyproteins in BHK-21 cells. Cells were infected with SFV6, SFV6-6/74, or SFV6-6/6 at an MOI of 10, starved for 30 min at 3 h p.i. in methionine-cysteine-free medium, and then pulsed with 50 μCi of [35S]methionine-cysteine mixture for 15 min. Cells were collected immediately after pulse or after a 45-min chase. Synthesized ns polyproteins, their processing intermediates, and mature nsPs were immunoprecipitated using the indicated combinations of antibodies. Positions of nsPs and ns polyproteins are shown at both sides of the panel. For all panels, data from one of three reproducible experiments are shown.
FIG 4.

Neurovirulence of SFV6, A774wt, and viruses harboring two copies of nsP3. (A) Groups (n = 37) of 4- to 5-week-old mice were inoculated i.p. with the indicated virus (5,000 PFU/mouse) and monitored for survival for 10 days. (B) Groups (n = 7) of 4- to 5-week-old mice were inoculated i.p. with the indicated virus (5,000 PFU/mouse) and monitored for survival for 10 days. (C and D) Virus titers in the blood of infected animals on PID 1 (C) and on PID 3 (D). (E) Virus titers in brain tissue on PID 3. Each symbol represents an individual mouse; the line depicts the mean titer of each group. The dotted line depicts the limit of detection. Statistical significance was determined using the Mann-Whitney test. Only statistically significant differences are indicated: **, P < 0.01; ****, P < 0.0001. (F) Detection of viruses with two copies of nsP3 in brains of mice at the time of the clinical endpoint of the experiment. Total RNA was isolated from brain homogenates, reverse transcribed, and analyzed using PCR. The left panels show schematic representations of PCR products. Obtained PCR products were analyzed on a 1% agarose gel (right panels).
Immunoblot analysis.
For immunoblot analysis, BHK-21 cells were infected with P0 stocks of A774wt, A774wt-6/74, A774wt-6/6, SFV6, SFV6-6/74, and SFV6-6/6 at an MOI of 1. At 8 h p.i., cells were lysed using SDS gel loading buffer (100 mM Tris-HCl [pH 6.8], 4% SDS, 20% glycerol, 200 mM dithiothreitol, and 0.2% bromophenol blue). Proteins were separated via 10% SDS-PAGE, transferred to nitrocellulose membranes, and detected using antisera against nsP1, nsP2, nsP3, and nsP4 (developed in-house) as primary antisera and horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibodies as secondary antibodies (LabAs Ltd.). β-Actin was used as a loading control and was detected with mouse monoclonal antibody against β-actin (Santa Cruz Biotechnology) and HRP-conjugated goat anti-mouse antibody (LabAs Ltd.). Amersham enhanced chemiluminescence (ECL) Western blotting detection reagents (GE Healthcare Life Sciences) were used to develop the blots. The ImageQuant RT ECL imager was used to visualize the proteins, and ImageQuant TL software (GE Healthcare) was used to analyze and quantify the signals.
Immunofluorescence microscopy.
For indirect immunofluorescence microscopy, BHK-21 cells were grown on coverslips in 35-mm dishes. At 50% confluence, the cells were infected with SFV6 or with SFV6-6/6 at an MOI of 10. At 4 h p.i., the cells were washed with PBS, fixed with 4% paraformaldehyde, quenched with 50 mM NH4Cl, permeabilized with 0.1% Triton X-100 for 2 min at room temperature, and treated with rabbit anti-nsP1, anti-nsP2, anti-nsP3, or anti-nsP4 antisera and mouse monoclonal antibody against double-stranded RNA (dsRNA; J2; Scicons). Incubation with primary antibodies was followed by incubation with secondary anti-mouse antibodies conjugated to Alexa Fluor 488 and anti-rabbit antibodies conjugated to Alexa Fluor 568 (Invitrogen). The samples were analyzed using a Carl Zeiss LSM710 confocal microscope.
Metabolic labeling and immunoprecipitation.
The ns polyprotein processing was studied using pulse-chase analysis as described previously (38). Briefly, BHK-21 cells were infected at an MOI of 10. At 3 h p.i., cells were starved in methionine- and cysteine-free DMEM (Life Technologies) for 30 min and then labeled with 50 μCi of [35S]methionine-cysteine mixture (PerkinElmer) for 15 min. After this, cells were collected (pulse) or chased in the presence of excess unlabeled methionine and cysteine for 45 min. Cells were lysed, and viral proteins were captured by immunoprecipitation with antibodies against the ns proteins and precipitated with protein A–Sepharose CL-4B (Sigma-Aldrich). Obtained samples were resolved by 8% SDS-PAGE and visualized with a Typhoon imager. The images were analyzed and the signals quantified with ImageQuant TL software (GE Healthcare).
Mouse infections.
Mouse infections were performed as previously described (34). Four- to 5-week-old BALB/c mice were used (Harlan Laboratories or in-house breeding). Experiments were either conducted at the University of Edinburgh (animals treated and housed in the Hugh Robson Building Animal Unit, College of Medicine and Veterinary Medicine, United Kingdom) or at the University of Tartu under specific-pathogen-free and environmentally enriched conditions with food and water supplied ad libitum. All experimental studies were approved by the University of Edinburgh Ethical Review Committee or by the Estonian National Board of Animal Experiments.
With one exception, indicated below, mortality due to the SFV infection was analyzed using groups of mice (n = 7). To compare mortality resulting from infection with A774wt, A774wt-6/74, and A774wt-6/6, large groups of mice (n = 37, with control groups of SFV6-infected and PBSA-inoculated mice [n = 7]) were used. (In our experiments, PBSA was PBS supplemented with 0.75% BSA.) Mice were inoculated i.p. with 5,000 PFU of virus in 100 μl of PBSA. The animals were monitored twice daily for up to 10 days. Mice were sacrificed upon reaching well-defined terminal endpoints of disease, and the brains of these mice were sampled. None of the PBSA-inoculated mice died during the monitoring period, and no virus was detected in samples collected from these mice; therefore, the PBSA mice are not shown in the corresponding figures.
Additional groups (n = 5) of mice were infected as described above and were euthanized on either postinoculation day (PID) 1 or 3. Blood and brain samples were collected for further analysis. Virus titers in both the blood and the brain homogenates were determined by plaque assay on BHK-21 cells as previously described (6).
Statistical analysis.
Statistical analysis was carried out using GraphPad Prism software. Data were analyzed with the nonparametric Mann-Whitney test for mouse studies. A comparison resulting in a P value of <0.05 was considered a statistically significant difference.
RESULTS
Low infectivity of the SFV rA774 clone is caused by a single amino acid difference from the A7(74) consensus sequence.
To investigate the basis of the different phenotypes of SFV strains, biological stocks of L10 and A7(74) were analyzed using high-throughput sequencing. Consensus sequences of L10 and A7(74) had 26 nonsynonymous differences in the ns region: 14 in nsP3 (including the deletion of amino acid residues 1723 to 1730 and an Arg1812-to-opal substitution), 6 in nsP1, 4 in nsP2, and 2 in nsP4 (Table 1).
TABLE 1.
Amino acid differences between ns polyproteins of L10 and A7(74)
| Gene | Amino acid positiona | Amino acid in straine |
|
|---|---|---|---|
| L10 | A7(74) | ||
| nsP1 | 237 | C | S |
| 308 | Y | H | |
| 387 | I | V | |
| 427 | R | K | |
| 484 | V | A | |
| 534b | H | R | |
| nsP2 | 679 | V | I |
| 1052c | V | E | |
| 1216 | F | Y | |
| 1258 | S | N | |
| nsP3 | 1347d | I | I |
| 1406 | A | G | |
| 1537 | F | L | |
| 1585 | N | D | |
| 1723 | G | — | |
| 1724 | I | — | |
| 1725 | A | — | |
| 1726 | D | — | |
| 1727 | L | — | |
| 1728 | A | — | |
| 1729 | A | — | |
| 1730 | D | — | |
| 1778 | A | T | |
| 1785 | L | F | |
| 1812 | R | * | |
| nsP4 | 1974 | D | E |
| 2429 | R | K | |
Numbering of amino acid residues is based on the ns polyprotein of L10 (GenBank accession number KP271965); the sequence of the ns polyprotein of SFV6 is identical to that of L10.
