

Abstract
DLC1 is a RhoGAP-containing tumor suppressor and many of DLC1’s functions are absolutely dependent on its RhoGAP activity. Through its RhoGAP domain, DLC1 inhibits the activity of RhoA GTPase, which regulates actin cytoskeleton networks and dis/assembly of focal adhesions. Tensin1 (TNS1) is a focal adhesion molecule that links the actin cytoskeleton to integrins and forms signaling complexes through its multiple binding domains. Here, we report that TNS1 enhances RhoA activity in a DLC1-dependent manner. This is accomplished by binding to DLC1 through TNS1’s C2, SH2, and PTB domains. Point mutations at these three sites disrupt TNS1’s interaction with DLC1 as well as its effect on RhoA activity. The biological relevance of this TNS1-DLC1-RhoA signaling axis is investigated in TNS1 knockout (KO) cells and mice. Endothelial cells isolated from TNS1 KO mice or those silenced with TNS1 siRNA show significant reduction in proliferation, migration, and tube formation activities. Concomitantly, the RhoA activity is down-regulated in TNS1 KO cells and this reduction is restored by further silencing of DLC1. Furthermore, the angiogenic process is compromised in TNS1 KO mice. These studies demonstrate that TNS1 binds to DLC1 and fine-tunes its RhoGAP activity toward RhoA and that the TNS1-DLC1-RhoA signaling axis is critical in regulating cellular functions that lead to angiogenesis.
Keywords: tensin, DLC1, RhoA, focal adhesion, angiogenesis
1. Introduction
Deleted in liver cancer 1 (DLC1, also known as ARHGAP7 and STARD12 in human and p122RhoGAP in rat) was first isolated from rat brain as a phospholipase C delta 1 binding protein [1] and then was independently cloned by subtractive hybridization as a gene homozygously deleted in a human hepatocellular carcinoma [2]. Together with its gene locus at human chromosome 8p22, a region frequently deleted in liver cancer, DLC1 was considered as a potential tumor suppressor in liver [3–5]. Further studies showed that DLC1 expression was lost or down-regulated in various cancers including liver, breast, lung, brain, stomach, colon, and prostate cancers due to either genomic deletion or aberrant DNA methylation [3, 4]. Mutations that altered the expression or function of DLC1 were detected in pancreas [6], colon, and prostate cancers [7]. These results suggest that DLC1 may function as a tumor suppressor in tissues other than liver [3, 4]. DLC1 contains a SAM, a RhoGAP, and a START domain. Its RhoGAP domain suppresses the activity of RhoA GTPase by converting the GTP-bound active RhoA to GDP-bound inactive form. RhoA mediates cell adhesion, shape, and migration through its roles in modulating actin cytoskeleton networks and focal adhesion turnover [8]. The essential role of the RhoGAP in DLC1’s tumor suppression and other activities is well established. Without a functional RhoGAP domain, DLC1 is not able to regulate cell shape, proliferation, adhesion, and migration [4]. Therefore, tight control of its RhoGAP activity is a critical task of DLC1.
Tensin is a focal adhesion family with four members: tensin1 (TNS1), tensin2 (TNS2), tensin3 (TNS3), and C-terminal tensin-like (cten) [9]. TNS1 is the founding member of the family. It interacts with actin filaments and regulates actin polymerization [10]. Tensin contains a PTB (phosphotyrosine binding) domain that binds to the NPXY motif in β-integrin and a SH2 (Src homology 2) domain that binds to tyrosine phosphorylated proteins, including Axl, EGFR, Src, Fak, p130cas, and paxillin [9, 11–13]. TNS2 (160 kD) and TNS3 (180 kD) have very similar domain structures and molecular masses with those of TNS1 (220 kD) [11, 14, 15]. Cten, on the other hand, is a much smaller protein (80 kD) and only shares the SH2 and PTB domains with other tensins [16]. Previously, we have discovered that the SH2 domains of all tensins bind to DLC1 in a phosphorylation-independent fashion and that this interaction is essential for recruiting DLC1 to focal adhesion sites and for DLC1’s tumor suppression activity [17]. However, the effect of this interaction on DLC1’s RhoGAP activity is not well understood.
