

Abstract
Background
Effectiveness of the immune defense formed by the genotype often determines the predisposition to cancer. Nitric oxide (NO) produced by macrophages is an important element in this defense.
Material/Methods
We hypothesized that genetic characteristics of NO generation systems can predetermine the vulnerability to tumor development. The study was conducted on mice of 2 genetic substrains – C57BL/6J and C57BL/6N – with Ehrlich ascites carcinoma (EAC). NO production in the tumor was changed using ITU, an iNOS inhibitor; c-PTIO, a NO scavenger; and SNP, a NO donor. Macrophage NO production was estimated by nitrite concentration in the culture medium. iNOS content was measured by Western blot analysis. Macrophage phenotype was determined by changes in NO production, iNOS level, and CD markers of the phenotype.
Results
The lifespan of C57BL/6N mice (n=10) with EAC was 25% longer (p<0.01) than in C57BL/6J mice (n=10). Decreased NO production 23% reduced the survival duration of C57BL/6N mice (p<0.05), which were more resistant to tumors. Elevated NO production 26% increased the survival duration of C57BL/6J mice (p<0.05), which were more susceptible to EAC. Both the NO production and the iNOS level were 1.5 times higher in C57BL/6N than in C57BL/6J mice (p<0.01). CD markers confirmed that C57BL/6N macrophages had the M1 and C57BL/6J macrophages had the M2 phenotype.
Conclusions
The vulnerability to the tumor development can be predetermined by genetic characteristics of the NO generation system in macrophages. The important role of NO in anti-EAC immunity should be taken into account in elaboration of new antitumor therapies.
MeSH Keywords: Carcinoma, Ehrlich Tumor; Macrophages; Nitric Oxide
Background
Understanding molecular and cellular mechanisms of carcinogenesis and justification of new, effective approaches to restriction of tumor growth are critical challenges of modern medicine. Vulnerability to tumor development is determined by multiple factors, including genetic predisposition [1–3], which, in turn, is determined by the capability of an individual or a species genotype to form an antitumor immune defence. One of important factors of such a defence is nitric oxide (NO) and effectiveness of the NO-generating system [4–7]. In the immune system, NO is generated by macrophages.
Based on these facts, we hypothesized that the individual or species vulnerability to tumors could be predetermined by genetic characteristics of the macrophage NO-generating system. The study goal was to test this hypothesis. The hypothesis was tested on mouse substrains C57BL/6J and C57BL/6N. C57BL/6J mice had the a, H-2b (http://andreevka.msk.ru/index.htm) genotype and C57BL/6N mice had the MHC HAPLOTYPE H-2b (http://www.spf-animals.ru) genotype. More detailed characteristics of these strains are provided on the Jackson Laboratory (https://www.jax.org) and TACONIC (http://www.taconic.com) websites.
Three main objectives were designed to achieve the study goal: 1) to evaluate susceptibility of different mouse substrains, C57BL/6J and C57BL/6N, to development of Ehrlich ascites carcinoma (EAC); 2) to study the role of NO in different susceptibility of these mouse substrains to development of tumor using an NO scavenger, an inducible NO synthase (iNOS) inhibitor, and a NO donor; and 3) to identify the macrophage phenotype and to evaluate the effectiveness of NO generation in macrophages from mice of different substrains.
Material and Methods
Experimental animals
Experiments were performed on 2 genetically different substrains of C57BL/6 mice: C57BL/6J and C57BL/6N. C57BL/6J mice were obtained from the vivarium “Andreevka” (http://andreevka.msk.ru/index.htm), and C57BL/6N – from the vivarium “Pushchino” (http://www.spf-animals.ru). All experiments were designed and performed in accordance with the WHO guidance for biomedical research in animals (http://www.cioms.ch/publications/guidelines/1985_texts_of_guidelines.htm). Mice of both substrains were 8–9-week-old males weighing 20–24 g. The protocol of experiments was approved by the University Ethics Committee. All mice in experimental groups died because of tumor progression.
EAC
Tumor growth was initiated by an injection of EAC cells, which were obtained from the N.N. Blokhin Russian Cancer Research Center (Moscow, Russia). Mice were injected intraperitoneally with 250,000 tumor cells diluted in 0.2 ml saline. All mice were weighed daily until their death. Resistance of mice to EAC was assessed by survival duration after the injection of tumor cells and by changes in the animal weight reflecting accumulation of ascitic fluid in the peritoneal cavity.
