

ABSTRACT
The E4-ORF1 protein encoded by human adenovirus stimulates viral replication in human epithelial cells by binding and activating cellular phosphatidylinositol 3-kinase (PI3K) at the plasma membrane and cellular Myc in the nucleus. In this study, we showed that E4-ORF1 hijacks the tyrosine kinase activities of cellular epidermal growth factor receptor (EGFR) and insulin receptor (InsR)/insulin-like growth factor receptor 1 (IGF1R), as well as the lipid kinase activity of PI3K, to mediate constitutive Myc protein expression. We additionally demonstrated that EGFR contributes to constitutive Myc expression through the capacity of E4-ORF1 to induce ligand-independent EGFR activation and stimulation of the Ras/Mek/Erk pathway, the latter activity of which was conserved by human adenoviruses. Results further suggested that EGFR normally forms a complex with the cellular PDZ protein Discs Large 1 (Dlg1), a component of the Dlg1:E4-ORF1:PI3K ternary complex that mediates E4-ORF1-induced PI3K activation, and that E4-ORF1 binds the Dlg1:EGFR complex and promotes the association of EGFR with InsR and IGF1R. In addition to its role in constitutive Myc expression, InsR/IGF1R also negatively regulates EGFR autophosphorylation and EGFR-mediated Ras/Mek/Erk signaling, and data suggested that E4-ORF1 binding to Dlg1 antagonizes these activities. Collectively, our findings suggest that in human epithelial cells, E4-ORF1 targets EGFR, InsR/IGF1R, and PI3K at the plasma membrane to activate cytosolic signaling pathways that sustain Myc protein levels in the nucleus. We postulate that E4-ORF1-induced constitutive Myc expression functions to ensure the formation of nuclear E4-ORF1:Myc complexes, which have been shown to activate Myc and to enhance adenovirus replication.
IMPORTANCE While human adenoviruses primarily produce self-limited acute infections in humans, these agents are associated with life-threatening diseases in immunocompromised patients and in otherwise healthy individuals infected with certain virulent serotypes. The adenovirus E4-ORF1 protein enhances viral replication by activating the cellular lipid kinase PI3K and the cellular transcription factor Myc. Here we report that E4-ORF1 usurps the functions of the cellular tyrosine kinase receptors EGFR and InsR/IGF1R, as well as PI3K, to sustain Myc protein expression in cells. Furthermore, sustained Myc expression depended on E4-ORF1-induced ligand-independent EGFR activation that stimulated Ras/Mek/Erk signaling, a function found to be conserved by human adenoviruses. Given the established roles of PI3K, the Ras/Mek/Erk pathway, and Myc in the adenovirus life cycle, our findings may aid in the development of safer, more effective therapeutic strategies to treat severe adenovirus infections as well as improved adenovirus vectors for use in vaccination and gene and cancer therapy.
INTRODUCTION
Over 60 human adenovirus (Ad) serotypes are classified into seven subgroups (subgroups A through G) based on oncogenic, hemagglutination, and virion structural protein properties as well as genome sequence similarity (1). Ad is a human pathogen primarily associated with mild, self-limited respiratory diseases but additionally causes gastroenteritis, conjunctivitis, cystitis, and skin rashes (1). Certain viral serotypes, such as Ad type 14 (Ad14), can cause life-threatening symptoms that require hospitalization for up to 40% of otherwise healthy individuals (2), and Ad infections of neonates and immunosuppressed patients are associated with high morbidity and mortality rates (3). However, current treatments for severe Ad infections are controversial and carry significant risks for adverse events (1). Ad is also exploited as a vector for vaccination and gene and cancer therapy as well as a tool of discovery to reveal fundamental mechanisms of the cell and mechanisms for cancer development (1).
The Ad early region 4 open reading frame 1 gene (E4-ORF1) encodes a 14-kDa protein required for optimal viral replication (4–6). In humans, subgroup D Ad9 is associated with eye infections (1), whereas Ad9 inoculation into experimental animals elicits estrogen-dependent mammary tumors (7, 8). The first known function of E4-ORF1 was its role as the primary determinant of Ad9-induced tumorigenesis (9–11), and the capacity to transform human epithelial cells is a conserved function of Ad E4-ORF1 genes (12). While E4-ORF1 evolved from a cellular dUTPase that codes for an enzyme of nucleotide metabolism, and E4-ORF1 is predicted to share the dUTPase protein fold, it lacks dUTPase catalytic activity (13, 14). Instead, E4-ORF1 acquired two functions, the activation of cellular class IA phosphatidylinositol 3-kinase (PI3K) and the cellular transcription factor Myc (4, 6, 15). E4-ORF1-mediated oncogenic cellular transformation is reported to depend on PI3K activation, whereas E4-ORF1-mediated enhancement of Ad replication is reported to depend on both PI3K and Myc activation (4, 6, 12, 15, 16).
Two crucial elements in the 125-residue E4-ORF1 polypeptide are domain 2 (D2), defined by 7 centrally located residues, and the PDZ domain-binding motif (PBM), situated at the extreme carboxyl terminus (17, 18). PI3K activation dually depends on D2 residues and the PBM, whereas Myc activation relies on at least one D2 residue but not the PBM (6, 15, 17–19). While the D2 element mediates binding to unidentified cellular factors and also possibly Myc (6, 18), the PBM interacts with a select group of cellular PDZ proteins to exert two distinct functions: (i) disruption of tight junctions and loss of apicobasal cell polarity induced by the E4-ORF1 monomer sequestering the tight junction-associated cellular PDZ proteins MUPP1, PATJ, MAGI-1, and ZO-2 in the cytoplasm and (ii) PI3K activation induced by the E4-ORF1 homotrimer interacting with the adherens junction-associated cellular PDZ protein Discs Large 1 (Dlg1), forming the Dlg1:E4-ORF1:PI3K ternary complex that translocates cytoplasmic PI3K to the plasma membrane (12, 13, 15, 16, 19–23). In the cytoplasm, the Dlg1:E4-ORF1:PI3K ternary complex elevates PI3K protein levels and, upon trafficking to the membrane, stimulates the PI3K/Akt/mTOR pathway to increase protein translation (12, 16, 19). E4-ORF1 also localizes to the nucleus, where it forms the E4-ORF1:Myc complex, which elevates Myc protein levels and induces the expression of specific Myc-responsive genes that stimulate anabolic glucose metabolism and nucleotide biosynthesis (6). Consequently, the ability of E4-ORF1 to activate PI3K and Myc during an Ad infection augments virion production by increasing viral protein expression and viral DNA replication, respectively (4–6).
Epidermal growth factor receptor (EGFR) as well as insulin receptor (InsR) and the related insulin-like growth factor 1 receptor (IGF1R) consist of extracellular, transmembrane, and tyrosine kinase-containing cytoplasmic domains. The extracellular domain binds receptor ligands, which are epidermal growth factor (EGF) family proteins for EGFR and insulin or insulin-like growth factor 1 (IGF-1) and IGF-2 for InsR/IGF1R (24, 25). In their inactive state at the cell surface, EGFR and InsR/IGF1R exist as monomers and covalently linked dimers, respectively. Ligand binding induces conformational changes in the receptors and additionally the dimerization of EGFR, triggering the activation of tyrosine kinase activity and autophosphorylation of the cytoplasmic domain. The phosphorylated tyrosine residues act as docking sites for membrane recruitment of effector proteins that stimulate a variety of signaling cascades, such as the Ras/Mek/Erk and PI3K/Akt/mTOR pathways, which promote protein synthesis, cell growth and survival, and cell cycle progression (26, 27). Transphosphorylation of EGFR and InsR/IGF1R and their synergistic coactivation of common effectors, activities known as receptor cross talk, have also been reported (28). Furthermore, proteins encoded by the Ad early region 1A gene (E1A) and the Ad early region 3 gene (E3) downregulate EGFR and/or InsR/IGF1R (29, 30), and the Ad type 36 E4-ORF1 protein blocks InsR-mediated tyrosine phosphorylation of insulin receptor substrate 1 (31), although the functions of these effects during the Ad life cycle are not known. Also notable are studies showing that treatment of E4-ORF1-expressing rodent fibroblasts with a PI3K inhibitor drug exposes the capacity of E4-ORF1 to activate the Ras/Mek/Erk pathway and that an undetermined Ad5 gene activates this pathway at the late phase of infection in human epithelial cells (19, 32).
Here we report that Ad9 E4-ORF1 dysregulates EGFR and InsR/IGF1R in MCF10A cells, a nontransformed human mammary epithelial line that retains the growth and morphological properties of normal epithelial cells. Results indicated that E4-ORF1 hijacks the functions of these receptor tyrosine kinases, and also PI3K, to promote constitutive Myc expression. The latter activity was mediated in part by E4-ORF1 inducing D2-dependent, ligand-independent EGFR activation and stimulation of Ras/Mek/Erk signaling. In addition, E4-ORF1 targeted a Dlg1:EGFR complex that exists in normal cells and promoted the association of EGFR with InsR and IGF1R, resulting in a postulated E4-ORF1:Dlg1:EGFR:InsR:IGF1R quinary complex, where InsR/IGF1R dampened both EGFR autophosphorylation and Ras/Mek/Erk signaling and where PBM-dependent E4-ORF1 binding to Dlg1 partially relieved these inhibitory effects. We postulate that E4-ORF1-induced constitutive Myc protein expression ensures the formation of optimal quantities of E4-ORF1:Myc complexes, which, by activating Myc in the nucleus, enhance Ad virion production in infected cells (6).