In this report, we have established that TNS1 negatively regulates DLC1’s RhoGAP activity toward RhoA through its multiple interactions. The biological significance of this TNS1-DLC1-RhoA signaling axis is demonstrated by showing abnormal cellular function and angiogenic response in TNS1 knockout (KO) endothelial cells and mice.
2. Materials and methods
2.1 Mice
TNS1 KO mice were produced by our group [18]. All animal procedures were performed according to UC Davis guidelines for the care and use of laboratory animals.
2.2 Cell culture and reagents
HUVECs were cultured in endothelial cell growth medium (ScienCell). 293T cells were maintained in DMEM/high glucose with 10% fetal bovine serum. Lipofectamine-2000 (Invitrogen) was used for transfections. TNS1 siRNA and DLC1 siRNA were purchased from Sigma-Aldrich.
2.3 Plasmid constructions and mutagenesis
The full-length or truncated cDNA fragments encoding human TNS1 residues 75 to 310, 75 to 210, and 211 to 310 were subcloned in frame into mammalian expression vector pTomato or pEGFP (Clontech). The site-specific mutation of 244M (GD244AA), 252M (HK252AA), SH2M (R1488A), PTBM (FFRR1656AAAA) was introduced into TNS1 cDNA by site-directed mutagenesis. All constructs were verified by DNA sequencing.
2.4 Isolation of endothelial cells from mouse lung tissues
Lung tissues from three 7–10 day old mice were removed aseptically and rinsed in ice-cold DMEM. After removal of the larger blood vessels, the lungs were minced into ~1×2-mm squares and digested with 1.5mg/ml collagenase A (Roche), at 37°C for 45 minutes on a rotator. The cellular digest was filtered through sterile 70μm nylon mesh and the mesh washed with 15ml 10% FBS-DMEM media to stop digestion. Cells were centrifuged at 400xg for 10 minutes, resuspended in 0.1% BSA/PBS, incubated with anti-CD31-antibody (BD) coated Dynabeads (Invitrogrn), and then separated in a magnetic field. After washing away of unbound cells, remaining cells were seeded on 2% gelatin coated plates cultured with Endothelial cell medium (ScienCell). After 3–5 days, cells were separated again by anti-ICAM2 antibody (BD) coated Dynabeads. The cells were plated at a concentration of 300,000 cells/ml into gelatin-coated T-25 flasks, and subsequently split 1:2 at each passage. After 3 passages, cells were immortalized using lentivirus expressing SV40 large T antigen (Genecopoeia).
2.5 RhoA activity assay
Cells were washed with ice-cold PBS and lysed. Equal protein amounts of cell lysates were incubated with 30ug glutathione S-transferase–Rho binding domain of rhotekin (GST-RBD) beads (Cytoskeleton) for 1h at 4°C. The beads were then washed twice with washing buffer and bound Rho proteins were analyzed by immunoblotting using an anti-RhoA antibody (Cell signaling).
2.6 Migration assay
Cells (50,000 cells) were added to the upper chamber of a transwell insert (Corning) with growth factor free media. Endothelial cell growth medium (500μL) was added to the lower chamber. After 6h, cells were fixed and stained. Cells that had migrated to the bottom surface were visualized microscopically and photographed.
2.7 In vitro tube formation assay
Growth factor–reduced Matrigel (BD Biosciences) was used to coat a 48-well plate (150μL/well) and endothelial cells (50,000 cells/well) were seeded with media (300μL). After 4h of incubation, capillary–like structures were scored by measuring the lengths of tubules per field in each well at ×100 magnification with ImageJ software (NIH).