The P4 position of the 1/2 site.
Amino acid residue 515 of nsP2.
A Val residue in the rA774 clone.
—, for positions 1723 to 1730, designates amino acid residues absent in the sequence of A7(74) ns polyprotein. Up to position 1585 (included), amino acid positions in the L10 and A7(74) ns polyproteins are identical. *, opal termination codon.
ICA revealed that the efficiency of rescue of infectious virus from pCMV-rA774 was below 1,000 PFU/μg, compared to >100,000 PFU/μg for pCMV-SFV4 (Fig. 1A). As rescued viruses replicated to comparable titers, it was concluded that the original rA774 clone contained a mutation(s) that drastically reduced the recombinant virus rescue efficiency; however, during virus rescue and propagation, this defect(s) reverted and/or was compensated by second-site mutations. Comparison of the consensus sequence of A7(74) with that of rA774 (33) revealed a single nonsynonymous difference in codon 1347 of the open reading frame of the ns polyprotein. This change leads to the change of amino acid residue 11 of nsP3 from Ile (consensus) to Val (rA774). Introduction of the Val1347-to-Ile substitution resulted in an over-100-fold increase in the efficiency of infectious A774 virus rescue; in that context, the construct designated pCMV-A774wt was similar to pCMV-SFV6 (Fig. 1B). Furthermore, sequencing of virus stocks rescued from cells transfected with in vitro transcripts of the original rA774 clone revealed that these viruses always contained the Ile1347 codon. It was also observed that virus stocks rescued from the rA774 clone tended to display inconsistent phenotypes: some stocks had a mild temperature-sensitive phenotype, while stocks rescued in parallel experiments did not (unpublished data). These variations could be the result of different second-site mutations, which often occur during the rescue and propagation of SFV variants with compromised infectivity (18, 39, 40). Thus, rA774 contained a mistake that remained undetected until methods for the identification of consensus sequences became available. Therefore, it was necessary to verify the conclusions reached by using viruses rescued from the rA774 infectious clone.
Duplication of native nsP3-encoding sequences results in genetic instability.
Analysis of chimeras between SFV4 and rA774 demonstrated that nsP3 is the main virulence determinant of SFV (33). Thus, either nsP3 of SFV4 is the factor responsible for virulence or, alternatively, nsP3 of rA774 represents a factor that restricts SFV virulence. The observation that dissimilar combinations of substitutions, introduced into nsP3 of an avirulent construct, all result in virulent viruses (32) also suggests that virulence may originate from the loss of a protective function(s) of nsP3. Thus, it remained unclear which phenotype caused by nsP3, virulence or avirulence, is dominant. To address this question directly, viruses carrying duplicated nsP3 regions were constructed and analyzed.
The original panel of these viruses was based on the duplication of homologous (controls) or the addition of heterologous nsP3-encoding sequences into pCMV-SFV4 or pCMV-rA774 backbones. All these constructs, including pCMV-rA774-4/74 and pCMV-SFV4-4/74, had high efficiencies of infectious virus rescue (Fig. 1A), indicating that two copies of nsP3 were tolerated by the virus and that a defect, caused by the Ile1347Val mutation in nsP3 of rA774, was recessive. All rescued viruses grew to high titers; compared to SFV4 and rA774, only small, 3- to 5-fold reductions of titers of P0 stock were observed (Fig. 1A). Subsequent genetic analysis revealed that the obtained stocks mostly contained viruses with a single copy of nsP3. Sequence analysis confirmed that recombination had occurred between two copies of the nsP3 coding sequences. All deletions maintained the reading frame for the ns polyprotein, and the majority of them were precise, resulting in an nsP3 of the correct size. Among the progeny of constructs carrying heterologous nsP3 sequences, different recombination points were found. Attempts to overcome the problem by undergoing multiple cycles of plaque purification and propagation of plaque-purified stocks at different MOIs were unsuccessful, indicating that copy choice recombination between identical or nearly identical sequences [e.g., sequences encoding nsP3 of SFV4 and A7(74) are 96% identical] was highly efficient and led to viruses with a single copy of nsP3 that had clear growth advantages.
Recombination between two copies of nsP3-encoding sequences can be reduced by introduction of synonymous changes.
To increase recombinant genome stability, numerous synonymous changes were introduced into the sequence encoding nsP3 of SFV6. The region with altered codon usage was used as the first (upstream) copy of nsP3 in the recombinant virus genomes. The second copy of the nsP3-encoding sequence was the nonmodified sequence originating from A774wt or SFV6. This order of nsP3 regions was selected to ensure that the recombinant viruses would express both nsP3 proteins at the same level (the opal termination codon in the region encoding nsP3 of A774wt would lead to a drastic reduction of expression of the downstream copy of nsP3).
The ICA revealed that the efficiency of infectious virus rescue from pCMV-A774wt-based constructs was 5.3 × 104 to 8.4 × 104 PFU/μg, whereas values between 1.1 × 105 and 1.5 × 105 PFU/μg were obtained for pCMV-SFV6-based constructs. Thus, the efficiencies of recombinant rescues viruses were similar to those of the parental clones (Fig. 1B). Titers of P0 virus stocks, collected at 24 h posttransfection, were 7.1 × 108 PFU/ml and 4.8 × 108 PFU/ml for A774wt and SFV6, respectively. Titers of A774wt-6/74 and A774wt-6/6 were approximately 5-fold lower, and titers of SFV6-6/74 and SFV6-6/6 were approximately 10-fold lower than their respective parental viruses (Fig. 1B); such differences were also maintained for P1 to P5 stocks (data not shown). Single-step and multistep growth curves, obtained for the rescued viruses, confirmed that viruses carrying two copies of nsP3 had a delay in accumulation of infectious progeny and reached approximately 1-log-lower titers than did A774wt and SFV6 (Fig. 1C and D). Similar reductions in titers and changes in growth kinetics have been previously observed for genetically stable SFV reporter viruses with essentially the same design (41). Reduced titers most likely originate from the larger genome sizes and serve as indirect evidence for the genetic stability of rescued recombinant viruses.
Immunofluorescence analysis, performed using antibodies against dsRNA and SFV ns proteins, revealed that the localization of ns proteins in BHK-21 cells infected with SFV6 (Fig. 2A) was very similar to that of ns proteins in SFV6-6/6-infected cells (Fig. 2B); ns proteins of both of the viruses displayed localization similar to that reported for ns proteins of SFV4 (42, 43). In addition to replicase organelles (indicated by dsRNA staining), nsP1 was also detected at the plasma membrane, and nsP2 was diffuse in the cytoplasm and, to a lesser extent, in the nucleus (Fig. 2A and B). nsP3 staining was punctate. Some of these proteins were colocalized by dsRNA staining and thus represented replicase organelles (Fig. 2A and B). Other structures lacked dsRNA staining. The analysis of their compositions was beyond the scope of the current study, though it can be speculated that some of these may represent stress granules, which are characteristic of SFV-infected cells at early stages of infection (44) and/or other nsP3-containing complexes found in alphavirus-infected cells (45). nsP4 was also detected both in dsRNA-positive replicase organelles and diffusely in the cytoplasm (Fig. 2A and B). These data confirmed that addition of an extra copy of nsP3 did not appear to interfere with the localization of virus replicase organelles or individual ns proteins.
FIG 2.