MATERIALS AND METHODS
Plasmids, antibodies, and inhibitors.
Plasmid pBABE-puro or -blasti harboring wild-type (wt) Ad9 E4-ORF1, mutant V125A, mutant H93A, or RasV12 cDNA; plasmid pSUPER.retro.puro harboring Dlg1 short hairpin RNA (shRNA) or matched scrambled shRNA; and rabbit polyclonal antiserum to Ad9 E4-ORF1 were described previously (9, 15, 16, 18, 19). Commercial antibodies to EGFR, InsRβ, IGF1Rβ, Akt, phospho-Akt(Ser473), p42/44 mitogen-activated protein kinase (MAPK) (Erk1/2), and phospho-p42/44 MAPK(Thr202/Tyr204) (P-Erk1/2) (Cell Signaling Technology, Beverly, MA); phosphotyrosine PY20, SAP97 (human Dlg1), and c-Myc (Santa Cruz Biotechnology, Santa Cruz, CA); or phosphotyrosine 4G10 and actin (EMD Millipore, Billerica, MA) were purchased, as were the inhibitor drugs AG1478 and AG1024 (Calbiochem Biochemicals, San Diego, CA) or LY294002 (LY) and U0126 (Cell Signaling Technology, Beverly, MA), all of which were dissolved in dimethyl sulfoxide (DMSO).
Cells, retroviral vectors, and adenoviruses.
Human MCF10A mammary epithelial cells (ATCC) were maintained in complete medium consisting of Dulbecco's modified Eagle's medium (DMEM)–F-12 medium (Life Technologies, Carlsbad, CA) supplemented with 5% horse serum (Life Technologies, Carlsbad, CA), 20 ng/ml EGF (Peprotech, Rocky Hill, NJ), 10 μg/ml insulin, 100 μg/ml hydrocortisone, 1 ng/ml cholera toxin, and 20 μg/ml gentamicin (Sigma-Aldrich, St. Louis, MO). Minimal medium was defined as DMEM–F-12 medium containing gentamicin. Retroviral vectors were produced by transfection of the pBABE and/or pSUPER plasmid into Phoenix-ampho cells (ATCC) using TransIT-LT1 transfection reagent (Mirus Bio, Madison, WI). Cell lines were generated by transduction of MCF10A cells with retroviral vectors followed by selection in complete medium containing 2 μg/ml puromycin and/or 25 μg/ml blasticidin. For experiments, pools of selected cells passaged five times or less were cultured in complete medium containing 5 ng/ml EGF, and an identical number of cells was plated for comparisons of different cell lines and treatments. Inhibitor treatment of cells cultured in complete medium was performed by the direct addition of the drug(s) to conditioned medium followed by incubation for the indicated times. For inhibitor treatment in minimal medium, cells cultured in complete medium were washed three times with minimal medium followed by either incubation in minimal medium containing the drug(s) for 1 h or 3 h or incubation in minimal medium alone for 2 h, at which time the drug(s) was added directly to minimal medium for an additional 1 h. Each drug treatment and matched control DMSO vehicle contained an identical volume of DMSO. Ad12, Ad3, Ad5 (wt300), and Ad9 (Hicks) were propagated and titrated in human A549 cells, as described previously (10).
Cell extract preparation, immunoprecipitations, and immunoblotting.
Extracts were prepared by lysis of cells in ice-cold radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris-HCl [pH 8.0], 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS) containing protease inhibitors (2 mM phenylmethylsulfonyl fluoride [PMSF], 20 μg/ml each of leupeptin and aprotinin) and phosphatase inhibitors (50 mM NaF, 10 mM sodium pyrophosphate, 1 mM sodium orthovanadate) (17). Protein concentrations in extracts were determined by using a bicinchoninic acid (BCA) protein estimation kit (Pierce Biotechnology, Rockford, IL) and an Epoch microplate spectrophotometer with Gen5 data analysis software (BioTek, Winooski, VT). Immunoprecipitations (IPs) were conducted with protein G-Sepharose beads (GE Healthcare Life Sciences, Piscataway, NJ) or protein A/G magnetic beads (Life Technologies, Carlsbad, CA) (33). Proteins in extracts (30 or 50 μg) or in immunoprecipitates were resolved by SDS-PAGE, transferred onto a polyvinylidene difluoride (PVDF) membrane, and immunoblotted, as described previously (33). Immunoblotted protein bands were developed with Amersham ECL Prime (GE Healthcare Life Sciences, Cleveland, OH), imaged with a Biospectrum 810 imaging system (UVP, Upland, CA), and quantified with VisionWorksLS software (UVP, Upland, CA). Differences in the amounts of a given immunoblotted protein between samples were determined after normalization to each respective actin band.
Statistical analyses.
Microsoft Excel was used to calculate means and standard deviations (SD). R statistical software was used to evaluate statistical significance with the Student t test or analysis of variance (ANOVA) with Tukey's post hoc analysis.
RESULTS
E4-ORF1 activates the Ras/Mek/Erk pathway.
We recently reported proteomic analyses that identified PI3K as a direct E4-ORF1 cellular target (12). Interestingly, EGFR was also among the candidate E4-ORF1-interacting proteins revealed by that study. This observation together with the fact that, in rodent fibroblasts, E4-ORF1 has the capacity to activate Ras/Mek/Erk signaling (19), a key EGFR effector pathway, prompted us to examine whether E4-ORF1 dysregulates EGFR in cells.
We initiated this work by comparing the levels of activated, phosphorylated Erk1 and Erk2 (P-Erk1/2) in MCF10A cells stably transduced with either the pBABE retroviral vector carrying wt Ad9 E4-ORF1 (wtORF1 cells) or control empty pBABE (vector cells). Considering the requirement for the E4-ORF1 PBM and/or D2 element in E4-ORF1-induced PI3K and Myc activation (6, 15, 18), MCF10A cells stably transduced with pBABE harboring the Ad9 E4-ORF1 V125A PBM mutant (V125A cells) or the H93A D2 mutant (H93A cells) were also included in the experiment (15, 18). Control immunoblots of extracts from the four MCF10A lines showed comparable E4-ORF1 expression levels in wtORF1, V125A, and H93A cells (P = 0.35; n = 3 independent experiments) (Fig. 1A, compare lanes 2 to 4 to lanes 6 to 8) and also a dramatically larger amount of activated, phosphorylated PI3K effector Akt (P-Akt) in wtORF1 cells than in vector, V125A, and H93A cells (Fig. 1A, compare lane 2 to lanes 1, 3, and 4, and compare lane 6 to lanes 5, 7, and 8), reflecting the dual D2/PBM dependence of E4-ORF1-induced PI3K activation (15, 18). We also observed that in all four cell lines, the EGFR doublet seen at 24 h post-cell plating changed to a single slower-migrating band at 72 h post-cell plating (Fig. 1A, compare lanes 1 to 4 to lanes 5 to 8). Because an antiphosphotyrosine antibody blot revealed lower tyrosine-phosphorylated EGFR (PY-EGFR) levels at 72 h than at 24 h in all four cell lines (data not shown), the slower-migrating EGFR at 72 h post-cell plating was not due to increased tyrosine phosphorylation and was more likely due to an undetermined posttranslational modification(s). More importantly, at 24 h post-cell plating in complete medium, 1.9-fold-higher levels of activated, phosphorylated Erk1 and Erk2 (P-Erk1/2) were detected in wtORF1 cells than in vector, V125A, and H93A cells (Fig. 1A, compare lane 2 to lanes 1, 3, and 4). From 24 h to 72 h post-cell plating, P-Erk1/2 levels in control vector cells exhibited an expected precipitous drop due to normal downregulation of growth factor receptor signaling (Fig. 1A, compare lane 1 to lane 5) (34). Over the same time period, P-Erk1/2 levels likewise precipitously dropped in H93A cells (Fig. 1A, compare lane 4 to lane 8) yet remained stable in wtORF1 and V125A cells (Fig. 1A, compare lane 2 to lane 6, and compare lane 3 to lane 7). At 72 h post-cell plating, these differential effects resulted in 7.2-fold- or 2.3-fold-higher P-Erk1/2 levels in wtORF1 or V125A cells, respectively, than in H93A and vector cells (Fig. 1A, compare lanes 6 and 7 to lanes 5 and 8). Moreover, treatment of wtORF1 cells with the Mek1/2-specific inhibitor drug U0126 (20 μM) (35, 36), but not the control DMSO vehicle, returned the elevated P-Erk1/2 levels to those of vector cells (Fig. 1B, compare lane 3 to lanes 1, 2, and 4). Thus, in MCF10A cells cultured in complete medium, E4-ORF1 stimulates Ras/Mek/Erk signaling and does so by a primarily D2-dependent mechanism enhanced by the PBM, differing from the absolute dual D2/PBM dependence of E4-ORF1-induced PI3K activation.
FIG 1.

