

ABSTRACT
Coxsackievirus A16 (CVA16) is one of the major etiological agents of hand, foot, and mouth disease (HFMD) in children. The host defense mechanisms against CVA16 infection remain almost entirely unknown. Unlike previous observations with enterovirus 71 (EV71) infection, here we show that gamma interferon (IFN-γ) or invariant NK T cell deficiency does not affect disease development or the survival of CVA16-infected mice. In contrast, type I interferon receptor deficiency resulted in the development of more severe disease in mice, and the mice had a lower survival rate than wild-type mice. Similarly, a deficiency of Toll-like receptor 3 (TLR3) and TRIF, but not other pattern recognition receptors, led to the decreased survival of CVA16-infected mice. TLR3-TRIF signaling was indispensable for the induction of type I interferons during CVA16 infection in mice and protected young mice from disease caused by the infection. In particular, TRIF-mediated immunity was critical for preventing CVA16 replication in the neuronal system before disease occurred. IFN-β treatment was also found to compensate for TRIF deficiency in mice and decreased the disease severity in and mortality of CVA16-infected mice. Altogether, type I interferons induced by TLR3-TRIF signaling mediate protective immunity against CVA16 infection. These findings may shed light on therapeutic strategies to combat HFMD caused by CVA16 infection.
IMPORTANCE Hand, foot, and mouth disease (HFMD) is a major threat to public health in the Asia-Pacific region. Both CVA16 and EV71 are major pathogens that are responsible for HFMD. The majority of research efforts have focused on the more virulent EV71, but little has been done with CVA16. Thus far, host immune responses to CVA16 infection have not yet been elucidated. The present study discovered an initial molecular mechanism underlying host protective immunity against CVA16 infection, providing the first explanation for why CVA16 and EV71 cause different clinical outcomes upon infection of humans. Therefore, different therapeutic strategies should be developed to treat HFMD cases caused by these two viruses.
INTRODUCTION
Coxsackievirus A16 (CVA16) is a single-stranded positive RNA virus and belongs to the Enterovirus genus of the Picornaviridae family. CVA16 is one of the major agents causing hand, foot, and mouth disease (HFMD). CVA16 was first identified by Sickles and colleagues in 1951 (1). The first large HFMD outbreak caused by CVA16 occurred in England in 1994 (2). Later, from 1999 to 2006, a large outbreak occurred in Taiwan (3). Surveillance data showed that CVA16 was the predominantly circulating virus during three HFMD outbreaks in 2002, 2005, and 2007, respectively, in Singapore (4). Both CVA16 and another major etiological pathogen, enterovirus 71 (EV71), contributed to the large outbreak in Taiwan in 1998 (5). Similarly, the cocirculation of CVA16 with EV71 was found to result in the current large epidemic of HFMD in China (6, 7). According to the proceedings of the National Health and Family Planning Commission in China, a total of 2,781,712 HFMD cases were reported in 2014. CVA16 infection is generally responsible for almost 50% of all confirmed cases (6). Therefore, it is of great interest to understand the pathogenesis of CVA16 infection to better manage large outbreaks.
Although CVA16 infection occasionally causes fatal myocarditis and pneumonitis (8, 9), EV71 infection is more frequently associated with severe central nervous system (CNS) complications (5, 10–13). Therefore, the majority of research efforts have focused on EV71 infection or EV71-associated HFMD; thus far, little is known about the pathogenesis of CVA16 infection. In particular, the mechanisms underlying why CVA16 infection causes a relatively milder disease than EV71 infection remain elusive.
Accumulating evidence supports the notion that infections with different enterovirus strains may trigger distinct innate immune responses. As RNA viruses, their viral RNA components may be recognized upon infection by the pattern recognition receptors (PRRs) of host cells, such as Toll-like receptor 3 (TLR3), TLR7, TLR8, RIG-I, and MDA5 molecules, thereby triggering innate immune responses, such as the production of type I interferons (IFNs). Infections with coxsackievirus B1 (CVB1) and CVB5 can stimulate the production of cytokines, including type I IFNs, in a TLR7-dependent manner (14), whereas the production of type I IFNs induced by CVB3 and poliovirus infections relies on the MDA5 and TLR3 pathways, respectively (15, 16). In contrast, signaling through TLR3 and RIG-I is blocked by the 2C and 3C proteases of EV71, resulting in the production of very small amounts of type I IFNs by host cells in vitro and in vivo upon infection (17–20). A study with genetically mutated mice demonstrated that type I IFNs appear to be dispensable for controlling EV71 infection in mice (21). Rather, invariant natural killer T (iNKT) cells were later found to be crucial antiviral effector cells to protect young mice from EV71 infection. EV71 infection led to the activation of iNKT cells dependent on signaling in macrophages through TLR3 but not other TLRs (22). CVA16 is closely related to EV71 in terms of genomic similarity (23), and it is possible that CVA16 infection, like EV71 infection, may also activate TLR3 signaling and lead to similar innate immune responses.
In the present study, we infected genetically mutant mice with a clinical CVA16 isolate and studied the host immune response to the infection. We found that type I IFNs mediate protective immunity against CVA16 infection in young mice. TLR3 was indispensable for the induction of type I IFNs in CVA16-infected mice, but other PRRs were not. This TLR3 dependence is similar to that of EV71 infection, but both gamma IFN (IFN-γ) and iNKT cells are dispensable for protective immunity against CVA16 infection.
MATERIALS AND METHODS
Ethics statement on animal subjects.
All animal experiments were performed in strict accordance with the regulations in the Guide for the Care and Use of Laboratory Animals issued by the Ministry of Science and Technology of the People's Republic of China (24). All efforts were made to minimize pain and suffering. The protocol was approved by the Institutional Animal Care and Use Committee of the Institut Pasteur of Shanghai (permit number A2014001).
Mice.