Chemicals modifying NO production and content
To change NO production in the tumor area, we used S- (2-aminoethyl) isothiourea (ITU, 10 mg/kg, i.p.) (Cat. # 270-029M050, Alexis Corp., USA), a selective iNOS inhibitor; ([2-4-carboxyphenyl) -4,4,5,5-tetramethylimidazoline-1-oxil-3-oxide] (cPTIO, 8 mmol/kg) (cat. # C7912, Invitrogen, USA). To change NO content in the tumor area, we used the NO trap, which cannot penetrate the cell [8]; and sodium nitroprusside (SNP, 10 μmol/kg) (cat. # S13755389.0100, DiaM, Russia), a NO donor [9]. The chemicals were administered at 3, 7 and 11 days after tumor inoculation. This timing corresponded to the lag-, log- and terminal phases of EAC growth, respectively.
Evaluation of macrophage NO-generating system
To assess the macrophage NO production, peritoneal macrophages were isolated from mice using a standard method described by Zhang et al. [10]. After isolation, macrophages were placed in wells of flat-bottomed 48-well culture plates in RPMI-1640 medium with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C and 5% CO2. Macrophages were placed in culture wells by 0.5×106 cells per well in 0.5 ml of the medium. Macrophage basal NO production was evaluated after 24 hours of culturing. Stimulated NO production was evaluated after stimulation of macrophages with 500 ng/ml lipopolysaccharide (LPS) (cat. # L2654, Sigma-Aldrich, USA) for 24 hours. Macrophage NO production was evaluated by nitrite concentration, which was measured spectrophotometrically in the culture medium using the Griess reaction [11]. The iNOS content was measured using a standard Western blot analysis [12] with anti-iNOS (iNOS) rabbit polyclonal antibodies (cat. # KAS – N 0001, Assay Designs, USA).
Determination of macrophage phenotype
Phenotype was determined by macrophage NO production and iNOS content and by cell-surface markers of the M1 phenotype, CD80, and the M2 phenotype, CD206 [13]. The CD markers were measured by flow cytometry using monoclonal antibodies to CD80 (Beckman Coulter, cat. # 12-0801-82, USA) and CD206 (Beckman Coulter, cat. # FAB2535P, USA) [13]. Preparation of macrophage samples for analysis was performed according to the manufacturer’s instructions. Content of M1 and M2 CD markers on macrophages was expressed as percent.
Increased production of NO, expression of iNOS, and content of M1 phenotype CD markers indicated formation of the M1 phenotype, whereas decreased production of NO, expression of iNOS, and increased content of M2 phenotype CD markers indicated formation of the M2 phenotype [13].
Animal experiments were performed in duplicate, and cell culture experiments were performed in triplicate. Experimental protocol is presented in Figure 1.
Figure 1.

Experimental protocol.
Statistical analyses were performed using analysis of variance followed by Student-Newman-Keuls test. Data are presented as means (M) with standard errors of the mean (±SEM). Differences were considered statistically significant at p<0.05.
Theory background
Macrophages can exhibit their secretory activity as a pro-inflammatory M1 phenotype or an anti-inflammatory M2 phenotype. The M1 phenotype produces more NO than the M2 phenotype [13]. The M1 phenotype is known to possesess a marked capacity for restricting tumors, whereas the M2 phenotype, in contrast, can promote tumor growth [13]. Ability of the organism to form specific phenotypes of cells is predetermined by the genotype. These facts support our hypothesis and justify its experimental verification.
Results
C57BL/6N mice are more resistant to EAC than C57BL/6J mice
After injection of EAC cells, the survival duration of C57BL/6J mice was 15.6±0.6 days, whereas C57BL/6N mice survived for 19.5±0.5 days (p <0.01), i.e., by 25% longer than C57BL/6J mice. The toxic effect of EAC was evident as progressive accumulation of ascetic fluid in the abdominal cavity in both substrains, which resulted in gaining body weight. However, for 11 days of tumor growth, the body weight of C57BL/6N mice increased by 6.5±1.1%, whereas the body weight of C57BL/6J mice increased by 12.7±1.8% (p<0.01), i.e., almost twice as much as in C57BL/6N mice.
