

ABSTRACT
We recently discovered that desmoglein 2 (DSG2) is a receptor for human adenovirus species B serotypes Ad3, Ad7, Ad11, and Ad14. Ad3 is considered to be a widely distributed human pathogen. Ad3 binding to DSG2 triggers the transient opening of epithelial junctions. Here, we further delineate the mechanism that leads to DSG2-mediated epithelial junction opening in cells exposed to Ad3 and recombinant Ad3 fiber proteins. We identified an Ad3 fiber knob-dependent pathway that involves the phosphorylation of mitogen-activated protein (MAP) kinases triggering the activation of the matrix-metalloproteinase ADAM17. ADAM17, in turn, cleaves the extracellular domain of DSG2 that links epithelial cells together. The shed DSG2 domain can be detected in cell culture supernatant and also in serum of mice with established human xenograft tumors. We then extended our studies to Ad14 and Ad14P1. Ad14 is an important research and clinical object because of the recent appearance of a new, more pathogenic strain (Ad14P1). In a human epithelial cancer xenograft model, Ad14P1 showed more efficient viral spread and oncolysis than Ad14. Here, we tested the hypothesis that a mutation in the Ad14P1 fiber knob could account for the differences between the two strains. While our X-ray crystallography studies suggested an altered three-dimensional (3D) structure of the Ad14P1 fiber knob in the F-G loop region, this did not significantly change the fiber knob affinity to DSG2 or the intracellular signaling and DSG2 shedding in epithelial cancer cells.
IMPORTANCE A number of widely distributed adenoviruses use the epithelial junction protein DSG2 as a receptor for infection and lateral spread. Interaction with DSG2 allows the virus not only to enter cells but also to open epithelial junctions which form a physical barrier to virus spread. Our study elucidates the mechanism beyond virus-triggered junction opening with a focus on adenovirus serotype 3. Ad3 binds to DSG2 with its fiber knob domain and triggers intracellular signaling that culminates in the cleavage of the extracellular domain of DSG2, thereby disrupting DSG2 homodimers between epithelial cells. We confirmed this pathway with a second DSG2-interacting serotype, Ad14, and its recently emerged strain Ad14P1. These new insights in basic adenovirus biology can be employed to develop novel drugs to treat adenovirus infection as well as be used as tools for gene delivery into epithelial tissues or epithelial tumors.
INTRODUCTION
We recently discovered that desmoglein 2 (DSG2) is a receptor for human adenovirus species B serotypes Ad3, Ad7, Ad11, and Ad14 (1–3). DSG2 is a calcium-binding transmembrane glycoprotein belonging to the cadherin protein family. In epithelial cells of the respiratory, gastrointestinal, and urinary tracts, DSG2 is a component of the cell-cell adhesion structure (4). It is well established that in addition to maintaining cell adhesion, DSG2 is also involved in intracellular signaling (5). Its cytoplasmic tail interacts with a series of proteins, including plakoglobin and plakophilins, that are in direct contact with regulators of cell adhesion and intercellular junctions/cell morphology (6). Plakoglobin and plakophilins not only interact with intermediate filaments but can also localize to the nucleus and bind to transcription factors (e.g., the T cell factor/lymphoid enhancer factor [TCF/LEF]) (7) or DNA binding proteins (e.g., p53) (8), thus influencing gene expression and cell cycling. While the signaling cascade mediated by the adherens junction protein β-catenin and the regulation of classical E-cadherin-mediated cell-cell interactions are relatively well studied, little is known about signaling mediated by desmosomal proteins. It has been reported that epidermal growth factor receptor (EGFR) activation triggers tyrosine phosphorylation of DSG2 and plakoglobin and subsequent modulation of cell-cell interaction (9, 10), in part through the activation of matrix metalloprotease (MMP) cleavage of DSG2 homodimers between neighboring epithelial cells (11).
In the present study, we focus on studying intracellular signaling triggered by the DSG2-interacting serotypes Ad3 and Ad14. Ad3 is considered to be a widely distributed human pathogen. Studies from the United States and Europe show that Ad3 infections occur more often in adolescents and adults (12–15), while studies from Asia indicate that Ad3 is prevalent in young children, often causing severe respiratory symptoms (16–18). Ad14 is an important research and clinical object because of the recent appearance of a new strain (Ad14P1). Never previously documented in the United States, Ad14P1 was first reported in March and April 2006 during routine surveillance at several U.S. military recruit training centers (13). During March to June of the following year, a total of 140 additional cases of confirmed Ad14P1 respiratory illness were reported in patients in Oregon, Washington, and Texas. Thirty-eight percent of these patients were hospitalized, including 17% who were admitted to intensive care units; 5% of patients died. Outbreaks of Ad14P1 infection were subsequently detected in other five bases and in civilian populations in Washington (19), Oregon (20), Alaska (21), Wisconsin, and Pennsylvania (22, 23), as well as in Canada (24), China (25), and South Korea (26). At this point, the molecular basis for the high pathogenicity and/or virulence of Ad14P1 is unclear.
To study intracellular signaling triggered by viruses, we employed recombinant subviral particles and recombinant fiber knob domains, i.e., the moiety within the viral capsid that interacts with DSG2. During Ad infection, the penton base and fiber proteins are produced in excess and assemble in the cytosol to form fiber-penton base hetero-oligomers called pentons (27, 28). In the case of Ad3, Ad14, and Ad14P1, 12 pentons self-assemble into dodecamers (penton-dodecahedra, or PtDd) with a diameter of ∼30 nm (29, 30). Recently, we demonstrated that the ability of Ad3 to produce PtDd is critically important for virus spread in epithelial tumors and most likely also during Ad3 infection of the airway tract in humans (30). PtDd also form in insect cells during overexpression of recombinant Ad3 penton base and fiber (31). Insect cells that express only the Ad3 penton base assemble penton base dodecahedra (BsDd). Recombinant PtDd and BsDd produced in insect cells can be purified by density gradient ultracentrifugation. To study intracellular signaling, in addition to Ad3 virus, we used recombinant PtDd and BsDd. Furthermore, we produced recombinant Ad3 fiber knob domains in Escherichia coli. High-affinity DSG2 binding and efficient competition of Ad3 infection required that several fiber knobs were in a specific spatial constellation (32). This was achieved by linking the coding sequence of the Ad3 fiber knob to a K-coil domain. A protein that contained the K-coil domain and Ad3 fiber knob was called JO1 (33). JO1 can be purified by affinity chromatography. We are currently developing JO1 derivatives for clinical applications to increase the penetration of chemotherapy drugs in epithelial tumors (34–36).
Previously, we showed that the binding of Ad3 virions, PtDd, and JO1 to DSG2 triggers intracellular signaling that culminates in the transient opening of intercellular junctions in epithelial cells. Using mRNA expression arrays and quantitative reverse transcription-PCR (qRT-PCR), we found that 12 h after the incubation of polarized breast cancer epithelial cells with PtDd, 430 genes were upregulated and that 352 genes were downregulated compared to results for incubation with BsDd. Altered gene expression indicated the activation of a pathways such as phosphatidylinositol (PI), mitogen-activated protein kinase (MAPK), Wnt, adherens junctions, focal adhesion, and regulation of actin cytoskeleton signaling pathways (3). We also showed that downregulation of DSG2 by small interfering RNA (siRNA) inhibited MAPK signaling (3, 37).
In the present study, we further delineated the mechanism that leads to DSG2-mediated epithelial junction opening in cells exposed to Ad3, PtDd, and JO1. We identified an Ad3 fiber knob-dependent pathway that involves the phosphorylation of MAPK and the activation of the matrix metalloproteinase ADAM17. ADAM17 in turn cleaves the extracellular domain (ECD) of DSG2 that links epithelial cells together. The shed DSG2 domain can be detected in cell culture supernatant and also in serum of mice with established human xenograft tumors after exposure to Ad3 virus, PtDd, or JO1. We then extended our studies to Ad14 and Ad14P1. We performed X-ray crystallography and three-dimensional (3D) structure comparison of the Ad14 and Ad14P1 fiber knobs and found differences in the conformation of the F-G loop of the fiber knob domain. These structural differences, however, did not change the affinity of the Ad14P1 fiber knob to DSG2 nor the intracellular signaling and DSG2 shedding mediated by Ad14P1 virus.
MATERIALS AND METHODS
Adenoviruses.
Ad3 virus (GB strain) and Ad14 were obtained from the ATCC. Ad14P1 virus, strain Portland 2971/2007, was provided by the Centers for Disease Control and Prevention (Atlanta, GA) (38). Propagation, [methyl-3H]thymidine labeling, purification, and titers of Ads were determined as described elsewhere (39). Ad3-green fluorescent protein (GFP) is a wild-type (wt) Ad3-based vector containing a cytomegalovirus (CMV)-GFP expression cassette inserted into the E3 region (3). Viral particle (VP) concentrations were determined spectrophotometrically by measuring the optical density at 260 nm (OD260). Titers of PFU were determined using 293 cells as described elsewhere (40). The VP-to-PFU ratio was 20:1 for all virus preparations.
Proteins.
Recombinant human DSG2 protein was from Leinco Technologies, Inc. (St. Louis, MO). The Ad3, Ad14, and Ad14P1 fiber knobs and JO1 were produced in E. coli with N-terminal 6×His tags using the pQE30 expression vector (Qiagen, Valencia, CA) and purified by Ni-nitrilotriacetic acid (NTA)-agarose chromatography as described elsewhere (41). Recombinant Ad3 penton-dodecahedra (PtDd) and base dodecahedra (BsDd) were produced in insect cells and purified as described previously (31).
Antibodies.
The following antibodies were used: mouse monoclonal antibody (MAb) anti-DSG2 (clone 6D8; Cell Sciences, Canton, MA), mouse MAb anti-DSG2 (clone AH12.2; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), polyclonal anti-DSG2 antibody (AF947; R&D Systems), anti-proDSG2 (clone 20G1; provided by James K. Wahl III, University of Nebraska Medical Center [3]), rabbit MAb anti-p38 (clone D13E1; Cell Signaling Technology, Danvers, MA), rabbit MAb anti-pp38 (clone D3F9; Cell Signaling Technology, Danvers, MA), polyclonal rabbit anti-ADAM17 antibody (Millipore), and mouse MAb anti-β-actin (clone Ac-74; Sigma).
Inhibitors.
The MAPK inhibitor U-0216 was from Sigma, and the ADAM17 inhibitor TAPI-0 was from EMD Millipore Corp. (Billerica, MA).
Cell lines.
293, HeLa, and A549 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin (P/S), 2 mM glutamine (Glu), and 1× minimal essential medium (MEM) nonessential-amino-acid solution (Invitrogen, Carlsbad, CA). Colon cancer HT29 cells (ATCC CCL-248) were cultured in a 1:1 mixture of Ham's F12 medium and DMEM with 10% FBS, Glu, and P/S. SKOV3-ip1 cells were cultured in McCoys medium supplemented with 10% FBS, Glu, and P/S.
Western blotting.