E4-ORF1 activates the Ras/Mek/Erk pathway. (A) E4-ORF1 increases Erk1/2 activation in MCF10A cells cultured in complete medium (CM) by a primarily D2-dependent mechanism. Cell extracts prepared from the indicated MCF10A lines at 24 h or 72 h postplating in complete medium were immunoblotted for the indicated proteins. (B) E4-ORF1-induced Erk1/2 activation in MCF10A cells cultured in complete medium requires Mek1/2. The indicated MCF10A lines cultured in complete medium were treated with DMSO or U0126 (20 μM) for 5.5 h, when cell extracts were prepared and immunoblotted for the indicated proteins. (C) Erk1/2 activation is a conserved function of human adenoviruses. Extracts prepared from MCF10A cells that were mock infected or infected with the indicated Ad at a multiplicity of infection of 1 for 48 h were immunoblotted for the indicated proteins. (D) Mek1/2 is required for Erk1/2 activation induced by Ad9 infection and RasV12. MCF10A cells infected with Ad9, as described above for panel C, or RasV12 cells were treated with DMSO or U0126 (20 μM) for 5.5 h, when cell extracts were prepared and immunoblotted for the indicated proteins. (E) E4-ORF1 also induces D2-dependent Erk1/2 activation in MCF10A cells incubated in serum and growth factor-depleted medium. The indicated MCF10A lines cultured in complete medium were incubated in minimal medium (MM) for the indicated times, when cell extracts were prepared and immunoblotted for the indicated proteins.
During an infection of human epithelial cells, Ad5 activates the Ras/Mek/Erk pathway (32), so we wanted to determine whether this activity is shared by other human adenoviruses. Compared to mock-infected MCF10A cells, MCF10A cells infected with representative human Ad serotypes from subgroups A, B, C, and D (Ad12, Ad3, Ad5, and Ad9) displayed high levels of P-Akt, consistent with previously reported findings (4, 12, 16), and also of P-Erk1/2 (Fig. 1C, compare lanes 2 to 5 to lane 1). E4-ORF1 expression was detected only in Ad9-infected cells because the Ad9 E4-ORF1 antibody fails to cross-react with E4-ORF1 proteins from Ad subgroups A, B, and C (Fig. 1C, compare lane 5 to lanes 2 to 4) (12). Furthermore, the Mek1/2 inhibitor U0126, but not DMSO, abolished the elevated P-Erk1/2 levels in not only positive-control MCF10A cells stably transduced by the RasV12 oncogene (RasV12 cells) (16) but also Ad9-infected MCF10A cells (Fig. 1D, compare lane 2 to lane 1, and compare lane 4 to lane 3), similar to results reported previously for Ad5-infected cells (32). Therefore, the activation of the Ras/Mek/Erk pathway is a conserved function of human adenoviruses, similar to the activation of the PI3K/Akt/mTOR pathway (12, 15).
Given that normal downregulation of growth factor receptor signaling heightened the detection of Erk1/2 activation induced by E4-ORF1 (Fig. 1A), we hypothesized that this E4-ORF1 activity is mediated by a serum- and growth factor-independent mechanism. To test this idea, we compared P-Erk1/2 levels in vector, wtORF1, V125A, and H93A cells incubated in minimal medium for 0 h, 1 h, 2 h, or 3 h. Surprisingly, replacement of conditioned medium with minimal medium caused a rapid rise in P-Erk1/2 levels in wtORF1 and V125A cells but not in vector and H93A cells (Fig. 1E, compare lanes 5 to 8 and 9 to 12 to lanes 1 to 4 and 13 to 16). This observation may indicate that a factor secreted by MCF10A cells into conditioned medium stimulates autocrine/paracrine signaling that dampens E4-ORF1-induced Erk1/2 activation. Additionally, despite V125A cells having lower P-Erk1/2 levels than those in wtORF1 cells before the switch to minimal medium at 0 h (Fig. 1A and E, compare lane 5 to lane 9), the elevated P-Erk1/2 levels achieved by the two cell lines after 1 h of incubation in minimal medium were statistically the same after normalization to actin levels (1.3-fold ± 0.35-fold difference [mean ± SD]; P = 0.29; n = 3 independent experiments) (Fig. 1E, compare lane 6 to lane 10). The slightly elevated actin levels seen in wtORF1 cells were not reproducible (Fig. 1E, compare lanes 5 and 6 to lanes 1 and 2), however, as quantification of data from multiple experiments revealed no statistically significant difference between vector and wtORF1 cells (P = 0.25; n = 5 independent experiments). After 2 h or 3 h of incubation in minimal medium, P-Erk1/2 levels moderately declined in wtORF1 and V125A cells, although in V125A cells, the decline was 3-fold or 2-fold greater, respectively (Fig. 1E, compare lanes 6 to 8 to lanes 10 to 12). These data indicate that E4-ORF1 promotes D2-dependent and serum- and growth factor-independent activation of the Ras/Mek/Erk pathway and that the PBM augments this activity, perhaps by antagonizing inhibitory autocrine/paracrine signaling induced by a factor in conditioned medium.
EGFR mediates E4-ORF1-induced activation of the Ras/Mek/Erk pathway.
To test the hypothesis that EGFR mediates Erk1/2 activation induced by E4-ORF1, we employed the EGFR-specific inhibitor drug AG1478 that blocks EGFR tyrosine kinase activity (37). A control experiment verified that treatment of vector cells with AG1478 (0.5 μM), but not DMSO, inhibits acute EGF stimulation-induced autophosphorylation of EGFR (PY-EGFR) and elevation of P-Erk1/2 levels, without affecting EGFR, Erk1/2, or P-Akt levels (Fig. 2A, compare lane 2 to lane 1). More importantly, in wtORF1 cells either cultured in complete medium or incubated for 1 h in minimal medium, AG1478 but not DMSO decreased the elevated P-Erk1/2 levels to those of control vector cells (Fig. 2B, compare lane 4 to lanes 1 to 3, and C, compare lane 2 to lane 1). In contrast, in wtORF1 cells incubated for 1 h in minimal medium, the PI3K-specific inhibitor drug LY294002 (LY) (100 μM), but not DMSO, dramatically reduced P-Akt levels, as expected (12, 16), but did not affect P-Erk1/2 levels (Fig. 2C, compare lane 4 to lane 3). These results suggest that E4-ORF1 promotes ligand-independent EGFR activation to mediate Ras/Mek/Erk signaling.
FIG 2.

EGFR mediates E4-ORF1-induced Erk1/2 activation. (A) The EGFR inhibitor AG1478 blocks EGF-induced EGFR autophosphorylation and Erk1/2 activation. Vector cells preincubated in minimal medium for 1 h were stimulated with EGF (5 ng/ml) mixed with either DMSO or AG1478 (1478) (0.5 μM) for 15 min, when cell extracts were prepared and immunoblotted for the indicated proteins. PY-EGFR detected with antiphosphotyrosine antibody PY20 comigrated with EGFR. (B) EGFR activity is required for E4-ORF1-induced Erk1/2 activation in MCF10A lines cultured in complete medium (CM). The indicated MCF10A lines cultured in complete medium were treated with DMSO or AG1478 (1478) (0.5 μM) for 1 h, when extracts were prepared and immunoblotted for the indicated proteins. (C) EGFR is also required for E4-ORF1-induced Erk1/2 activation in MCF10A cells incubated in serum and growth factor-depleted medium. wtORF1 cells cultured in complete medium were incubated in minimal medium (MM) containing either DMSO, LY294002 (LY) (100 μM), or AG1478 (0.5 μM) for 1 h, when cell extracts were prepared and immunoblotted for the indicated proteins. PY-EGFR was detected as described above for panel A.
To determine whether E4-ORF1 also augments ligand-dependent activation of EGFR and Ras/Mek/Erk signaling, we measured PY-EGFR and P-Erk1/2 levels in vector and wtORF1 cells at 15 and 30 min post-EGF stimulation and then calculated the fold increases in the protein levels relative to their levels in each respective cell line prior to EGF stimulation at 0 min. Increases in PY-EGFR levels were comparable in both cell lines at 15 and 30 min post-EGF stimulation (Fig. 3, compare lanes 1 to 3 to lanes 4 to 6), whereas increases in P-Erk1/2 levels were 1.8-fold higher in vector cells than in wtORF1 cells at 15 min post-EGF stimulation or were comparable in both cell lines at 30 min post-EGF stimulation (Fig. 3, compare lanes 1 to 3 to lanes 4 to 6). We conclude that E4-ORF1 does not enhance EGF-induced EGFR activation or stimulation of Ras/Mek/Erk signaling in MCF10A cells.
FIG 3.
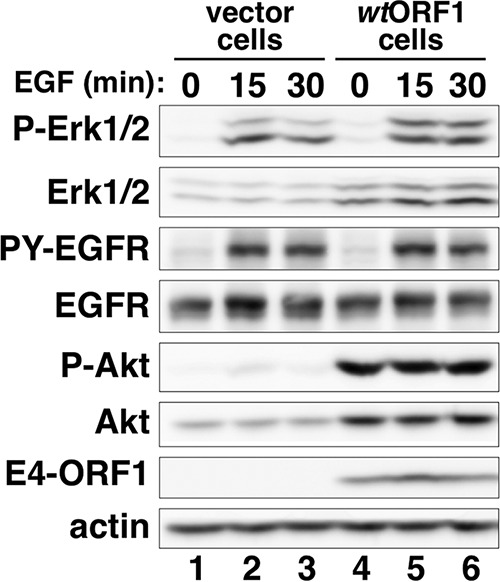
E4-ORF1 does not enhance EGF-dependent EGFR and Erk1/2 activation. The indicated MCF10A lines cultured in complete medium were incubated in minimal medium containing EGF (5 ng/ml) for the indicated times, when cell extracts were prepared and immunoblotted for the indicated proteins. PY-EGFR was detected as described in the Fig. 2A legend.