2.8 Aortic ring assay
Thoracic aortas from TNS1 WT or KO mice were dissected and transferred to ice-cold PBS. The fat tissue was removed and 1-mm-long aortic rings were sectioned and embedded in growth factor–reduced Matrigel. Rings were cultured with 500 μL of conditioned media for 8 days and the outgrowth of endothelial tubes was counted.
2.9 Matrigel plug assay
Growth factor-reduced matrigel (250 ul, BD Biosciences) containing 60 U/ml heparin (Sigma- Aldrich) was subcutaneously injected into the mice (8 weeks old). After 5 days, matrigel plugs were harvested and embedded in OCT and frozen sections were processed for immunohistochemical staining using CD31 antibody. CD31 expression levels were scored by a blinded reviewer (AW) as 0 (no or low expression) or 1 (middle or high expression) per image. Scores from 8 non-overlapping images per sectioned slide were added. Therefore the possible scores were from 0 to 8. The final results were the averages of a total of 8 non-continuous sectioned slides per plug sample and 8 plugs per group.
3 Results
3.1 Tensins enhance RhoA activities only in the presence of DLC1
To examine whether tensins regulate DLC1 RhoGAP activity toward its major target RhoA, we used HEK293T human kidney epithelial cells, which do not express endogenous TNS1 and DLC1, for assays (fig 1). Overexpression of TNS1 as tomato-TNS1 (Tom-TNS1) fusion protein alone in HEK293T showed no effect on the activity of RhoA as indicated by the Rho-GTP level, whereas DLC1 (as GFP-DLC1) overexpression alone decreased the level of Rho-GTP. Nonetheless, the reduction effect of DLC1 was abolished by co-expression of TNS1 and DLC1, demonstrating that TNS1 regulates RhoA activity in a DLC1-dependent fashion. Other tensin members such as TNS2, TNS3, and cten were tested for the similar function. Only expression of TNS2 or TNS3, but not cten, enhanced RhoA activities in the presence of DLC1 (fig 1), suggesting that a larger N-terminal fragment, which is not presented in cten, may be required for suppressing DLC1’s RhoGAP activity.
Figure 1. TNS1, TNS2, and TNS3, but not cten, regulate RhoA activity through DLC1.

(A) Cell lystaes from HEK293 transfected with indicated constructs were used to determine RhoA activities by rhotekin pull down assays and shown as Rho-GTP levels. Whole cell lysates were immunoblotted (IB) with antibodies against RhoA or tomato (Tom) to show the expression levels of endogenous RhoA and recombinant fusion proteins. (B) Bar graph shows quantification of Rho-GTP level. Shown are means ± SD for a minimum of three repetitions per transfection condition. TNS1, TNS2, and TNS3 significantly reduced RhoGAP activity of DLC1 when they were individually compared to control (Tom). P value was calculated compared to Tom control by Student’s t-test. *, P < 0.05.
3.2 The C2 domain of TNS1 binds to DLC1
DLC1 binds to various tensin members at three separate regions: the NTR (N-terminal region), SH2, and PTB domains. While our previous studies had established the essential residues within SH2 and PTB domains for their binding activities [17, 19], the interaction between TNS1-NTR and DLC1, and the critical residues in the NTR for DLC1-binding were not determined. We first demonstrated that TNS1-NTR (aa 75-310) indeed interacted with the SAM domain in the N-terminus of DLC1 by co-immunoprecipitation assays (fig 2Aa). Further truncation analysis showed that the C-terminal region of TNS1-NTR containing aa 211-310 (aka C2 domain) was responsible for the binding to DLC1-SAM (fig 2Ab). Since the SAM domain of DLC1 is also known to interact with the tumor suppressor PTEN within a region containing a C2 domain [20], we analyzed the C2 regions of human TNS1, TNS2, TNS3 and PTEN by sequence alignments (fig 2B). Several conserved sites were identified as potentially critical residues involving DLC1-SAM and C2 interaction. We mutated and tested two of them in TNS1: 244M (244GD to AA, also called C2 mutant) and 252M (252HK to AA). As shown in fig 2Ac, while TNS1(211-310) and TNS1(211-310)252M still interacted with DLC1-SAM, the binding was abolished in TNS1(211-310)244M. A schematic diagram showing the domain structure and mutation sites is shown in fig 2C.