C57BL/6 mice were obtained from the Shanghai Laboratory Animal Center (SLAC). IFN-α/β receptor (IFNAR)-knockout (IFN-α/βR−/−) mice with the strain 129 background were purchased from B&K Universal and have been backcrossed onto the C57BL/6 background for more than six generations. The TLR3−/− mice used in the assays whose results are presented in Fig. 2 were kindly provided by Richard Flavell and have been backcrossed onto the C57BL/6 background for more than eight generations. The TLR3 −/− mice with the 129 background used in the assays whose results are presented in Fig. 3 were kindly provided by Ming Wang. C57BL/6J-Ticam1Lps2/J (TRIF−/−) mice and B6.129S7-Ifngr1tm1AgtNJU (IFN-γR−/−) mice were purchased from the Model Animal Research Center of Nanjing University. B6.129S1-Tlr7tm1Flv/J (TLR7−/−) mice with the C57BL/6 background were purchased from The Jackson Laboratory. MAVS-knockout (MAVS−/−) mice with the strain 129 background were kindly gifted by James Z. Chen. All mice were kept under specific-pathogen-free (SPF) conditions in the SLAC. Infection was performed in containment isolators under SPF conditions. Animals were infected intraperitoneally (i.p.) with the dose of CVA16 indicated below in 50 μl of RPMI 1640 medium. For IFN-β treatment, infected mice were additionally injected i.p. with 3 × 104 units of IFN-β (Sino Biological) per mouse per day or saline on days 1, 2, and 3 postinfection. The clinical scores of the mice after CVA16 infection were as follows: 0, healthy; 1, ruffled hair and hunched back; 2, limb weakness; 3, paralysis in 1 limb; 4, paralysis in both hind limbs; and 5, death.
FIG 2.
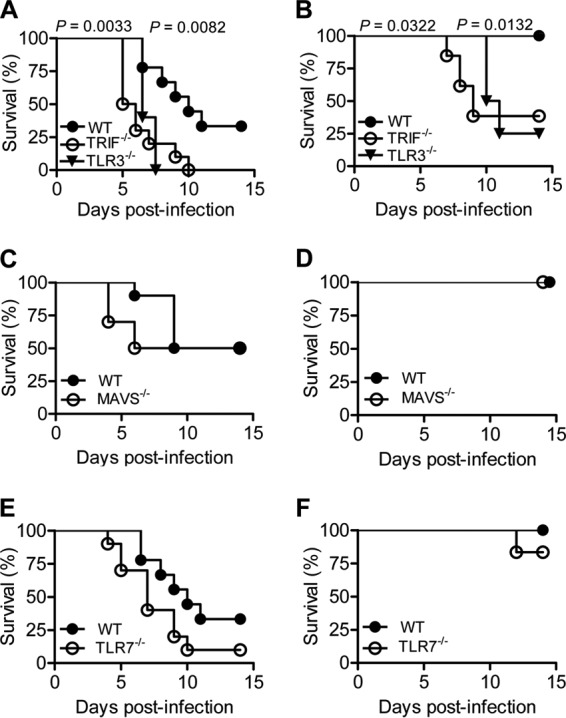
Effects of TLR3, TLR7, MAVS, and TRIF deficiency on survival of CVA16-infected young mice. (A and B) Survival rates of 14-day-old WT (n = 9 and 5, respectively), TLR3−/− (n = 5 and 8, respectively), and TRIF−/− (n = 10 and 13, respectively) C57BL/6 mice infected with 1.5 × 104 PFU CVA16 (A) or 1.5 × 103 PFU CVA16 (B). (C and D) Survival rates of 14-day-old WT (n = 10 and 5, respectively) and MAVS−/− (n = 10 and 5, respectively) mice with a strain 129 background infected with 1.5 × 104 PFU CVA16 (C) or 1.5 × 103 PFU CVA16 (D). (E and F) Survival rates of 14-day-old WT (n = 9 and 5, respectively) and TLR7−/− (n = 10 and 6, respectively) C57BL/6 mice infected with 1.5 × 104 PFU CVA16 (E) or 1.5 × 103 PFU CVA16 (F).
FIG 3.

Role of TRIF-mediated signaling in IFN-β expression. (A and B) Fourteen-day-old WT or TRIF-deficient C57BL/6 mice were infected i.p. with 1.5 ×104 PFU CVA16, and then the IFN-β expression levels in the indicated tissues of infected mice were measured by qRT-PCR on days 2 (A) and 4 (B). (C and D) WT and TLR3-deficient BMDCs (C) and BMMs (D) from mice with a strain 129 background were infected with CVA16 at a multiplicity of infection of 10 (1.0 × 106 PFU/1.0 × 105 cells) for the indicated times, and the expression of IFN-β was measured by qRT-PCR. (E to G) WT and TRIF-deficient BMDCs (E), BMMs (F), and MEFs (G) from mice with a C57BL/6 background were infected with CVA16 at a multiplicity of infection of 10 (1.0 × 106 PFU/1.0× 105 cells) for the indicated times, and the expression of IFN-β was measured by qRT-PCR. Positive controls consisted of BMDCs, BMMs, and MEFs stimulated with poly(I·C) (pI:C). (H) The viral loads in infected cells from WT mice with a C57BL/6 background were examined by qRT-PCR at 0 and 6 h postinfection. (I) BMDCs and BMMs from WT mice with a C57BL/6 background were infected with live or UV-inactivated CVA16 at a multiplicity of infection of 10 (1.0 × 106 PFU/1.0 × 105 cells) for the indicated times, and the expression of IFN-β was measured by qRT-PCR. Data are shown as the mean ± SEM and are representative of those from three independent experiments (n = 3, respectively). ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Cell lines and virus.