Dlg1 and E4-ORF1 associate with a tyrosine-phosphorylated 170-kDa protein that comigrates with EGFR in a PBM-dependent fashion and with a tyrosine-phosphorylated 120-kDa protein in a dual-D2/PBM-dependent fashion.
To assess whether E4-ORF1 binds EGFR in cells, we subjected extracts from the four MCF10A lines to immunoprecipitation (IP) with antibody to Dlg1 or E4-ORF1 and then tested for coimmunoprecipitation (coIP) of EGFR and PY-EGFR. The Dlg1 IP was included because the E4-ORF1 PBM, which mediates binding to Dlg1, augments E4-ORF1 D2-dependent EGFR and Erk1/2 activation (Fig. 1A and E). Despite comparable expression levels of E4-ORF1 and Dlg1 in wtORF1, H93A, and V125A cells (Fig. 4A, lanes 2 to 4), the Dlg1 IP resulted in coIP of E4-ORF1 only from wtORF1 and H93A cells (Fig. 4A, compare lanes 6 and 7 to lanes 5 and 8), and the reciprocal E4-ORF1 IP led to coIP of Dlg1 again only from wtORF1 and H93A cells (Fig. 4A, compare lanes 10 and 11 to lanes 9 and 12), reflecting PBM-dependent formation of E4-ORF1:Dlg1 complexes by wt E4-ORF1 and the H93A D2 mutant but not the V125A PBM mutant (18).
FIG 4.

E4-ORF1 and Dlg1 associate with tyrosine-phosphorylated proteins p170 and p120. (A) E4-ORF1 and Dlg1 bind to tyrosine-phosphorylated 170-kDa (PY-p170) and 120-kDa (PY-p120) cellular proteins. Cell extracts (400 μg of protein) from the indicated MCF10A lines were subjected to IP with either Dlg1 or E4-ORF1 antibody. Recovered proteins and cell extracts were immunoblotted for the indicated proteins. The expanded antiphosphotyrosine blot showing PY-p170 and PY-p120 covers the molecular mass range from 200 kDa to 100 kDa. PY-p170 and PY-p120 were detected with antiphosphotyrosine antibody 4G10. (B) The level of PY-p170, but not that of PY-p120 or PY-p95, is diminished by the EGFR inhibitor AG1478. Cell extracts (1 mg of protein) from the indicated MCF10A lines were subjected to IP with E4-ORF1 antibody. Recovered proteins and cell extracts were immunoblotted for the indicated proteins. PY-p170, PY-p120, and PY-p95 were detected with antiphosphotyrosine antibody PY20.
Significantly, the Dlg1 IP resulted in coIP of EGFR from vector, wtORF1, H93A, and V125A cells (Fig. 4A, lanes 5 to 8), suggesting that Dlg1:EGFR complexes normally exist in MCF10A cells. Moreover, for wtORF1 and H93A cells only, IP of Dlg1 or E4-ORF1 followed by blotting with antiphosphotyrosine antibody revealed coIP of a tyrosine-phosphorylated 170-kDa protein doublet (PY-p170) that comigrated with EGFR (Fig. 4A, compare lanes 6 and 7 to lanes 5 and 8, and compare lanes 10 and 11 to lanes 9 and 12), and treatment with the EGFR inhibitor AG1478 abolished the PY-p170 doublet (Fig. 4B, compare lane 6 to lane 5). These data suggest the formation of E4-ORF1:Dlg1:EGFR complexes, where PBM-mediated binding of wt E4-ORF1 or H93A to Dlg1 promotes EGFR autophosphorylation to produce PY-EGFR. The fact that we detected coIP of PY-EGFR, but not EGFR (data not shown), with E4-ORF1 suggests that E4-ORF1 associates with a small fraction of Dlg1:EGFR complexes in MCF10A cells and that the phosphotyrosine antibody is more sensitive than the EGFR antibody in immunoblots. The IP of Dlg1 or E4-ORF1 additionally led to coIP of a tyrosine-phosphorylated 120-kDa protein (PY-p120) from only wtORF1 cells (Fig. 4A, compare lane 6 to lanes 5, 7, and 8, and compare lane 10 to lanes 9, 11, and 12). Furthermore, antiphosphotyrosine antibody blots for the entire molecular mass range of proteins recovered by IP of Dlg1 or E4-ORF1 indicated that PY-p170 and PY-p120, as well as PY-p95 (see below), were the only tyrosine-phosphorylated proteins detected in wtORF1 cells but not vector cells (data not shown). Contrary to PY-p170, PY-p120 was unaffected by AG1478 treatment (Fig. 4B, compare lane 6 to lane 5), suggesting that wt E4-ORF1 instead induces EGFR-independent and dual-D2/PBM-dependent tyrosine phosphorylation of this unidentified cellular factor in E4-ORF1:Dlg1:EGFR complexes. The IP assays described above employed cells stimulated with fresh culture medium 15 min prior to cell extract preparation to assess the effects of acute growth factor stimulation on the detection of tyrosine-phosphorylated proteins in complexes; however, similar results were also obtained without this stimulation (data not shown). The data suggest that E4-ORF1 PBM-dependent EGFR autophosphorylation alone is insufficient to activate Erk1/2, as evidenced by the H93A D2 mutant retaining the former but not the latter activity (Fig. 1A and E and 4). However, because the V125A PBM mutant did not detectably increase EGFR autophosphorylation and also showed a reduced capacity to activate Erk1/2 (Fig. 1A and E and 4), we postulate that E4-ORF1 PBM-dependent EGFR autophosphorylation functions to augment E4-ORF1 D2-dependent EGFR and Erk1/2 activation.
E4-ORF1 promotes the association of EGFR with InsR and IGF1R, which dampen EGFR autophosphorylation and stimulation of Ras/Mek/Erk signaling.
Given our findings showing ligand-independent EGFR activation by E4-ORF1 and previous reports of cross talk between EGFR and InsR or the related IGF1R (Fig. 1, 2, and 4) (28), we also investigated whether the latter two receptors influence E4-ORF1-induced, EGFR-mediated Ras/Mek/Erk signaling. This effort was initiated by examining whether EGFR interacts with InsR or IGF1R in MCF10A cells. Interestingly, IP of EGFR led to coIP of InsR from wtORF1 and V125A cells but not vector and H93A cells (Fig. 5, compare lanes 6 and 7 to lanes 5 and 8) and also led to coIP of IGF1R only from wtORF1 cells (Fig. 5, compare lane 6 to lanes 5, 7, and 8). Thus, in a D2- or dual-D2/PBM-dependent fashion, E4-ORF1 promotes EGFR to form a complex with InsR or IGF1R, respectively. Moreover, IP of E4-ORF1 from wtORF1 but not vector cells led to coIP of a tyrosine-phosphorylated 95-kDa protein (PY-p95) that comigrated with IGF1R but was unaffected by the EGFR inhibitor AG1478 (Fig. 4B, compare lane 6 to lanes 4 and 5), suggesting that E4-ORF1 also associates with activated IGF1R.
FIG 5.
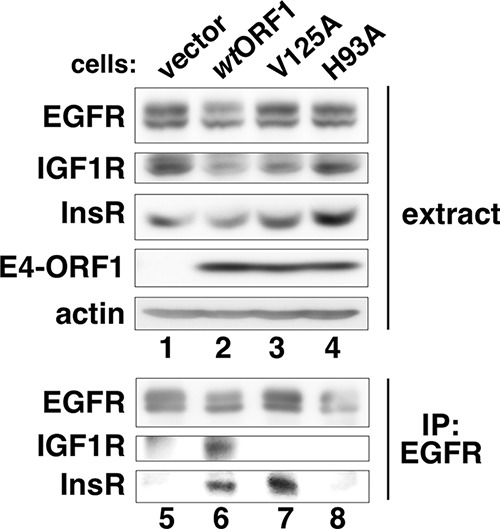
E4-ORF1 promotes association of EGFR with InsR and IGF1R. Cell extracts (350 μg of protein) from the indicated MCF10A lines were subjected to IP with EGFR antibody. Recovered proteins and cell extracts were immunoblotted for the indicated proteins.
To explore the effects of InsR/IGF1R on E4-ORF1-induced, EGFR-dependent Ras/Mek/Erk signaling, we utilized the InsR/IGF1R-specific inhibitor drug AG1024, which blocks the tyrosine kinase activities of InsR and IGF1R with 50% inhibitory concentrations (IC50s) of 57 μM and 7 μM, respectively (38). A control experiment confirmed that in MCF10A cells, AG1024 (50 μM) but not DMSO or the EGFR inhibitor AG1478 blocks the ability of acute insulin stimulation to elevate the levels of activated, autophosphorylated InsR (PY-InsR) and PY-IGF1R (Fig. 6A, compare lane 3 to lanes 1 and 2) (39). Furthermore, AG1024 or AG1478 reduced P-Akt levels strongly or modestly, respectively, or reduced P-Erk1/2 levels moderately or strongly, respectively (Fig. 6A, compare lanes 2 and 3 to lane 1), suggesting that in MCF10A cells acutely stimulated with insulin, InsR/IGF1R directly mediates the majority of Akt activation, whereas positive cross talk from InsR/IGF1R to EGFR mediates the majority of Erk1/2 activation.
FIG 6.