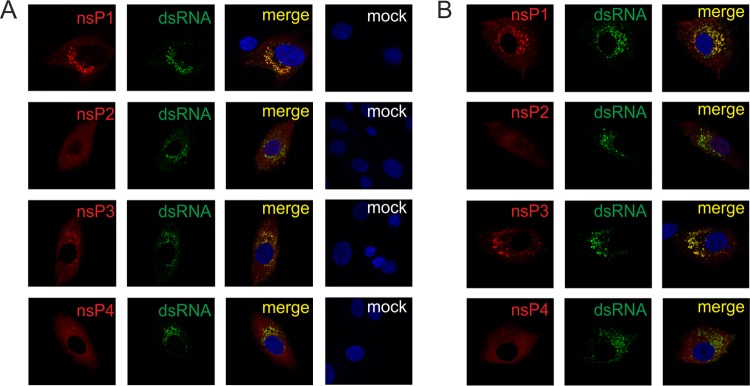
Localization of ns proteins in BHK-21 cells infected with SFV6 (A) and SFV6-6/6 (B). Cells were infected at an MOI of 10, fixed at 4 h p.i., permeabilized, and probed with antibodies for dsRNA (green) and nsP1, nsP2, nsP3, and nsP4 of SFV (red). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (for clarity, shown only on merge panels). Merged images of mock-infected cells (mock) stained using the same antibodies are shown for comparison.
The genetic homogeneity of P0 stocks of A774wt-6/74, A774wt-6/6, SFV6-6/74, and SFV6-6/6 was confirmed by RT-PCR analysis of RNA isolated from corresponding P0 stocks. All viruses maintained the upstream copy of nsP3 (Fig. 3A). In addition, RT-PCR with primers corresponding to the 3′ region of the nsP2-encoding sequence and complementary to the 5′ region of the nsP4-encoding sequence did not result in any products that indicated recombination or deletions (data not shown). However, following five passages in BHK-21 cells (MOI, 5), RT-PCR analysis revealed the presence of viruses which had apparently lost one copy of nsP3 in the P3 stock (Fig. 3A). The amount of such genomes was relatively low and did not increase during the next two passages (Fig. 3A), indicating that at least under the conditions we used, viruses that had lost one nsP3 copy had no significant growth advantage over A774wt-6/74, A774wt-6/6, SFV6-6/74, or SFV6-6/6. Thus, these recombinant viruses have similar and relatively high genetic stability in vitro. To minimize the effect of small, but detectable, genetic instabilities, subsequent experiments were performed using P0 stocks of recombinant viruses.
Western blot analysis revealed that A774wt-6/74, A774wt-6/6, SFV6-6/74, and SFV6-6/6 expressed somewhat more nsP3 than did A774wt or SFV6 (Fig. 3B). However, the significance of this observation is unclear, as the amounts of other ns proteins, produced by different viruses, also exhibited variations. The differences were most obvious for nsP4, partly due to the presence of an opal termination codon in the region encoding nsP3 of A774wt, A774wt-6/74, and SFV6-6/74 (Fig. 3B). Surprisingly, relatively high levels of nsP4 were also detected with SFV6-6/74-infected cells. Finally, the P34 polyprotein, normally undetectable in Western blot assays, was detected in A774wt-6/6-infected cells, and to a lesser extent in SFV6-6/6-infected cells (Fig. 3B).
An extra copy of nsP3 has little effect on ns polyprotein processing but increases stability of nsP4.
Changes in ns polyprotein processing can affect replicase complex formation and the phenotype of the virus. Therefore, processing of P12334 polyproteins, encoded by SFV6-6/74 and SFV6-6/6, was analyzed in a pulse-chase experiment. Processing of P1234 of parental SFV6 followed the pattern previously described for SFV4 (38, 39). In pulsed samples, the P1234, P123, P12, and P34 polyproteins were detected. After a 45-min chase, their amounts were reduced, and the amounts of free nsP1, nsP2, and nsP3 were correspondingly increased. In contrast, due to its degradation in the ubiquitin/proteasome pathway (26), the amounts of free nsP4 decreased during the chase (Fig. 3C).
Large polyproteins, presumably P12334 and/or P1233, were detectable in pulse samples from SFV6-6/74- and SFV6-6/6-infected cells (Fig. 3C). Similar to SFV6, P123, P12, and P34 were the major processing intermediates, indicating that the presence of an extra copy of nsP3 did not cause major defects in patterns of ns polyprotein processing. Expression of two nsP3 proteins with slightly different mobilities was clearly detected for SFV6-6/74 (Fig. 3C); combined, their ratio to nsP2 was approximately 2.5-fold higher than the ratio of nsP3 to nsP2 in SFV6-infected cells. This slightly exceeded the predicted 2-fold increase and could indicate increased stability of nsP3. In the case of SFV6-6/6-infected cells, a band corresponding to the P33 polyprotein was detected (Fig. 3C). This polyprotein was not detected for SFV6-6/74, probably due to its more efficient processing. As significant amounts of nsP3 of SFV6-6/6 were present in the form of P33, the ratio of individual nsP3 to nsP2 was not increased. Interestingly, it was noted that in sharp contrast to SFV6-infected cells, the amounts of free nsP4 in SFV6-6/74- and SFV6-6/6-infected cells not only did not decrease but actually increased during chase by approximately 2.7-fold (Fig. 3C). This clearly indicated that nsP4, expressed by these viruses, was somehow protected from degradation. In contrast to the immunoblotting analysis (Fig. 3B), pulse-chase analysis failed to reveal any increase in the amounts of P34 polyprotein in SFV6-6/6-infected cells (Fig. 3C). This could indicate that at early stages of infection (the pulse-chase experiment was performed at 3.5 h p.i.), the processing of P34 was not affected. In contrast, later in infection (immunoblotting was performed at 8 h p.i.), processing of P34 of SFV6-6/6 slowed down; a similar phenomenon has been reported for the processing of P34 of Sindbis virus (SINV) (46).
Neurovirulence is the dominant function of nsP3 of SFV6.
To find out which phenotype caused by nsP3—virulent (SFV6) or avirulent (A774wt)—is dominant, the neurovirulence of A774wt was first compared to that of A774wt-6/74 and A774wt-6/6. As the virus termed rA774-V11I, which is actually identical to A774wt, is slightly neurovirulent (32), large groups of mice were needed to acquire data sufficient for statistical analysis. Four- to 5-week-old BALB/c mice (n = 37) were inoculated i.p. with 5,000 PFU of each of these viruses. Because at this dose SFV6 is universally lethal for mice (34), both the SFV6 and PBSA control groups were smaller (n = 7). Animals were monitored for up to 2 weeks for signs of encephalitis and mortality (Fig. 4A). All mice inoculated with SFV6 reached a clinical endpoint and were euthanized by PID 4; in contrast, only three of the A774wt-infected mice developed disease during the monitoring period. Thus, A774wt (which killed 3 mice out of 37) and rA774-V11I (which killed 1 mouse out of 10) (32) indeed possess similar (low, but detectable) neurovirulence. Both A774wt-6/74, which killed 15 mice, and A774wt-6/6, which killed 19 mice (Fig. 4A), were significantly (P = 0.0003) more virulent than parental A774wt but less virulent than SFV6 (P < 0.0001). The difference between the virulence of A774wt-6/74 and A774wt-6/6 was not statistically significant (P = 0.287). These data clearly showed that the insertion of nsP3 of SFV6 into the A774wt genome increased the virulence of the recombinant viruses. Thus, the neurovirulent phenotype caused by the presence of nsP3 of SFV6 was found to be dominant.