Mini-Protean precast gels (Bio-Rad, Hercules, CA) with a 4 to 15% polyacrylamide gradient were used. A total of 1 μg of protein mixed with 2× loading buffer (10 mM Tris-HCl, pH 6.8, 200 mM dithiothreitol [DTT], 4% SDS, 20% glycerol, 0.2% bromophenol blue) was loaded per lane. Samples were either boiled (B) for 5 min or loaded unboiled (UB). The following running buffer was used: 25 mM Tris, pH 8.3, 0.192 M glycine, and 0.1% SDS. After electrophoresis, proteins were transferred to nitrocellulose and incubated with the corresponding antibodies. For detection of DSG2 binding, filters were incubated with recombinant human DSG2 protein and anti-DSG2 antibody 6D8 as described previously (3). Other Western blotting experiments used a primary mouse monoclonal antibody followed by an anti-mouse IgG horseradish peroxidase (HRP) conjugate. Selected Western blots were scanned and quantified using ImageJ, version 1.32, software (National Institutes of Health, Bethesda, MD).
DSG2 ELISA.
An enzyme-linked immunosorbent assay (ELISA) was performed using a goat polyclonal anti-DSG2 antibody (R&D Systems, Minneapolis, MN) and the mouse monoclonal antibody 6D8 directed against ECD3 (AbD Serotec, Raleigh, NC). The detection limit of the DSG2 ELISA was 0.5 ng/ml.
Anti-Ad14 fiber knob antibody ELISA.
A rabbit polyclonal antibody against the Ad14 fiber knob was used as a capture antibody, followed by recombinant Ad14 or Ad14P1 fiber knob and a 1:200 dilution of human serum. Binding of human IgG antibodies was detected using an anti-human IgG-HRP conjugate. Pooled normal human serum (NHS) was used for comparison (lot 16923; Innovative Research, Novi, MI).
MS/MS.
Confluent A549 cells were cultured in serum-free 293 cell medium (Freestyle 293; Invitrogen, Carlsbad, CA) supplemented with 1 μg/ml PtDd for 11 h. Culture medium was then collected and centrifuged to remove cells. Magnetic Dynabeads protein G (Invitrogen, Dynal AS, Norway) were washed with phosphate-buffered saline (PBS) twice and incubated with mouse MAb anti-DSG2 (clone 6D8; Cell Sciences) at room temperature for 50 min. After unbounded antibody was washed away, the beads were incubated with A549 medium for 3 h at 4°C with gentle shaking. Beads were then washed with PBS, and target proteins were eluted with 0.1 M citric acid (pH 3.0) and run on an SDS-PAGE gel. The bands of interest were cut and subjected to mass spectrometry analysis. Tandem mass spectrometry (MS/MS) was performed by Martin Sadilek at the University of Washington, Department of Chemistry MS Core, as described previously (42).
Metabolic protein labeling.
A549 cells (80% confluence) were cultured in 10-cm dishes in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS), 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were rinsed with PBS and labeling medium (RPMI 1640 medium, methionine and cysteine free; Gibco) supplemented with 1% dialyzed FCS. Cells were incubated in 5 ml of labeling medium supplemented with 250 μCi of [35S]Met-Cys Redivue labeling mix (PerkinElmer) and with either PBS (control), 1 μg/ml PtDd, or wt Ad3 (multiplicity of infection [MOI], 200 PFU/cell). After culture at 37°C for 2 h, 5 ml of chase medium (labeling medium supplemented with 500 μg/ml cysteine-HCl and 100 μg/ml methionine) was added, and cells were cultured for another 2 h before harvesting.
Immunoprecipitation of DSG2 protein was carried out based on the manufacturer's instructions (Pierce cross-linking immunoprecipitation kit; Thermo Scientific). Briefly, 10 μg of DSG2 monoclonal antibody (clone 6D8; AbD Serotec) per reaction mixture was bound to protein A/G plus agarose at room temperature for 60 min and then cross-linked for another 60 min at room temperature. Cells were lysed with 0.5 ml of immunoprecipitation buffer from the kit. Cell lysates and culture supernatant were precleared by incubation with the control agarose resin for 2 h at 4°C and then incubated with DSG2 MAb-cross-linked agarose resin overnight at 4°C. The protein complex-coupled resin was then collected, washed, and eluted. Samples were mixed with 5× loading buffer (0.3 M Tris-HCl, pH 6.8, 5% SDS, 50% glycerol, 100 mM DTT) and loaded onto 4 to 15% Mini-Protean precast gels (Bio-Rad).
To visualize 35S-labeled DSG2 protein, a polyacrylamide gel was fixed in fixation solution (7% acetic acid and 25% methanol in water) for 30 min after electrophoresis, incubated in enhancer solution (EN3HANCE; PerkinElmer) for 60 min, and then soaked in soaking solution (1% glycerol and 5% polyethylene glycol [PEG] 8000 in water) for 30 min. The gel was dried and exposed to X-ray film.
For Western blotting, proteins were transferred to nitrocellulose after electrophoresis and incubated with DSG2 monoclonal antibody (6D8) and HRP-conjugated goat anti-mouse IgG antibody (BD Pharmingen).
Virus attachment competition assays.
HeLa cells were detached from culture dishes by incubation with Versene and washed with PBS. A total of 105 cells per tube were resuspended in 50 μl of ice-cold adhesion buffer (DMEM supplemented with 2 mM MgCl2, 1% FBS, and 20 mM HEPES) containing recombinant DSG2 protein (6 μg/ml) and incubated on ice for 1 h. Then, 3H-labeled wild-type virus was added in adhesion buffer at a multiplicity of infection (MOI) of 8,000 viral particles (VPs) per cell to a final volume of 100 μl. After 1 h of incubation on ice, cells were pelleted and washed twice with 0.5 ml of ice-cold PBS. After the last wash, the supernatant was removed, and the cell-associated radioactivity was determined by a scintillation counter. The number of VPs bound per cell was calculated by using the virion-specific radioactivity and the number of cells.
Phosphokinase proteome array.
Activation of signaling pathways was assessed by using a Proteome Profiler Human Phospho-Kinase Array kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instruction. A549 cells were cultured in 10-cm tissue culture dishes in DMEM with 10% FBS until they reached 80 to 90% confluence. Cells were then treated with virus or proteins as specified in the figure legends. Treated cells were rinsed with cold phosphate-buffered saline (PBS) and immediately solubilized in lysis buffer at 4°C for 30 min. Following centrifugation at 14,000 × g for 5 min, supernatants were transferred into a clean test tube, and sample protein concentrations were determined. Five hundred micrograms (total protein) of lysate was diluted and incubated per array set. Array data were developed on films following exposure to chemiluminescent reagents and analyzed by ImageQuant TL (GE Healthcare).
MAPK reporter system.
Cell lines were transduced with a MAPK/ extracellular signal-regulated kinase-luciferase (ERK-Luc) reporter lentivirus (Cignal Lenti assay, catalog number CLS010; Qiagen). Cells were selected with G418 (0.7 mg/ml) for 4 weeks to obtain stable clones. Luciferase expression was measured using the Promega luciferase assay system. The number of relative light units (RLU) was normalized to the total protein concentration in the sample. The data are expressed at RLU/milligram of protein.
In vivo imaging.
In vivo luciferase imaging was performed on an IVIS (In Vivo Imaging System) Lumina Series II (PerkinElmer, Inc., Waltham, MA). Mice with subcutaneous A549-SRE-Luc (where SRE is serum response element) tumors were imaged as follows. Animals were injected intraperitoneally (i.p.) with 15 mg/ml luciferin in PBS at 150 mg/kg. Five minutes later animals were transferred to anesthesia induction chambers, and animals were induced for 3 min. Animals were then transferred to the IVIS imaging chamber. At 10 min after substrate injection, the imaging procedure was started. Two sequences of five 1-min exposures were recorded at small binning and an F-stop of 1. For the first sequence the primary tumor site was shielded; for the second sequence the shielding was removed. For analysis, regions of interest were put over the tumor site, and the total flux (photons per second summed over the area of the region of interest) was measured using Living Image, version 4.0, Software (PerkinElmer, Inc.).
Ad14 crystallization.
Crystallization conditions for Ad14 and Ad14P1 fiber knob were screened using the service of the High Throughput Screening Lab at Hauptman Woodward Medical Research Institute. Crystallization for the Ad14P1 knob variant was performed using the hanging-drop method. Crystals were grown using a reservoir solution of 29% PEG 3.350–5% 1-butyl-2,3-dimethylimidazolium tetrafluoroborate (C9H17BF4N2) in 0.09 M bis-tris propane buffer, pH 7.8, and a protein solution of 15 mg/ml. Improvement in crystal diffraction was performed using the Hampton II detergent additives screen. The best hit was obtained by microseeding of a crushed crystal under the above-described condition supplemented by 0.1% isopropyl-β-d-thiogalactopyranoside (IPTG). Crystals were frozen using a cryoprotectant composed of 85% reservoir and 15% glycerol (vol/vol). Data collection was performed at 100 K on ID23-2 of the European Synchrotron Radiation Facility (ESRF) using the EDNA pipeline. Data were indexed and scaled using XDS/XSCALE, and the structure was solved by molecular replacement (PDB 3F0Y) with the program PHASER. The model was built using COOT (29). Refinements were carried out using Buster (43), version 2.10.2 (Global Phasing, Ltd., Cambridge, United Kingdom) and Phenix (8), and statistics are given in Table 1. PyMol software was used to generate all 3D structure figures and align the structures of Ad3 (1H7Z), Ad14 fiber knob (3F0Y), and Ad14P1 (this publication). All structures are displayed as cartoons with the F-G loop colored uniquely.
TABLE 1.
X-ray crystallography data
| Parameter | Value for the parameterb |
|---|---|
| Data collection | |
| Space group | P63 |
| Cell dimensions | |
| a, b, c (Å) | 112.414, 112.414, 79.538 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å) | 48.68–3.2 (3.3–3.2) |
| Rsym or Rmerge | 17.9 (104) |
| I/σI | 9.3 (1.6) |
| Completeness (%) | 99.7 (98.4) |
| Redundancy | 6.8 (6.75) |
| Refinement | |
| Resolution (Å) | 48–3.2 |
| No. of reflections | 9,523 (923) |
| Rwork/ Rfree | 0.20 (0.26) |
| No. of atoms | |
| Protein | 4,868 |
| Ligand/ion | 0 |
| Water | 0 |
| B factor | |
| Protein | 83.2 |
| RMS deviationsa | |
| Bond length (Å) | 0.011 |
| Bond angle (°) | 1.44 |
RMS, root mean square.
Values in parentheses refer to the highest-resolution shell.
Surface plasmon resonance.
Acquisitions were done on a BIAcore 3000 instrument. HBS-P (GE-Healthcare) supplemented with 2 mM CaCl2 was used as running buffer in all experiments at a flow rate of 5 μl/min. Immobilization on a CM5 sensorchip (BIAcore) was performed using either recombinant DSG2 (Leinco) or the Ad14 fiber knobs at 10 μg/ml in 10 mM acetate buffer, pH 4.5, injected for 10 min on an ethyl(dimethylaminopropyl) carbodiimide (EDC)/N-hydroxysuccinimide (NHS)-activated flow cell. A control flow cell was activated by EDC/NHS and inactivated by ethanolamine. DSG2 was injected on the immobilized fiber knob at 167 nM, and in the reverse experiment the fiber knob was injected at 167 nM on the immobilized DSG2 for a 3-min association followed by a 2.5-min dissociation time. The signal was automatically subtracted from the background of the ethanolamine-deactivated EDC/NHS flow cell.
