

Abstract
Despite the ubiquitous role of ATP Binding Cassette (ABC) importers in nutrient uptake, only the E. coli maltose and vitamin B12 ABC transporters have been structurally characterized in multiple conformations relevant to the alternating access transport mechanism. To complement our previous structure determination of the E. coli MetNI methionine importer in the inward facing conformation (Kadaba et al. (2008) Science 321, 250–253), we have explored conditions stabilizing the outward facing conformation. Using two variants, the Walker B E166Q mutation with ATP+EDTA to stabilize MetNI in the ATP-bound conformation and the N229A variant of the binding protein MetQ, shown in this work to disrupt methionine binding, a high affinity MetNIQ complex was formed with a dissociation constant measured to be 27 nM. Using wild type MetQ containing a co-purified methionine (for which the crystal structure is reported at 1.6 Å resolution), the dissociation constant for complex formation with MetNI is measured to be ~40-fold weaker, indicating that complex formation lowers the affinity of MetQ for methionine by this amount. Preparation of a stable MetNIQ complex is an essential step towards the crystallographic analysis of the outward facing conformation, a key intermediate in the uptake of methionine by this transport system.
Keywords: ABC transporter, methionine transporter, methionine-binding protein, MetNI, MetQ, L-methionine, transinhibition
INTRODUCTION
Many different transporters are encoded in the genomes of microorganisms to accommodate the diversity of molecules they need to acquire from the environment. For example, ~10% of the E. coli genome has been classified as participants in transport processes, of which approximately half are ATP-binding cassette (ABC) transporters (Blattner et al., 1997; Linton and Higgins, 1998). ABC transporters consist of four domains: two transmembrane domains (TMDs) that form the translocation pathway, and two nucleotide-binding domains (NBDs) that use the energy from ATP binding and hydrolysis to fuel transport (reviewed in (Ames et al., 1992; Higgins, 1992; Holland et al., 2003; Locher, 2008; Oldham et al., 2008; Rees et al., 2009)). The transport mechanism utilizes an alternating access model (Widdas, 1952; Jardetsky, 1966) that involves the interconversion of two distinct conformations during the transport cycle, inward facing and outward facing, where the translocation pathway is open to the cytoplasm and periplasm, respectively. ABC importers require an additional component for transport, namely substrate-binding proteins (SBP), located in the periplasm of Gram-negative bacteria or tethered to the membrane in Gram-positive bacteria and archaea, to bind and deliver substrates to their cognate transporter (Ames et al., 1992; Wilkinson and Verschueren, 2003; Berntsson et al., 2010). Structures of transporters in complex with their binding proteins (Hollenstein et al., 2007; Hvorup et al., 2007; Oldham et al., 2007; Korkhov et al., 2012) have been instrumental in defining basic features of the transport cycle, including the two best structurally characterized ABC importers, the Type I maltose uptake system, MalFGK2 (Oldham and Chen, 2011) and the Type II vitamin B12 uptake system, BtuCD (Korkhov et al., 2014). Despite the common nucleotide-dependent engine driving translocation, binding protein - transporter interactions can vary significantly, reflecting mechanistic diversity between various ABC importers (Lewinson et al., 2010).
In this work, we focus on the Escherichia coli high affinity methionine uptake system (Kadner, 1974), comprising the MetNI transporter and its cognate binding-protein, MetQ (Gál et al., 2002; Merlin et al., 2002). Currently, only the inward facing conformational state of MetNI has been solved crystallographically (Kadaba et al., 2008; Johnson et al., 2012). To build a comprehensive model for MetNI transport, we sought to identify conditions under which a stable transporter-binding protein complex can form. A challenge in working with E. coli MetNI is that while methionine is the transported ligand, it also functions as an allosteric effector inhibiting methionine uptake at sufficiently high intracellular concentrations (Kadner, 1975). This phenomenon of transinhibition is mediated by methionine functioning as a noncompetitive inhibitor of ATPase activity (Yang and Rees, 2015) by binding to a cytoplasmic regulatory domain of the ABC subunit MetN to stabilize MetNI in an inward facing conformation incapable of hydrolyzing ATP (Kadaba et al., 2008; Johnson et al., 2012). Further complicating the analysis is the observation of a co-purifying methionine in crystal structures of methionine binding SBPs (Deka et al., 2004; Williams et al., 2004; Yang et al., 2009; Yu et al., 2011), including the E. coli MetQ structure (Kadaba, 2008).
Here we demonstrate that formation of a high affinity MetNIQ complex requires an ATP-bound state of MetNI and an unliganded form of MetQ. Preparation of this complex is facilitated by the identification of mutations in MetQ that abolish methionine binding. The influence of methionine on complex formation between ATP-stabilized MetNI and MetQ is quantitated by titrating fluorescently labeled MetQ with varying concentrations of MetNI. From these measurements, dissociation constants of 1100 ± 300 and 27 ± 9 nM are found for methionine-bound and methionine free MetQ, respectively, establishing that ligand-free MetQ can form a stable complex with the ATP-bound form of MetNI. Our isolation of a stable MetNIQ complex is an essential step towards the crystallographic analysis of the outward facing conformation, a key intermediate in the uptake of methionine by this transporter.