Rhabdomyosarcoma (RD) cells were cultured in Dulbecco's modified Eagle's medium (HyClone) supplemented with 10% fetal calf serum (FCS). Mouse fibroblast L929 cells were grown in RPMI 1640 medium (HyClone) with 10% FCS. The CVA16 clinical strain was prepared as described previously (25) and passaged in RD cells for five generations. The virus, which was used in all experiments, was purified and concentrated by centrifugation at 10,000 × g for 20 min at 4°C with 10% polyethylene glycol 8000. Purified virus was titrated in a plaque assay using L929 cell monolayers in 96-well plates. For UV inactivation, a live CVA16 stock solution was spread on the dish surface and exposed to UV light with no cover (at a distance of 60 cm and a wavelength of 250 to 270 nm) for 1 h at room temperature.
BMDCs, BMMs, and MEFs.
Bone marrow-derived dendritic cells (BMDCs) and bone marrow-derived macrophages (BMMs) were generated as described previously (22, 26). In brief, harvested bone marrow cells were cultured for 6 days in RPMI 1640 medium supplemented with 10% FCS. Four percent of the supernatant from granulocyte-macrophage colony-stimulating factor-expressing J5 cells and 50 ng/ml interleukin-4 (Peprotech) were added to the culture medium to induce BMDCs. Ten percent of the L929 supernatant was added to the culture medium to induce BMMs. Mouse embryonic fibroblasts (MEFs) were generated from pregnant mice on day 14 after mating, as described previously (27).
Flow cytometry.
Cells were washed and incubated in staining buffer (phosphate-buffered saline [PBS], 0.3% bovine serum albumin, 0.1% sodium azide) containing anti-CD16/CD32 for 10 min at 4°C and then stained with fluorophore-conjugated mouse PBS57-loaded/unloaded CD1d tetramers, anti-T cell receptor β (anti-TCRβ), and anti-CD69 (eBioscience) for 20 min. After washing twice with staining buffer, data were collected on an LSRII flow cytometer (BD Biosciences). The data were analyzed using FlowJo software (Tree Star).
Quantitative reverse transcription-PCR (qRT-PCR).
Total RNA was isolated from CVA16-infected or uninfected cells with the TRIzol reagent (Invitrogen) or from tissues with an RNAprep pure tissue kit (Tiangen), and cDNAs were prepared from 1 μg of total RNA with random hexamer primers and superscript reverse transcriptase with a FastQuant reverse transcription kit (Tiangen) according to the manufacturer's procedure. cDNAs were used as the templates for PCR amplification using a SuperReal preMix Plus (SYBR green I) kit (Tiangen) and an ABI 7900HT Fast real-time PCR system (Applied Biosystems). The primers used were as follows: CVA16 forward and reverse (5′-GAACCATCACTCCACACAGGAG-3′ and 5′-GTACCTGTGGTGGGCATTG-3′, respectively), GAPDH (glyceraldehyde-3-phosphate dehydrogenase) forward and reverse (5′-CCCACTAACATCAAATGGGG-3′ and 5′-CCTTCCACAATGCCAAAGTT-3′, respectively), IFN-β forward and reverse (5′-AGCTCCAAGAAAGGACGAACAT-3′ and 5′-GCCCTGTAGGTGAGGTTGATCT-3′, respectively), IFN-non-α4 forward and reverse (5′-TCTGATGCAGCAGGTGGG-3′ and 5′-AGGGCTCTCCAGAYTTCTGCTCTG-3′, respectively), and IFN-α4 forward and reverse (5′-CCTGTGTGATGCAGGAACC-3′ and 5′-TCACCTCCCAGGCACTGA-3′, respectively). The levels of expression of the RNAs of interest were normalized to the level of GAPDH expression.
Histopathological staining.
Tissues from euthanized mice were fixed in 4% neutral buffered paraformaldehyde and embedded in paraffin. Paraffin-embedded sections were stained with hematoxylin and eosin (H&E).
Statistical analyses.
Statistical analyses for continuous data were performed with Prism (version 5) for Windows software (GraphPad Software Inc.) using two-tailed Student's t tests. Statistically significant differences in mouse survival were determined by analysis by the Gehan-Breslow-Wilcoxon test. Statistically significant differences in clinical scores between two groups were determined by two-way analysis of variance plus Bonferroni's posttest. P values of less than 0.05 were considered significant. Graphs were produced and statistical analyses were performed using GraphPad Prism software.
RESULTS
Type I IFNs are essential for the protection of young mice against CVA16 infection.
Our previous study revealed that iNKT cells play a critical role in the control of EV71 infection in young mice (22); thus, we wondered whether iNKT cells play a similar role in CVA16 infection. We first sought to examine the activation of iNKT cells in wild-type (WT) 14-day-old mice after CVA16 infection at a dose of 2 × 105 PFU/mouse through i.p. injection. As shown in Fig. 1A, CVA16 infection, unlike EV71 infection, did not increase the level of CD69 expression on iNKT cells (Fig. 1A), suggesting that CVA16 infection does not significantly activate iNKT cells. Although CD1d-deficient mice infected with 1.5 × 104 PFU/mouse of CVA16 developed disease slightly earlier, their overall survival rate was not significantly different from that of WT mice (Fig. 1B). These observations imply that iNKT cells are dispensable for the control of CVA16 infection in young mice.
FIG 1.