The extremely clear virulent phenotype of SFV6 (Fig. 4A) permitted the use of smaller groups of mice (n = 7) in subsequent experiments that were aimed at detecting the phenotypes of recombinant viruses. In the second half of this experiment, mice were inoculated i.p. with 5,000 PFU of SFV6, SFV6-6/74, or SFV6-6/6 and monitored for up to 2 weeks for signs of encephalitis and mortality. Again, all mice inoculated with SFV6 reached a clinical endpoint and were euthanized by PID 5. SFV6-6/74 and SFV6-6/6 killed five and four out of seven mice, respectively (Fig. 4B). Thus, insertion of a copy of nsP3 of A774wt or an extra copy of nsP3 of SFV6 into the full genome of SFV6 did reduce but did not eliminate the virulence of SFV6.
For the next part of the experiment, groups of mice (n = 5) were inoculated i.p. with A774wt, SFV6, A774wt-6/74, A774wt-6/6, SFV6-6/74, and SFV6-6/6, and virus titers in the blood were determined on PID 1 or PID 3; on PID 3, virus titers in brain tissue were also determined. Both A774wt and SFV6 produced high-level viremia on PID 1 (Fig. 4C). On PID 3, the blood titers for A774wt were higher than those for SFV6 (Fig. 4D), and both viruses were detected in the brain (Fig. 4E). Brain tissue titers of SFV6 were, however, significantly higher (P < 0.01) than those of A774wt, indicating that only the virulent strain, SFV6, replicated efficiently and spread rapidly throughout the brain (Fig. 4E).
It was evident from the data obtained from the first part of the experiment that mice infected with viruses encoding two copies of nsP3 succumbed to infection later than mice infected with SFV6 (Fig. 4A and B). This was probably caused by their slower replication due to the larger genome size. Indeed, on PID 1, blood titers of these viruses were significantly (P < 0.0001) lower than the titers of A774wt and SFV6 (Fig. 4C). By PID 3, this difference was less prominent (Fig. 4D). Consistent with reduced viremia, the brain titers of A774wt-6/74, A774wt-6/6, SFV6-6/74, and SFV6-6/6 on PID 3 were all significantly lower (P < 0.0001) than those of the less-virulent A774wt (Fig. 4E). Thus, at that time point, the replication of A774wt-6/74, A774wt-6/6, SFV6-6/74, and SFV6-6/6 in the brain was very limited; this was especially evident from the prominent (100,000-fold or larger) difference between their average titers and those with SFV6 (P < 0.0001) (Fig. 4E). However, by the time animals reached the terminal endpoint of the experiment and were euthanized (PID 7 to 10), the average titers of A774wt-6/74 in the brain reached 8.25 × 105 PFU/g. At the same endpoint, the titers of A774wt-6/6, SFV6-6/74, and SFV6-6/6 were even higher (between 107 and 109 PFU/g), which was similar to the titers of SFV6 in the brains of infected mice at the clinical endpoint. Note that the few mice that developed encephalitis upon A774wt infection had comparable brain titers as well. These data clearly indicate that there is a correlation between encephalitis development and efficient SFV replication in the brain.
Finally, the in vivo genetic stability of viruses harboring an extra copy of nsP3 was analyzed. For this, total RNA from brains of animals that reached the clinical endpoint of the experiment (PID 7 to 10) was extracted and analyzed using RT-PCR. Using primers which amplify only the nsP3/nsP3 junction region, clear and abundant RT-PCR products were obtained for samples from animals infected wtih A774wt-6/74, A774wt-6/6, or SFV6-6/74. Weaker and less clear product was also obtained for SFV6-6/6 infected brain but, as expected, not for SFV6- or A774wt-infected brains (Fig. 4F). RT-PCR, performed with primers also used for analysis of in vitro stability of recombinant viruses, confirmed this finding. The abundance of RT-PCR product, corresponding to the genomes with two nsP3 copies, varied greatly, being highest for A774wt-6/6 and lowest for SFV6-6/74 (Fig. 4F). Whether or not it reflects different genetic stabilities of these viruses is unclear as, in contrast to RNA purified from virus stocks, the RNA purified from brain homogenates contains not only packed genomes but also all replicating and nonreplicating RNAs made during virus infection. Furthermore, due to the limited amount of collected material, only one animal per group was analyzed using this assay; therefore, we cannot exclude variation of genetic heterogeneity (or homogeneity) of viral genomes in brains of individual infected animals. RT-PCR products, similar to these from genomes harboring single nsP3, were observed for in vivo samples of A774wt-6/74, A774wt-6/6, SFV6-6/74, and SFV6-6/6; however, their amount was smaller than in samples obtained from A774wt- or SFV6-infected mice (Fig. 4F). Thus, it can be concluded that the genetic stability of viruses with two copies of nsP3 is sufficient to be maintained in brains as late as PID 7 to PID 10; furthermore, at least for A774wt-6/6, the genome that harbored two nsP3 copies was still dominant at the time of infection. Most importantly, these findings demonstrated that the viruses with two nsP3 copies were able not only to reach the brain but also to replicate and accumulate in brain tissues.
Swapping of nsP3 regions of SFV6 and A774wt results in virulent viruses.
Replacement of the nsP3 region of rA774 with its counterpart from SFV4 results in virulent virus (33). However, the phenotype of virus with nsP3 of SFV6 in the A774wt backbone is not known. To address this question, the recombinants A774wt-6 and SFV6-74 were constructed (Fig. 5A). Swapping of nsP3 regions did not affect the efficiency of rescue of recombinant viruses from the obtained constructs, and both recombinant viruses grew to titers similar to those of the parental viruses (Fig. 5A).
Consistent with the results of an earlier study (33), A774wt-6 had a virulent phenotype and killed 6 out of 7 infected mice by PID 6 (Fig. 5B). Similar to A774wt, A774wt-6 produced a high-titer viremia on PID 1 which was reduced by PID 3 (Fig. 5C and D). Compared to A774wt, A774wt-6 produced significantly (P = 0.008) higher titers in the brain on PID 3 (Fig. 5E). These data are consistent with the role of nsP3 as a virulence factor for SFV infection. Surprisingly, SFV6-74 was as virulent as parental SFV6 and killed all infected mice with essentially the same kinetics (Fig. 5B). This virus produced a very high-titer viremia on PID 1 (Fig. 5C) and reached high brain titers (average, 1.23 × 108 PFU/g) by the time mice reached the clinical endpoint (PID 5 to 6). Interestingly, brain titers of A774wt-6 and SFV6-74 on PID 3 were lower than these of SFV6 (Fig. 5E). The difference was smaller (average, 8.83 × 108 PFU/g for SFV6 compared to 2.37 × 108 PFU/μg for recombinants) but significant (P = 0.02) at the time when animals reached the clinical endpoint. In conclusion, the results of this experiment demonstrated clearly that other virulence determinants are present outside the nsP3 of SFV6. Furthermore, these determinants individually (regardless of the type of nsP3) are sufficient to produce a virus with a virulent phenotype.
Functionally coupled differences in the P4 position of the 1/2 site and the S4 subsite of nsP2 represent major determinants of SFV virulence.
As sequences of the nsP4 proteins of A7(74) and L10 are nearly identical (Table 1), the differences in virulence of A774wt and SFV6-74 are most likely the result of differences in the nsP1 (6 nonsynonymous changes) and/or nsP2 (4 nonsynonymous changes) region(s). A parallel study, performed in our laboratory, revealed that the His-to-Arg substitution in the P4 position of the 1/2 site of SFV4 was poorly tolerated, and substitutions at this region sometimes resulted in compensatory changes at amino acid residue 515 of nsP2 (V. Lulla, K. Rausalu, S. Saul, A. Merits, and A. Lulla, unpublished data). Functional linkage between position P4 of the 1/2 site (residue 534 of ns polyprotein) and amino acid residue 515 of nsP2 (residue 1052 of the ns polyprotein), which is part of the protease S4 subsite, was also predicted by Russo et al. (47). Therefore, it has been proposed that amino acid residues 534 and 1052, which are His and Val in L10 and SFV6 but Arg and Glu in A7(74) and A774wt (Table 1), may represent additional determinants for SFV virulence.