InsR/IGF1R dampens EGFR autophosphorylation and Erk1/2 activation. (A) AG1024 inhibits insulin-induced InsR/IGF1R autophosphorylation and activation of Akt and Erk1/2. Vector cells preincubated in minimal medium (MM) for 1 h were stimulated with insulin (10 μg/ml) mixed with DMSO, AG1478 (1478) (0.5 μM), or AG1024 (1024) (50 μM) for 15 min, when cell extracts were prepared and immunoblotted for the indicated proteins. PY-InsR and PY-IGF1R detected with antiphosphotyrosine antibody 4G10 comigrated with InsR and IGF1R, respectively. (B) InsR/IGF1R dampens EGFR autophosphorylation and Erk1/2 activation in MCF10A cells cultured in complete medium (CM). The indicated MCF10A lines cultured in complete medium were treated with DMSO or AG1024 (50 μM) for 1 h, when cell extracts were prepared and immunoblotted for the indicated proteins. PY-EGFR was detected as described in the Fig. 2A legend. (C) InsR/IGF1R inhibits EGFR autophosphorylation in MCF10A cells incubated in serum and growth factor-depleted medium. The indicated MCF10A lines cultured in complete medium were incubated in minimal medium containing DMSO or AG1024 (50 μM) for 1 h, when cell extracts were prepared and immunoblotted for the indicated proteins. PY-EGFR was detected as described in the Fig. 2A legend. (D) InsR/IGF1R dampens D2-dependent E4-ORF1-induced Erk1/2 activation in MCF10A cells incubated in serum and growth factor-depleted medium. The indicated MCF10A lines cultured in complete medium were incubated in minimal medium containing DMSO or AG1024 (50 μM) for 1 h, when cell extracts were prepared and immunoblotted for the indicated proteins. (E) InsR/IGF1R dampens EGFR-mediated Erk1/2 activation induced by E4-ORF1. wtORF1 cells cultured in complete medium were incubated in minimal medium containing DMSO or either AG1478 (0.5 μM) or AG1024 (50 μM) alone or in combination for 1 h, when cell extracts were prepared and immunoblotted for the indicated proteins. PY-EGFR was detected as described in the Fig. 2A legend.
In contrast, in vector and wtORF1 cells cultured in complete medium, AG1024 but not DMSO instead elevated the levels of both PY-EGFR and P-Erk1/2 (Fig. 6B, compare lane 2 to lane 1, and compare lane 4 to lane 3), and H93A and V125A cells yielded similar results (data not shown). In vector, wtORF1, V125A, and H93A cells incubated for 1 h in minimal medium, AG1024 likewise elevated PY-EGFR levels (Fig. 6C, compare lane 1 to lane 2, lane 3 to lane 4, lane 5 to lane 6, and lane 7 to lane 8); however, under these conditions, AG1024 increased P-Erk1/2 levels in wtORF1 and V125A cells but not in vector and H93A cells (Fig. 6D, compare lanes 3 to 6 to lanes 1, 2, 7, and 8, and E, compare lane 3 to lane 1). Moreover, in wtORF1 cells incubated for 1 h in minimal medium, the EGFR inhibitor AG1478 ablated the AG1024-induced elevation of both PY-EGFR and P-Erk1/2 levels (Fig. 6E, compare lane 3 to lane 4).
In addition to providing further support for the idea that E4-ORF1 promotes ligand-independent activation of EGFR and Erk1/2 by a mechanism that requires the D2 element, the data shown in Fig. 6B to E reveal negative cross talk from InsR/IGF1R to EGFR in MCF10A cells cultured for an extended time in complete medium or incubated in minimal medium. This finding is consistent with data from a previous report showing that IGF1R downregulates the activation of EGFR and the Ras/Mek/Erk pathway (40) but differs from the positive cross talk from InsR/IGF1R to EGFR observed in MCF10A cells acutely stimulated with insulin (Fig. 6A). Specifically, our results indicate that InsR/IGF1R negatively regulates EGFR (i) by suppressing EGFR autophosphorylation and (ii) by reducing the capacity of EGFR activated by ligand or by E4-ORF1 to stimulate Ras/Mek/Erk signaling. The first conclusion is supported by data in Fig. 6B, C, and E showing that AG1024 increases PY-EGFR levels in all four cell lines either cultured in complete medium or incubated in minimal medium. The second conclusion is supported by data in Fig. 6B, D, and E showing that AG1024 increases P-Erk1/2 levels only in cell lines incubated with or expressing an EGFR-activating stimulus (i.e., ligand, wt E4-ORF1, or the V125A mutant). Because Erk1/2 was not activated in vector and H93A cells incubated in minimal medium (Fig. 1E), and AG1024 treatment elevated PY-EGFR but not P-Erk1/2 levels (Fig. 6C and D, lanes 1, 2, 7, and 8), we additionally conclude that like E4-ORF1 PBM-dependent EGFR autophosphorylation (Fig. 4A), AG1024-induced EGFR autophosphorylation alone is insufficient to trigger EGFR activation capable of stimulating Ras/Mek/Erk signaling. Also like E4-ORF1 PBM-dependent EGFR autophosphorylation, we postulate that AG1024-induced EGFR autophosphorylation functions to enhance E4-ORF1 D2-dependent EGFR-mediated Erk1/2 activation.
E4-ORF1 promotes constitutive, serum- and growth factor-independent Myc expression.
Thai et al. reported previously that Ad5 E4-ORF1 binds and activates Myc, as well as elevates Myc protein levels, to increase viral DNA replication by stimulating nucleotide biosynthesis (6). Moreover, previous studies showed that the activation of EGFR or IGF1R, as well as the Ras/Mek/Erk or PI3K/Akt/mTOR pathway, upregulates Myc expression (41–43). These findings prompted us to investigate whether Ad9 E4-ORF1 likewise dysregulates Myc protein expression in MCF10A cells and, if so, whether EGFR, InsR/IGF1R, Mek1/2, and/or PI3K is required for this activity. We first compared total Myc protein levels in vector, wtORF1, V125A, and H93A cells either cultured in complete medium or incubated in minimal medium. At 24 h post-cell plating in complete medium, Myc expression was 2.3-fold ± 0.23-fold lower in wtORF1 cells than in vector, V125A, and H93A cells (P = 0.013; n = 3 independent experiments) (Fig. 7A, compare lane 5 to lanes 1, 9, and 13). Nonetheless, upon further normal downregulation of growth factor receptor signaling at 72 h post-cell plating in complete medium, Myc expression was instead 1.8-fold higher in wtORF1 cells than in vector cells (Fig. 7B, compare lane 2 to lane 1). Moreover, at 24 h post-cell plating in complete medium, the cell lines shown in Fig. 7A were incubated in minimal medium for 1 h, 2 h, or 3 h, at which time the Myc expression levels dropped precipitously to nearly undetectable levels in vector, V125A, and H93A cells (Fig. 7A, compare lanes 4, 12, and 16) yet were sustained in wtORF1 cells at 13-fold- ± 4.9-fold-higher levels than in vector cells (P = 0.016; n = 3 independent experiments) (Fig. 7A, compare lane 8 to lane 4). These data importantly show that by a dual-D2/PBM-dependent mechanism, Ad9 E4-ORF1 promotes constitutive and serum- and growth factor-independent Myc protein expression.
FIG 7.

E4-ORF1 promotes constitutive Myc protein expression in MCF10A cells by a mechanism that requires the activities of EGFR, InsR/IGF1R, and PI3K. (A) E4-ORF1 prevents Myc downregulation in MCF10A cells incubated in serum and growth factor-depleted medium. The indicated MCF10A lines cultured for 24 h in complete medium (CM) were incubated in minimal medium (MM) for the indicated times, when cell extracts were prepared and immunoblotted for the indicated proteins. (B) E4-ORF1 also prevents Myc downregulation in MCF10A cells cultured for 72 h in complete medium. Extracts of the indicated MCF10A lines cultured for 72 h in complete medium were immunoblotted for the indicated proteins. (C) InsR/IGF1R, EGFR, PI3K, and Mek1/2 activities are required for E4-ORF1-induced constitutive Myc expression in MCF10A cells incubated in serum and growth factor-depleted medium. wtORF1 cells cultured in complete medium were incubated in minimal medium containing DMSO, AG1024 (1024) (50 μM), AG1478 (1478) (0.5 μM), LY294002 (LY) (100 μM), or U0126 (10 μM) for 3 h, when cell extracts were prepared and immunoblotted for the indicated proteins. (D) The activities of InsR/IGF1R and PI3K, but not EGFR, are required for Myc expression in MCF10A cells cultured in complete medium. The indicated MCF10A lines cultured in complete medium were treated with DMSO, AG1024 (50 μM), AG1478 (0.5 μM), LY294002 (100 μM), or U0126 (10 μM) for 1 h, when cell extracts were prepared and immunoblotted for the indicated proteins. (E) AG1478 or U1026 treatment for 1 h reduces E4-ORF1-induced constitutive Myc expression in wtORF1 cells incubated in serum and growth factor-depleted medium. Extracts of wtORF1 cells cultured in complete medium were incubated for 3 h in minimal medium containing DMSO, AG1478 (0.5 μM), or U0126 (10 μM) during the last 1 h of incubation, at which time the cell extracts were prepared and immunoblotted for the indicated proteins. (F) E4-ORF1-induced constitutive Myc expression depends on Dlg1. At 72 h post-cell plating in complete medium, extracts of wtORF1 cells expressing the Dlg1 shRNA (+) or control scrambled shRNA (−) were prepared and immunoblotted for the indicated proteins.
Constitutive Myc expression induced by E4-ORF1 depends on EGFR and InsR/IGF1R activity as well as PI3K activation mediated by the Dlg1:E4-ORF1:PI3K ternary complex.