RESULTS AND DISCUSSION
To better understand the mechanism of the methionine transport system, we sought to identify conditions under which the MetNI transporter forms a stable complex with the cognate periplasmic binding protein MetQ. Previous studies demonstrated that wild type MetNI and wild type MetQ interact weakly in the absence of nucleotide (Kd = 7.4 × 10−5 M) and a stable complex under these conditions could not be detected by gel filtration chromatography (Lewinson et al., 2010). Based on studies of the maltose system (Chen et al., 2001), we hypothesized that the MetNI transporter binds MetQ with the highest affinity in an ATP-bound state. To maintain the ATP-bound state, an ATPase deficient form was generated by substitution of Glu 166 in the Walker B motif of MetN with Gln (E166Q), in combination with the use of the chelator EDTA with ATP to remove the catalytically essential Mg2+. Mutation of the Walker B Glu is commonly used to inhibit hydrolysis, but not binding, of ATP by ABC transporters (Moody et al., 2002). ATPase assays show that the E166Q mutation results in an ~20-fold reduction in ATPase activity (data not shown). All subsequent experiments in this study were conducted using this mutant transporter, denoted E166Q MetNI, in the presence of saturating amounts of ATP+EDTA (1 mM ATP and 1 mM EDTA) to mimic the ATP-bound state.
We first tested for complex formation by incubating E166Q MetNI with wild type MetQ and saturating amounts of ATP+EDTA at different ratios of transporter to binding protein (Fig. 1A). Mixtures of either 1:1.25 or 1:6 (MetNI: MetQ) were injected onto a gel filtration column (Fig. 1A), and the peak fraction was analyzed by blue native PAGE (Fig. 1B) (BN PAGE (Reisinger and Eichacker, 2006)) to evaluate the extent of complex formation. Despite an excess of MetQ, incomplete complex formation was observed in both cases, as established by the slight shift in the peaks of MetNI after incubation with its binding protein at different molar ratios (Fig. 1A), shown by blue and green traces compared to that of MetNI alone (black trace). This finding is confirmed by the presence of two bands on BN PAGE (Fig. 1B, lanes 2 and 3), the upper one corresponding to the MetNIQ complex and the lower one to free transporter by comparison to E166Q MetNI alone (Fig. 1B, lane 1).
Figure 1. Detection of MetNIQ complex formation.

A. Size-exclusion chromatography for detection of complex formation. Reactions of E166Q MetNI (40 μM) and MetQ at the indicated molar ratios in the presence of 1 mM ATP and 1 mM EDTA were injected onto a size exclusion column. Incomplete complex formation was shown by a slight shift in E166Q MetNI (EQ) peak after incubation with wt MetQ (wt) at different ratios, 1:1.25 (blue trace) and 1:6 (green trace) (MetNI:MetQ), compared to the peak of MetNI alone (black trace). Stable complex formation was shown by a distinct shift in E166Q MetNI (EQ) peak after incubation with N229A MetQ (NA) at a ratio of 1:1.25 (MetNI: MetQ) (red traces), compared to the peak of MetNI alone (black traces). B. BN PAGE of the highest A280 eluate peak from the size-exclusion chromatography shown in A. The upper arrow corresponding to the MetNIQ complex band and the lower arrow to MetNI band. Lane 1: E166Q MetNI alone; lanes 2 and 3: wild type MetQ added to E166Q MetNI at different ratios, 1:1.25 and 1:6 (MetNI:MetQ); lane 4: N229A MetQ added to E166Q MetNI at 1:1.25 (MetNI:MetQ) ratio.
Wild type MetQ contains a co-purifying methionine identified in the crystal structures of MetQ homologs S. aureus lipoprotein-9 (PDB entry 1P99, (Williams et al., 2004)), T. pallidium Tp32 (PDB entry 1XS5, (Deka et al., 2004)), E. coli MetQ (Kadaba, 2008), N. meningitides (PDB 3IR1, (Yang et al., 2009)), V. vulnificus (PDB 3K2D, (Yu et al., 2011)) and unpublished structures with PDB entries 3TQW, 3UP9, 4EF1, 4GOT, 4IB2, 4K3F, 4QHQ, 4QYM, and 4Q5T. The basic structure and binding site for L-methionine are illustrated in Fig. 2 for E. coli MetQ refined at 1.6 Å resolution (PDB entry 4YAH, see Methods). An extensive hydrogen bond/salt bridge network between protein side chains and the methionine α-amino and carboxyl groups likely contributes to the tight binding of ligand to MetQ. Groups interacting with the α-carboxyl group of methionine include the side chains of Arg144, Asn202, and a buried water, while the α-amino group interacts with the side chains of Glu42 and Thr204 and two buried waters. Significantly, the side chain of Asn229 forms two hydrogen bonds (one each) to the α-amino and carboxyl groups of the L-methionine. The side chain of the bound L-methionine fits in a pocket surrounded by the aromatic side chains of Tyr69, Phe86, His88, and Tyr91. Aromatic interactions with the methionine side chain are not uncommon (Pal and Chakrabarti, 2001), particularly the interaction observed with the ligand SD packed against the plane of His88 such that the CG-SD bond is oriented towards the NE2 atom of His88.