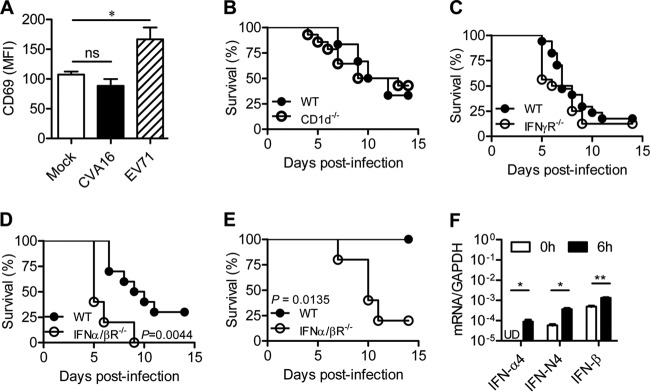
Role of IFNs and iNKT cells in protective immunity against CVA16 infection. (A) CD69 expression on iNKT cells of CVA16-infected mice. Fourteen-day-old C57BL/6 mice were injected i.p. with a mock treatment (n = 5) or 2 × 105 PFU CVA16 (n = 5) or EV71 (n = 5). Splenocytes from mock-infected or infected mice were separated at 16 h postinjection and stained with TCRβ, CD1d tetramer, CD69, and DAPI (4′,6-diamidino-2-phenylindole). The levels of CD69 expression are shown for live CD1d tetramer-positive TCRβ-positive gated cells. MFI, mean fluorescence intensity. (B) Survival rates of 14-day-old WT (n = 6) and CD1d−/− (n = 14) C57BL/6 mice infected with 1.5 × 104 PFU CVA16. (C) Survival rates of 14-day-old WT (n = 17) and IFN-γR−/− (n = 16) C57BL/6 mice infected with 1.5 × 104 PFU CVA16. (D and E) Survival rates of 14-day-old WT (n = 5 and 11, respectively) and IFN-α/βR−/− (n = 5 and 5, respectively) C57BL/6 mice infected with 1.5 × 104 PFU CVA16 (D) or 1.5 × 103 PFU CVA16 (E). (F) BMMs from mice with a WT C57BL/6 background were infected with CVA16 at a multiplicity of infection of 10 (1.0 × 106 PFU/1.0 × 105 cells) for the indicated times, and the expression of IFN-α4, IFN-non-α4 (IFN-N4), and IFN-β was measured by qRT-PCR. Data are shown as the mean ± SEM and are representative of those from three independent experiments. ns, not significant; *, P < 0.05; **, P < 0.01; UD, undetectable.
Given that type I and/or II IFNs are indispensable for mice to control enterovirus infections (21, 28), we first sought to examine the protective role of IFNs in CVA16 infection by challenging 14-day-old IFN-α/βR−/− and IFN-γR−/− mice, which lack the receptors for type I IFNs and IFN-γ, respectively. After infection with CVA16 at 1.5 × 104 PFU/mouse, 90% of the WT mice displayed symptoms, including ruffled hair, hunched back, and limb weakness, starting from day 4 postinfection, and 75% died of paralysis within 12 days. One hundred percent of IFN-γR−/− mice became ill starting from day 3 postinfection, but the mortality rate of IFN-γR−/− mice was not significantly different from that of WT mice (Fig. 1C), suggesting that IFN-γ does not play a large role in host protection against CVA16 infection. In contrast, IFN-α/βR−/− mice that were infected with CVA16 at the same dosage died earlier and had a significantly (P = 0.0044) reduced survival rate (Fig. 1D). With infection with CVA16 at a lower dosage, 80% of IFN-α/βR−/− mice died within 11 days, but all the infected WT mice survived (Fig. 1E). These observations imply that type I IFNs play a protective role in CVA16 infection. Thus, in contrast to EV71 infection (21), type I IFNs are important in protecting mice from CVA16 infection, but IFN-γ is not.
We next tested whether CVA16 infection induced type I IFN expression. We infected WT BMMs with CVA16 and analyzed their expression of type I IFNs. The infection significantly increased the expression levels of all the tested IFNs, including IFN-α4, IFN-non-α4, and IFN-β (Fig. 1F). Altogether, induction of type I IFNs is involved in protective immunity against CVA16 infection.
The TLR3-TRIF pathway plays a critical role in eliciting innate immunity to protect against CVA16 infection.
We next investigated which PRR is critical in protecting young mice against CVA16 infection. We infected 14-day-old WT and TLR3-, TRIF-, TLR7-, and MAVS-knockout mice with CVA16 at a dose of 1.5 × 104 or 1.5 × 103 PFU. At the higher dosage of infection, all the mice deficient for TLR3 and TRIF exhibited paralysis starting from day 4, died before 14 days postinfection, and had significantly (P = 0.0082 and 0.0033, respectively) lower survival rates than WT mice (Fig. 2A). At the lower dosage of infection, only 30% of WT mice had symptoms, and none of the infections was fatal. In contrast, 100% of mice deficient in TLR3 and TRIF displayed hind limb weakness or paralysis, resulting in decreased survival rates (25% and 37.5%, respectively) (Fig. 2B). These observations suggest that TLR3-TRIF signaling is essential for eliciting immune responses to control CVA16 infection in young mice.
Although CVA16-infected MAVS−/− mice developed symptoms slightly earlier with the higher dosage of CVA16 infection, their survival rates did not differ from those of WT mice, regardless of the infection dosage (Fig. 2C and D), suggesting that neither RIG-I nor the MDA5-MVAS pathway is indispensable for mediating innate immunity against CVA16 infection in young mice. At the higher dose of CVA16 infection, TLR7−/− mice tended to die earlier than WT mice, but their overall survival rate was not significantly (P = 0.0994 and 0.3613, for the higher and lower dose, respectively) different from that of infected WT mice (Fig. 2E and F). Altogether, TLR3 but not TLR7 or MAVS is critical for protective immunity against CVA16 infection in young mice.
TRIF signaling is required to trigger IFN-β production.
Next, we investigated whether TRIF signaling affects the production of type I IFNs. We infected WT and TRIF−/− mice with 1.5 × 104 PFU of CVA16 and examined IFN-β expression in different tissues. On day 2 postinfection, the levels of IFN-β expression in all tested tissues of TRIF−/− mice tended to be diminished relative to those in wild-type mice; in particular, the levels in skeletal muscle, small intestine, kidney, and lung tissues were about 50- to 1,000-fold, on average, lower than those in the tissues of WT mice (Fig. 3A). The dramatic reduction in IFN-β levels in skeletal muscle, small intestine, and lung was not observed on day 4 postinfection (Fig. 3B). In contrast, IFN-β levels in the heart, spinal cord, and brain of TRIF−/− mice decreased by about 200-fold relative to those in WT mice (Fig. 3B). Noticeably, the IFN-β reduction was followed by the progression of CVA16 infection to fatal disease (Fig. 2A). These results suggest that TLR3-TRIF signaling is required for IFN-β induction in the neuronal system to prevent fatality in CVA16-infected mice.