Therefore, C57BL/6N mice were significantly more resistant to the development of EAC than C57BL/6J mice, as shown by the parameters of survival duration and accumulation of ascetic fluid in the peritoneal cavity.
Increased NO production prolongs while reduced NO production shortens survival of mice with EAC
Changed NO production in the tumor growth zone influenced the lifespan of mice (Figure 2). The decrease in NO induced by the NO trap, PTIO, or the iNOS inhibitor, ITU, significantly shortened the survival duration of the highly resistant C57BL/6N mice with EAC by 23% (p<0.05) and the low resistant C57BL/6J mice with EAC by 17% (p>0.05). At the same time, the increase in NO induced by the NO donor, SNP, significantly prolonged the survival of low resistant C57BL/6J mice with EAC by 26% (p<0.05) and non-significantly prolonged the survival of highly resistant C57BL/6J mice with EAC by 10% (p>0.05). However, none of these NO modulators influenced peritoneal accumulation of ascetic fluid (data not shown).
Figure 2.
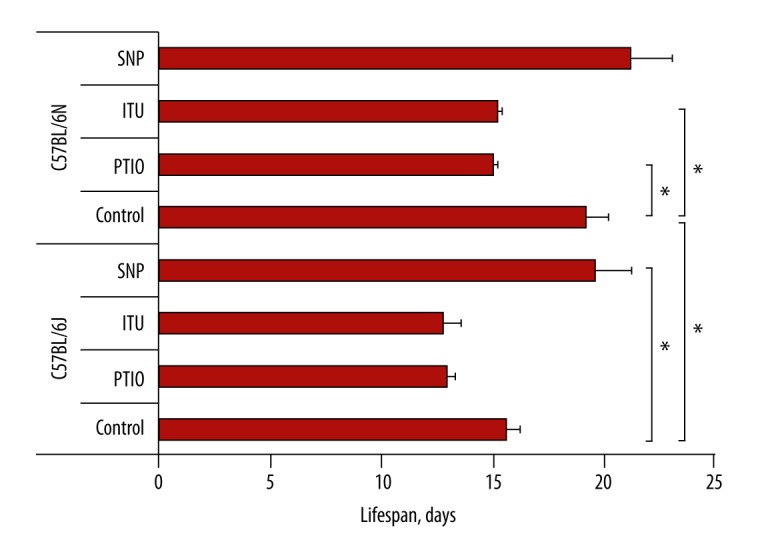
Effects of changes in NO level in the tumor zone on mouse lifespan. A NO scavenger (PTIO) or an iNOS inhibitor (ITU) was used for decreasing the NO level. A NO donor (SNP) was used for increasing the NO level. NO – nitric oxide; PTIO – ([2-4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxil-3-oxide]), a NO scavenger; ITU – (S-(2-aminoethyl isothiourea), an iNOS inhibitor; SNP – (sodium nitroprusside), a NO donor; C57BL/6N and C57BL/6J – mouse substrains. Bars show lifespan in days. * Significant difference between mouse substrains, p<0.05.
These results are consistent with our hypothesis about an important role of NO in genetically determined mechanisms responsible for the life expectancy of mice after the onset of tumor development.
Macrophages of highly resistant to tumor C57BL/6N mice have more powerful NO generating system and a more distinct M1 phenotype than macrophages of low resistant to tumor C57BL/6J mice
Both basal and stimulated NO production was approximately 1.5 times higher in macrophages of highly tumor-resistant C57BL/6N mice than in macrophages of low resistant to tumor C57BL/6J mice (Table 1). This difference in macrophage NO production was consistent with a higher content of iNOS in macrophages of C57BL/6N mice compared with C57BL/6J mice (data in the insert of Table 1).
Table 1.
Basal and stimulated NO production, iNOS content and phenotype CD markers in macrophages isolated from highly tumor-resistant C57BL/6N substrain and lowly tumor-resistant C57BL/6J substrain.
| Substrain | Survival after tumor induction, days | Indices of NO generation system | M1 marker, CD 80 | M2 marker, CD206 | |
|---|---|---|---|---|---|
| C57BL/6N | 19.5+0.5 | Nitrite, μmol | 80.6±5.6% | 52.7±4.9% | |
| Basal conditions | Stimulated conditions | ||||
| 45.7±1.34 | 74.4±1.26 | ||||
| iNOS | |||||
| Basal conditions | Stimulated conditions | ||||

|

|
||||
| C57BL/6J | 15.6±0.6** | Nitrite, μmol | 4.0±0.3%** | 58.8±4.7% | |
| Basal conditions | Stimulated conditions | ||||
| 29.6±1.43** | 52.2±1.28** | ||||
| iNOS | |||||
| Basal conditions | Stimulated conditions | ||||

|

|
||||
Immunograms of Western blot analysis are presented as inserts in the table; intensity of the black line reflects iNOS content in macrophages.