Figure 2. The N-terminal TNS1 binds to DLC1 through its C2 domain.

(A) HEK293T cells transfected with indicated constructs were immunoprecipitated (IP) and then immunobloted (IB) with listed antibodies (upper panel). Whole cell lysates were analyzed for the expression of GFP fusion proteins (lower panel). (B) Sequence alignment of C2 domains in the human TNS1, TNS2, TNS3 and PTEN. 244GD and 252HR are conserved (arrows) in all C2 domains. (C) Domain structures of TNS1 and DLC1 proteins are included.
3.3 TNS1 triple-mutant is unable to bind to DLC1 and has no effect on RhoA activity
To characterize each binding site in the context of full length proteins, DLC1 was co-expressed with TNS1, TNS1-C2 mutant, TNS1-SH2 mutant, TNS1-PTB mutant, or TNS1 triple-mutant for binding assays (fig 3A). The interaction of DLC1 to TNS1 was reduced with each mutation and only when all three sites (C2, SH2, and PTB) were mutated, was the interaction totally disrupted. With this TNS1 triple-mutant, we investigated whether TNS1’s effect on RhoA activity required its physical interaction with DLC1. As shown in fig 3B, lacking DLC1-TNS1 interaction, TNS1 triple-mutant has no effect on RhoA activity, demonstrating that binding of TNS1 to DLC1 negatively regulates its RhoGAP activity toward RhoA.
Figure 3. TNS1 interaction with DLC1 is essential for its regulation on DLC1’s RhoGAP activity toward RhoA.

(A) HEK293T cells co-transfected GFP-DLC1 with tomato vector (Tom), Tom-TNS1, or Tom-TNS1 mutants (C2M, SH2M, PTBM, or triple-mutant-C2+SH2+PTBM), were subjected to IP with anti-GFP and IB with anti-tomato antibodies. (B) HEK293T cells co-transfected GFP-DLC1 with Tom, Tom-TNS1, or Tom-TNS1-triple mutant were analyzed for RhoA activities. Bar graphs show quantification of binding ability (left) or Rho-GTP level (right). Shown are means ± SD for a minimum of three repetitions per transfection condition. P value was calculated compared to Tom control by Student’s t-test. *, P < 0.05.
3.4 Tensin1-DLC1-RhoA signaling axis is critical for cellular functions in endothelial cells and angiogenesis in mice
To investigate the TNS1-DLC1-RhoA signaling axis in endothelial cells and its biological significance, we isolated and analyzed endothelial cells from TNS1 KO or WT mouse lung tissues. As predicted, the Rho-GTP level was markedly reduced in TNS1 KO endothelial cells (fig 4A). The Rho-GTP reduction was restored by further silencing of DLC1 in TNS1 KO cells (fig 4A). These results were reproducible in human umbilical vascular endothelial cells (HUVEC). Silencing of TNS1 in HUVEC suppressed the Rho-GTP level and further knockdown of DLC1 restored RhoA activity (fig 4B). Functional analysis of these cells showed that lack of TNS1 reduced endothelial cell proliferation (fig 5A), migration (fig 5B), and tube formation (fig 5C). Similar results were observed in human endothelial cells. Silencing of TNS1 in HUVEC also led to decreased proliferation, migration, and tube formation (fig 5).
Figure 4. Lack of TNS1 expression reduces the level of active RhoA, which could be restored by further silencing of DLC1 in mouse endothelial cells.