Animal studies.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Washington, Seattle, WA (protocol number 3108-01). Mice were housed in specific-pathogen-free facilities. Immunodeficient (CB17) mice (strain name, NOD.CB17-Prkdcscid/J) were obtained from the Jackson Laboratory. A549 and SKOV3-ip1 tumors were established by injection of the corresponding tumor cells into the mammary fat pad (1:1 with Matrigel) of CB17 mice. When tumors reached a volume of ∼500 mm3, Ad3-GFP at a dose of 2 × 109 PFU or JO1 (2 mg/kg) was injected into the tail vein. Female CB17 mice were injected subcutaneously with A549-SRE-Luc cells at the mammary fat pad. After tumors formed, the mice were injected with luciferin at a concentration of 30 mg/kg (Caliper, Hopkinton, MA, USA). The mice were imaged with a Xenogen IVIS-200 at three time points, i.e., 0 h, 3 h, and 6 h post-JO1 injection. The luciferase signal was detected at fixed exposure times and intervals between the images (30-s exposure, 3-min intervals for a 10-min total). The relative optical intensity (ROI) of the luciferase signal was derived by selecting the tumor area with the ROI tool, and total ROI was taken from three images. The average ROIs were normalized to the tumor volume for each mouse.
Virus titering in tumor lysates.
Tumors were collected and homogenized in PBS with a Qiagen Tissue Ruptor (Valencia, CA). Samples were subjected to three cycles of freeze-thawing. The lysates were clarified by centrifugation at 12,000 × g for 2 min, and the supernatants were titrated by limiting dilution assay on 293 cells as described earlier (30).
Statistical analysis.
All results are expressed as means ± standard deviations (SD). Two-way analysis of variance (ANOVA) for multiple testing was applied. Animal numbers and P values are indicated in the figure legends.
Protein structure accession number.
The structure of the adenovirus 14 P1 mutant knob domain, Ad14P1, has been deposited in the Protein Data Bank under accession number 4ZDG (RCSB identification code D_1000209055).
RESULTS
Shedding of the DSG2 extracellular domain triggered by Ad3 virus, PtDd, and JO1 in vitro and by Ad3 and JO1 in vivo.
DSG2 forms homodimers between neighboring epithelial cells through binding of its extracellular domains (ECDs) (Fig. 1A). Cleavage of these ECDs is considered one of the mechanisms that results in the transient opening of epithelial junctions (11). To study ECD shedding we used two monoclonal antibodies (6D8 and AH12.2) against different ECDs as well as an antibody (20H1) against the DSG2 propeptide domain that is cleaved during exposure of DSG2 on the cell surface (Fig. 1B). We then exposed human epithelial lung cancer A549 cells to Ad3 virus and analyzed DSG2 by Western blotting at different time points (Fig. 1C). Antibodies 6D8 and AH12.2 detected the full-length DSG2 form with a molecular mass of ∼130-kDa as well as a smaller product (∼80 kDa) in cells before the exposure to Ad3 (Fig. 1C, pre lane), indicating that these cells produce a cleaved form of DSG2 that is recognized by the anti-ECD antibodies 6D8 and AH12.2. The intensity of the cleaved band increased within 1 h upon Ad3 exposure and then gradually declined to intensities that were lower than those of pretreatment levels by 8 h after exposure. The antibody that recognized the propeptide domain (20G1) did not detect the cleaved DSG2 form. Notably, the 80-kDa band was only readily detectable when confluent cells with intercellular junctions were used for the assay, indicating that the cleaved form remains cell associated or trapped in junctions. The 80-kDa band was not detected by Western blotting in cell lysates of proliferating A549 cells (data not shown).
FIG 1.

DSG2 shedding in vitro in tissue cultures. (A) DSG2 is localized in desmosomal junctions at the lateral side of epithelial cells. The extracellular domain of DSG2 forms a homodimer between two neighboring cells. On the cytoplasmic side, DSG2 binds to cytoskeletal proteins such as plakoglobin, plakophilins 1 to 4, and the intermediate filament binding protein desmoplakin. The desmin/keratin intermediate filaments link the DSG2 transmembrane protein complex to the cell membrane. (B) Structure of DSG2. The extracellular part of DSG2 contains four domains (ECD1 to ECD4), followed by an extracellular anchor domain, the transmembrane domain, and the intracellular domain. Monoclonal antibodies (20G1, AH12.2, and 6D8) against different ECDs are listed. The predicted molecular masses of the cell-associated and shed portion of DSG2 are indicated. (C) DSG2 Western blot of cell lysates using anti-DSG2 MAbs. Confluent lung cancer A549 cells were treated with Ad3 at an MOI of 200 PFU/cell. Cells were collected at different time points and subjected to Western blotting with anti-DSG2 MAbs 6D8, AH12.2, and 20G1. Molecular mass makers are shown on the right side of the blots. The full-length DSG2 band runs at a molecular mass of ∼130 kDa. A dominant small band at ∼80 kDa is detected with 6D8 and AH12.2 MAbs but not with MAb 20G1. (D) DSG2 immunoprecipitation/Western blotting of cell supernatants. A549 cells were incubated with Ad3 (200 PFU/cell) or PtDd (1 μg/ml). Culture supernatant was collected 4 h later, and shed DSG2 was pulled down using 6D8 antibodies/protein A/G-agarose. Pulled-down proteins were analyzed by Western blotting with 6D8 antibodies. (E) Metabolic labeling for detection of de novo-produced DSG2 in cell lysates and culture supernatant. Cells were exposed to Ad3 and PtDd as described for panel E in the presence of [35S]methio nine-serine for 2 h, followed by a 2-h incubation in chase medium. Cell lysates and supernatant were immunoprecipitated with anti-DSG2 6D8 antibody/protein A/G-agarose and separated by polyacrylamide gel electrophoresis. The right panel shows a phosphorimager exposure of the supernatant signals. (F) Concentration of DSG2 in supernatant of A549 cells measured at different time points by ELISA. A549 cells were treated with Ad3 virus (200 PFU/cell), purified recombinant penton-dodecahedral particles (PtDd) or penton base-dodecahedral particles (BsDd) (1 μg/ml). Culture supernatant was collected at the indicated time points and used for ELISAs. Shown is the relative increase in DSG2 levels compared to pretreatment (time point 0) levels (n = 3). The differences in results for BsDd versus the untreated cells (control) (at all time points), for Ad3 versus PtDd at 8 h, and for PtDd versus JO1 at 8 h were not significant. For results with Ad3 versus those with the control at 8 h, P < 0.01; for PtDd results versus those with BsDd at 12 h, P < 0.01. In Ad3-treated samples, the first signs of cytopathic effect were observed at 6 h. (G) Immunofluorescence analysis of A549 cells. Six hours after the addition of Ad3 (200 PFU/cell) or PtDd (1 μg/ml) cells were stained with fluorescein isothiocyanate-labeled 6D8 antibody. DSG2 signals are green. Scale bar, 50 μm. Note that the immunofluorescence analysis was performed on nonpermeabilized cells and therefore does not detect DSG2 that is not presented on the membrane. (H) Results of tandem mass spectrometry analysis of shed DSG2. A549 cells were incubated with PtDd as described for panel D. Eight hours later, shed DSG2 was pulled down using 6D8 antibodies. Pulled-down proteins were then subjected to MS/MS. The triangles indicate the top peptides identified by MS/MS.
To demonstrate that the cleaved form is shed from the cells, we immunoprecipitated culture supernatant collected 4 h after adding Ad3 virus or PtDd with antibody 6D8 (Fig. 1D). Western blotting of precipitated material revealed a dominant band in the range of 80 kDa which was more intense in cells incubated with PtDd and Ad3 than in untreated cells (control).
To evaluate whether the 80-kDa band is a product of de novo synthesis of a smaller DSG2 species that is triggered by PtDd or Ad3, proteins were metabolically labeled with [35S]methionine-serine. DSG2 was immunoprecipitated with 6D8 antibodies from cell lysates and culture supernatant 4 h after the addition of PtDd or Ad3 and analyzed by polyacrylamide gel electrophoresis. Autoradiography of precipitated proteins showed full-length DSG2 (130 kDa) and, to a lesser degree, a band in the range of 80 kDa in the cell lysates (Fig. 1E). Weak signals in the range of 80 kDa were also detected after longer exposure in immunoprecipitated supernatant proteins, indicating background cleavage of de novo-synthesized DSG2 (Fig. 1E, right panel). Importantly, the intensity of the 80-kDa band in cell lysates and the supernatant did not increase after PtDd and Ad3 incubation to a degree seen in the Western blot shown in Fig. 1C and D. We therefore concluded that PtDd and Ad3 triggered shedding of the 80-kDa protein rather than its de novo synthesis.
To better quantitate DSG2 shedding, we measured DSG2 concentrations in the supernatant of A549 cells by ELISA using the 6D8 antibody (Fig. 1F). Shed DSG2 accumulated over time in the supernatant of untreated cells (control). Upon addition of Ad3, DSG2 concentrations increased, reaching a plateau at ∼5 h. Ad3-treated cells developed virus-associated cytopathic effects after 8 h and were therefore not followed any longer. A similar kinetic was seen in cells exposed to recombinant Ad3 PtDd. BsDd, which did not contain Ad3 fibers, had no effect on DSG2 shedding compared to results in control cells, indicating that shedding is triggered by the fiber. In support of this, we found increased DSG2 shedding upon incubation with recombinant Ad3 fiber knobs (JO1) although the kinetics of shedding appeared to be slower than that with PtDd. An immunofluorescence study with 6D8 antibodies on nonpermeabilized A549 cells exposed to Ad3 or PtDd showed the disappearance of DSG2 from the cell membrane, which further corroborates DSG2 shedding (Fig. 1G). To study structure details of shed DSG2, we used tandem mass spectrometry (MS/MS). DSG2 protein from supernatant of PtDd-incubated A549 cells was pulled down using 6D8 antibodies and protein G Dynabeads. The pulled-down material was run on an SDS polyacrylamide gel. The band in the area of 80 kDa was cut out from the gel, trypsin digested, and subjected to MS/MS. The identified peptides are shown in Fig. 1H as triangles. All hits were located in the extracellular domain or the extracellular anchor near the transmembrane domain. This suggests that cleavage most likely occurs within the extracellular anchor domain.
To support our observation, we performed a similar study in another epithelial cancer cell line, ovarian cancer SKOV3 cells, in which exposure to Ad3 also increased the concentration of DSG2 in the culture supernatant significantly more than in untreated control cells (Fig. 2).
FIG 2.