Figure 2. The crystal structure of E. coli L-methionine-bound MetQ.
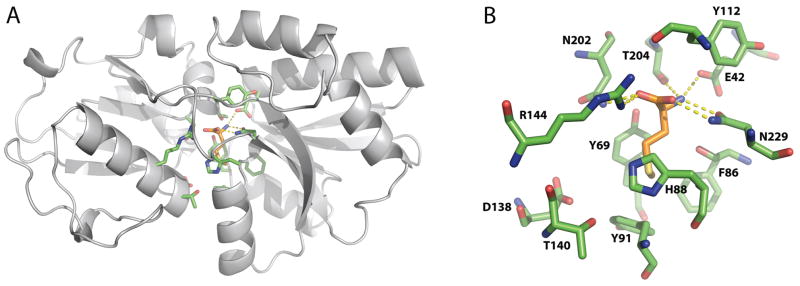
A. Ribbon diagram of L-methionine-bound E. coli MetQ. B. The L-methionine binding site in MetQ. The side chains of the interacting residues are colored green, and the L-methionine ligand is colored grey. Hydrogen bonds between the α-amino and carboxyl groups of L-methionine and surrounding MetQ residues are represented by yellow dashes.
To evaluate the contribution of L-methionine binding to MetQ to the stability of the MetNIQ complex, it is necessary to prepare ligand-free MetQ. While methionine can be removed from MetQ by an unfolding/refolding process (Deka et al., 2004), we developed an alternative approach of using alanine-scanning mutagenesis to identify MetQ mutants that disrupt the binding of L-methionine. Residues in the MetQ binding pocket in contact with methionine were mutated to alanine (Fig. 2B and Fig. 3), with the exception of Glu42 that was substituted with Gln (E42Q) since the E42A mutant aggregated. To test for L-methionine binding, we unfolded MetQ in 6 M guanidine-HCl to release any co-purified L-methionine (Deka et al., 2004), and then refolded the protein in the presence of L-selenomethionine. Selenium content was measured by inductively coupled plasma mass spectrometry (ICP-MS), and the molar ratio of selenium to MetQ was calculated (Fig. 3). Mutation of Glu42 to Gln (E42Q) and Asn229 to Ala (N229A) significantly decreased the binding affinity of MetQ to selenomethionine, while substitutions at other residues had relatively little effect on selenomethionine binding. The E42Q and N229A MetQ mutants, identified in this screen as unable to bind selenomethionine (and presumably methionine), were then tested for complex formation with E166Q MetNI in the presence of ATP+EDTA. The E42Q mutant was unable to form a complex with its cognate transporter (data not shown). When N229A MetQ was incubated with E166Q MetNI in the presence of ATP+EDTA, however, a stable complex formation was indicated by a distinct shift in the MetNI gel filtration peak after the reaction, shown by the red trace (Fig. 1A). This stoichiometric complex formation was also observed by BN PAGE (Fig. 1B, lane 4). This result contrasts with the mixture of free and complexed species observed when E166Q MetNI is incubated with wild type liganded MetQ under comparable conditions (Fig. 1B, lanes 2 and 3). Based on these results, the N229A MetQ variant was used in this study to mimic methionine-free MetQ.
Figure 3. Screening for substrate-binding-deficient MetQ mutants.
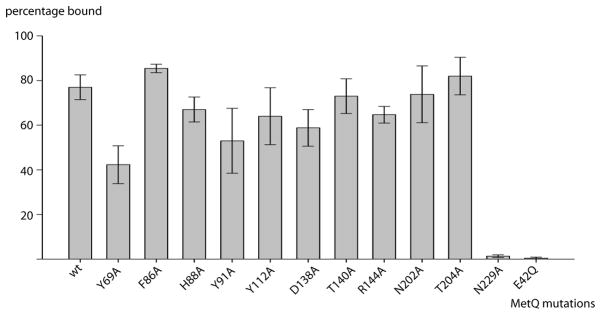
Alanine-scanning mutants of MetQ were analyzed for selenium content by ICP-MS after exchange of native substrate with L-selenomethionine (see text). The horizontal and vertical axes denote the MetQ mutations and fraction of MetQ bound by selenomethionine, respectively. Error bars represent the standard deviation of duplicate samples.
To quantify the binding affinity of E166Q MetNI for MetQ in either the bound or unliganded state, we measured the dissociation constants for complex formation from titration curves conducted with fluorescently labeled MetQ on a Monolith NT.115 Microscale Thermophoresis Instrument (NanoTemper Technologies GmbH). Wild type and N229A MetQ were labeled with Cy3-maleimide and titrated with varying concentrations of E166Q MetNI in the presence of ATP+EDTA. The fraction of MetQ in complex with E166Q MetNI was calculated from fluorescent intensity values (see Methods). The affinity of L-methionine-bound wild type MetQ for E166Q MetNI was ~40-fold weaker than that for unliganded N229A MetQ to E166Q MetNI (Fig. 4). The calculated Kd of 1100 ± 300 nM for wild type MetQ represents a lower limit for the affinity, as saturating amounts of E166Q MetNI were prone to aggregation. The Kd for N229A MetQ was determined to be 27 ± 9 nM. This difference in affinity is qualitatively consistent with our gel filtration (Fig. 1A) and BN PAGE results (Fig. 1B).