Significant difference between mouse substrains, p<0.01.
Therefore, macrophages of highly resistant to EAC C57BL/6N mice generate more basal and stimulated NO than macrophages of low resistant to EAC C57BL/6J mice.
High NO production and iNOS content are markers of the proinflammatory M1 macrophage phenotype [13]. Based even on this fact alone, the macrophage phenotype of C57BL/6N mice with high NO production can be defined as a more distinct M1 phenotype compared with macrophages of C57BL/6J mice with lower NO production.
Macrophages from C57BL/6N and C57BL/6J mice expressed equal amounts of CD206, the M2 phenotype marker (Table 1). However, macrophages from C57BL/6N mice expressed 20 times more CD80, the M1 phenotype marker, than C57BL/6J mouse macrophages (Table 1). Therefore, the CD marker analysis has confirmed that macrophages from C57BL/6N mice possess a more distinct M1 phenotype than macrophages from C57BL/6J mice.
Discussion
Results of the study are consistent with the initial hypothesis that susceptibility of a body or species to development of tumor is predetermined by genetic characteristics of NO generating systems. NO plays a dual role in carcinogenesis. On the one hand, NO can induce apoptosis of tumor cells by inhibiting synthesis of anti-apoptotic Bcl-2 and increasing expression of proapoptotic Bax and p53 [14], by inhibiting enzymes of glycolysis [15], which is the major pathway of energy production in tumor cells [16] and, thereby. NO exerts anti-tumor effects. On the other hand, NO can restrict development of apoptosis in tumor cells by nitrosylation of caspases [17,18] and/or activating HSP70 synthesis [19,20], and, thereby, NO can exert pro-tumor effects. The protumor effect of NO can be also due to NO-dependent inhibition of mitochondrial respiratory chain complex IV [21] and subsequent reprogramming of the normal cell phenotype to the tumor phenotype [22]. Local concentrations of NO and other free radicals in the tumor area determine interactions of NO and its metabolites with DNA, the tumor suppressor p53, and other activators and inhibitors of apoptosis in tumor cells. These interactions, in turn, form the pro- or anti-tumor effect of NO [7].
In our experiments, both the NO scavenger and the iNOS inhibitor decreased survival in both C57BL/6J and C57BL/6N mice inoculated with tumor cells, while administration of the NO donor prolonged the life of mice (Figure 2). Therefore, in this case, NO exerted an anti-tumor effect.
During the immune response, macrophages are the major source of synthesized NO. Macrophages can limit tumor growth, not only due to NO production [23,24], but also through other mechanisms, such as increased production of proinflammatory cytokines [24], free radicals activity [23] or tumor antigen presentation, and formation of Th1 and cytotoxic lymphocytes [25,26]. However, the fact that selective inhibition of macrophage iNOS reduces the lifespan of mice with EAC (Figure 2) indicates the important role of macrophage NO in the antitumor defense.
In this study, we found that macrophages of the highly resistant C57BL/6N substrain possess a more expressed pro-inflammatory M1 phenotype than macrophages of the low resistant C57BL/6J substrain. These data are consistent with the idea that the M1 macrophage phenotype has anti-tumor properties as distinct from the M2 phenotype [27–31].
Experimental data of this study did show what determines the genetically higher potency of NO-generating systems in macrophages of C57BL/6N mice compared with macrophages of C57BL/6J mice. However, since both substrains have a common origin (line C57BL/6), the difference between the NO synthesis mechanisms is likely related with minor differences in genotypes of these substrains.
Genetic studies showed that genotypes of C57BL/6J and C57BL/6N mice have 2 important differences.