To further corroborate the dependence of IFN-β induction on TLR3-TRIF signaling, we also examined IFN-β expression in TLR3- or TRIF-deficient BMDCs and BMMs. CVA16 infection led to the induction of IFN-β in WT BMDCs and BMMs of about 100- and 50-fold, respectively (Fig. 3C to F). However, TLR3 or TRIF deficiency abolished IFN-β induction in both BMDCs and BMMs (Fig. 3C to F), suggesting that TLR3-TRIF signaling is required for IFN-β induction in these innate immune cells.
In contrast to the findings for BMDCs and BMMs, IFN-β expression was not induced at all in MEFs that were infected with CVA16, as IFN-β levels in CVA16-infected and uninfected cells were not significantly different. TRIF deficiency did not affect IFN-β levels either (Fig. 3G). In contrast to the significant increase in the viral loads in CVA16-infected BMDCs and BMMs, the viral loads did not significantly change in MEFs after infection (Fig. 3H). These findings, together with evidence that UV-inactivated CVA16 failed to induce IFN-β expression (Fig. 3I), suggest that CVA16 replication is required for IFN-β expression triggered by TLR3-TRIF signaling.
IFN-β treatment prevents severe disease in CVA16-infected TRIF-deficient mice.
The reduction in the level of IFN-β in TRIF-deficient mice led to us wonder whether the administration of IFN-β could be effective in treating CVA16-infected TRIF-deficient mice. We infected TRIF-deficient mice with 1.5 × 103 or 1.5 × 104 PFU/mouse of CVA16 and then treated the infected mice with recombinant IFN-β at a dosage of 3 × 104 units per day per mouse for 3 days starting at 24 h postinfection. The treatment did not significantly affect the survival rate of TRIF-deficient mice infected with CVA16 at the high dosage (data not shown). However, the treatment significantly (P < 0.0001) lowered the clinical disease score (Fig. 4A) and also significantly (P = 0.0066) improved the survival of TRIF-deficient mice infected with CVA16 at the low dosage (Fig. 4B). Thus, these results suggest that supplemental type I IFNs can compensate for the diminished protective immunity due to a defect in TLR3-TRIF signaling.
FIG 4.
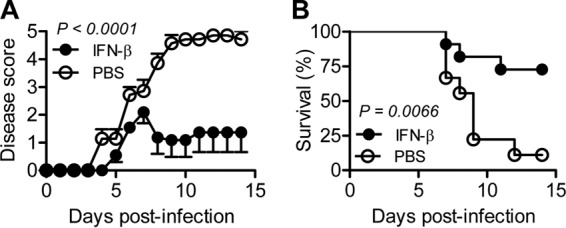
Therapeutic effects of IFN-β on CVA16-infected TRIF-deficient mice. Fourteen-day-old TRIF-deficient C57BL/6 mice were infected i.p. with 1.5 × 103 PFU CVA16 and then injected i.p. with 3 × 104 units of IFN-β per mouse per day (n = 11) or saline (n = 9) on days 1, 2, and 3 postinfection. The clinical scores (A) and survival rates (B) are shown.
TRIF signaling protects young mice from invasion of CVA16 into the neuronal system.
To reveal the protective mechanism mediated by TLR3-TRIF signaling in CVA16 infection, we performed H&E staining of tissues from PBS-injected WT mice and CVA16-infected WT or TRIF−/− mice. Compared to the pathology in the PBS-treated group, skeletal muscle tissues in both CVA16-infected WT and TRIF−/− mice displayed similarly severe necrosis (Fig. 5A, white arrow) and myonecrosis associated with leukocyte infiltration (Fig. 5A, black arrow). In contrast, the spinal cord (anterior horn area) and brain (the brain stem reticular formation) of TRIF−/− mice showed much more severe damage than the spinal cord and brain of WT mice, including genuine basophilic necrotic neurons (Fig. 5A, black arrow), neuropil vacuolation and neuronal degeneration or loss (Fig. 5A, white arrow), and neuronal granulovacuolar changes (Fig. 5A, black arrowhead). These observations suggest that TLR3-TRIF signaling can prevent CVA16 invasion in the mouse CNS but not skeletal muscle.
FIG 5.

Pathological changes and viral loads in CVA16-infected WT and TRIF-deficient mice. Fourteen-day-old WT or TRIF-deficient C57BL/6 mice were infected i.p. with 1.5 × 104 PFU CVA16. (A) The infected mice were euthanized at 4 days postinfection, and paraffin-embedded sections of skeletal muscle, spinal cord (anterior horn area), and brain (the brain stem reticular formation) were examined at ×200, ×200, and ×400 magnifications with H&E staining. White arrow (skeletal muscle), necrosis; black arrow (skeletal muscle), myonecrosis associated with leukocyte infiltration; white arrow (spinal cord and brain), neuropil vacuolation and neuronal degeneration or loss; black arrow (spinal cord and brain), genuine basophilic necrotic neurons; black arrowhead (spinal cord and brain), neuronal granulovacuolar change. (B and C) The viral loads in the indicated tissues of infected mice were measured by qRT-PCR on days 2 (B) and 4 (C). Data are shown as the mean ± SEM and are representative of those from three independent experiments (n = 3). ns, not significant; ***, P < 0.001; UD, undetectable.
We also examined the viral loads in different tissues of WT and TRIF−/− mice on days 2 and 4 postinfection with CVA16. Although the virus was detectable in all the tested tissues in both types of mice on day 2 postinfection, TRIF deficiency tended to increase the viral loads in spinal cord and brain tissues (Fig. 5B). Noticeably, the virus was detectable only in skeletal muscle, spinal cord, and brain tissues of both types of mice on day 4 postinfection. Although the viral loads in the skeletal muscle of TRIF−/− mice were comparable to those in the skeletal muscle of WT mice, the viral loads in the spinal cord and brain of TRIF−/− mice increased dramatically by an average of 50 and 100 times, respectively (Fig. 5C). In summary, in young mice TRIF signaling is essential for protective immunity against the invasion of CVA16 into the nervous system.