To verify this hypothesis, viruses designated A774wt-HV (Arg534His plus Glu1052Val) and SFV6-74-RE (His534Arg plus Val1052Glu) were constructed. Rescue of viruses from corresponding recombinant plasmids occurred with high efficiency, and rescued viruses grew to high titers (Fig. 6A). In an in vivo experiment, A774wt killed one mouse on PID 5, while all mice infected with SFV6 were dead by PID 4 (Fig. 6B). A774wt-HV was highly virulent, killing 6 mice out of 7 by PID 6 (Fig. 6B). In contrast, SFV6-74-RE was clearly less pathogenic than SFV6-74 and killed only 2 mice out of 7 (compare Fig. 6B and 5B). Both A774wt-HV and SFV6-74-RE caused high-titer viremia on PID 1 (Fig. 6C), which was reduced by approximately 3 logs on PID 3 (Fig. 6D). The brain tissue titers on PID 1 (Fig. 6E) correlated with the viremia (Fig. 6C) and therefore were not the result of virus replication in the brain. A774wt-HV was clearly able to initiate replication in the brain; due to its average titer in the brain tissue on PID 3 that was 5 times higher than on PID 1 (P = 0.008) (compare Fig. 6E and F), and by the time mice reached the clinical endpoint the titers had reached 1.05 × 107 PFU/g on average. In contrast, the brain tissue titers of SFV6-74-RE on PID 3 had, compared to PID 1, dropped by approximately 1 log, and the observed difference was statistically significant (P = 0.02) (compare Fig. 6E and F). These data confirmed that amino acid residues 534 and 1052 of the ns polyprotein affect SFV virulence.
Naturally occurring differences in the P4 position of the 1/2 site and the S4 subsite of nsP2 affect the processing of the ns polyprotein.
Pulse-chase experiments were used to determine the effect(s) of substitutions that altered the virulence of SFV with regard to ns polyprotein processing. The ratio of label incorporated into the P123 polyprotein to that present in fully cleaved nsP2 (for simplicity, designated the P123:nsP2 ratio) was determined for A774wt, A774wt-HV, SFV6-74, and SFV6-74-RE. In the case of A774wt, cleavage of the 1/2 site was fast: the average ratio of P123 to nsP2 was 4.6:1 in pulsed samples (Fig. 7A and B); during the 45-min chase, P123 was almost completely processed and its amounts became too low for reliable quantification (Fig. 7A). SFV6-74-RE displayed a very similar pattern, with a P123-to-nsP2 ratio of 2.1:1 in pulsed samples. Interestingly, for these viruses, P34 polyprotein was clearly also visible in chased samples (Fig. 7A), indicating that the Glu515 residue of nsP2 reduced its ability to process the P34 precursor. In contrast, virulent A774wt-HV and SFV6-74 had considerably higher P123-to-nsP2 ratios in pulse samples: 9.8:1 for the former and 11.6:1 for the latter. Consistent with this, P123 was easily detected in chased samples for these viruses (Fig. 7A and B). In contrast, P34 could not be detected in chased samples for these viruses, indicating that nsP2 containing the Val515 residue efficiently cleaved this substrate.
FIG 7.

Changes at the P4 position of the 1/2 site and at the S4 subsite residue of nsP2 alter the speed of ns polyprotein processing in infected cells. (A) BHK-21 cells were infected at an MOI of 10 with A774wt, A774wt-HV, SFV6-74, or SFV6-74-RE. At 3 h p.i., cells were starved in methionine-cysteine-free medium and then pulsed with 50 μCi of [35S]methionine-cysteine mixture for 15 min. Cells were collected immediately after pulse or after 45-min chase. Synthesized ns polyproteins, their processing intermediates, and mature nsPs were immunoprecipitated using antibodies against the indicated proteins. The positions of P123, P34, and nsP2 are indicated with arrows at the left side of the panel. Data from one of three reproducible experiments are shown. (B) Quantification of data obtained in three independent pulse-chase experiments. The ratio of P123 to nsP2 was determined as the ratio of label present in the corresponding products. Columns represent the averages of three experiments; error bars represent standard errors of the means.
Both the 1/2 and 3/4 cleavages are related to replicase complex formation (38, 48); however, their significance is different. Even a major reduction in 3/4 site cleavage efficiency has little or no effect on the infectivity of the SFV genome (38), while acceleration of 1/2 site cleavage efficiency reduces the infectivity of mutant SFV genomes and triggers selection of compensatory mutations (Lulla et al., unpublished). These data, together with the findings of others (49), strongly indicate that the in vivo phenotype of SFV correlates with differences in 1/2 site cleavage.
DISCUSSION
A previous study demonstrated that the different virulence phenotypes of SFV4 and rA774 are caused by differences in their ns regions. It was also established that nsP3 is important for SFV virulence (33). However, the Val1347-to-Ile substitution, reported here as the reason behind low infectivity of the rA774 clone, was previously erroneously considered to represent a natural sequence difference between SFV4 and A7(74); the correct A7(74) molecular clone (rA774-V11I) has actually been constructed and to some degree even analyzed (32). Comparison of the results obtained using the flawed constructs (rA774 and clones based on it) and correct constructs (rA774-V11I and other constructs that contain the Ile1347 codon) is extremely difficult and confusing. To note, pSP6-SFV4, another clone used in previous studies (32, 33), exhibits a similar problem (34).
In this study, the consensus sequence of A7(74) was determined using high-throughput sequencing, and determinants of virulence residing in the ns region of SFV were studied using new consensus clones of avirulent (A774wt) and virulent (SFV6) SFV constructs. It was observed that A774wt was, in contrast to original avirulent A7(74) (4, 29), slightly virulent and killed (based on combined data from three experiments) 5 out of 51 mice. This is consistent with previous data for rA774-V11I (32) and with data from in vivo experiments that demonstrated that SFV6 is slightly more virulent than the parental L10 strain (34). Taken together, this indicates that slightly enhanced virulence may be a common property of consensus clones of SFV (and possibly other alphaviruses). SFV6 and A774wt correspond to the most common (and possibly the fittest) sequences of the L10 and A7(74) strains. Unlike native isolates, which consist of quasispecies with different properties (34), P0 viruses rescued from respective molecular clones have little sequence variation and, if they represent consensus sequences, may have higher fitness and virulence.
Viruses containing duplicated sequences provide useful research tools. Duplication of genes of RNA viruses, however, represents a significant challenge. nsP3 duplication increases the size of the SFV genome by approximately 12.5%, slowing down its replication and reducing its titer. It also creates an unnatural processing site between two nsP3 regions in the P12334 polyprotein. Such manipulation is tolerated (41), but the biological properties of the virus may be affected. Combined, these factors demanded the construction and the use of multiple controls. Finally, as nsP3 coding sequences of A774wt and SFV6 have an identity of 96%, they can recombine during plasmid propagation and/or during virus replication (copy choice recombination). The failure to obtain stable stocks of viruses with two native copies of nsP3-encoding regions by plaque purification indicated that the extra copy of nsP3 was lost due to copy choice recombination. Introduction of numerous synonymous mutations, which reduced the similarity of the two nsP3-encoding sequences to a nonsignificant level, greatly reduced (though did not completely eliminate) the instability of the recombinant genomes (Fig. 3A). The obtained viruses expressed an extra copy of nsP3, but the processing of ns polyprotein did not exhibit major defects (Fig. 3C). The differences (aside from the 2.5-times-larger amount of nsP3 produced by SFV6-6/74) in cell culture experiments were slower growth (Fig. 1C and D), reduced virus titers (Fig. 1B), and the unexpected stabilization of nsP4 (Fig. 3C).