First, C57BL/6J mice, as distinct from C57BL/6N, have a small deletion in the nicotinamide nucleotide transhydrogenase (NNT) gene and, therefore, the NNT protein is not synthesized [32]. NNT is a mitochondrial proton pump encoded by nuclear DNA. The lack of NNT leads to impaired glucose homeostasis and decreased insulin secretion [32]. In macrophages, as shown by Ripoll et al. [33], NNT plays the role of regulator in inflammatory responses, free radicals production, and NO synthesis.
Second, Mekada et al. [34] and Zurita et al. [35] found differences in DNA single-nucleotide polymorphism of these substrains.
Differences in mechanisms of NO generation and susceptibility to tumor growth may be associated with the absence/presence of NNT protein and/or differences in the single-nucleotide polymorphism. This assumption requires thorough investigation. However, the fact that NO plays an important role in the prolonged survival under EAC, the most aggressive form of cancer, makes it promising to develop new approaches to anti-tumor treatment by manipulating NO in the tumor.
Conclusions
Data of this study and the literature show that the NNT gene and protein and the differences in single-nucleotide polymorphism found in different C57BL/6 substrains should also be considered in developing new methods of antitumor treatment.
Footnotes
Source of support: The study was supported by the State through the Russian Ministry of Education and Science (Agreement dated June 17, 2014 No. 14.604.21.0020, unique identifier for applied research, RFMEFI60414X0020)
References
- 1.Demichelis F, Stanford JL. Genetic predisposition to prostate cancer: Update and future perspectives. Urol Oncol. 2015;33(2):75–84. doi: 10.1016/j.urolonc.2014.04.021. [DOI] [PubMed] [Google Scholar]
- 2.Cybulski C, Nazarali S, Narod SA. Multiple primary cancers as a guide to heritability. Int J Cancer. 2014;135(8):1756–63. doi: 10.1002/ijc.28988. [DOI] [PubMed] [Google Scholar]
- 3.Lochhead P, Chan AT, Nishihara R, et al. Etiologic field effect: reappraisal of the field effect concept in cancer predisposition and progression. Mod Pathol. 2015;28(1):14–29. doi: 10.1038/modpathol.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bellin D, Asai S, Delledonne M, Yoshioka H. Nitric oxide as a mediator for defense responses. Mol Plant Microbe Interact. 2013;26(3):271–77. doi: 10.1094/MPMI-09-12-0214-CR. [DOI] [PubMed] [Google Scholar]
- 5.Buijs N, Luttikhold J, Houdijk AP, van Leeuwen PA. The role of a disturbed arginine/NO metabolism in the onset of cancer cachexia: a working hypothesis. Curr Med Chem. 2012;19(31):5278–86. doi: 10.2174/092986712803833290. [DOI] [PubMed] [Google Scholar]
- 6.Korde Choudhari S, Sridharan G, et al. Nitric oxide and oral cancer: a review. Oral Oncol. 2012;48(6):475–83. doi: 10.1016/j.oraloncology.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Engin AB. Dual function of nitric oxide in carcinogenesis, reappraisal. Curr Drug Metab. 2011;12(9):891–99. doi: 10.2174/138920011797470092. [DOI] [PubMed] [Google Scholar]
- 8.Akaike T, Maeda H. Quantitation of nitric oxide using 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (PTIO) Methods Enzymol. 1996;268:211–21. doi: 10.1016/s0076-6879(96)68023-9. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, He Q, Yan X, Cai Y, Chen J. Effect of exogenous nitric oxide on sperm motility in vitro. Biol Res. 2014;47(1):44. doi: 10.1186/0717-6287-47-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;Chapter 14(Unit 14):1. doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Redente EF, Dwyer-Nield LD, Barrett BS, et al. Lung tumor growth is stimulated in IFN-gamma–/– mice and inhibited in IL-4Ralpha–/– mice. Anticancer Res. 2009;29(12):5095–101. [PMC free article] [PubMed] [Google Scholar]
- 12.Smith FS, Titheradge MA. Nitric oxide protocols. Methods Mol Biol. 1998;100:171–80. doi: 10.1385/1-59259-749-1:171. [DOI] [PubMed] [Google Scholar]
- 13.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 14.Zeini M, Través PG, López-Fontal R, et al. Specific contribution of p19(ARF) to nitric oxide-dependent apoptosis. J Immunol. 2006;177(5):3327–36. doi: 10.4049/jimmunol.177.5.3327. [DOI] [PubMed] [Google Scholar]