Figure 4. Dissociation constants between MetNI and MetQ.

The affinity of MetNI for MetQ was measured by titration curves using fluorescently labeled MetQ in the presence or absence of bound methionine (wild type or N229A MetQ, respectively). All experiments contained 1 mM ATP and 1 mM EDTA. 101 nM of Cy3-labeled wild type MetQ was titrated with 1.95 nM – 32 μM non-labeled E166Q MetNI (closed squares). 99 nM of Cy3-labeled N229A MetQ was titrated with 1.95 nM – 4 μM non-labeled MetNI E166Q (open circles). The dissociation constants calculated from these data for wild type (methionine-bound) and N229A (methionine-free) MetQ binding to E166Q MetNI are Kd = 1100 ± 300 nM and 27 ± 9 nM, respectively. Error bars represent standard error from three independent measurements.
We have identified conditions promoting stable complex formation between MetNI and MetQ through the use of two mutations (the E166Q variant in the Walker B motif of MetN that reduces ATP hydrolysis, and the N229A variant of MetQ that disrupts methionine binding) and the presence of ATP+EDTA to reduce the concentration of Mg2+ critical for ATP hydrolysis. These conditions should stabilize the ATP-bound form of MetNI, corresponding to the outward facing conformation of a Type I ABC transporter (Oldham and Chen, 2011). An unexpected feature of the MetNI system is that the use of ligand free MetQ resulted in stable complex formation, in contrast to the maltose transporter system where formation of a stable transporter-SBP complex required the presence of the transported substrate (Chen et al., 2001; Oldham et al., 2007). In the absence of ATP, a “pre-translocation” species has also been crystallized with substrate-loaded SBP and an inward facing conformation of the transporter for the molybdate (Hollenstein et al., 2007) and maltose (Oldham and Chen, 2011) transporters. While the corresponding complex between liganded SBP and the methionine transporter is substantially less stable (Kd ~ 7.5 × 10−5 M (Lewinson et al., 2010)) than the complex in the presence of ATP measured in this work (Kd ~ 10−6 M), at the protein concentrations (~10−4 M) used in crystallization trials, perhaps this state of the MetNIQ complex could also be crystallized under appropriate conditions.
The role of transported ligand in complex formation is of great mechanistic interest. For complex formation between MetNI and MetQ, the difference in dissociation constants between unliganded (27 nM) and liganded (1100 nM) MetQ implies that the binding of methionine to MetQ in complex with MetNI is ~40-fold weaker than the binding of methionine to MetQ alone. Energetically, this observation is consistent with a model where the SBP must release ligand for transport to occur. The detailed energetics will certainly depend on the transporter and assay conditions; for example, while liganded maltose binding protein has been reported to more efficiently stimulate ATP hydrolysis of maltose transporter reconstituted into proteoliposomes (Davidson et al., 1992), ligand-free maltose binding protein binds 5-fold more tightly than liganded binding protein to maltose transporter in nanodiscs with AMPPNP (Kd ~ 80 nM and ~400 nM in the absence or presence of maltose, respectively (Bao and Duong, 2012)). An additional complication with the MetNI transporter is that methionine is both a transported ligand and an allosteric inhibitor of transport, and these effects will need to be distinguished from the contributions of the liganded state of MetQ to the observed kinetics. The development of a detailed molecular transport mechanism will require analysis of conformational dynamics of the type recently described for the GlnPQ uptake system (Gouridis et al., 2015). The structural analysis of stable forms of ABC transporters such as the MetNIQ complex detailed here will provide crucial reference states for these future mechanistic studies.
MATERIALS AND METHODS
Cloning, expression and purification of MetQ
The metNIQ operon was amplified from Escherichia coli K-12 genomic DNA and cloned into a zero blunt pCR-4 TOPO vector as described in (Kadaba et al., 2008). The metQ gene, lacking the stop codon, was subcloned using oligonucleotides that included NdeI and XhoI restriction-enzyme sites, and then ligated into the pET21b(+) vector with a C-terminal 6x-histidine tag. The cloned plasmids were expressed separately in E. coli BL21 (DE3) gold cells (EMD) at 37 °C in Terrific Broth medium with 100 μg ml−1 ampicillin. The cells were induced at OD600 ~4.0 with 0.4 mM IPTG for 2 hours, then harvested and stored at −80 °C.