We next determined whether E4-ORF1-induced constitutive Myc expression in wtORF1 cells incubated in minimal medium for 3 h requires the tyrosine kinase activity of EGFR or InsR/IGF1R, the protein kinase activity of Mek1/2, or the lipid kinase activity of PI3K. The results revealed that Myc expression is dramatically reduced in wtORF1 cells incubated for 3 h in minimal medium containing an inhibitor of EGFR (AG1478), InsR/IGF1R (AG1024), Mek1/2 (U0126), or PI3K (LY) but not DMSO (Fig. 7C, compare lanes 2 to 4 to lane 1, and compare lane 6 to lane 5). In contrast, in both vector and wtORF1 cells cultured in complete medium, Myc expression was unaffected by 1 h of treatment with AG1478 or U0126, similarly to DMSO (Fig. 7D, compare lanes 1 and 3, lanes 5 and 8, lanes 9 and 10, and lanes 11 and 12), but was diminished by 1 h of treatment with either AG1024 or LY (Fig. 7D, compare lanes 2 and 4 to lane 1, and compare lanes 6 and 8 to lane 5). The latter negative results with AG1478 and U1026 in cell lines cultured in complete medium are specific because constitutive Myc expression in wtORF1 cells incubated for 3 h in minimal medium was also diminished by 1 h of treatment with either AG1478 or U0126 but not DMSO (Fig. 7E, compare lane 2 to lane 1, and compare lane 4 to lane 3). These data demonstrate specific roles for EGFR and Ras/Mek/Erk signaling in constitutive Myc protein expression induced by E4-ORF1. Thus, while Myc expression sustained by serum and growth factors in MCF10A lines requires the activities of InsR/IGF1R and PI3K but not the activities of EGFR or Erk1/2, E4-ORF1-induced constitutive Myc expression requires all four activities. These findings may indicate that to sustain Myc expression in MCF10A lines cultured in complete medium, EGFR and Ras/Mek/Erk signaling are functionally redundant with other undetermined growth factor receptors and downstream signaling pathways. However, in wtORF1 cells cultured in minimal medium, the absence of serum growth factors precludes the activation of other growth factor receptors, making E4-ORF1-induced EGFR and Ras/Mek/Erk activation essential to sustain Myc expression.
Given the requirement of PI3K for E4-ORF1-induced constitutive Myc expression (Fig. 7C) and the established role of Dlg1 in E4-ORF1-induced PI3K activation (12, 16, 19), we reasoned that E4-ORF1-induced constitutive Myc expression likewise depends on Dlg1. Our data revealed that E4-ORF1-induced constitutive Myc expression in wtORF1 cells, at 72 h post-plating in complete medium (Fig. 7B), is diminished by shRNA-mediated Dlg1 depletion but not by the control scrambled shRNA and also that Dlg1 depletion reduces P-Akt levels, as expected, without affecting P-Erk1/2 levels (Fig. 7F, compare lane 2 to lane 1). In addition to showing that Dlg1 is not absolutely required for E4-ORF1-induced Erk1/2 activation, the results indicate that E4-ORF1-induced constitutive Myc expression depends on PI3K activation mediated by the Dlg1:E4-ORF1:PI3K ternary complex. Because Dlg1 normally functions to reduce Myc expression by negatively regulating PI3K activation (44), we postulate that E4-ORF1 promotes Myc expression, in part, by converting Dlg1 from a negative to a positive regulator of PI3K activation.
DISCUSSION
Novel findings reported in this study demonstrate that the Ad9 E4-ORF1 protein promotes constitutive Myc protein expression in human MCF10A cells by a mechanism requiring the tyrosine kinase activities of EGFR and InsR/IGF1R and the lipid kinase activity of PI3K. A model for the molecular mechanism of this novel E4-ORF1 function is illustrated in Fig. 8 (steps 1 to 8).
FIG 8.

Hypothesized model for E4-ORF1-induced constitutive Myc protein expression mediated by EGFR, InsR/IGF1R, and PI3K. (A) In normal MCF10A cells, EGFR and Dlg1 form a Dlg1:EGFR complex at the plasma membrane. When activated by ligand, EGFR autophosphorylates tyrosine (Y) residues that directly mediate Ras/Mek/Erk signaling (black) and also those that augment Ras/Mek/Erk signaling (white). InsR/IGF1R activated by ligand specifically downregulates EGFR autophosphorylation on white Y residues that augment Ras/Mek/Erk signaling. (B) E4-ORF1 assembles two different protein complexes at the plasma membrane to activate three separate signaling pathways. E4-ORF1 forms the E4-ORF1:Dlg1:EGFR:InsR:IGF1R quinary complex by binding to the Dlg1:EGFR complex and promoting EGFR dimerization and EGFR association with InsR and IGF1R. In the quinary complex, the E4-ORF1 D2 element promotes EGFR autophosphorylation on black Y residues to activate Ras/Mek/Erk signaling, whereas the E4-ORF1 PBM via binding to Dlg1 enhances the latter signaling by antagonizing InsR/IGF1R-mediated suppression of EGFR autophosphorylation on white Y residues. An additional consequence of E4-ORF1 forming the quinary complex is activation of InsR/IGF1R (denoted by asterisks) and undetermined downstream signaling pathways. E4-ORF1 also binds both Dlg1 and PI3K to form the Dlg1:E4-ORF1:PI3K ternary complex, which activates the PI3K/Akt/mTOR pathway. (C) The three signaling pathways (Ras/Mek/Erk, InsR/IGF1R, and PI3K/Akt/mTOR) activated by E4-ORF1 converge in the nucleus to promote constitutive Myc expression, which enhances Ad replication by ensuring the formation of sufficient quantities of E4-ORF1:Myc complexes to stimulate anabolic glucose metabolism and nucleotide biosynthesis. See the text for details about steps 1 to 8 in the model.
In step 1, Dlg1 and EGFR normally exist as a Dlg1:EGFR complex in human epithelial cells (Fig. 4). In step 2, after the acute phase of EGF stimulation, negative cross talk from constitutively dimeric InsR/IGF1R downregulates EGFR autophosphorylation and Ras/Mek/Erk signaling (Fig. 2C and 6B to E). One possibility is that InsR/IGF1R induces EGFR phosphorylation on the inhibitory tyrosine residues Y992, Y1045, and/or Y1173, which suppress Erk1/2 signaling by recruiting cellular phosphatases or ubiquitin ligase to EGFR (45). Because an inhibitory factor(s) in conditioned medium may dampen EGFR activation and signaling (Fig. 1E), we hypothesize that this factor is the InsR/IGF1R ligand IGF-1 or -2, the latter of which is expressed in MCF10A cells (46). In step 3, E4-ORF1 binds the Dlg1:EGFR complex (Fig. 4). Either D2 or the PBM may suffice to mediate the association of E4-ORF1 with the Dlg1:EGFR complex because, as discussed below for subsequent steps, D2 induces EGFR to activate Erk1/2 (Fig. 1A and E), and the PBM induces EGFR autophosphorylation (Fig. 4A), supporting the idea that D2- or PBM-mediated E4-ORF1 binding to the Dlg1:EGFR complex can promote EGFR dimerization and activation of EGFR tyrosine kinase activity. In a D2- or dual-D2/PBM-dependent fashion, E4-ORF1 also causes EGFR to associate with InsR or IGF1R, respectively (Fig. 5), resulting in the formation of a postulated E4-ORF1:Dlg1:EGFR:InsR:IGF1R quinary complex. In step 4, in a D2-dependent fashion, E4-ORF1 triggers ligand-independent EGFR activation that stimulates Ras/Mek/Erk signaling (Fig. 1A, B, and E and 2C), presumably by inducing EGFR autophosphorylation on tyrosine residues Y1068, Y1086, and/or Y1148, which is known to stimulate this pathway (45). Because E4-ORF1 interacts with only a small fraction of Dlg1:EGFR complexes in cells (Fig. 4A), the level of D2-dependent EGFR autophosphorylation is presumably below our level of detection, as we failed to detect coIP of PY-EGFR with the V125A PBM mutant (Fig. 4A), which induced EGFR-dependent Erk1/2 activation (Fig. 1A and E and 6D). In step 5, E4-ORF1 PBM-dependent binding to Dlg1 in quinary complexes also triggers EGFR autophosphorylation (Fig. 4). Because the PBM is required for EGFR autophosphorylation and augmentation of D2-dependent Erk1/2 activation (Fig. 1A and E and 4), we postulate that, similar to the InsR/IGF1R inhibitor AG1024, the PBM antagonizes InsR/IGF1R-mediated EGFR suppression, thereby increasing EGFR autophosphorylation in quinary complexes and, in so doing, enhancing the capacity of D2-dependent EGFR activation to stimulate Ras/Mek/Erk signaling. PBM-dependent EGFR autophosphorylation alone is insufficient to activate Erk1/2 (Fig. 1A and E, 4, and 6D), so this phosphorylation presumably occurs on EGFR tyrosine residues distinct from those that mediate Ras/Mek/Erk signaling. The association of E4-ORF1 with only a small fraction of Dlg1:EGFR complexes also would explain our detection of increased PY-EGFR levels in quinary complexes but not in total extracts of wtORF1 and H93A cells versus vector and V125A cells (Fig. 1E, 3, and 6C and data not shown). For step 6, we hypothesize that in Ad-infected cells, E4-ORF1 likewise activates EGFR, which in turn stimulates Ras/Mek/Erk signaling, a conserved Ad function required for optimal viral protein expression and virion production (Fig. 1C) (32). Similarly, the Dlg1:E4-ORF1:PI3K ternary complex activates the PI3K/Akt/mTOR pathway (12, 16), which in Ad-infected cells enhances cell cycle progression, viral protein translation, and virion production (4, 5). As Dlg1 is present in both ternary and quinary complexes, we plan to investigate whether quinary complexes associate with a subset of ternary complexes to form a single supramolecular signaling complex. In step 7, E4-ORF1-induced signaling from EGFR/Ras/Mek/Erk, PI3K/Akt/mTOR, and undetermined InsR/IGF1R effector pathways additionally acts in concert to mediate constitutive Myc protein expression (Fig. 7) (6). Considering the shared dual D2/PBM dependence of E4-ORF1-induced (i) EGFR associations with IGF1R, (ii) p120 tyrosine phosphorylation, and (iii) constitutive Myc expression (Fig. 4, 5, and 7A), we envision that E4-ORF1 within quinary complexes activates IGF1R by inducing autophosphorylation on tyrosine residues (Fig. 4B) and that activated IGF1R in turn phosphorylates p120, which contributes to constitutive Myc expression. For step 8, we hypothesize that E4-ORF1-induced constitutive Myc protein expression ensures the formation of optimal numbers of nuclear E4-ORF1:Myc complexes, which stimulate anabolic glucose metabolism and nucleotide biosynthesis, thereby enhancing Ad DNA replication and virion production in infected cells (6). Future studies will investigate the roles for these novel E4-ORF1 activities in Ad-infected normal human epithelial cells. As EGFR, InsR, IGF1R, and Myc are dysregulated in many human cancers (47–49), it will also be important to determine whether these cellular factors contribute to E4-ORF1-induced cellular transformation and tumorigenesis.