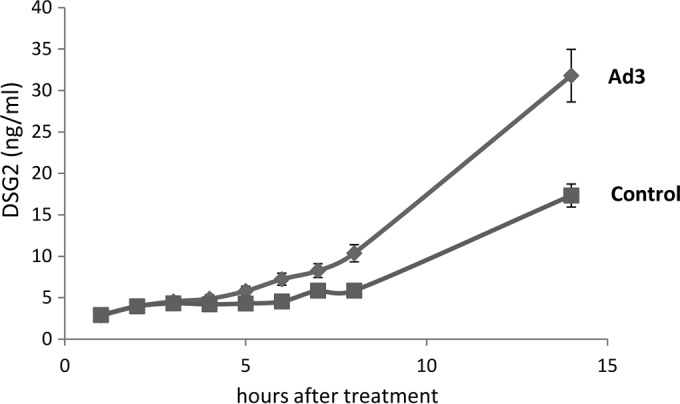
DSG2 concentrations in the supernatant of SKOV3 cells incubated with Ad3 at an MOI of 200 PFU/cell. The experiment was performed as described in the legend of Fig. 1F. Control values are those from untreated cells (n = 3).
To test whether Ad3 has a similar effect in vivo, we used mice with human xenograft tumors derived from colon cancer HT29 and ovarian cancer SKOV3 cells (Fig. 3A). Notably, mouse DSG2 is not recognized by Ad3 or Ad3 derivatives (3). The presence of human tumors in mice resulted in detectable levels of human DSG2 in serum, most likely due to background shedding from tumor cells that we also observed in vitro. The serum DSG2 concentration in mice before Ad3 injection ranged from 10 to 20 ng/ml for the HT29 tumor model (Fig. 3B, HT29, mock). Mice were then intravenously injected with Ad3. Serum was collected at the time points indicated in the figure legend, and DSG2 concentrations were measured by ELISA. Because preinjection DSG2 levels varied, they were subtracted for each animal. In both the HT29 and SKOV3 models, serum DSG2 levels increased at 24 h after Ad3 injection and remained high during the time of observation (72 h). This could be due to the half-life of DSG2 in serum or to the fact that Ad3 replicates in human tumor cells and produces PtDd, which in turn could trigger more DSG2 shedding. The latter is supported by the finding that, upon JO1 injection, serum DSG2 levels also peaked at 24 h but declined later (Fig. 3B, SKOV3-JO1). DSG2 cleavage in Ad3-injected animals could also be observed by Western blotting in lysates of tumors (Fig. 3C). As seen with confluent epithelial cancer in culture, part of the shed DSG2 remains tumor associated, potentially trapped in epithelial junctions. Another part of shed DSG2 is released into the blood circulation.
FIG 3.

DSG2 shedding in vivo in mice with tumors derived from human cancer cell lines. (A) Experimental design. Immunodeficient CB17 SCID/beige mice with established subcutaneous human xenograft tumors (∼500 mm3) were injected intravenously with Ad3 virus (2 × 109 PFU per mouse) or JO1 (2 mg/kg). Preinjection serum samples (0 h) and samples harvested at 3, 6, 24, 48, and 72 h after Ad3 injection were analyzed by ELISA for DSG2 ECD concentrations. (B) Serum DSG2 levels in mice with xenograft tumors derived from SKOV3 or HT29 cells. The left upper panel shows background DSG2 serum levels in mice with HT29 tumors. In all other panels, background (preinjection) DSG2 levels were subtracted from the levels measured after Ad3 or JO1 injection (n = 5). *, P < 0.01. (C) DSG2 Western blot of tumor lysates after Ad3 injection. Mice with HT29- or SKOV3-derived xenograft tumors were injected with Ad3, and tumors were harvested 24 h later. 6D8 antibody was used as a probe. DSG2 band signals were normalized to β-actin signals and are expressed as a percentage of combined (DSG2) intensities (according to the table below the blot). Each lane represents an individual tumor.
In conclusion, Ad3 and specifically the Ad3 fiber knob trigger the shedding of the extracellular part of DSG2 from epithelial cancer cells in culture and in xenograft tumor models.
Activation of MAPK signaling by Ad3.
To study intracellular signaling, we exposed A549 cells to Ad3 and lysed cells 2.5 h later for analysis by a Proteome Profiler antibody array. This array is able to detect relative levels of phosphorylation of 43 different kinase phosphorylation sites (Fig. 4A). The 2.5-h time point for Ad3 signaling was selected to avoid potential intracellular signaling triggered by viral gene expression, which starts as early as 6 h after Ad infection (44). Signal intensities were quantitated and normalized to signals from untreated cells (taken as 100%). The analysis shows significant increases in the activation of members of the mitogen-activated protein kinase (MAPK) pathway, including p38a, ERK1/2, Jun N-terminal protein kinase (JNK), EGFR, Akt, and CREB as well as signaling molecules that are activated by MAPK, including c-Jun, p27, and STAT3. The activation of Chk-2, a protein kinase that is activated in response to DNA damage, was unexpected but could be related to the cellular response to Ad3 infection.
FIG 4.

Analysis of phosphokinase activation after incubation of A549 cells with Ad3 and JO1. (A) A549 cells were incubated with Ad3 at an MOI of 100 PFU/cell for 1 h on ice to allow for attachment. Virus was then removed, and cells were incubated for 2.5 h at 37°C. Cell lysates were subjected to hybridization on filters containing antibodies that are able to detect relative levels of phosphorylation of 43 different kinase phosphorylation sites (Proteome Profiler antibody array; R&D Systems). Signals were quantitated and plotted, taking the signals from mock-infected cells as 100%. *, P < 0.01 (n = 3 independent experiments). (B) A549 cells were incubated with 1 μg/ml of JO1 and harvested at 2.5, 8, and 24 h later. As a control, A549 cells were incubated with Ad35 fiber knob (1 μg/ml) and harvested at 2.5 h. JO1 signals were normalized to Ad35 fiber knob signals (taken as 100%) (n = 3). eNOS, endothelial nitric oxide synthase.
Ad3 interaction with cells is complex and can involve capsid proteins other than the fiber, for example, penton base RGD-containing domains that bind to αv integrins. To dissect this, we studied phosphokinase activation upon incubation of cells with JO1, e.g., the dimerized Ad3 fiber knob. To further streamline our analysis, we compared JO1-triggered signaling with that occurring after incubation of cells with a recombinant Ad35 fiber knob, which binds to CD46 and not to DSG2 (45). Figure 4B shows JO1 signals normalized to Ad35 fiber knob signals (taken as 100%). Overall, JO1 triggered activation of members of the MAPK pathway at 2.5 h similar to that seen with Ad3. At 4 h most of the MAPK signals returned to background levels. Interestingly, JO1 also activated β-catenin, however, with a slower kinetics than MAPK members. We also observed changes in phosphokinase activation that were not seen with Ad3, including less intense p70(S6), p27, and Lck signals and stronger activation of STAT3. As expected, there was no Chk-2 activation when cells were incubated with JO1. Overall, the study with JO1 indicates prolonged activation of signaling pathways, specifically of kinases that are downstream of MAPK, including STAT3 and β-catenin. A similar MAPK pathway activation pattern was detected in another cell line (HT29) after incubation with JO1 (data not shown). The study also shows that the fiber knob and its interaction with DSG2 are the central triggers in activating MAPK signaling. Because MAPK signaling appeared to be the dominant effect, in subsequent studies we attempted to validate the activation of this pathway. The first validation assay involved Western blotting for p38 and its phosphorylated form, pp38 (Fig. 5A and B). The pp38 signal increased within 45 min after exposure to Ad3, declined by 3 h, and then increased again (Fig. 5A). The second wave of p38 phosphorylation was absent when cells were incubated with PtDd (Fig. 5B) or JO1 (data not shown), indicating that it is stimulated by virus moieties or functions other than the fiber knob. The second validation assay involved a Cignal Lenti SRE reporter system that monitors the activity of serum response factor (SRF)-mediated signal transduction pathways. The ternary complex factors, TCR and Elk-1, form a complex with the SRF over the serum response element (SRE) and activate gene expression. The Elk-1 protein is phosphorylated by MAPK, causing increased DNA binding, ternary complex formation, and transcriptional activation of target genes. The SRE reporter therefore measures the activation of the SRF and the MAPK signal transduction pathway. We generated A549 cells that expressed luciferase under SRE/MAPK control (Fig. 5C). Exposure of these cells to Ad3 increased luciferase expression levels to a degree seen with the potent MAPK pathway stimulator EGF (Fig. 5D). This reporter cell line also allowed us to study MAPK activation in vivo in xenograft A549-SRE-Luc tumors. Mice with established tumors were injected intravenously with JO1, and luciferase expression was measured by in vivo imaging after injection of the luciferase substrate, luciferin (Fig. 5E). In all four animals studied, the luciferase signal was significantly increased at 6 h after JO1 injection. It then declined to background levels by day 3.
FIG 5.

Validation of MAPK activation by Ad3. (A and B) Western blot analysis of p38. A549 cells were exposed to Ad3 (200 PFU/cell) (A) or JO1 (1 μg/ml) (B) as described in the legend of Fig. 4. Cells were harvested, lysed, and analyzed by Western blotting using MAbs against total p38 and phosphorylated p38 (pp38) pre, before exposure to Ad3. (C to E) Studies with a MAPK-dependent reporter system. The expression cassette of the SRE/MAPK-Luc reporter lentivirus vector is diagrammed in panel C. Luciferase expression is under the control of a minimal CMV promoter and tandem repeats of the SRE transcriptional response element (SRE-TATA). hPGK, human phosphoglycerate kinase promoter; PuroR, puromycin resistance gene; cppt, central polypurine tract. Luciferase expression was determined in A549 cells that were modified with the SRE/MAPK-reporter lentivirus vector (D). Cells were treated with EGF (200 ng/ml) or infected with Ad3 at an MOI of 200 PFU/cell for 3 h. Luciferase expression was measured 48 h later and expressed as RLU per milligram of total protein (n = 3). *, P < 0.01. In vivo imaging of luciferase expression after JO1 injection is shown in panel E. A549 cells marked with the SRE/MAPK reporter system were injected subcutaneously into CB17 SCID/beige mice. When tumors reached a volume of ∼500 mm3, mice were intravenously injected with JO1 (2 mg/kg) and imaged for luciferase expression before injection (0 h) and at 6 h and 3 days after Ad injection. Normalized signals (flux/ROI) from four individual animals (1 to 4) are shown.
In summary, these studies demonstrate that Ad3, through its fiber knob, triggers the activation of MAPK pathways.
Ad3 activates MAPK and ADAM17, which in turn leads to DSG2 shedding.
Previous studies on human tumor cell lines have shown that DSG2 is subject to shedding via matrix metalloproteases (MMPs), including the ADAM17 (a disintegrin and metalloprotease) member of transmembrane sheddases (11, 46). These studies also established a functional link between EGF-MAPK stimulation and ADAM17 activation. We therefore analyzed the effect of Ad3 exposure on ADAM17. ADAM17 activation occurs by proteolytic cleavage of the inactive form containing a propeptide (pro-ADAM17) (47). Incubation of A549 cells with Ad3 resulted in an increase of the ratio of ADAM17 to pro-ADAM17, indicating activation of this MMP (Fig. 6A, left panel). The Ad3 fiber-less BsDd had no effect on ADAM17 activation. This process was blocked by the MMP/sheddase inhibitor TAPI-0 (11) and by the MAPK inhibitor U-0126 (Fig. 6A, right panel). This suggests that Ad3 triggers the activation of ADAM17 and that this involves MAPK as a functional link.