For purification of mature MetQ, periplasmic extract was prepared by resuspending 10 g of cell paste in 10 ml of 40% sucrose, 10 mM Tris-HCl pH 7.5, and 1 mM EDTA, and stirred for one hour at room temperature. The cells were then shocked by addition of 500 ml of ice-cold water. After stirring for 10 min, buffer components were added to a final concentration of 25 mM Tris pH 7.5, 150 mM NaCl, and 17 mM imidazole. The resulting suspension was then centrifuged at 37,500 × g for 30 min to remove all cellular debris from the periplasmic extract. MetQ was isolated from the periplasmic extract by immobilized metal ion affinity chromatography using a 5 ml Ni-NTA column (GE). The affinity column was washed with 25 mM Tris pH 7.5, 150 mM NaCl, and 17 mM imidazole for 10 column volumes. Protein was then eluted from the column using 25 mM Tris pH 7.5, 100 mM NaCl and 150 mM imidazole. Peak fractions were dialyzed against 25 mM Tris pH 7.5 and 150 mM NaCl overnight, and then concentrated to 30 mg ml−1 using a 10kD MWCO concentrator (Millipore).
The full-length protein cloned for this work, including the signal sequence and the C-terminal 6x-his tag, has the sequence:
| 1 | MAFKFKTFAAVGALIGSLALVGCGQDEKDPNHIKVGVIVGAEQQVAEVAQKVAKDKYGLD |
| 61 | VELVTFNDYVLPNEALSKGDIDANAFQHKPYLDQQLKDRGYKLVAVGNTFVYPIAGYSKK |
| 121 | IKSLDELQDGSQVAVPNDPTNLGRSLLLLQKVGLIKLKDGVGLLPTVLDVVENPKNLKIV |
| 181 | ELEAPQLPRSLDDAQIALAVINTTYASQIGLTPAKDGIFVEDKESPYVNLIVTREDNKDA |
| 241 | ENVKKFVQAYQSDEVYEAANKVFNGGAVKGWLEHHHHHH |
The cleaved signal sequence is highlighted in bold, so that the purified protein used in this study consists of residues 17 to 279, of which residues 29–273 (underlined) are observed in the electron density.
Purification of MetNI
Purification of wild type and mutant E166Q MetNI was carried out as described (Johnson et al., 2012) with the one modification that buffer containing 20 mM TAPS pH 8.5, 100 mM NaCl and 0.025% DDM (with varying amounts of imidazole) was used in all purification steps (the original protocol used 250 mM NaCl and different detergents).
MetQ crystallization and structure determination
Crystallization conditions for MetQ were screened with varying concentrations of protein from 15 mg ml−1 to 60 mg ml−1. Crystals were obtained using a hanging drop vapor diffusion method at 20° C, by mixing 1 μl of protein to 1 μl of the following well solution: 24% PEG 4000, 4% PEG 400, 0.1 M sodium citrate pH 5.6, and 0.2 M ammonium acetate at 30 mg ml−1of protein, similar to the conditions originally described (Kadaba, 2008). Initial thin needle crystals appeared within a week but crystals were not large enough for data collection. Both microseeding and macroseeding were used to obtain larger crystals, reaching a final size 0.5 × 0.2 × 0.1 mm3 in a week. The best crystal was obtained by macroseeding with a precipitant 20% PEG 4000, 3% PEG 400, 0.1 M sodium citrate pH 5.6, and 0.2 M ammonium acetate. Diffraction data to 1.6 Å resolution were collected at the Stanford Synchrotron Radiation Lightsource (SSRL) on beamline 12-2. The data was processed using xia2 (Winter, 2010) using XDS (Kabsch, 2010), XSCALE, and AIMLESS (Evans, 2006), with data collection and processing statistics listed in Table 1. Molecular replacement was performed using Phenix (Adams et al., 2010) from the previously solved 1.8 Å resolution MetQ structure (Kadaba, 2008) that was in turn solved by molecular replacement from PDB entry 1P99 (Williams et al., 2004), a Gly-Met-binding protein. The structure was refined with Phenix to 1.6 Å resolution to a final R/Rfree of 0.154/0.183. Coordinates and structure factors have been deposited in the Protein Data Bank of the Research Collaboratory for Structural Bioinformatics, with ID 4YAH.
Table 1.
Data processing and refinement statistics
| Data Processing Statistics | MetQ |
|---|---|
| Space group | C2221 |
| Unit cell dimensions (a, b, c) (Å) | 59.7, 87.4, 112.9 |
| Wavelength (Å) | 1.00 |
| Resolution (Å)a | 24.5-1.60 (1.64 – 1.60) |
| Unique reflections | 38,555 |
| Redundancy | 4.1 |
| Completeness (%) | 98.4 (96.1) |
| I/σ | 11.5(1.11) |
| Rmerge | 0.07 (0.952) |
| Refinement Statistics | PDB ID 4YAH |
| Resolution (Å)a | 24.5 – 1.60 (1.64 – 1.60) |
| R-work | 0.154 (0.360) |
| R-free | 0.183 (0.409) |
| Average B factor (Å) | 27.7 |
| RMSD bond length (Å) | 0.008 |
| RMSD bond angle (°) | 1.21 |
| Ramachandran plot (favored, allowed, outliers, %) | 98, 2, 0 |
Numbers in parentheses represent data in the highest resolution shell
Screening of substrate-binding deficient MetQ mutants
Individual residues located in the substrate-binding pocket of MetQ were substituted with alanine by site-directed mutagenesis using the Quickchange Mutagenesis kit (Stratagene), with the exception of Glu42, which was substituted with glutamine to avoid aggregation. To evaluate the binding ability of each mutant for substrate, L-methionine was exchanged with L-selenomethionine, and the amount of selenium was measured using inductively coupled plasma mass spectrometry (ICP-MS).