Due to their less efficient replication, viruses harboring two copies of nsP3 produced average blood titers of approximately 104 PFU/ml on PID 1 (Fig. 4C). However, on PID 3 double-nsP3 viruses were detected in the brains of 7 out of 20 mice, indicating that low viremia did not prevent entry into the brain (Fig. 4E). Once in the brain, all viruses replicated to high titers. In addition, RT-PCR analysis revealed that viruses that replicated in the brain had maintained the extra copy of nsP3, though different recombinants were also detected (Fig. 4F). The virulent phenotype of SFV6-6/74 indicated that the nsP3 of A774wt was unable to reduce efficiently the virulence of SFV6 (a small reduction could be attributed to slower replication and lower titers due the general increase in the size of the genome). Consistent with this, both A774wt-6/74 and A774wt-6/6 killed approximately 50% of infected mice (Fig. 4A). Thus, the presence of nsP3 from SFV6 converted A774wt to a virulent virus regardless of the presence (A774wt-6/74) or absence (A774wt-6/6) of nsP3 from the avirulent strain (Fig. 4A). Consistent with previously published results (33), it was also found that A774wt-6 was virulent (Fig. 5B). Thus, neurovirulence caused by nsP3 of SFV6 clearly represents a dominant phenotype (Fig. 5). Interestingly, however, SFV6-74 resulting from a reciprocal swap also killed all the infected mice. These data confirmed the existence of another virulence determinant(s) in the ns region of SFV6 and revealed that the nsP3 of A774wt could not eliminate virulence properties caused by these determinants.
Mutation in nsP1, located close to the 1/2 site of the ns polyprotein, in a virulent strain of SINV results in the acceleration of P123 processing and in an avirulent phenotype (49). Here, we describe that residues at the P4 position of the 1/2 site and at position 515 of nsP2 are crucial for SFV virulence. Indeed, if these positions were occupied by His and Val residues, then the virus had a virulent phenotype regardless of the origin of nsP3. The presence of Glu515 in nsP2 (as in A774wt and SFV6-74-RE) also slowed down the processing of the 3/4 site (Fig. 7A), indicating that this amino acid residue reduced the basal protease activity of nsP2. Most likely this reduction, caused by the Val515-to-Glu substitution, compensated for the presence of the Arg residue (the most favorable residue for fast processing) in the P4 position of the 1/2 site. Indeed, without reduction of nsP2 activity, the 1/2 site with the P4 Arg residue is cleaved too quickly, severely compromising the infectivity of corresponding virus (Lulla et al., unpublished).
It is less obvious how the accelerated processing of the 1/2 site and/or reduced protease activity of nsP2 affects the in vivo phenotype of the virus. One possibility is that faster processing of the 1/2 site provides less time for the proper formation of replication complexes and may reduce the amount of functional replicase complexes in infected cells. As avirulent viruses produced high titers both in cell culture and in the blood of infected mice (Fig. 4C, 5C, and 6C), such a defect must be cell type/tissue specific and reduce or block virus replication in mature neurons. Indeed, SINV mutants with reduced virulence have defects in neuronal replication (50). Consistent with this, A774wt and SFV6 replicated similarly in undifferentiated cells of a rat neuronal cell line, but only SFV6 was capable of efficient replication in differentiated cells (our unpublished data). Another possibility is that the Val515-to-Glu mutation, which reduced the protease activity of nsP2 (Fig. 7), may hamper the cytotoxic properties of the protein. This, however, is unlikely because protease activity is not required for the cytotoxic effects of nsP2 (51, 52). Finally, it is possible that the reduced protease activity of nsP2 and/or altered kinetics of P123 processing affect other virus-cell interactions, such as the induction of interferon production and/or interference with interferon signaling. Indeed, for SINV, accelerated processing of the 1/2 site diminished the ability of the virus to reduce STAT1/STAT2 phosphorylation (53). Separate studies are needed to reveal the mechanisms of how exactly altered processing of ns polyprotein and/or the protease activity of nsP2 affects the in vitro and in vivo properties of SFV.
The mechanism(s) underlying how nsP3 affects the virulence of SFV remains even more obscure. The most obvious difference between L10 and A7(74) is the opal termination codon close to the 3′ end of the nsP3 coding sequence of the latter (Table 1). However, the effect of the termination codon alone on SFV virulence is rather limited (33). For other alphaviruses, such as O'nyong-nyong virus, the opal termination codon even enhances virus infectivity (54). For Chikungunya virus, most of the field isolates contain a termination codon, while laboratory-adapted strains based on the same isolates often do not. Thus, it is likely that both variants of the genome, with the terminator and without it, may coexist in natural isolates of many alphaviruses. As the effects of the termination codon are limited, the avirulence/virulence determinant in nsP3 of SFV likely has a more complicated nature. nsP3 is important for the binding of RNA (22) and ADP-ribose and poly(ADP-ribose) (50, 55–57) and interacts with multiple host components (23, 44, 58). Alteration(s) in these activities may result in an altered phenotype for SFV. Separate studies are clearly needed to reveal the mechanism(s) by which nsP3 is involved in the neurovirulence of SFV. Similarly, it is a topic of considerable interest to determine if and how nsP3 is involved in the virulence of other alphaviruses.
The analysis of ns polyprotein processing revealed that duplication of the nsP3 region resulted in profound stabilization of nsP4 (Fig. 3C). This effect was not simply caused by the addition of an extra processing unit into the ns polyprotein of SFV, as the nsP4 of viruses carrying enhanced green fluorescent protein (or any other marker) between the nsP3 and nsP4 regions (41) is not stabilized. Rapid degradation of individual nsP4 and its stability in formed replicase complexes has been known for decades. However, the mechanisms responsible for the latter have not been revealed. Our data strongly suggest that nsP3 is actively involved in this phenomenon, and if so, its role in nsP4 stabilization represents an important and previously unknown function of nsP3. Taking into account the role of nsP3 in the formation and relocalization of alphavirus replicase organelles, it can be assumed that stabilization may occur via the inclusion of excessive amounts of nsP4 into replicase complexes, by the formation of excessive amounts of replicase complexes and/or some other complexes of nsP3 and nsP4. Detailed analyses of the composition of SFV replicase organelles (8) and/or other complexes made by ns proteins of SFV are needed to distinguish between these possibilities. Similarly, if and how possible changes in virus replicase complexes affect SFV replication and virulence represent important topics for further studies.
In conclusion, the consensus clones SFV A7(74) and L10 were found to be useful tools for the analysis of determinants of SFV virulence. A large amount of data obtained using previously developed but imperfect tools was confirmed. The virulence of SFV was found to be dependent on fine-tuned functions of ns proteins, such as the basic protease activity of nsP2 and efficiency of P123 processing. As the properties of nsP3 and processing of P123 are also important for the neurovirulence of SINV, they may represent common determinants for the pathogenicity of Old World alphaviruses in general.
ACKNOWLEDGMENTS
This work was supported by institutional research funding (IUT20-27) of the Estonian Ministry of Education and Research, the Estonian Science Foundation (grant 9400), the European Union through the European Regional Development Fund (grant agreement 10.1-9/1008) and via the Center of Excellence in Chemical Biology, and by Biotechnology and Biological Sciences Research Council (BBSRC) Institute Strategic Project grants to The Roslin Institute and The Pirbright Institute (BBB/E/I/00001735). Sequencing was carried out by Edinburgh Genomics, The University of Edinburgh. Edinburgh Genomics is partly supported through core grants from NERC (R8/H10/56), MRC (MR/K001744/1), and BBSRC (BB/J004243/1).