Our findings reveal two distinct InsR/IGF1R functions: (i) downregulation of EGFR autophosphorylation and EGFR-mediated Ras/Mek/Erk signaling and (ii) upregulation of Myc expression. We envision that E4-ORF1 antagonizes the first function to promote the second by redirecting InsR/IGF1R activity to quinary complexes, where activated InsR/IGF1R acquires the capacity to stimulate an undetermined signaling pathway(s) that contributes to constitutive Myc expression. While E4-ORF1 promoted the association of both InsR and IGF1R with EGFR, E4-ORF1 associated with PY-IGF1R but not PY-InsR (Fig. 4B and data not shown). These findings may indicate that E4-ORF1 activates InsR kinase activity without a concomitant increase in InsR autophosphorylation, similar to the effect observed by overexpression of certain caveolin subtypes in cells (50).
Numerous pathogenic human viruses usurp EGFR or the Ras/Mek/Erk pathway to facilitate virus entry, replication, assembly, host immune surveillance evasion, inflammation, pathogenesis, and transmission (29, 51). In Ad-infected cells, the immediate early Ad E1A transcriptional activator downregulates EGFR, an effect hypothesized to isolate virus-infected cells from host-specific signals such as the EGFR-dependent induction of the neutrophil chemoattractant interleukin-8 (IL-8) (29, 52). In contrast, our results showed that Ad E4-ORF1, the expression of which depends on E1A in Ad-infected cells, binds and activates EGFR (Fig. 2B and C and 4), revealing opposing functional effects of these two Ad proteins on EGFR. Furthermore, Ad5 induces biphasic activation of the Ras/Mek/Erk pathway, with the first phase being triggered at the initial step of infection by virion binding to host αV integrin coreceptors at the cell surface and the second phase being induced at the late stage of infection by an undetermined Ad protein (32, 53). In this study, we demonstrated that the second phase of Ras/Mek/Erk signaling is a conserved function of human Ad (Fig. 1C and D) and also that E4-ORF1 expression alone in cells is sufficient to activate this pathway in an EGFR-dependent manner (Fig. 1A and E and 2B and C). A future study will investigate whether E4-ORF1 represents the undetermined viral protein responsible for the second phase of Ras/Mek/Erk signaling required for optimal Ad protein expression and virion production in Ad-infected cells (32).
Multiple DNA virus proteins, including Ad E1A and E4-ORF1, are reported to increase Myc expression (6, 54–57). Here we showed that E4-ORF1 sustains Myc protein expression in human epithelial cells cultured in serum and growth factor-depleted medium and that this novel E4-ORF1 function is mediated by the combined activities of EGFR, InsR/IGF1R, and PI3K (Fig. 7). While the role of E4-ORF1-induced constitutive Myc expression in the Ad life cycle was not determined, it may be pertinent that during the late phase of infection, Ad shuts off host gene expression to promote high levels of late viral protein synthesis by blocking the export of cellular mRNAs from the nucleus and their translation in the cytoplasm (1). Thus, an intriguing possibility is that host shutoff in Ad-infected normal human epithelial cells abolishes cellular Myc expression and that E4-ORF1 functions in part to sustain Myc expression under these conditions, thereby allowing the formation of E4-ORF1:Myc complexes that enhance viral DNA replication and virion production.
ACKNOWLEDGMENTS
This work was supported by NIH grant R01CA58541 (R.T.J.) and by predoctoral fellowship CPRIT RP101499 from the Baylor College of Medicine Comprehensive Cancer Training Program (K.K.).
We thank Andrew Rice and Susan Marriott for their helpful critiques of the manuscript.
REFERENCES
- 1.Wold WS, Ison MG. 2013. Adenoviruses, p 1732–1767. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Centers for Disease Control and Prevention. 2007. Acute respiratory disease associated with adenovirus serotype 14—four states, 2006-2007. MMWR Morb Mortal Wkly Rep 56:1181–1184. [PubMed] [Google Scholar]
- 3.Echavarria M. 2008. Adenoviruses in immunocompromised hosts. Clin Microbiol Rev 21:704–715. doi: 10.1128/CMR.00052-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Shea C, Klupsch K, Choi S, Bagus B, Soria C, Shen J, McCormick F, Stokoe D. 2005. Adenoviral proteins mimic nutrient/growth signals to activate the mTOR pathway for viral replication. EMBO J 24:1211–1221. doi: 10.1038/sj.emboj.7600597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Shea CC, Choi S, McCormick F, Stokoe D. 2005. Adenovirus overrides cellular checkpoints for protein translation. Cell Cycle 4:883–888. doi: 10.4161/cc.4.7.1791. [DOI] [PubMed] [Google Scholar]
- 6.Thai M, Graham NA, Braas D, Nehil M, Komisopoulou E, Kurdistani SK, McCormick F, Graeber TG, Christofk HR. 2014. Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab 19:694–701. doi: 10.1016/j.cmet.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Javier R, Raska K Jr, Macdonald GJ, Shenk T. 1991. Human adenovirus type 9-induced rat mammary tumors. J Virol 65:3192–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Javier R, Raska K Jr, Shenk T. 1992. Requirement for the adenovirus type 9 E4 region in production of mammary tumors. Science 257:1267–1271. doi: 10.1126/science.1519063. [DOI] [PubMed] [Google Scholar]
- 9.Javier RT. 1994. Adenovirus type 9 E4 open reading frame 1 encodes a transforming protein required for the production of mammary tumors in rats. J Virol 68:3917–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas DL, Schaack J, Vogel H, Javier R. 2001. Several E4 region functions influence mammary tumorigenesis by human adenovirus type 9. J Virol 75:557–568. doi: 10.1128/JVI.75.2.557-568.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas DL, Shin S, Jiang BH, Vogel H, Ross MA, Kaplitt M, Shenk TE, Javier RT. 1999. Early region 1 transforming functions are dispensable for mammary tumorigenesis by human adenovirus type 9. J Virol 73:3071–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar M, Kong K, Javier RT. 2014. Hijacking Dlg1 for oncogenic phosphatidylinositol 3-kinase activation in human epithelial cells is a conserved mechanism of human adenovirus E4-ORF1 proteins. J Virol 88:14268–14277. doi: 10.1128/JVI.02324-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung SH, Weiss RS, Prasad BVV, Javier RT. 2008. Functionally distinct monomers and trimers produced by a viral oncoprotein. Oncogene 27:1412–1420. doi: 10.1038/sj.onc.1210784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiss RS, Lee SS, Prasad BV, Javier RT. 1997. Human adenovirus early region 4 open reading frame 1 genes encode growth-transforming proteins that may be distantly related to dUTP pyrophosphatase enzymes. J Virol 71:1857–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frese KK, Lee SS, Thomas DL, Latorre IJ, Weiss RS, Glaunsinger BA, Javier RT. 2003. Selective PDZ protein-dependent stimulation of phosphatidylinositol 3-kinase by the adenovirus E4-ORF1 oncoprotein. Oncogene 22:710–721. doi: 10.1038/sj.onc.1206151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kong K, Kumar M, Taruishi M, Javier RT. 2014. The human adenovirus E4-ORF1 protein subverts discs large 1 to mediate membrane recruitment and dysregulation of phosphatidylinositol 3-kinase. PLoS Pathog 10:e1004102. doi: 10.1371/journal.ppat.1004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee SS, Weiss RS, Javier RT. 1997. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci U S A 94:6670–6675. doi: 10.1073/pnas.94.13.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chung SH, Frese KK, Weiss RS, Prasad BV, Javier RT. 2007. A new crucial protein interaction element that targets the adenovirus E4-ORF1 oncoprotein to membrane vesicles. J Virol 81:4787–4797. doi: 10.1128/JVI.02855-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frese KK, Latorre IJ, Chung SH, Caruana G, Bernstein A, Jones SN, Donehower LA, Justice MJ, Garner CC, Javier RT. 2006. Oncogenic function for the Dlg1 mammalian homolog of the Drosophila discs-large tumor suppressor. EMBO J 25:1406–1417. doi: 10.1038/sj.emboj.7601030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Latorre IJ, Roh MH, Frese KK, Weiss RS, Margolis B, Javier RT. 2005. Viral oncoprotein-induced mislocalization of select PDZ proteins disrupts tight junctions and causes polarity defects in epithelial cells. J Cell Sci 118:4283–4293. doi: 10.1242/jcs.02560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glaunsinger BA, Lee SS, Thomas M, Banks L, Javier R. 2000. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene 19:5270–5280. doi: 10.1038/sj.onc.1203906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glaunsinger BA, Weiss RS, Lee SS, Javier R. 2001. Link of the unique oncogenic properties of adenovirus type 9 E4-ORF1 to a select interaction with the candidate tumor suppressor protein ZO-2. EMBO J 20:5578–5586. doi: 10.1093/emboj/20.20.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SS, Glaunsinger B, Mantovani F, Banks L, Javier RT. 2000. Multi-PDZ domain protein MUPP1 is a cellular target for both adenovirus E4-ORF1 and high-risk papillomavirus type 18 E6 oncoproteins. J Virol 74:9680–9693. doi: 10.1128/JVI.74.20.9680-9693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siddle K. 2011. Signalling by insulin and IGF receptors: supporting acts and new players. J Mol Endocrinol 47:R1–R10. doi: 10.1530/JME-11-0022. [DOI] [PubMed] [Google Scholar]