To exchange L-methionine with L-selenomethionine, individual MetQ mutants immobilized onto Ni-NTA columns were unfolded by washing with 12 column volumes of denaturing buffer containing 6 M guanidine-HCl, 25 mM Tris pH 7.5, 150 mM NaCl at the rate of 1.5 ml min−1 to remove bound L-methionine. To refold MetQ, guanidine-HCl was slowly removed by flowing renaturation buffer (2 mM L-selenomethionine, 25 mM Tris pH 7.5, 150 mM NaCl) over the column at 1 ml min−1 for 90 min. The column was further washed with 10 column volumes of renaturation buffer to ensure reloading of MetQ with L-selenomethionine. This was followed by 12 column volumes of wash buffer (25 mM Tris pH 7.5 and 150 mM NaCl) to remove unbound L-selenomethionine. Finally, MetQ was eluted in 25 mM Tris pH 7.5, 150 mM NaCl, and 150 mM imidazole, and run over a size-exclusion column (Superdex 200 16/60, GE Healthcare) equilibrated in 25 mM Tris, pH 7.5 and 150 mM NaCl. The monodisperse peak was collected and concentrated to 20 mg ml−1 using an Amicon 10-kD MWCO concentrator (Millipore).
The amount of bound L-selenomethionine was measured by quantitation of the selenium content of acid-digested protein samples using ICP-MS. A final concentration of 0.1 μM of protein was digested with 2% trace metal grade nitric acid (Fluka) at 70 °C overnight. Samples were centrifuged at 30,000 × g for 10 min using a bench top centrifuge to remove particulate matter. The supernatant was then diluted to a final volume of 5 ml in double distilled H2O and measured for Se78 with an HP 4500 ICP-MS instrument. L-selenomethionine powder stock (Anatrace) was used to prepare standards. Each sample was measured in duplicate in two independent experiments. Error bars represent standard deviation. Data were processed and graphed using Microsoft Excel and Sigmaplot 11.0, respectively.
Qualitative determination of complex formation
MetNI and MetQ concentrations were measured using a Nanodrop 2000 Spectrophotometer (Thermo Scientific), according to their calculated extinction coefficients for a 1 mg ml−1 solution at 280 nm of 0.64 and 0.73, respectively. Complex formation was assessed by mixing E166Q MetNI transporter and its cognate binding protein, MetQ, at a molar ratio of 1:1.25 (MetNI: MetQ) in the presence of 1 mM ATP and 1 mM EDTA. The mixture, containing 40 μM MetNI, 50 μM MetQ, 1 mM ATP, 1 mM EDTA, 20 mM TAPS pH 8.5, 100 mM NaCl, 0.025% DDM, and 5 mM β-mercaptoethanol, was incubated for 1 hour at room temperature and ultracentrifuged at 200,000 × g for 20 min to remove any aggregation. Samples were injected onto a Superdex S200 10/300 sizing column (GE Healthcare) equilibrated with buffer containing 20 mM TAPS pH 8.5, 100 mM NaCl, 0.5 mM ATP, 1 mM EDTA, 0.3% Cymal-5, and 5 mM β-mercaptoethanol. The absorbance reading at 280 nm and the retention volume of the peak were used to follow complex formation. MetNIQ complex formation was verified by analyzing the highest peak by BN PAGE (Reisinger and Eichacker, 2006) using 4–20% Criterion gels (Bio-Rad). Two independent experiments were conducted per condition.
Measurement of MetNIQ complex dissociation constant
The E. coli MetQ sequence contains one cysteine residue at amino acid position 23. Purified wild type MetQ and N229A MetQ were labeled with Cy3-maleimide (GE Healthcare), according to the manufacturer’s instructions, in buffer containing 25 mM Tris pH 7.5, 150 mM NaCl, and 10 mM TCEP pH 7.5. Unreacted dye was removed using a Superdex 200 10/300 GL column (GE Healthcare) equilibrated with the aforementioned buffer condition. The label to protein ratio was determined using a NanoDrop 2000 Spectrophotometer (Thermo Scientific) at 550 and 280 nm, and a labeling efficiency of 10% was calculated. Fluorescence measurements were performed on a Monolith NT.115 Instrument (NanoTemper Technologies GmbH) at 25 °C using standard capillaries. Varying concentrations (ranging from 1.95 nM to 32 μM) of unlabeled E166Q MetNI in ATP buffer (1 mM ATP, 1 mM EDTA, 20 mM TAPS pH 8.5, 100 mM NaCl, and 0.3 % Cymal-5) were titrated against a constant concentration of MetQ (101 nM of wild type MetQ or 99 nM of N229A MetQ). Fluorescent intensity as a function of E166Q MetNI concentration was normalized to fraction bound by averaging the intensity values for the lowest three concentrations of E166Q MetNI per experiment. This background intensity was subtracted from all concentrations to yield corrected intensity values. The corrected intensity values were then divided by the corrected intensity at the highest concentration of E166Q MetNI to normalize intensity values to fraction bound values. Dissociation constants for wild type MetQ or N229A MetQ binding to E166Q MetNI were calculated using the Kd Fit function of the NanoTemper Analysis 1.5.41 software. Three independent MST measurements per condition were conducted, and error bars represent standard error of the mean.