DISCUSSION
CVA16 and EV71, two major etiological agents of HFMD, are genetically similar to each other, and their nonstructural and conservative structural proteins share similar functions (23). In terms of the severity of HFMD caused by these two pathogens, CVA16 is, interestingly, much less virulent than EV71 (5, 29). It remains unclear how CVA16 infection differently elicits immune responses and causes pathogenesis in the host. The present study revealed that TLR3 deficiency caused young mice to be more vulnerable to CVA16 infection, as was previously observed with EV71 infection (22). However, inconsistent with their protective role against EV71 infection (22), iNKT cells are dispensable for protecting the host against CVA16 infection. Rather, type I IFNs are required for protective immunity against CVA16 infection in young mice. In addition, the induction of type I IFNs both in vivo and in vitro depended on the TLR3-TRIF signaling pathway. Furthermore, IFN-β treatment could significantly reduce disease development in TRIF-deficient mice that were infected with a low dosage of CVA16. Collectively, type I IFNs play a critical role in host protective immunity against CVA16 infection, which differs from their role in EV71 infection (21). The difference in host immune responses to EV71 and CVA16 infections may explain the distinct clinical outcomes of patients with HFMD caused by the two viruses.
The dependence on type I IFNs but not IFN-γ and iNKT cells for protection against CVA16 infection is unexpected and distinct from the observations of EV71 infection (21, 22). However, our observations are consistent with the findings reported by the Tien group that CVA16 and EV71 infections have opposing effects on the response of rhabdomyosarcoma (RD) cells to type I IFNs (30). Specifically, CVA16 infection normally enhances the signaling triggered by type I IFNs, but EV71 infection represses it (30). The opposing effects may reflect a difference in viral tropisms in the same types of cells or different cell types. Evidently, patients with severe HFMD caused by CVA16 infection display myocarditis and pneumonitis (8, 9) but rarely display CNS complications, which are frequently caused by EV71 infection (5, 29). Clinical CVA16 isolates are capable of infecting young mice without adaptation (25), but EV71 frequently requires adaptation to acquire the capability to infect young mice (22, 31–33). Thus, the molecular and genetic basis that contributes to the different tropisms of CVA16 infection merits further investigation.
Our study revealed that a decreased induction of IFN-β in spinal cord and brain at a late time postinfection (day 4) due to TRIF deficiency was associated with increased CVA16 replication and a more severe CNS pathology (Fig. 3A and B and 5A to C). This finding indicates that the induction of IFN-β in the CNS by TLR3-TRIF signaling may prevent neuronal pathology by directly inhibiting CVA16 replication. Although this hypothesis needs to be further investigated with tissue-specific TLR3- or TRIF-knockout mice, it was indirectly supported by the evidence that CVA16 infection caused both autophagy (34) and apoptosis of neural cells in vitro (35). Therefore, it is plausible that higher viral loads in the brains of CVA16-infected TRIF-knockout mice may cause neuronal cell death and thereby contribute to neuronal pathogenesis.
Our present study revealed that the signaling triggered by TLR3 but not other PRRs plays a major role in the induction of type I IFNs and protective immunity against CVA16 infection. The following reasons can probably explain it. First, a study of poliovirus has suggested that uncoating of nonenveloped virus depends on acidification of early endosomes (36). The structure study also revealed that the entry of CVA16 is highly dependent on endocytosis (37). It is also known that TLR3 is mainly located in intracellular compartments, such as the endosome (38), while RIG-I-like receptors (RLRs), including RIG-I and MDA5, are expressed in the cytoplasm (39), suggesting that TLR3 is mainly involved in sensing CVA16 infection during innate immune responses, whereas RLRs are not. Second, TLR7 is mainly expressed in plasmacytoid dendritic cells (pDCs), rare cells in vivo, while TLR3 is expressed by more abundant conventional dendritic cells and macrophages as well as nonimmune cells, including fibroblasts and epithelial cells. It is also possible that mouse pDCs do not express the molecules that capture CVA16 particles or mediate CVA16 infection or replication. Finally, our experiments revealed that viral replication is needed to trigger IFN-β expression, while single-stranded RNA (ssRNA) containing inactivated CVA16 was not effective at the induction of IFN-β expression (Fig. 3H and I). These results also do not support the possibility that ssRNA sensors, including TLR7, play an important role in IFN-β expression. Nevertheless, the detailed mechanisms merit further investigation.
The differences in the host immune responses to EV71 and CVA16 infections suggest that different mechanisms are involved in the pathogenesis of HFMD caused by infections with these viruses. In addition to EV71 and CVA16, other enteroviruses, such as CVA4, CVA6, CVA10, and CVB1 to CVB5, are also associated with the disease (29, 40). Therefore, this also implies that therapeutic strategies for different forms of HFMD must be developed, given that their mechanisms of pathogenesis are likely dramatically different due to the different causative pathogens. In particular, the efficacy of type I IFNs as a treatment in patients with CVA16 infections most likely will be different from that in patients with EV71 infection.
Collectively, the present study demonstrated that type I IFNs play a critical role in host defense against CVA16 infection. TLR3-TRIF signaling was indispensable for the induction of type I IFNs during CVA16 infection in mice, as deficiencies in both TLR3 and TRIF increased disease severity in and mortality of CVA16-infected mice. Whether deficiency in the TLR3 or type I IFN signaling pathways contributes to the disease severity in HFMD patients with CVA16 infection requires further genetic and immunological studies.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (grant numbers 31270951 and 81172807), the National Basic Research Program of China (973 Programs, grant number 2011CB504903), and the Total Foundation.
PBS57-loaded CD1d and unloaded mouse CD1d were provided by the NIH Tetramer Facility at Emory University.