REFERENCES
- 1.Allsopp TE, Fazakerley JK. 2000. Altruistic cell suicide and the specialized case of the virus-infected nervous system. Trends Neurosci 23:284–290. doi: 10.1016/S0166-2236(00)01591-5. [DOI] [PubMed] [Google Scholar]
- 2.Fazakerley JK, Cotterill CL, Lee G, Graham A. 2006. Virus tropism, distribution, persistence and pathology in the corpus callosum of the Semliki Forest virus-infected mouse brain: a novel system to study virus-oligodendrocyte interactions. Neuropathol Appl Neurobiol 32:397–409. doi: 10.1111/j.1365-2990.2006.00739.x. [DOI] [PubMed] [Google Scholar]
- 3.Gates MC, Sheahan BJ, O'Sullivan MA, Atkins GJ. 1985. The pathogenicity of the A7, M9 and L10 strains of Semliki Forest virus for weanling mice and primary mouse brain cell cultures. J Gen Virol 66:2365–2373. doi: 10.1099/0022-1317-66-11-2365. [DOI] [PubMed] [Google Scholar]
- 4.Fazakerley JK, Pathak S, Scallan M, Amor S, Dyson H. 1993. Replication of the A7(74) strain of Semliki Forest virus is restricted in neurons. Virology 195:627–637. doi: 10.1006/viro.1993.1414. [DOI] [PubMed] [Google Scholar]
- 5.Fragkoudis R, Tamberg N, Siu R, Kiiver K, Kohl A, Merits A, Fazakerley JK. 2009. Neurons and oligodendrocytes in the mouse brain differ in their ability to replicate Semliki Forest virus. J Neurovirol 15:57–70. doi: 10.1080/13550280802482583. [DOI] [PubMed] [Google Scholar]
- 6.Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev 58:491–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fazakerley JK. 2002. Pathogenesis of Semliki Forest virus encephalitis. J Neurovirol 8(Suppl 2):S66–S74. [DOI] [PubMed] [Google Scholar]
- 8.Varjak M, Saul S, Arike L, Lulla A, Peil L, Merits A. 2013. Magnetic fractionation and proteomic dissection of cellular organelles occupied by the late replication complexes of Semliki Forest virus. J Virol 87:10295–10312. doi: 10.1128/JVI.01105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shirako Y, Strauss JH. 1994. Regulation of Sindbis virus RNA replication: uncleaved P123 and nsP4 function in minus-strand RNA synthesis, whereas cleaved products from P123 are required for efficient plus-strand RNA synthesis. J Virol 68:1874–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salonen A, Vasiljeva L, Merits A, Magden J, Jokitalo E, Kaariainen L. 2003. Properly folded nonstructural polyprotein directs the Semliki Forest virus replication complex to the endosomal compartment. J Virol 77:1691–1702. doi: 10.1128/JVI.77.3.1691-1702.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahola T, Lampio A, Auvinen P, Kaariainen L. 1999. Semliki Forest virus mRNA capping enzyme requires association with anionic membrane phospholipids for activity. EMBO J 18:3164–3172. doi: 10.1093/emboj/18.11.3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lulla V, Sawicki DL, Sawicki SG, Lulla A, Merits A, Ahola T. 2008. Molecular defects caused by temperature-sensitive mutations in Semliki Forest virus nsP1. J Virol 82:9236–9244. doi: 10.1128/JVI.00711-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cruz CC, Suthar MS, Montgomery SA, Shabman R, Simmons J, Johnston RE, Morrison TE, Heise MT. 2010. Modulation of type I IFN induction by a virulence determinant within the alphavirus nsP1 protein. Virology 399:1–10. doi: 10.1016/j.virol.2009.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deuber SA, Pavlovic J. 2007. Virulence of a mouse-adapted Semliki Forest virus strain is associated with reduced susceptibility to interferon. J Gen Virol 88:1952–1959. doi: 10.1099/vir.0.82264-0. [DOI] [PubMed] [Google Scholar]
- 15.Rikkonen M, Peranen J, Kaariainen L. 1994. ATPase and GTPase activities associated with Semliki Forest virus nonstructural protein nsP2. J Virol 68:5804–5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vasiljeva L, Merits A, Auvinen P, Kaariainen L. 2000. Identification of a novel function of the Alphavirus capping apparatus RNA 5′-triphosphatase activity of Nsp2. J Biol Chem 275:17281–17287. doi: 10.1074/jbc.M910340199. [DOI] [PubMed] [Google Scholar]
- 17.Das PK, Merits A, Lulla A. 2014. Functional cross-talk between distant domains of Chikungunya virus non-structural protein 2 is decisive for its RNA-modulating activity. J Biol Chem 289:5635–5653. doi: 10.1074/jbc.M113.503433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lulla A, Lulla V, Merits A. 2012. Macromolecular assembly-driven processing of the 2/3 cleavage site in the alphavirus replicase polyprotein. J Virol 86:553–565. doi: 10.1128/JVI.05195-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fazakerley JK, Boyd A, Mikkola ML, Kaariainen L. 2002. A single amino acid change in the nuclear localization sequence of the nsP2 protein affects the neurovirulence of Semliki Forest virus. J Virol 76:392–396. doi: 10.1128/JVI.76.1.392-396.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merits A, Vasiljeva L, Ahola T, Kaariainen L, Auvinen P. 2001. Proteolytic processing of Semliki Forest virus-specific non-structural polyprotein by nsP2 protease. J Gen Virol 82:765–773. doi: 10.1099/0022-1317-82-4-765. [DOI] [PubMed] [Google Scholar]
- 21.Tamm K, Merits A, Sarand I. 2008. Mutations in the nuclear localization signal of nsP2 influencing RNA synthesis, protein expression and cytotoxicity of Semliki Forest virus. J Gen Virol 89:676–686. doi: 10.1099/vir.0.83320-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shin G, Yost SA, Miller MT, Elrod EJ, Grakoui A, Marcotrigiano J. 2012. Structural and functional insights into alphavirus polyprotein processing and pathogenesis. Proc Natl Acad Sci U S A 109:16534–16539. doi: 10.1073/pnas.1210418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neuvonen M, Kazlauskas A, Martikainen M, Hinkkanen A, Ahola T, Saksela K. 2011. SH3 domain-mediated recruitment of host cell amphiphysins by alphavirus nsP3 promotes viral RNA replication. PLoS Pathog 7:e1002383. doi: 10.1371/journal.ppat.1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vihinen H, Ahola T, Tuittila M, Merits A, Kaariainen L. 2001. Elimination of phosphorylation sites of Semliki Forest virus replicase protein nsP3. J Biol Chem 276:5745–5752. doi: 10.1074/jbc.M006077200. [DOI] [PubMed] [Google Scholar]
- 25.Rubach JK, Wasik BR, Rupp JC, Kuhn RJ, Hardy RW, Smith JL. 2009. Characterization of purified Sindbis virus nsP4 RNA-dependent RNA polymerase activity in vitro. Virology 384:201–208. doi: 10.1016/j.virol.2008.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Groot RJ, Rumenapf T, Kuhn RJ, Strauss EG, Strauss JH. 1991. Sindbis virus RNA polymerase is degraded by the N-end rule pathway. Proc Natl Acad Sci U S A 88:8967–8971. doi: 10.1073/pnas.88.20.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McIntosh BM, Worth CB, Kokernot RH. 1961. Isolation of Semliki Forest virus from Aedes (Aedimorphus) argenteopunctatus (Theobald) collected in Portuguese East Africa. Trans R Soc Trop Med Hyg 55:192–198. doi: 10.1016/0035-9203(61)90025-6. [DOI] [PubMed] [Google Scholar]
- 28.Amor S, Scallan MF, Morris MM, Dyson H, Fazakerley JK. 1996. Role of immune responses in protection and pathogenesis during Semliki Forest virus encephalitis. J Gen Virol 77:281–291. doi: 10.1099/0022-1317-77-2-281. [DOI] [PubMed] [Google Scholar]
- 29.Atkins GJ. 1983. The avirulent A7 strain of Semliki Forest virus has reduced cytopathogenicity for neuroblastoma cells compared to the virulent L10 strain. J Gen Virol 64:1401–1404. doi: 10.1099/0022-1317-64-6-1401. [DOI] [PubMed] [Google Scholar]