Acknowledgments
A Vietnam International Education Development scholarship from the Vietnam Ministry of Education and Training scholarship to P.T.N, an NSF Graduate Fellowship to Q.W.L., and support of NIH Predoctoral Training Grant T32 GM07737 to N.S.K., are gratefully acknowledged. We thank Allen Lee for generating the original MetNIQ constructs, Christoph Müller for MetNIQ discussions, and Dr. Jens Kaiser and Dr. Nathan Dalleska for assistance with crystallography and ICP-MS, respectively. We gratefully acknowledge the Gordon and Betty Moore Foundation and the Beckman Institute for their generous support of the Molecular Observatory at Caltech and the staff at Beamline 12–2, Stanford Synchrotron Radiation Lightsource (SSRL), for their assistance with data collection. Use of the SSRL, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). This project benefited from the use of instrumentation made available by the Caltech Environmental Analysis Center. This work was supported in part by NIH GM045162.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames GF, Mimura CS, Holbrook SR, Shyamala V. Traffic ATPases: a superfamily of transport proteins operating from Escherichia coli to humans. Adv Enzymol Relat Areas Mol Biol. 1992;65:1–47. doi: 10.1002/9780470123119.ch1. [DOI] [PubMed] [Google Scholar]
- Bao H, Duong F. Discovery of an auto-regulation mechanism for the maltose ABC transporter MalFGK2. PLoS One. 2012;7:e34836. doi: 10.1371/journal.pone.0034836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berntsson RPA, Smits SHJ, Schmitt L, Slotboom DJ, Poolman B. A structural classification of substrate-binding proteins. FEBS Lett. 2010;584:2606–2617. doi: 10.1016/j.febslet.2010.04.043. [DOI] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, ColladoVides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Chen J, Sharma S, Quiocho FA, Davidson AL. Trapping the transition state of an ATP-binding cassette transporter: evidence for a concerted mechanism of maltose transport. Proc Natl Acad Sci USA. 2001;98:1525–1530. doi: 10.1073/pnas.041542498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson AL, Shuman HA, Nikaido H. Mechanism of maltose transport in Escherichia coli: Transmembrane signaling by periplasmic binding proteins. Proc Natl Acad Sci USA. 1992;89:2360–2364. doi: 10.1073/pnas.89.6.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deka RK, Neil L, Hagman KE, Machius M, Tomchick DR, Brautigam CA, Norgard MV. Structural evidence that the 32-kilodalton lipoprotein (Tp32) of Treponema pallidum is an L-methionine-binding protein. J Biol Chem. 2004;279:55644–55650. doi: 10.1074/jbc.M409263200. [DOI] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr. 2006;D62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Gál J, Szvetnik A, Schnell R, Kálmán M. The metD D-methionine transporter locus of Escherichia coli is an ABC transporer gene cluster. J Bact. 2002;184:4930–4932. doi: 10.1128/JB.184.17.4930-4932.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouridis G, Schuurman-Wolters GK, Ploetz E, Husada F, Vietrov R, de Boer M, Cordes T, Poolman B. Conformational dynamics in substrate-binding domains influences transport in the ABC importer GlnPQ. Nat Struct Mol Biol. 2015;22:57–64. doi: 10.1038/nsmb.2929. [DOI] [PubMed] [Google Scholar]
- Higgins CF. ABC transporters: from microorganisms to man. Annu Rev Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- Holland IB, Cole SPC, Kuchler K, Higgins CF, editors. ABC proteins: From Bacteria to Man. London: Academic; 2003. [Google Scholar]
- Hollenstein K, Frei DC, Locher KP. Structure of an ABC transporter in complex with its binding protein. Nature. 2007;446:213–216. doi: 10.1038/nature05626. [DOI] [PubMed] [Google Scholar]
- Hvorup RN, Goetz BA, Niederer M, Hollenstein K, Perozo E, Locher KP. Asymmetry in the structure of the ABC transporter binding protein complex BtuCD-BtuF. Science. 2007;317:1387–1390. doi: 10.1126/science.1145950. [DOI] [PubMed] [Google Scholar]
- Jardetsky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- Johnson E, Nguyen PT, Yeates TO, Rees DC. Inward facing conformations of the MetNI methionine ABC transporter: Implications for the mechanism of transinhibition. Prot Sci. 2012;21:84–96. doi: 10.1002/pro.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. 2010;D66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadaba NS. PhD Thesis, Chemistry. California Institute of Technology; Pasadena, CA: 2008. Structural studies of the E. coli methionine ABC transporter and its cognate binding protein. [Google Scholar]