FIG 6.

Inhibition of DSG2 shedding by MAPK and ADAM17 inhibitors. (A) Analysis of ADAM17. The left panel shows activation of ADAM17. A549 cells were incubated with BsDd (1 μg/ml) (as a negative control) or Ad3 virus (MOI of 200 PFU/cell). Cells were washed once with PBS at 11 h after incubation, lysed, and subjected to Western blotting with polyclonal anti-ADAM17 antibodies. The arrows indicate pre-ADAM17 and the processed form of ADAM17. Pixel densities of bands were measured by GE Healthcare's ImageQuant software. The numbers below each lane are the ratios of signals from the ADAM17 band to signals from the pro-ADAM17 band. The right panel shows the inhibition of ADAM17 by the ADAM17 inhibitor TAPI-0 (final concentration, 50 μM) or the MAPK inhibitor U-0216 (final concentration, 10 mM), added alone or together with Ad3 virus. Cells were harvested 11 h later. Dimethyl sulfoxide (DMSO) is a solvent for both inhibitors and was therefore used as a control. β-Actin served as a loading control. (B) DSG2 concentrations in A549 culture supernatant. Culture supernatant was collected 11 h after treatment, and DSG2 concentrations were measured by ELISA (n = 3). *, P < 0.01. (C) DSG2 concentrations in SKOV3 cell culture supernatants. Cells were treated as described for panel C. The differences in the amounts of shedding triggered by PBS, Ad3, PtDd, and BsDd in the presence of TAPI-0 in SKOV3 cells were not significant.
We performed DSG2 shedding studies in the presence of MAPK and ADAM17 inhibitors to consolidate the functional chain: Ad3 binding to DSG2 → activation of MAPK → activation of ADAM17 → cleavage and shedding of DSG2. We measured shed DSG2 in the culture supernatant of A549 cells (Fig. 6B) and SKOV3 cells (Fig. 6C). Both U-0126 and TAPI-0 reduced the shedding from A549 cells (Fig. 6B and C, PBS). Shedding was blocked more by TAPI-0 than by U-0126. Ad3 and PtDd, but not BsDd, increased the amount of shed DSG2. Incubation with U-0126 or TAPI-0 decreased DSG2 levels to control (PBS) levels. In SKOV3 cells, U-0126 did not block background DSG2 shedding; however, it blocked shedding triggered by Ad3 or PtDd. TAPI-0 had a more profound inhibitory effect than U-0126. The only partial blocking of DSG2 shedding by U-0126 suggests that pathways other than MAPK are involved in shedding.
X-ray crystallography and 3D structure modeling of the Ad14P1 fiber knob.
Based on epidemiological data, the newly emerged strain, Ad14P1, is considered more pathogenic/virulent than the parental strain (Ad14) (22–24). In an attempt to experimentally support this, we studied the effect of Ad14 and Ad14P1 on tumor growth in a xenograft model with epithelial tumors derived from A549 cells (Fig. 7). Intratumoral injection of Ad14P1 resulted in a greater attenuation of tumor growth than Ad14 injection. This was most likely due to better virus replication and/or intratumoral spread (Fig. 7A). While the infectious particle titers of the two viruses were comparable at day 2 after injection, more Ad14P1 virus was detectable at day 7 after injection (Fig. 7B). The genomes of the two viruses differ by an insertion in the E1A gene and a small deletion in the fiber knob gene (48). In this study, we tested the hypothesis that the more efficient Ad14P1 production in xenograft tumors and, potentially, its higher pathogenicity are linked to the mutation within the Ad14P1 fiber knob. The deletion (ΔK250-E251, or KE) in the fiber knob is located within the F-G loop directly adjacent to the G β-sheet and could theoretically change the structure of the F-G loop and its proximity to the neighboring knob monomer (Fig. 8A) (38). Potential structural differences in the DSG2-interacting fiber knob domain(s) of Ad14P1 could influence binding to DSG2 and subsequent intracellular signaling and thus account for the differences in Ad14 and Ad14P1 pathologies. We therefore delineated the atomic structure of the Ad14P1 fiber knob. As shown in Fig. 8B and C, the two-amino-acid deletion breaks the short alpha helix present in the Ad14 structure. The loop exhibits no defined secondary structure and is quite flexible, as denoted by higher than average B-factors (Table 1). Structural overlays with Ad3 and Ad14 show the multiple conformations possible for the F-G loop (Fig. 8C and D). Each structure exposes different residues, which likely influences protein-protein interactions mediated by this region of the structure. We also tested 78 human serum samples for IgG antibodies specific to recombinant Ad14 and Ad14P1 fiber knob and found that the levels of antibodies in a given serum sample were different in the majority of cases (Fig. 9). Although very speculative, this could be the result of different immunoreactivity due to structural differences. Clearly, other factors could also account for this.
FIG 7.
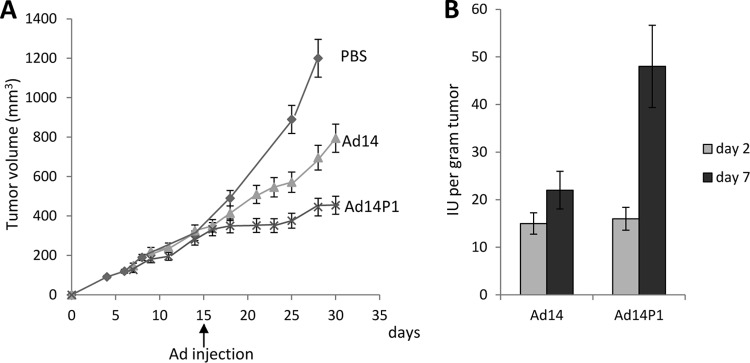
Spread of Ad14 and Ad14P1 in epithelial xenograft tumors derived from A549 cells. When subcutaneous tumors reached a volume of ∼300 mm3 at day 15 after tumor cell inoculation, 2 × 109 PFU of the Ad14 and Ad14P1 viruses was injected intratumorally. (A) Tumor volumes. Differences in tumor volumes between Ad14 and Ad14P1 are significantly different (P < 0.01) from day 21 on (n = 5). (B) In a second study, established tumors with a volume of ∼300 mm3 were injected intratumorally with 2 × 109 PFU of the Ad14 and Ad14P1 viruses. Tumors were harvested at day 2 and day 7 after Ad injection, homogenized, freeze-thawed, and subjected to titering for infectious viral particles (IU) by limiting dilution assay (n = 3).
FIG 8.

Structure of Ad14P1 fiber knob compared to the structures of Ad14 (3F0Y) and Ad3 (1H7Z). (A) Amino acid sequence of a section of the Ad14 and Ad14P1 fiber knobs. Beta-sheets F, G, H, and I are shaded in gray. The two-amino-acid deletion in the F-G loop of Ad14P1 is indicated by an arrow. (B) Overlay of the structures of Ad14 (3F0Y) and Ad14P1. Ad14P1 is shown in dark blue with the F-G loop in cyan. Ad14 is shown in gray with the F-G loop highlighted in yellow. The KE deletion in Ad14P1 breaks the alpha helix as shown. (C) Close-up of the F-G loop with Ad14P1 shown in cyan and Ad14 shown in gray (top). In the bottom panel, residues for the F-G loop are shown and labeled with numbering as per that of Ad14P1. (D) Overlay of the structures of Ad3 (1H7Z), Ad14 (3F0Y), and Ad14P1. Structures are shown as cartoons with partial transparency. Ad3 is shown in light green with the F-G loop in bright green, Ad14 is shown in gray with the F-G loop in gray, and Ad14P1 is shown in dark blue with the F-G loop in cyan. (E) Close-up of the F-G loops for Ad3, Ad14, and Ad14P1 colored as described for panel C.
FIG 9.

Level of antibodies against Ad14 and Ad14P1 fiber knobs in human serum. Serum samples from 78 ovarian cancer patients (x axis) were diluted 1:50 and subjected to ELISA to measure Ad14- and Ad14P1-specific IgG antibodies. OD450 readings were compared to those from pooled normal human serum (NHS) (taken as 1.0).
Structural changes with the F-G loop of Ad14P1 fiber knob do not significantly change the binding to DSG2.
To study potential functional consequences of the changed structure of the Ad14P1 fiber knob, we tested the attachment of Ad14 and Ad14P1 virus to HeLa cells using recombinant DSG2 as a competitor (Fig. 10A). Attachment of both viruses was blocked to a similar degree by recombinant DSG2, whereas DSG2 did not influence the binding of the coxsackievirus-adenovirus receptor (CAR)-interacting Ad5. The latter finding is in agreement with our previous studies showing that Ad5 does not bind to DSG2 (41). Furthermore, we did not observe differences between Ad14 and Ad14P1 fiber knob binding using a Western blot assay (Fig. 10B). In agreement with previous studies, this assay showed that only trimeric fiber knobs bind to DSG2 and not monomeric forms. A more sensitive and quantitative assay for measuring fiber knob-DSG2 interactions is surface plasmon resonance using DSG2 and fiber knobs as analytes (Fig. 10C and D). According to the conformational change in the F-G loop structure, it would be conceivable that DSG2 recognition by Ad14P1 fiber knob would be affected. In a first attempt, recombinant DSG2 was immobilized on the sensor chip surface, and fiber knobs from Ad3, Ad14, and Ad14P1 were used as analytes. Even though a slight difference was observed between the Ad3 and the two different Ad14 knobs, very similar profiles were observed for the two Ad14 knobs, suggesting that the KE deletion in Ad14P1 and the resulting structure changes were not involved in DSG2 binding (Fig. 10C). In order to ascertain that this result would not be due to an avidity effect, a reverse experiment in which the fiber knobs were immobilized and the recombinant DSG2 was used as the analyte was performed. Again, the profiles of the different sensorgrams were very similar, thus confirming that the Ad14 knob interaction with DSG2 was not affected by the structural changes induced by the KE deletion (Fig. 10D). This conclusion is further supported by an Ad3 virus infection assay in the presence of increasing concentrations of Ad5, Ad3, Ad14, and Ad14P1 fiber knobs, which also did not reveal significant differences between the DSG2-interacting fiber knobs (Fig. 10E).
FIG 10.