- 25.Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A. 2001. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr Relat Cancer 8:11–31. doi: 10.1677/erc.0.0080011. [DOI] [PubMed] [Google Scholar]
- 26.Roskoski R., Jr 2012. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 66:105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 27.Cantley LC. 2002. The phosphoinositide 3-kinase pathway. Science 296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 28.Xu AM, Huang PH. 2010. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res 70:3857–3860. doi: 10.1158/0008-5472.CAN-10-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng K, Kitazato K, Wang Y. 2014. Viruses exploit the function of epidermal growth factor receptor. Rev Med Virol 24:274–286. doi: 10.1002/rmv.1796. [DOI] [PubMed] [Google Scholar]
- 30.Kuivinen E, Hoffman BL, Hoffman PA, Carlin CR. 1993. Structurally related class I and class II receptor protein tyrosine kinases are down-regulated by the same E3 protein coded for by human group C adenoviruses. J Cell Biol 120:1271–1279. doi: 10.1083/jcb.120.5.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rogers PM, Fusinski KA, Rathod MA, Loiler SA, Pasarica M, Shaw MK, Kilroy G, Sutton GM, McAllister EJ, Mashtalir N, Gimble JM, Holland TC, Dhurandhar NV. 2008. Human adenovirus Ad-36 induces adipogenesis via its E4 orf-1 gene. Int J Obes (Lond) 32:397–406. doi: 10.1038/sj.ijo.0803748. [DOI] [PubMed] [Google Scholar]
- 32.Schumann M, Dobbelstein M. 2006. Adenovirus-induced extracellular signal-regulated kinase phosphorylation during the late phase of infection enhances viral protein levels and virus progeny. Cancer Res 66:1282–1288. doi: 10.1158/0008-5472.CAN-05-1484. [DOI] [PubMed] [Google Scholar]
- 33.Weiss RS, Javier RT. 1997. A carboxy-terminal region required by the adenovirus type 9 E4 ORF1 oncoprotein for transformation mediates direct binding to cellular polypeptides. J Virol 71:7873–7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sorkin A, Waters CM. 1993. Endocytosis of growth factor receptors. Bioessays 15:375–382. doi: 10.1002/bies.950150603. [DOI] [PubMed] [Google Scholar]
- 35.Li Q, Mattingly RR. 2008. Restoration of E-cadherin cell-cell junctions requires both expression of E-cadherin and suppression of ERK MAP kinase activation in Ras-transformed breast epithelial cells. Neoplasia 10:1444–1458. doi: 10.1593/neo.08968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, Cheng C. 2011. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest 121:1064–1074. doi: 10.1172/JCI44540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burdick AD, Davis JW II, Liu KJ, Hudson LG, Shi H, Monske ML, Burchiel SW. 2003. Benzo(a)pyrene quinones increase cell proliferation, generate reactive oxygen species, and transactivate the epidermal growth factor receptor in breast epithelial cells. Cancer Res 63:7825–7833. [PubMed] [Google Scholar]
- 38.Parrizas M, Gazit A, Levitzki A, Wertheimer E, LeRoith D. 1997. Specific inhibition of insulin-like growth factor-1 and insulin receptor tyrosine kinase activity and biological function by tyrphostins. Endocrinology 138:1427–1433. [DOI] [PubMed] [Google Scholar]
- 39.Gunton JE, Delhanty PJ, Takahashi S, Baxter RC. 2003. Metformin rapidly increases insulin receptor activation in human liver and signals preferentially through insulin-receptor substrate-2. J Clin Endocrinol Metab 88:1323–1332. doi: 10.1210/jc.2002-021394. [DOI] [PubMed] [Google Scholar]
- 40.Riedemann J, Sohail M, Macaulay VM. 2007. Dual silencing of the EGF and type 1 IGF receptors suggests dominance of IGF signaling in human breast cancer cells. Biochem Biophys Res Commun 355:700–706. doi: 10.1016/j.bbrc.2007.02.041. [DOI] [PubMed] [Google Scholar]
- 41.Grandori C, Cowley SM, James LP, Eisenman RN. 2000. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol 16:653–699. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- 42.Banskota NK, Taub R, Zellner K, Olsen P, King GL. 1989. Characterization of induction of protooncogene c-myc and cellular growth in human vascular smooth muscle cells by insulin and IGF-I. Diabetes 38:123–129. [DOI] [PubMed] [Google Scholar]
- 43.Hann SR. 2006. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin Cancer Biol 16:288–302. doi: 10.1016/j.semcancer.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 44.Sandoval GJ, Graham DB, Gmyrek GB, Akilesh HM, Fujikawa K, Sammut B, Bhattacharya D, Srivatsan S, Kim A, Shaw AS, Yang-Iott K, Bassing CH, Duncavage E, Xavier RJ, Swat W. 2013. Novel mechanism of tumor suppression by polarity gene discs large 1 (DLG1) revealed in a murine model of pediatric B-ALL. Cancer Immunol Res 1:426–437. doi: 10.1158/2326-6066.CIR-13-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang Y, Chang Y. 2011. Epidermal growth factor receptor (EGFR) phosphorylation, signaling and trafficking in prostate cancer, p 143–172. In Spiess PE. (ed), Prostate cancer—from bench to bedside. InTech, Rijeka, Croatia. [Google Scholar]
- 46.Vyas S, Asmerom Y, De Leon DD. 2006. Insulin-like growth factor II mediates resveratrol stimulatory effect on cathepsin D in breast cancer cells. Growth Factors 24:79–87. doi: 10.1080/08977190500366068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dang CV. 2012. MYC on the path to cancer. Cell 149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu T, Li C. 2010. Convergence between Wnt-beta-catenin and EGFR signaling in cancer. Mol Cancer 9:236. doi: 10.1186/1476-4598-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh P, Alex JM, Bast F. 2014. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: novel treatment strategies for cancer. Med Oncol 31:805. doi: 10.1007/s12032-013-0805-3. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto M, Toya Y, Schwencke C, Lisanti MP, Myers MG Jr, Ishikawa Y. 1998. Caveolin is an activator of insulin receptor signaling. J Biol Chem 273:26962–26968. doi: 10.1074/jbc.273.41.26962. [DOI] [PubMed] [Google Scholar]
- 51.Miller WE, Raab-Traub N. 1999. The EGFR as a target for viral oncoproteins. Trends Microbiol 7:453–458. doi: 10.1016/S0966-842X(99)01605-4. [DOI] [PubMed] [Google Scholar]
- 52.Jijon HB, Buret A, Hirota CL, Hollenberg MD, Beck PL. 2012. The EGF receptor and HER2 participate in TNF-alpha-dependent MAPK activation and IL-8 secretion in intestinal epithelial cells. Mediators Inflamm 2012:207398. doi: 10.1155/2012/207398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greber UF. 2002. Signalling in viral entry. Cell Mol Life Sci 59:608–626. doi: 10.1007/s00018-002-8453-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kadeppagari RK, Sankar N, Thimmapaya B. 2009. Adenovirus transforming protein E1A induces c-Myc in quiescent cells by a novel mechanism. J Virol 83:4810–4822. doi: 10.1128/JVI.02145-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fish K, Chen J, Longnecker R. 2014. Epstein-Barr virus latent membrane protein 2A enhances MYC-driven cell cycle progression in a mouse model of B lymphoma. Blood 123:530–540. doi: 10.1182/blood-2013-07-517649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang YW, Chang HS, Lin CH, Yu WC. 2007. HPV-18 E7 conjugates to c-Myc and mediates its transcriptional activity. Int J Biochem Cell Biol 39:402–412. doi: 10.1016/j.biocel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 57.Liu J, Martin HJ, Liao G, Hayward SD. 2007. The Kaposi's sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J Virol 81:10451–10459. doi: 10.1128/JVI.00804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]