- 30.Bradish CJ, Allner K, Maber HB. 1971. The virulence of original and derived strains of Semliki Forest virus for mice, guinea-pigs and rabbits. J Gen Virol 12:141–160. doi: 10.1099/0022-1317-12-2-141. [DOI] [PubMed] [Google Scholar]
- 31.Atkins GJ, Sheahan BJ, Liljestrom P. 1999. The molecular pathogenesis of Semliki Forest virus: a model virus made useful? J Gen Virol 80:2287–2297. doi: 10.1099/0022-1317-80-9-2287. [DOI] [PubMed] [Google Scholar]
- 32.Tuittila M, Hinkkanen AE. 2003. Amino acid mutations in the replicase protein nsP3 of Semliki Forest virus cumulatively affect neurovirulence. J Gen Virol 84:1525–1533. doi: 10.1099/vir.0.18936-0. [DOI] [PubMed] [Google Scholar]
- 33.Tuittila MT, Santagati MG, Roytta M, Maatta JA, Hinkkanen AE. 2000. Replicase complex genes of Semliki Forest virus confer lethal neurovirulence. J Virol 74:4579–4589. doi: 10.1128/JVI.74.10.4579-4589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferguson MC, Saul S, Fragkoudis R, Weisheit S, Cox J, Patabendige A, Sherwood K, Watson M, Merits A, Fazakerley JK. 2015. Ability of the encephalitic arbovirus Semliki Forest virus to cross the blood-brain barrier is determined by the charge of the E2 glycoprotein. J Virol 89:7536–7549. doi: 10.1128/JVI.03645-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ulper L, Sarand I, Rausalu K, Merits A. 2008. Construction, properties, and potential application of infectious plasmids containing Semliki Forest virus full-length cDNA with an inserted intron. J Virol Methods 148:265–270. doi: 10.1016/j.jviromet.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gorchakov R, Frolova E, Williams BR, Rice CM, Frolov I. 2004. PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J Virol 78:8455–8467. doi: 10.1128/JVI.78.16.8455-8467.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lulla A, Lulla V, Tints K, Ahola T, Merits A. 2006. Molecular determinants of substrate specificity for Semliki Forest virus nonstructural protease. J Virol 80:5413–5422. doi: 10.1128/JVI.00229-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lulla V, Karo-Astover L, Rausalu K, Merits A, Lulla A. 2013. Presentation overrides specificity: probing the plasticity of alphaviral proteolytic activity through mutational analysis. J Virol 87:10207–10220. doi: 10.1128/JVI.01485-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zusinaite E, Tints K, Kiiver K, Spuul P, Karo-Astover L, Merits A, Sarand I. 2007. Mutations at the palmitoylation site of non-structural protein nsP1 of Semliki Forest virus attenuate virus replication and cause accumulation of compensatory mutations. J Gen Virol 88:1977–1985. doi: 10.1099/vir.0.82865-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tamberg N, Lulla V, Fragkoudis R, Lulla A, Fazakerley JK, Merits A. 2007. Insertion of EGFP into the replicase gene of Semliki Forest virus results in a novel, genetically stable marker virus. J Gen Virol 88:1225–1230. doi: 10.1099/vir.0.82436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peranen J, Laakkonen P, Hyvonen M, Kaariainen L. 1995. The alphavirus replicase protein nsP1 is membrane-associated and has affinity to endocytic organelles. Virology 208:610–620. doi: 10.1006/viro.1995.1192. [DOI] [PubMed] [Google Scholar]
- 43.Peranen J, Rikkonen M, Liljestrom P, Kaariainen L. 1990. Nuclear localization of Semliki Forest virus-specific nonstructural protein nsP2. J Virol 64:1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Panas MD, Varjak M, Lulla A, Eng KE, Merits A, Karlsson Hedestam GB, McInerney GM. 2012. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol Biol Cell 23:4701–4712. doi: 10.1091/mbc.E12-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gorchakov R, Garmashova N, Frolova E, Frolov I. 2008. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J Virol 82:10088–10101. doi: 10.1128/JVI.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Groot RJ, Hardy WR, Shirako Y, Strauss JH. 1990. Cleavage-site preferences of Sindbis virus polyproteins containing the non-structural proteinase. Evidence for temporal regulation of polyprotein processing in vivo. EMBO J 9:2631–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Russo AT, Malmstrom RD, White MA, Watowich SJ. 2010. Structural basis for substrate specificity of alphavirus nsP2 proteases. J Mol Graph Model 29:46–53. doi: 10.1016/j.jmgm.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lemm JA, Rice CM. 1993. Roles of nonstructural polyproteins and cleavage products in regulating Sindbis virus RNA replication and transcription. J Virol 67:1916–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heise MT, White LJ, Simpson DA, Leonard C, Bernard KA, Meeker RB, Johnston RE. 2003. An attenuating mutation in nsP1 of the Sindbis-group virus S.A.AR86 accelerates nonstructural protein processing and up-regulates viral 26S RNA synthesis. J Virol 77:1149–1156. doi: 10.1128/JVI.77.2.1149-1156.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park E, Griffin DE. 2009. The nsP3 macro domain is important for Sindbis virus replication in neurons and neurovirulence in mice. Virology 388:305–314. doi: 10.1016/j.virol.2009.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akhrymuk I, Kulemzin SV, Frolova EI. 2012. Evasion of the innate immune response: the Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J Virol 86:7180–7191. doi: 10.1128/JVI.00541-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garmashova N, Gorchakov R, Frolova E, Frolov I. 2006. Sindbis virus nonstructural protein nsP2 is cytotoxic and inhibits cellular transcription. J Virol 80:5686–5696. doi: 10.1128/JVI.02739-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simmons JD, Wollish AC, Heise MT. 2010. A determinant of Sindbis virus neurovirulence enables efficient disruption of Jak/STAT signaling. J Virol 84:11429–11439. doi: 10.1128/JVI.00577-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Myles KM, Kelly CL, Ledermann JP, Powers AM. 2006. Effects of an opal termination codon preceding the nsP4 gene sequence in the O'Nyong-Nyong virus genome on Anopheles gambiae infectivity. J Virol 80:4992–4997. doi: 10.1128/JVI.80.10.4992-4997.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Egloff MP, Malet H, Putics A, Heinonen M, Dutartre H, Frangeul A, Gruez A, Campanacci V, Cambillau C, Ziebuhr J, Ahola T, Canard B. 2006. Structural and functional basis for ADP-ribose and poly(ADP-ribose) binding by viral macro domains. J Virol 80:8493–8502. doi: 10.1128/JVI.00713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malet H, Coutard B, Jamal S, Dutartre H, Papageorgiou N, Neuvonen M, Ahola T, Forrester N, Gould EA, Lafitte D, Ferron F, Lescar J, Gorbalenya AE, de Lamballerie X, Canard B. 2009. The crystal structures of Chikungunya and Venezuelan equine encephalitis virus nsP3 macro domains define a conserved adenosine binding pocket. J Virol 83:6534–6545. doi: 10.1128/JVI.00189-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park E, Griffin DE. 2009. Interaction of Sindbis virus non-structural protein 3 with poly(ADP-ribose) polymerase 1 in neuronal cells. J Gen Virol 90:2073–2080. doi: 10.1099/vir.0.012682-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Panas MD, Ahola T, McInerney GM. 2014. The C-terminal repeat domains of nsP3 from the Old World alphaviruses bind directly to G3BP. J Virol 88:5888–5893. doi: 10.1128/JVI.00439-14. [DOI] [PMC free article] [PubMed] [Google Scholar]