Ad14 and Ad14P1 binding to DSG2. (A) Attachment of 3H-labeled Ad14, Ad14P1, and Ad5 virus particles. HeLa cells were incubated without (white bars) or with (gray bars) 6 mg/ml recombinant human DSG2 protein on ice for 1 h, and then 8,000 VPs/cell of 3H-labeled virus was added to HeLa cells on ice. One hour later, unbound virus was washed away. Attachment of virus particles incubated with PBS was taken as 100% (n = 3). (B) Analysis of Ad14 and Ad14P1 fiber knob (Ad14K and Ad14P1K) binding to recombinant DSG2. Fiber knobs were run on a polyacrylamide gel and stained with Coomassie (left panel). A second gel was blotted and hybridized with recombinant human DSG2 followed by anti-DSG2 MAb and anti-mouse Ig-HRP (right panel). Lanes 1, trimeric (TM) Ad14 knob; lanes 2, monomeric (MM) Ad14 knob (after sample boiling); lanes 3, trimeric Ad14P1 knob; lanes 4, monomeric Ad14P knob. (C) Surface plasmon resonance analysis of Ad fiber knob interactions with immobilized DSG2. Ad3, Ad14, and Ad14P1 knobs were injected at the same concentrations (167 nM) for 3 min, followed by a 2.5-min dissociation time. Note that the differences between Ad14 and Ad14P1 values are within assay variability. (D) The reverse of the experiment shown in panel C in which the three different fiber knobs were immobilized, and DSG2 was used as the analyte (167 nM). (E) Competition of Ad3-GFP infection by recombinant Ad3, Ad5, Ad14, and Ad14P1 fiber knobs. HeLa cells were incubated with increasing concentrations of Ad fiber knob protein at 37°C for 60 min and then infected with Ad3-GFP virus at an MOI of 100 PFU/cell. Virus was removed 1 h later, and GFP expression was analyzed 18 h after infection. The right panels show representative immunofluorescence images from cells infected with Ad3-GFP in the presence of 50 μg/ml of fiber knobs.
Differences in the abilities of the Ad3 and Ad14 fiber knobs to block Ad3-GFP infection could be due to the fact that the Ad3 fiber knob contains an N-terminal 6×His tag while the Ad14 fiber knobs have C-terminal 6×His tags.
In summary, structural differences in the Ad14 and Ad14P1 fiber knobs did not result in measurable differences in DSG2 binding.
Comparison of intracellular signaling between Ad14 and Ad14P1.
As no difference in binding could be detected, it is conceivable that the two viruses differ in signaling. We therefore studied intracellular signaling triggered by Ad14 and Ad14P1 viruses in A549 cells in comparison to that of Ad3 virus. The plots show relative signals, with Ad3 signals taken as 100% (Fig. 11A), or a direct comparison between Ad14 and Ad14P1, with Ad14 signals taken as 100% (Fig. 11B). Overall, there are no remarkable differences between the three viruses, particularly in members of the MAPK pathway. Compared to Ad3, both Ad14 and Ad14P1 trigger strong activation of the stress-activated protein kinases c-Jun NH2-terminal kinase (JNK) and glycogen synthetase kinase 3α/β (GSK-3α/β), most likely as a result of virus entry. On the other hand, members of the Src family kinases (Yes and Fgr) and the Scr-interacting protein focal adhesion kinase (FAK) are less activated by Ad14 and Ad14P1. The Src pathway is involved in the control of many functions, including cell adhesion, growth, movement, and differentiation. The functional relevance of these pathways remains to be studied. Here, we focus on MAPK pathways in the context of DSG2 shedding. In the SRE/MAPK reporter assay, there was no significant difference in luciferase expression levels in cells exposed to Ad14 or Ad14P1 (Fig. 12A). Similar to Ad3, Ad14 and Ad14P1 activated ADAM17 (Fig. 12B). Analysis of DSG2 shedding upon exposure of A549 cells to Ad3, Ad14, and Ad14P1 showed slower shedding kinetics for Ad14 and Ad14P1 (Fig. 12C, 3 h versus 9 h). Furthermore, while for Ad3 the amount of shed DSG2 at 3 h correlated with the MOI of virus used, this effect was not obvious for Ad14 at 9 h. It is possible that an analysis at an earlier time would show a dose-response effect for Ad14 and Ad14P1. It can also not be excluded that with Ad14 and Ad14P a threshold of signaling activation is achieved at lower MOIs. In agreement with the signaling data, there was no difference in DSG2 shedding levels between Ad14 and Ad14P1.
FIG 11.

Intracellular signaling triggered by Ad14 and Ad14P1. (A) Analysis of phosphokinase activation after incubation of A549 cells with Ad3, Ad14, and Ad14P1 viruses. Conditions were as described in the legend of Fig. 4. The signals for Ad3 were taken as 100% (dotted lines). *, P < 0.01. (B) The signals for Ad14 were taken as 100% (dotted lines). *, P < 0.01.
FIG 12.
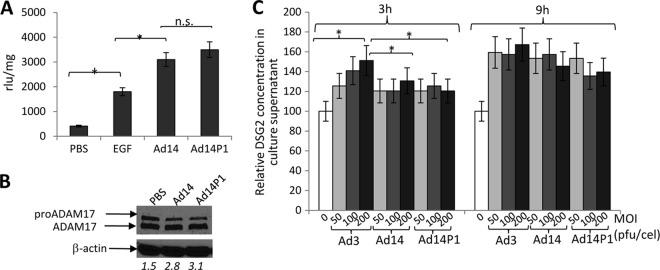
Intracellular signaling and DSG2 shedding triggered by Ad14 and Ad14P1. (A) SRE/MAPK reporter analysis of MAPK activation by Ad14 and Ad14P1. The conditions were as described in the legend of Fig. 5D. (B) Activation of ADAM17. A549 cells were incubated with Ad14 or Ad14P1 virus (MOI of 200 PFU/cell). ADAM17 was analyzed by Western blotting as described in the legend of Fig. 6A. The number below each lane is the ratio of the signal from the ADAM17 band to that from the pro-ADAM17 band. (C) DSG2 shedding triggered by Ad14 and Ad14P1 virus. A549 cells were exposed to Ad3, Ad14, and Ad14P1 at the indicated MOIs as described in the legend of Fig. 1F. Shown are relative DSG2 concentrations in the culture supernatant at 3 and 9 h. Pretreatment levels were taken at 100% (n = 3). *, P < 0.05; ns, not significant.
In conclusion, studies with Ad14 and Ad14P1 corroborated our data with Ad3 suggesting that DSG2-interacting Ads trigger MAPK activation that leads to DSG2 shedding. Although X-ray crystallography demonstrated structural differences in the fiber knobs of Ad14 and Ad14P1, this did not alter the binding to DSG2 and subsequent signaling.
DISCUSSION
Human adenoviruses have evolved mechanisms to efficiently breach the epithelial barrier to establish infection and disseminate. Two of the main adenovirus receptors, CAR and DSG2, are epithelial junction proteins. In polarized normal epithelial cells, DSG2 as well as CAR is sequestered in cell-cell junctions and is consequently inaccessible to the Ad from the apical surface. Although these receptors can be redirected to the apical surface by virus-stimulated macrophages and cytokine signaling (49), the initial round of Ad infection of polarized epithelium is inefficient. Therefore, both DSG2 and CAR are considered receptors supporting the lateral spread of de novo-produced virus (50, 51).
Our previous work has established that binding of Ad3 to DSG2 results in opening of epithelial junctions (3, 34, 35, 52). This process is further enhanced by the production of PtDd during Ad3 infection (30). PtDd are released from Ad-infected cells before virus-triggered cytolysis and can be found in the paracellular space in epithelial cell cultures. PtDd support spread of de novo-produced Ad3 by binding to DSG2 and subsequent junction opening. Our previous studies also indicated that Ad3- and Ad3 PtDd-mediated junction opening involves at least two mechanisms: (i) the activation of phosphokinase pathways and (ii) the downregulation of gene expression of junction/cytoskeletal proteins (3).
Another DSG2-interacting species B Ad is Ad14 and its recently emerged strain Ad14P1 (33). Like Ad3, Ad14 and Ad14P1 generate PtDd during infection (30). The basis of our studies on intracellular signaling triggered by these three viruses was the finding that they mediate the shedding of the DSG2 ECD, which can be detected in culture supernatant and serum of mice with xenograft tumors. ECD cleavage disrupts the DSG2 homodimers between two neighboring epithelial cells and thus probably contributes to Ad-mediated junction opening. We found that the following chain of events culminates in DSG2 ECD shedding: (i) binding of Ad3 via the fiber knob to DSG2, (ii) activation of MAPK, (iii) activation of ADAM17, and (iv) ECD cleavage. This does not exclude the possibility that other pathways are involved in DSG2 shedding.
DSG2 shedding is mediated by JO1, i.e., a dimerized trimeric Ad3 fiber knob (Fig. 1F and 3B). JO1 also triggered junction opening in a series of cancer cell lines and xenograft tumor models. Furthermore, an Ad35K++ fiber knob that does not bind to DSG2 did not trigger MAPK activation. In the data shown in Fig. 4B, the JO1-triggered signaling was normalized to that measured after Ad35K++ treatment (Fig. 4B).
We speculate that Ad3 binding to DSG2 triggers changes in the DSG2 conformation or clustering of several DSG2 molecules which, in turn, either directly or indirectly leads the phosphorylation of MAPK pathway members. Previous studies on the binding of Ad11 to CD46 showed that engagement by the Ad11 knob triggered a conformational change in the extracellular domain (ECD) of CD46, which resulted in the exposure of residues for binding that were hidden in the unligated receptor (53). Unlike Ad interaction with CD46 (53, 54), structural details on the Ad3 and PtDd interactions with DSG2 are still elusive. Our previous antibody competition studies indicate that the Ad3 fiber knob regions that interact with DSG2 are located in ECD3 and ECD4 (3). Attempts to cocrystallize DSG2 and Ad3 PtDd and resolve the structure of the complex are ongoing.
DSG2 shedding appears to be an integral part of epithelial cell biology (11). We found in sera from humans, human DSG2-transgenic mice, and macaques shed DSG2 ECD in the range of several hundred nanograms per milliliter (3, 36, 52). We also detected DSG2 ECD in the supernatant of cultured epithelial cancer cells and in the serum of mice with xenografts derived from human tumor cells (Fig. 1 to 3). Importantly, this process was greatly enhanced by Ad3, Ad14, Ad14P1, or Ad3 derivatives. With regard to “physiological” background from cancer cells, we speculate that the differences in the DSG2 concentrations in culture supernatants from A549 and SKOV3 cells are due to different degrees of MAPK and ADAM17 concentrations/activities in different cancer cell lines. The differences in shed DSG2 serum concentrations between individual mice with xenograft tumors could be due to different tumor volumes or vascularization. Preliminary studies of longitudinal serum samples from ovarian cancer patients indicate a potential correlation between DSG2 serum levels and tumor burden and recurrence (A. Lieber, personal communication).