REFERENCES
- 1.Sickles GM, Mutterer M, Feorino P, Plager H. 1955. Recently classified types of coxsackie virus, group A; behavior in tissue culture. Proc Soc Exp Biol Med 90:529–531. doi: 10.3181/00379727-90-22088. [DOI] [PubMed] [Google Scholar]
- 2.Bendig JW, Fleming DM. 1996. Epidemiological, virological, and clinical features of an epidemic of hand, foot, and mouth disease in England and Wales. Commun Dis Rep CDR Rev 6:R81–R86. [PubMed] [Google Scholar]
- 3.Chang LY. 2008. Enterovirus 71 in Taiwan. Pediatr Neonatol 49:103–112. doi: 10.1016/S1875-9572(08)60023-6. [DOI] [PubMed] [Google Scholar]
- 4.Ang LW, Koh BK, Chan KP, Chua LT, James L, Goh KT. 2009. Epidemiology and control of hand, foot and mouth disease in Singapore, 2001-2007. Ann Acad Med Singapore 38:106–112. [PubMed] [Google Scholar]
- 5.Chang LY, Lin TY, Huang YC, Tsao KC, Shih SR, Kuo ML, Ning HC, Chung PW, Kang CM. 1999. Comparison of enterovirus 71 and coxsackie-virus A16 clinical illnesses during the Taiwan enterovirus epidemic, 1998. Pediatr Infect Dis J 18:1092–1096. doi: 10.1097/00006454-199912000-00013. [DOI] [PubMed] [Google Scholar]
- 6.Chen X, Tan X, Li J, Jin Y, Gong L, Hong M, Shi Y, Zhu S, Zhang B, Zhang S, Zhang Y, Mao N, Xu W. 2013. Molecular epidemiology of coxsackievirus A16: intratype and prevalent intertype recombination identified. PLoS One 8:e82861. doi: 10.1371/journal.pone.0082861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu W, Wu S, Xiong Y, Li T, Wen Z, Yan M, Qin K, Liu Y, Wu J. 2014. Co-circulation and genomic recombination of coxsackievirus A16 and enterovirus 71 during a large outbreak of hand, foot, and mouth disease in central China. PLoS One 9:e96051. doi: 10.1371/journal.pone.0096051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang CY, Li Lu F, Wu MH, Lee CY, Huang LM. 2004. Fatal coxsackievirus A16 infection. Pediatr Infect Dis J 23:275–276. doi: 10.1097/01.inf.0000115950.63906.78. [DOI] [PubMed] [Google Scholar]
- 9.Legay F, Leveque N, Gacouin A, Tattevin P, Bouet J, Thomas R, Chomelt JJ. 2007. Fatal coxsackievirus A-16 pneumonitis in adult. Emerg Infect Dis 13:1084–1086. doi: 10.3201/eid1307.070295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang LY, Lin TY, Hsu KH, Huang YC, Lin KL, Hsueh C, Shih SR, Ning HC, Hwang MS, Wang HS, Lee CY. 1999. Clinical features and risk factors of pulmonary oedema after enterovirus-71-related hand, foot, and mouth disease. Lancet 354:1682–1686. doi: 10.1016/S0140-6736(99)04434-7. [DOI] [PubMed] [Google Scholar]
- 11.Chang LY, Huang LM, Gau SS, Wu YY, Hsia SH, Fan TY, Lin KL, Huang YC, Lu CY, Lin TY. 2007. Neurodevelopment and cognition in children after enterovirus 71 infection. N Engl J Med 356:1226–1234. doi: 10.1056/NEJMoa065954. [DOI] [PubMed] [Google Scholar]
- 12.Chang LY, Tsao KC, Hsia SH, Shih SR, Huang CG, Chan WK, Hsu KH, Fang TY, Huang YC, Lin TY. 2004. Transmission and clinical features of enterovirus 71 infections in household contacts in Taiwan. JAMA 291:222–227. doi: 10.1001/jama.291.2.222. [DOI] [PubMed] [Google Scholar]
- 13.Ho M, Chen ER, Hsu KH, Twu SJ, Chen KT, Tsai SF, Wang JR, Shih SR. 1999. An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. N Engl J Med 341:929–935. [DOI] [PubMed] [Google Scholar]
- 14.Chehadeh W, Alkhabbaz M. 2013. Differential TLR7-mediated expression of proinflammatory and antiviral cytokines in response to laboratory and clinical enterovirus strains. Virus Res 174:88–94. doi: 10.1016/j.virusres.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 15.Wang JP, Cerny A, Asher DR, Kurt-Jones EA, Bronson RT, Finberg RW. 2010. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J Virol 84:254–260. doi: 10.1128/JVI.00631-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oshiumi H, Okamoto M, Fujii K, Kawanishi T, Matsumoto M, Koike S, Seya T. 2011. The TLR3/TICAM-1 pathway is mandatory for innate immune responses to poliovirus infection. J Immunol 187:5320–5327. doi: 10.4049/jimmunol.1101503. [DOI] [PubMed] [Google Scholar]
- 17.Lei X, Liu X, Ma Y, Sun Z, Yang Y, Jin Q, He B, Wang J. 2010. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J Virol 84:8051–8061. doi: 10.1128/JVI.02491-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. 2011. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J Virol 85:8811–8818. doi: 10.1128/JVI.00447-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, Jin Q, Zhao Z. 2013. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog 9:e1003231. doi: 10.1371/journal.ppat.1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu ML, Lee YP, Wang YF, Lei HY, Liu CC, Wang SM, Su IJ, Wang JR, Yeh TM, Chen SH, Yu CK. 2005. Type I interferons protect mice against enterovirus 71 infection. J Gen Virol 86:3263–3269. doi: 10.1099/vir.0.81195-0. [DOI] [PubMed] [Google Scholar]
- 21.Liao CC, Liou AT, Chang YS, Wu SY, Chang CS, Lee CK, Kung JT, Tu PH, Yu YY, Lin CY, Lin JS, Shih C. 2014. Immunodeficient mouse models with different disease profiles by in vivo infection with the same clinical isolate of enterovirus 71. J Virol 88:12485–12499. doi: 10.1128/JVI.00692-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu K, Yang J, Luo K, Yang C, Zhang N, Xu R, Chen J, Jin M, Xu B, Guo N, Wang J, Chen Z, Cui Y, Zhao H, Wang Y, Deng C, Bai L, Ge B, Qin CF, Shen H, Yang CF, Leng Q. 2015. TLR3 signaling in macrophages is indispensable for the protective immunity of invariant natural killer T cells against enterovirus 71 infection. PLoS Pathog 11:e1004613. doi: 10.1371/journal.ppat.1004613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan YF, AbuBaker S. 2004. Recombinant human enterovirus 71 in hand, foot and mouth disease patients. Emerg Infect Dis 10:1468–1470. doi: 10.3201/eid1008.040059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ministry of Science and Technology. 2006. Guide for the care and use of laboratory animals. Ministry of Science and Technology, Beijing, People's Republic of China. http://www.most.gov.cn/fggw/zfwj/zfwj2006/200609/t20060930_54389.htm.