- 15.Brüne B, Mohr S, Messmer UK. Protein thiol modification and apoptotic cell death as cGMP-independent nitric oxide (NO) signaling pathways. Rev Physiol Biochem Pharmacol. 1996;127:1–30. doi: 10.1007/BFb0048263. [DOI] [PubMed] [Google Scholar]
- 16.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 17.Török NJ, Higuchi H, Bronk S, Gores GJ. Nitric oxide inhibits apoptosis downstream of cytochrome C release by nitrosylating caspase 9. Cancer Res. 2002;62(6):1648–53. [PubMed] [Google Scholar]
- 18.Maejima Y, Adachi S, Morikawa K, et al. Nitric oxide inhibits myocardial apoptosis by preventing caspase-3 activity via S-nitrosylation. J Mol Cell Cardiol. 2005;38(1):163–74. doi: 10.1016/j.yjmcc.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Malyshev IYu, Malugin AV, Golubeva LYu, et al. Nitric oxide donor induces HSP70 accumulation in the heart and in cultured cells. FEBS Lett. 1996;391(1–2):21–23. doi: 10.1016/0014-5793(96)00691-6. [DOI] [PubMed] [Google Scholar]
- 20.Murphy ME. The HSP70 family and cancer. Carcinogenesis. 2013;34(6):1181–88. doi: 10.1093/carcin/bgt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stefano GB, Kream RM. Nitric oxide regulation of mitochondrial processes: commonality in medical disorders. Ann Transplant. 2015;20:402–7. doi: 10.12659/AOT.894289. [DOI] [PubMed] [Google Scholar]
- 22.Davila AF, Zamorano P. Mitochondria and the evolutionary roots of cancer. Phys Biol. 2013;10(2):026008. doi: 10.1088/1478-3975/10/2/026008. [DOI] [PubMed] [Google Scholar]
- 23.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lum HD, Buhtoiarov IN, Schmidt BE, et al. Tumoristatic effects of anti-CD40 mAb-activated macrophages involve nitric oxide and tumour necrosis factor-alpha. Immunology. 2006;118(2):261–70. doi: 10.1111/j.1365-2567.2006.02366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Backer R, Schwandt T, Greuter M, et al. Effective collaboration between marginal metallophilic macrophages and CD8+ dendritic cells in the generation of cytotoxic T cells. Proc Natl Acad Sci USA. 2010;107(1):216–21. doi: 10.1073/pnas.0909541107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barrio MM, Abes R, Colombo M, et al. Human macrophages and dendritic cells can equally present MART-1 antigen to CD8(+) T cells after phagocytosis of gamma-irradiated melanoma cells. PLoS One. 2012;7(7):e40311. doi: 10.1371/journal.pone.0040311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mills CD, Thomas AC, Lenz LL, Munder M. Macrophage: SHIP of immunity. Front Immunol. 2014;5:620. doi: 10.3389/fimmu.2014.00620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 2014;6(3):1670–90. doi: 10.3390/cancers6031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galdiero MR, Bonavita E, Barajon I, et al. Tumor associated macrophages and neutrophils in cancer. Immunobiology. 2013;218(11):1402–10. doi: 10.1016/j.imbio.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 30.Hao NB, Lü MH, Fan YH, et al. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012;2012:948098. doi: 10.1155/2012/948098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–95. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toye AA, Lippiat JD, Proks P, et al. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia. 2005;48(4):675–86. doi: 10.1007/s00125-005-1680-z. [DOI] [PubMed] [Google Scholar]
- 33.Ripoll VM, Meadows NA, Bangert M, et al. Nicotinamide nucleotide transhydrogenase (NNT) acts as a novel modulator of macrophage inflammatory responses. FASEB J. 2012;26(8):3550–62. doi: 10.1096/fj.11-199935. [DOI] [PubMed] [Google Scholar]
- 34.Mekada K, Abe K, Murakami A, et al. Genetic differences among C57BL/6 substrains. Exp Anim. 2009;58(2):141–49. doi: 10.1538/expanim.58.141. [DOI] [PubMed] [Google Scholar]
- 35.Zurita E, Chagoyen M, Cantero M, et al. Genetic polymorphisms among C57BL/6 mouse inbred strains. Transgenic Res. 2011;20(3):481–89. doi: 10.1007/s11248-010-9403-8. [DOI] [PubMed] [Google Scholar]