The second objective of this study was to delineate potential mechanisms for the higher pathogenicity of Ad14P1 than that of the parental strain Ad14. Our data showing greater spread and oncolytic activity of Ad14P1 than of Ad14 in a human epithelial cancer model pointed toward better infectivity and/or replication of Ad14P1. The genomes of the two viruses differ by an insertion in E1A and a small deletion in the fiber knob (48). We tested the hypothesis that the mutation in the fiber knob would account for the observed differences. While our X-ray crystallography studies suggested differences in the structure of the Ad14P1 fiber knob in the F-G loop, this did not significantly change the knob affinity to DSG2 or the intracellular signaling and DSG2 shedding in epithelial cancer cells upon exposure to Ad14 and Ad14P1. In line with this is the outcome of an in vivo toxicity study. In an attempt to study potential pathological effects of Ad14 and Ad14P1, we injected these viruses intravenously into human DSG2-transgenic mice. These mice express human DSG2 in a pattern similar to that of humans, and binding of JO1 to DSG2 on mouse tumor cells that ectopically express human DSG2 triggers junction opening (52). The latter observation indicates that intracellular signaling in DSG2-transgenic mouse cells is similar to that in human cells. However, in contrast to human cells, mouse cells do not support productive adenovirus replication. Notably, human adenoviruses do not interact with the mouse orthologue for DSG2 (52). Our study did not reveal significant differences in blood parameters between the two viruses in blood cell counts (Fig. 13A). Both viruses triggered an increase in alanine aminotransferase (ALT; serum glutamic pyruvic transaminase [SGPT]) as a sign of liver inflammation (Fig. 13B). Overall, however, there were no significant differences in hematological parameters between the two Ad14 virus strains.
FIG 13.

Blood cell counts and chemistry in DSG2-transgenic mice injected with Ad14 and Ad14P1. Human DSG2-transgenic mice were intravenously injected with 2 × 109 PFU of Ad14 and Ad14P1, and blood samples were collected at the indicated days. (A) Blood cell counts. K/μl, x thousand cells per ml of blood. (B) Enzyme activity at day 7 after injection. U/liter, units per liter of blood. Normal ranges are indicated by gray boxes (n = 3). Alk phos, alkaline phosphatase; AST, aspartate aminotransferase; GOT, glutamic oxaloacetic transaminase; ALT, alanine aminotransferase; GPT, glutamic-pyruvic transaminase.
In summary, we conclude that the differences in the structures of the Ad14 and Ad14P1 fiber knobs do not change the viruses' effects on cells and that the insertion in the E1A gene might be the cause of greater viral spread and, potentially, the higher pathogenicity of Ad14P1. This speculation needs to be supported by introducing the E1A mutation into the genome of Ad14. Furthermore, the human DSG2-transgenic mouse model does not reflect a potentially higher pathogenicity of Ad14P1. We speculate that differences between the two viruses could become apparent in a more permissive animal model, e.g., using Syrian hamsters. However, at this point no human DSG2-transgenic hamster model is available.
A better understanding of structures and mechanisms of viral infection and spread in epithelial tissues is crucial for the development of new drugs that can interfere with these processes as well as for the development of potent prophylactic vaccines. Furthermore, most solid tumors are of epithelial origin and maintain epithelial junctions. DSG2 is overexpressed in most solid tumors (55–59). Considering that epithelial junctions are one of the factors that limit the dissemination of drugs and oncolytic viruses throughout the tumor, our studies of how PtDd trigger junction opening are also relevant for tumor therapy. Potentially, DSG2 shedding into the serum can also be used as a readout in clinical trials for the junction opening function of recombinant Ad3 derivatives that we plan to use clinically to improve drug penetration in epithelial tumors (36). Finally, our finding that Ad14P1 efficiently spreads in epithelial xenograft tumors makes this virus a potential platform for new oncolytic adenoviruses.
ACKNOWLEDGMENTS
The work was supported by NIH grants R01 CA080192 (A.L.) and R01 HLA078836 (A.L.), Pacific Ovarian Cancer Research Consortium/Specialized Program of Research Excellence in Ovarian Cancer grant P50 CA83636, a grant from the Department of Defense (A.L.), and a grant from BRIM Biotechnology, Inc. M.R. is a recipient of a fellowship award from the Deutscher Akademischer Austauschdienst.
We acknowledge the beam line staff on 23-2 at the European Synchrotron Radiation Facility. We thank Emilie Stermann for help with the Biacore graphics.
REFERENCES
- 1.Excoffon KJ, Bowers JR, Sharma P. 2014. 1. Alternative splicing of viral receptors: a review of the diverse morphologies and physiologies of adenoviral receptors. Recent Res Dev Virol 9:1–24. [PMC free article] [PubMed] [Google Scholar]
- 2.Lam E, Falck-Pedersen E. 2014. Unabated adenovirus replication following activation of the cGAS/STING-dependent antiviral response in human cells. J Virol 88:14426–14439. doi: 10.1128/JVI.02608-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang H, Li ZY, Liu Y, Persson J, Beyer I, Moller T, Koyuncu D, Drescher MR, Strauss R, Zhang XB, Wahl JK III, Urban N, Drescher C, Hemminki A, Fender P, Lieber A. 2011. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat Med 17:96–104. doi: 10.1038/nm.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chitaev NA, Troyanovsky SM. 1997. Direct Ca2+-dependent heterophilic interaction between desmosomal cadherins, desmoglein and desmocollin, contributes to cell-cell adhesion. J Cell Biol 138:193–201. doi: 10.1083/jcb.138.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harmon RM, Green KJ. 2013. Structural and functional diversity of desmosomes. Cell Commun Adhes 20:171–187. doi: 10.3109/15419061.2013.855204. [DOI] [PubMed] [Google Scholar]
- 6.Cowin P. 1994. Unraveling the cytoplasmic interactions of the cadherin superfamily. Proc Natl Acad Sci U S A 91:10759–10761. doi: 10.1073/pnas.91.23.10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maeda O, Usami N, Kondo M, Takahashi M, Goto H, Shimokata K, Kusugami K, Sekido Y. 2004. Plakoglobin (gamma-catenin) has TCF/LEF family-dependent transcriptional activity in beta-catenin-deficient cell line. Oncogene 23:964–972. doi: 10.1038/sj.onc.1207254. [DOI] [PubMed] [Google Scholar]
- 8.Aktary Z, Kulak S, Mackey J, Jahroudi N, Pasdar M. 2013. Plakoglobin interacts with the transcription factor p53 and regulates the expression of 14-3-3σ. J Cell Sci 126:3031–3042. doi: 10.1242/jcs.120642. [DOI] [PubMed] [Google Scholar]
- 9.Gaudry CA, Palka HL, Dusek RL, Huen AC, Khandekar MJ, Hudson LG, Green KJ. 2001. Tyrosine-phosphorylated plakoglobin is associated with desmogleins but not desmoplakin after epidermal growth factor receptor activation. J Biol Chem 276:24871–24880. doi: 10.1074/jbc.M102731200. [DOI] [PubMed] [Google Scholar]
- 10.Lorch JH, Klessner J, Park JK, Getsios S, Wu YL, Stack MS, Green KJ. 2004. Epidermal growth factor receptor inhibition promotes desmosome assembly and strengthens intercellular adhesion in squamous cell carcinoma cells. J Biol Chem 279:37191–37200. doi: 10.1074/jbc.M405123200. [DOI] [PubMed] [Google Scholar]
- 11.Klessner JL, Desai BV, Amargo EV, Getsios S, Green KJ. 2009. EGFR and ADAMs cooperate to regulate shedding and endocytic trafficking of the desmosomal cadherin desmoglein 2. Mol Biol Cell 20:328–337. doi: 10.1091/mbc.E08-04-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fox JP, Brandt CD, Wassermann FE, Hall CE, Spigland I, Kogon A, Elveback LR. 1969. The virus watch program: a continuing surveillance of viral infections in metropolitan New York families. VI. Observations of adenovirus infections: virus excretion patterns, antibody response, efficiency of surveillance, patterns of infections, and relation to illness. Am J Epidemiol 89:25–50. [DOI] [PubMed] [Google Scholar]
- 13.Metzgar D, Osuna M, Kajon AE, Hawksworth AW, Irvine M, Russell KL. 2007. Abrupt emergence of diverse species B adenoviruses at US military recruit training centers. J Infect Dis 196:1465–1473. doi: 10.1086/522970. [DOI] [PubMed] [Google Scholar]
- 14.Rebelo-de-Andrade H, Pereira C, Giria M, Prudencio E, Brito MJ, Cale E, Taveira N. 2010. Outbreak of acute respiratory infection among infants in Lisbon, Portugal, caused by human adenovirus serotype 3 and a new 7/3 recombinant strain. J Clin Microbiol 48:1391–1396. doi: 10.1128/JCM.02019-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeung R, Eshaghi A, Lombos E, Blair J, Mazzulli T, Burton L, Drews SJ. 2009. Characterization of culture-positive adenovirus serotypes from respiratory specimens in Toronto, Ontario, Canada: September 2007-June 2008. Virol J 6:11. doi: 10.1186/1743-422X-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong JY, Lee HJ, Piedra PA, Choi EH, Park KH, Koh YY, Kim WS. 2001. Lower respiratory tract infections due to adenovirus in hospitalized Korean children: epidemiology, clinical features, and prognosis. Clin Infect Dis 32:1423–1429. doi: 10.1086/320146. [DOI] [PubMed] [Google Scholar]
- 17.Lai CY, Lee CJ, Lu CY, Lee PI, Shao PL, Wu ET, Wang CC, Tan BF, Chang HY, Hsia SH, Lin JJ, Chang LY, Huang YC, Huang LM. 2013. Adenovirus serotype 3 and 7 infection with acute respiratory failure in children in Taiwan, 2010-2011. PLoS One 8:e53614. doi: 10.1371/journal.pone.0053614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li L, Phan TG, Nguyen TA, Kim KS, Seo JK, Shimizu H, Suzuki E, Okitsu S, Ushijima H. 2005. Molecular epidemiology of adenovirus infection among pediatric population with diarrhea in Asia. Microbiol Immunol 49:121–128. doi: 10.1111/j.1348-0421.2005.tb03711.x. [DOI] [PubMed] [Google Scholar]
- 19.Centers for Disease Control and Prevention (CDC). 2007. Acute respiratory disease associated with adenovirus serotype 14–four states, 2006-2007. MMWR Morb Mortal Wkly Rep 56:1181–1184. [PubMed] [Google Scholar]
- 20.Lewis PF, Schmidt MA, Lu X, Erdman DD, Campbell M, Thomas A, Cieslak PR, Grenz LD, Tsaknardis L, Gleaves C, Kendall B, Gilbert D. 2009. A community-based outbreak of severe respiratory illness caused by human adenovirus serotype 14. J Infect Dis 199:1427–1434. doi: 10.1086/598521. [DOI] [PubMed] [Google Scholar]