(A) Primary endothelial cells (mEndoCs) isolated from WT or TNS1 KO mice were lysed for RhoA activity assays (left panel). TNS1 KO cells infected with either shGFP or shDLC1 lentivirus were analyzed for RhoA activities (right panel). (B) HUVEC transfected with control (siCont), TNS1 siRNA (siTNS1), or siTNS1 together with siCont or siDLC1 were lysed to determine RhoA activities. Whole cell lysates were immunoblotted with TNS1, DLC1, and RhoA antibodies to assess TNS1 or DLC1 depletion in cells and total RhoA levels were served as loading controls. Bar graph shows quantification of Rho-GTP. Shown are means ± SD for a minimum of three repetitions per transfection condition. P value was calculated by one-way ANOVA with Tukey-Kramer post hoc test. *, P < 0.01
Figure 5. Loss of TNS1 reduces cell proliferation, migration, and tube formation.

TNS1 WT, KO mouse endothelial cells or HUVEC transfected with siCont or siTNS1 were used for studies. (A) The WST1 proliferation assay results of indicated cells from triplicate experiments. (B) Representative images of transwell cell migration of indicated cells and bar graphs of data from triplicate experiments. (C) Representative images of tube formation of indicated cells. Arrow indicates the tube-like structure. Bar graphs of data from triplicate experiments, means ± SD. P value was calculated by Student’s t-test. *, P < 0.05.
To examine the in vivo role of TNS1 in angiogenesis, matrigel plug assays were performed in TNS1 KO and WT mice. Five days post-injection, gel plugs were isolated and stained for CD31 positive blood vessels. Matrigel implanted in TNS1 KO mice consistently induced fewer vessel formation (fig 6A). In ex vivo aortic spouting assays, aortic rings isolated from TNS1 KO also grew significantly less angiogenic vessels (fig 6B). Together with results from analyzing endothelial cells, our studies have suggested that TNS1 mediates angiogenic response likely through the TNS1-DLC1-RhoA pathway in endothelial cells.
Figure 6. Lack of TNS1 reduces in vivo and ex vivo angiogenesis.
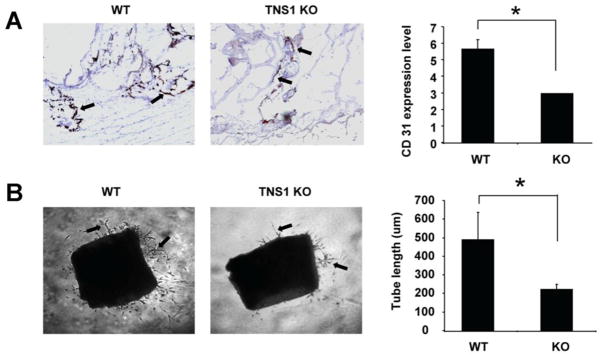
(A) Representative images of angiogenic response to Matrigel in TNS1 WT and KO mice. Gel plugs from KO showed significantly lower CD31-positive vessels than WT samples. Arrows indicate CD31-positive vessels. Bar graph shows quantification of vessel formation per sample. *P < 0.05. (B) Representative images of aortic ring explants placed in Matrigel. Arrows indicate the tube-like structures. Bar graph shows quantification of tube-like structure. Sprouting of aortic explants from TNS1 KO is significantly reduced. P value was calculated by Student’s t-test. *, P < 0.05.
4 Discussion
Our studies have established a novel function of TNS1, TNS2, and TNS3, but not cten, in promoting RhoA activity through its multi-interaction with DLC1. The interaction between tensins and DLC1 turns out to be complicated. It has been reported that the PTB of TNS2 [21], SH2 of TNS1, TNS2, TNS3 and cten [17, 22], and actin-binding domain (ABD) at the NTR of TNS3 [23] are able to bind DLC1. Our current studies have clarified and established minimal mutations to abolish the binding at each site. With the sequence similarity among tensins, it is very likely that C2 regions of TNS1, TNS2, and TNS3, as well as the SH2 and PTB of all tensins are able to interact with DLC1. Together with previous finding, tensin not only recruits DLC1 to focal adhesions [17, 22] but also modulates its RhoGAP activity. Disruption of the TNS1-DLC1-RhoA signaling axis leads to the reduction of proliferation, migration, and tube formation observed in TNS1 KO endothelial cells and angiogenic defects in TNS1 KO mice.