- Kadaba NS, Kaiser JT, Johnson E, Lee A, Rees DC. The high-affinity E. coli methionine ABC transporter: structure and allosteric regulation. Science. 2008;321:250–253. doi: 10.1126/science.1157987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadner RJ. Transport systems for L-methionine in Escherichia coli. J Bact. 1974;117:232–241. doi: 10.1128/jb.117.1.232-241.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadner RJ. Regulation of methionine transport activity in Escherichia coli. J Bact. 1975;122:110–119. doi: 10.1128/jb.122.1.110-119.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkhov VM, Mireku SA, Locher KP. Structure of AMP-PNP-bound vitamin B12 transporter BtuCD-F. Nature. 2012;490:367–372. doi: 10.1038/nature11442. [DOI] [PubMed] [Google Scholar]
- Korkhov VM, Mireku SA, Veprintsev DB, Locher KP. Structure of AMP-PNP-bound BtuCD and mechanism of ATP-powered vitamin B12 transport by BtuCD-F. Nat Struct Mol Biol. 2014;21:1097–1099. doi: 10.1038/nsmb.2918. [DOI] [PubMed] [Google Scholar]
- Lewinson O, Lee AT, Locher KP, Rees DC. A distinct mechanism for the ABC transporter BtuCD-BtuF revealed by the dynamics of complex formation. Nat Struct Mol Biol. 2010;17:332–338. doi: 10.1038/nsmb.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton KJ, Higgins CF. The Escherichia coli ATP-binding cassette (ABC) proteins. Mol Microbiol. 1998;28:5–13. doi: 10.1046/j.1365-2958.1998.00764.x. [DOI] [PubMed] [Google Scholar]
- Locher KP. Structure and mechanism of ATP-binding cassette transporters. Phil Trans R Soc B. 2008;364:239–245. doi: 10.1098/rstb.2008.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin C, Gardiner G, Durand S, Masters M. The Escherichia coli metD locus encodes an ABC transporter which includes Abc (MetN), YaeE (MetI), and YaeC (MetQ) J Bact. 2002;184:5513–5517. doi: 10.1128/JB.184.19.5513-5517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody JE, Millen L, Binns D, Hunt JF, Thomas PJ. Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters. J Biol Chem. 2002;277:21111–21114. doi: 10.1074/jbc.C200228200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham ML, Chen J. Crystal structure of the maltose transporter in a pretranslocation intermediate state. Science. 2011;332:1202–1205. doi: 10.1126/science.1200767. [DOI] [PubMed] [Google Scholar]
- Oldham ML, Davidson AL, Chen J. Structural insights into ABC transporter mechanism. Curr Opin Struct Biol. 2008;18:726–733. doi: 10.1016/j.sbi.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- Pal D, Chakrabarti P. Non-hydrogen bond interactions involving the methionine sulfur atom. J Biomolec Struct Dyn. 2001;19:115–128. doi: 10.1080/07391102.2001.10506725. [DOI] [PubMed] [Google Scholar]
- Rees DC, Johnson E, Lewinson O. ABC transporters: the power to change. Nat Rev Mol Cell Biol. 2009;10:218–227. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisinger V, Eichacker LA. Analysis of membrane protein complexes by blue native PAGE. Proteomics. 2006:6–15. doi: 10.1002/pmic.200600553. [DOI] [PubMed] [Google Scholar]
- Widdas WF. Inability of diffusion to account for placental glucose transfer in the sheep and consideration of the kinetics of a possible carrier transfer. J Physiol. 1952;118:23–39. doi: 10.1113/jphysiol.1952.sp004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson AJ, Verschueren KHG. Crystal structures of periplasmic solute-binding proteins in ABC transport complexes illuminate thier function. In: Holland IB, Cole SPC, Kuchler K, Higgins CF, editors. In ABC Proteins: From Bacteria to Man. London: Academic Press; 2003. p. 647. [Google Scholar]
- Williams WA, Zhang RG, Zhou M, Joachimiak G, Gornicki P, Missiakas D, Joachimiak A. The membrane-associated lipoprotein-9 GmpC from Staphylococcus aureus binds the dipeptide GlyMet via side chain interactions. Biochemistry. 2004;43:16193–16202. doi: 10.1021/bi048877o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. xia2: an expert system for macromolecular crystallography data reduction. J Appl Crystallogr. 2010;43:186–190. [Google Scholar]
- Yang JG, Rees DC. The allosteric regulatory mechanism of the Escherichia coli MetNI methionine ATP Binding Cassette (ABC) transporter. J Biol Chem. 2015;290 doi: 10.1074/jbc.M114.603365. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Wu ZH, Wang XY, Yang CT, Xu HL, Shen YQ. Crystal structure of lipoprotein GNA1946 from Neisseria meningitidis. J Struct Biol. 2009;168:437–443. doi: 10.1016/j.jsb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Yu S, Lee NY, Park SJ, Rhee S. Crystal structure of Toll-like receptor 2-activating lipoprotein IlpA from Vibria vulnificus. Proteins. 2011;79:1020–1025. doi: 10.1002/prot.22929. [DOI] [PubMed] [Google Scholar]