- 21.Esposito DH, Gardner TJ, Schneider E, Stockman LJ, Tate JE, Panozzo CA, Robbins CL, Jenkerson SA, Thomas L, Watson CM, Curns AT, Erdman DD, Lu X, Cromeans T, Westcott M, Humphries C, Ballantyne J, Fischer GE, McLaughlin JB, Armstrong G, Anderson LJ. 2010. Outbreak of pneumonia associated with emergent human adenovirus serotype 14—southeast Alaska, 2008. J Infect Dis 202:214–222. doi: 10.1086/653498. [DOI] [PubMed] [Google Scholar]
- 22.Carr MJ, Kajon AE, Lu X, Dunford L, O'Reilly P, Holder P, De Gascun CF, Coughlan S, Connell J, Erdman DD, Hall WW. 2011. Deaths associated with human adenovirus-14p1 infections, Europe, 2009-2010. Emerg Infect Dis 17:1402–1408. doi: 10.3201/1708.101760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kajon AE, Lu X, Erdman DD, Louie J, Schnurr D, George KS, Koopmans MP, Allibhai T, Metzgar D. 2010. Molecular epidemiology and brief history of emerging adenovirus 14-associated respiratory disease in the United States. J Infect Dis 202:93–103. doi: 10.1086/653083. [DOI] [PubMed] [Google Scholar]
- 24.Girouard G, Garceau R, Thibault L, Oussedik Y, Bastien N, Li Y. 2013. Adenovirus serotype 14 infection, New Brunswick, Canada, 2011. Emerg Infect Dis 19:119–122. doi: 10.3201/eid1901.120423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang L, An J, Xie Z, Dehghan S, Seto D, Xu W, Ji Y. 2013. Genome and bioinformatic analysis of a HAdV-B14p1 virus isolated from a baby with pneumonia in Beijing, China. PLoS One 8:e60345. doi: 10.1371/journal.pone.0060345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trei JS, Johns NM, Garner JL, Noel LB, Ortman BV, Ensz KL, Johns MC, Bunning ML, Gaydos JC. 2010. Spread of adenovirus to geographically dispersed military installations, May-October 2007. Emerg Infect Dis 16:769–775. doi: 10.3201/eid1605.091633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greber UF. 1998. Virus assembly and disassembly: the adenovirus cysteine protease as a trigger factor. Rev Med Virol 8:213–222. [DOI] [PubMed] [Google Scholar]
- 28.Trotman LC, Achermann DP, Keller S, Straub M, Greber UF. 2003. Non-classical export of an adenovirus structural protein. Traffic 4:390–402. doi: 10.1034/j.1600-0854.2003.00094.x. [DOI] [PubMed] [Google Scholar]
- 29.Fender P, Boussaid A, Mezin P, Chroboczek J. 2005. Synthesis, cellular localization, and quantification of penton-dodecahedron in serotype 3 adenovirus-infected cells. Virology 340:167–173. doi: 10.1016/j.virol.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 30.Lu ZZ, Wang H, Zhang Y, Cao H, Li Z, Fender P, Lieber A. 2013. Penton-dodecahedral particles trigger opening of intercellular junctions and facilitate viral spread during adenovirus serotype 3 infection of epithelial cells. PLoS Pathog 9:e1003718. doi: 10.1371/journal.ppat.1003718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fender P, Ruigrok RW, Gout E, Buffet S, Chroboczek J. 1997. Adenovirus dodecahedron, a new vector for human gene transfer. Nat Biotechnol 15:52–56. doi: 10.1038/nbt0197-52. [DOI] [PubMed] [Google Scholar]
- 32.Wang H, Li Z, Yumul R, Lara S, Hemminki A, Fender P, Lieber A. 2011. Multimerization of adenovirus serotype 3 fiber knob domains is required for efficient binding of virus to desmoglein 2 and subsequent opening of epithelial junctions. J Virol 85:6390–6402. doi: 10.1128/JVI.00514-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, Yumul R, Cao H, Ran L, Fan X, Richter M, Epstein F, Gralow J, Zubieta C, Fender P, Lieber A. 2013. Structural and functional studies on the interaction of adenovirus fiber knobs and desmoglein 2. J Virol 87:11346–11362. doi: 10.1128/JVI.01825-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beyer I, Cao H, Persson J, Song H, Richter M, Feng Q, Yumul R, van Rensburg R, Li Z, Berenson R, Carter D, Roffler S, Drescher C, Lieber A. 2012. Coadministration of epithelial junction opener JO-1 improves the efficacy and safety of chemotherapeutic drugs. Clin Cancer Res 18:3340–3351. doi: 10.1158/1078-0432.CCR-11-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beyer I, van Rensburg R, Strauss R, Li Z, Wang H, Persson J, Yumul R, Feng Q, Song H, Bartek J, Fender P, Lieber A. 2011. Epithelial junction opener JO-1 improves monoclonal antibody therapy of cancer. Cancer Res 71:7080–7090. doi: 10.1158/0008-5472.CAN-11-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richter M, Yumul R, Wang H, Saydaminova K, Ho M, May D, Baldessari A, Gough M, Drescher C, Urban N, Roffler S, Zubieta C, Carter D, Fender P, Lieber A. 2015. Preclinical safety and efficacy studies with an affinity enhanced epithelial junction opener and PEGylated liposomal doxorubicin. Mol Ther Methods Clin Dev 2:15005. doi: 10.1038/mtm.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamekura R, Kolegraff KN, Nava P, Hilgarth RS, Feng M, Parkos CA, Nusrat A. 2014. Loss of the desmosomal cadherin desmoglein-2 suppresses colon cancer cell proliferation through EGFR signaling. Oncogene 33:4531–4536. doi: 10.1038/onc.2013.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang H, Tuve S, Erdman DD, Lieber A. 2009. Receptor usage of a newly emergent adenovirus type 14. Virology 387:436–441. doi: 10.1016/j.virol.2009.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tuve S, Wang H, Ware C, Liu Y, Gaggar A, Bernt K, Shayakhmetov D, Li Z, Strauss R, Stone D, Lieber A. 2006. A new group B adenovirus receptor is expressed at high levels on human stem and tumor cells. J Virol 80:12109–12120. doi: 10.1128/JVI.01370-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shayakhmetov DM, Papayannopoulou T, Stamatoyannopoulos G, Lieber A. 2000. Efficient gene transfer into human CD34+ cells by a retargeted adenovirus vector. J Virol 74:2567–2583. doi: 10.1128/JVI.74.6.2567-2583.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H, Liaw YC, Stone D, Kalyuzhniy O, Amiraslanov I, Tuve S, Verlinde CL, Shayakhmetov D, Stehle T, Roffler S, Lieber A. 2007. Identification of CD46 binding sites within the adenovirus serotype 35 fiber knob. J Virol 81:12785–12792. doi: 10.1128/JVI.01732-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaggar A, Shayakhmetov D, Lieber A. 2007. Identifying functional adenovirus-host interactions using tandem mass spectrometry. Methods Mol Med 131:141–155. doi: 10.1007/978-1-59745-277-9_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blanc E, Roversi P, Vonrhein C, Flensburg C, Lea SM, Bricogne G. 2004. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr D Biol Crystallogr 60:2210–2221. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
- 44.Shenk T. 2001. Adenoviridae, p 2265–2328. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 4th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 45.Wang H, Liu Y, Li ZY, Fan X, Hemminki A, Lieber A. 2010. A recombinant adenovirus type 35 fiber knob protein sensitizes lymphoma cells to rituximab therapy. Blood 115:592–600. doi: 10.1182/blood-2009-05-222463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bech-Serra JJ, Santiago-Josefat B, Esselens C, Saftig P, Baselga J, Arribas J, Canals F. 2006. Proteomic identification of desmoglein 2 and activated leukocyte cell adhesion molecule as substrates of ADAM17 and ADAM10 by difference gel electrophoresis. Mol Cell Biol 26:5086–5095. doi: 10.1128/MCB.02380-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dassler K, Kaup M, Tauber R, Fuchs H. 2003. Mutational suppression of transferrin receptor shedding can be compensated by distinct metalloproteases acting on alternative sites. FEBS Lett 536:25–29. doi: 10.1016/S0014-5793(03)00004-8. [DOI] [PubMed] [Google Scholar]
- 48.Houng HS, Gong H, Kajon AE, Jones MS, Kuschner RA, Lyons A, Lott L, Lin KH, Metzgar D. 2010. Genome sequences of human adenovirus 14 isolates from mild respiratory cases and a fatal pneumonia, isolated during 2006-2007 epidemics in North America. Respir Res 11:116. doi: 10.1186/1465-9921-11-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lutschg V, Boucke K, Hemmi S, Greber UF. 2011. Chemotactic antiviral cytokines promote infectious apical entry of human adenovirus into polarized epithelial cells. Nat Commun 2:391. doi: 10.1038/ncomms1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bergelson JM. 2009. Intercellular junctional proteins as receptors and barriers to virus infection and spread. Cell Host Microbe 5:517–521. doi: 10.1016/j.chom.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 51.Walters RW, Freimuth P, Moninger TO, Ganske I, Zabner J, Welsh MJ. 2002. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell 110:789–799. doi: 10.1016/S0092-8674(02)00912-1. [DOI] [PubMed] [Google Scholar]
- 52.Wang H, Beyer I, Persson J, Song H, Li Z, Richter M, Cao H, van Rensburg R, Yao X, Hudkins K, Yumul R, Zhang XB, Yu M, Fender P, Hemminki A, Lieber A. 2012. A new human DSG2-transgenic mouse model for studying the tropism and pathology of human adenoviruses. J Virol 86:6286–6302. doi: 10.1128/JVI.00205-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Persson BD, Reiter DM, Marttila M, Mei YF, Casasnovas JM, Arnberg N, Stehle T. 2007. Adenovirus type 11 binding alters the conformation of its receptor CD46. Nat Struct Mol Biol 14:164–166. doi: 10.1038/nsmb1190. [DOI] [PubMed] [Google Scholar]
- 54.Bewley MC, Springer K, Zhang YB, Freimuth P, Flanagan JM. 1999. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 286:1579–1583. doi: 10.1126/science.286.5444.1579. [DOI] [PubMed] [Google Scholar]
- 55.Abbod MF, Hamdy FC, Linkens DA, Catto JW. 2009. Predictive modeling in cancer: where systems biology meets the stock market. Expert Rev Anticancer Ther 9:867–870. doi: 10.1586/era.09.47. [DOI] [PubMed] [Google Scholar]
- 56.Biedermann K, Vogelsang H, Becker I, Plaschke S, Siewert JR, Hofler H, Keller G. 2005. Desmoglein 2 is expressed abnormally rather than mutated in familial and sporadic gastric cancer. J Pathol 207:199–206. doi: 10.1002/path.1821. [DOI] [PubMed] [Google Scholar]
- 57.Harada H, Iwatsuki K, Ohtsuka M, Han GW, Kaneko F. 1996. Abnormal desmoglein expression by squamous cell carcinoma cells. Acta Derm Venereol 76:417–420. [DOI] [PubMed] [Google Scholar]
- 58.Schmitt CJ, Franke WW, Goerdt S, Falkowska-Hansen B, Rickelt S, Peitsch WK. 2007. Homo- and heterotypic cell contacts in malignant melanoma cells and desmoglein 2 as a novel solitary surface glycoprotein. J Investig Dermatol 127:2191–2206. doi: 10.1038/sj.jid.5700849. [DOI] [PubMed] [Google Scholar]
- 59.Trojan L, Schaaf A, Steidler A, Haak M, Thalmann G, Knoll T, Gretz N, Alken P, Michel MS. 2005. Identification of metastasis-associated genes in prostate cancer by genetic profiling of human prostate cancer cell lines. Anticancer Res 25:183–191. [PubMed] [Google Scholar]