- 25.Zhao H, Li HY, Han JF, Deng YQ, Zhu SY, Li XF, Yang HQ, Li YX, Zhang Y, Qin ED, Chen R, Qin CF. 2015. Novel recombinant chimeric virus-like particle is immunogenic and protective against both enterovirus 71 and coxsackievirus A16 in mice. Sci Rep 5:7878. doi: 10.1038/srep07878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weischenfeldt J, Porse B. 2008. Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protocols 2008:pdb.prot5080. doi: 10.1101/pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- 27.Jozefczuk J, Drews K, Adjaye J. 2012. Preparation of mouse embryonic fibroblast cells suitable for culturing human embryonic and induced pluripotent stem cells. J Vis Exp 2012:3854. doi: 10.3791/3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khong WX, Yan B, Yeo H, Tan EL, Lee JJ, Ng JK, Chow VT, Alonso S. 2012. A non-mouse-adapted enterovirus 71 (EV71) strain exhibits neurotropism causing neurological manifestations in a novel mouse model of EV71 infection. J Virol 86:2121–2131. doi: 10.1128/JVI.06103-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xing W, Liao Q, Viboud C, Zhang J, Sun J, Wu JT, Chang Z, Liu F, Fang VJ, Zheng Y, Cowling BJ, Varma JK, Farrar JJ, Leung GM, Yu H. 2014. Hand, foot, and mouth disease in China, 2008-12: an epidemiological study. Lancet Infect Dis 14:308–318. doi: 10.1016/S1473-3099(13)70342-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang W, Zhang L, Wu Z, Tien P. 2014. Differential interferon pathway gene expression patterns in rhabdomyosarcoma cells during enterovirus 71 or coxsackievirus A16 infection. Biochem Biophys Res Commun 447:550–555. doi: 10.1016/j.bbrc.2014.04.021. [DOI] [PubMed] [Google Scholar]
- 31.Chen CS, Yao YC, Lin SC, Lee YP, Wang YF, Wang JR, Liu CC, Lei HY, Yu CK. 2007. Retrograde axonal transport: a major transmission route of enterovirus 71 in mice. J Virol 81:8996–9003. doi: 10.1128/JVI.00236-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin YW, Chang KC, Kao CM, Chang SP, Tung YY, Chen SH. 2009. Lymphocyte and antibody responses reduce enterovirus 71 lethality in mice by decreasing tissue viral loads. J Virol 83:6477–6483. doi: 10.1128/JVI.00434-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang YF, Chou CT, Lei HY, Liu CC, Wang SM, Yan JJ, Su IJ, Wang JR, Yeh TM, Chen SH, Yu CK. 2004. A mouse-adapted enterovirus 71 strain causes neurological disease in mice after oral infection. J Virol 78:7916–7924. doi: 10.1128/JVI.78.15.7916-7924.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi Y, He X, Zhu G, Tu H, Liu Z, Li W, Han S, Yin J, Peng B, Liu W. 2015. Coxsackievirus A16 elicits incomplete autophagy involving the mTOR and ERK pathways. PLoS One 10:e0122109. doi: 10.1371/journal.pone.0122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Z, Yu J, Liu L, Wei Z, Ehrlich ES, Liu G, Li J, Liu X, Wang H, Yu XF, Zhang W. 2014. Coxsackievirus A16 infection induces neural cell and non-neural cell apoptosis in vitro. PLoS One 9:e111174. doi: 10.1371/journal.pone.0111174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brandenburg B, Lee LY, Lakadamyali M, Rust MJ, Zhuang X, Hogle JM. 2007. Imaging poliovirus entry in live cells. PLoS Biol 5:e183. doi: 10.1371/journal.pbio.0050183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren J, Wang X, Hu Z, Gao Q, Sun Y, Li X, Porta C, Walter TS, Gilbert RJ, Zhao Y, Axford D, Williams M, McAuley K, Rowlands DJ, Yin W, Wang J, Stuart DI, Rao Z, Fry EE. 2013. Picornavirus uncoating intermediate captured in atomic detail. Nat Commun 4:1929. doi: 10.1038/ncomms2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsumoto M, Funami K, Tanabe M, Oshiumi H, Shingai M, Seto Y, Yamamoto A, Seya T. 2003. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol 171:3154–3162. doi: 10.4049/jimmunol.171.6.3154. [DOI] [PubMed] [Google Scholar]
- 39.Loo YM, Gale M Jr. 2011. Immune signaling by RIG-I-like receptors. Immunity 34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yen FB, Chang LY, Kao CL, Lee PI, Chen CM, Lee CY, Shao PL, Wang SC, Lu CY, Huang LM. 2009. Coxsackieviruses infection in northern Taiwan—epidemiology and clinical characteristics. J Microbiol Immunol Infect 42:38–46. [PubMed] [Google Scholar]