DLC1’s RhoGAP domain negatively regulates RhoA activity and is essential for numerous functions including suppressing cell growth, adhesion, and migration. Therefore it requires tight and fine regulation in cells. Our DLC1 mutation screening study has identified several missense and nonsense mutations in prostate and colon cancer samples [7]. Missense mutations such as T301K and S308I, which are away from the RhoGAP domain, resulted in reducing RhoGAP activity of DLC1 and therefore weakening its tumor suppression activity. This finding offers the first clue that these sites or regions may be involved in allosteric regulation of DLC1’s RhoGAP domain. However, T301K/S308I mutations do not affect the interaction between DLC1 and TNS1 (unpublished data). It has then been suggested by Li’s group that the RhoGAP activity of DLC1 is inhibited by an intramolecular interaction with its SAM domain [23]. The binding of TNS3 ABD to DLC1’s SAM domain releases this inhibition, resulting in an increase in DLC1 RhoGAP activity. This model is further polished by Lowy’s lab with their discovery that DLC1 is a target of CDK5 serine/threonine kinase [24], which phosphorylates 4 serine sites (S120, S205, S422, and S509) in DLC1. In the absence of phosphorylation, the N-terminal region of DLC1 binds to its RhoGAP domain and keeps it in a closed and inactive confirmation. Phosphorylation by CDK5 reduces the binding and places DLC1 in an open confirmation, allowing stronger interaction with tensin and others [24]. In addition to CDK5, Low’s lab has recently shown that DLC1 is phosphorylated by MEK/ERK on T301 and S308 sites and the phosphorylation is critical for activation of its RhoGAP activity [25], which supports our cancer mutation screening results mentioned above. Our current finding has added a new spin on Li’s model in that binding of TNS1 (also TNS2 and TNS3) actually reduces the RhoGAP activity of DLC1, instead of increases as in their report. This provides another layer of fine control on DLC1’s RhoGAP activity. Although Lowy’s report indicates that depletion of TNS3 in multiple cell lines did not result in a substantial change in Rho-GTP levels [24], when carefully examining their fig S5B&D, silencing of TNS3 did lead to Rho-GTP level reduction in H1703, H157 and MCF10A, but not in A549, H358, and 293T [24]. Their Fig S5B in fact had confirmed that the former three lines were DLC1-positive and later three were DLC1-negative cell lines. Their data support our finding that tensin positively regulates RhoA activity in a DLC1-dependent fashion.
Highlight.
Tensin1 enhances RhoA activity by suppressing the RhoGAP activity of DLC1.
Tensin1 interacts with DLC1 through its C2, SH2, and the PTB domain.
The tensin1-DLC1-RhoA signaling axis is critical in regulating endothelial cell migration, proliferation, and angiogenesis.
Acknowledgments
This study was supported in part by NIH grants DK097291 to YPS and CA102537 and CA151366 to SHL.
Footnotes
The authors disclose no potential conflicts of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Homma Y, Emori Y. A dual functional signal mediator showing RhoGAP and phospholipase C-delta stimulating activities. The EMBO journal. 1995;14:286–291. doi: 10.1002/j.1460-2075.1995.tb07002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yuan BZ, Miller MJ, Keck CL, Zimonjic DB, Thorgeirsson SS, Popescu NC. Cloning, characterization, and chromosomal localization of a gene frequently deleted in human liver cancer (DLC-1) homologous to rat RhoGAP. Cancer Res. 1998;58:2196–2199. [PubMed] [Google Scholar]
- 3.Liao YC, Lo SH. Deleted in liver cancer-1 (DLC-1): a tumor suppressor not just for liver. The international journal of biochemistry & cell biology. 2008;40:843–847. doi: 10.1016/j.biocel.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Durkin ME, Yuan BZ, Zhou X, Zimonjic DB, Lowy DR, Thorgeirsson SS, Popescu NC. DLC-1:a Rho GTPase-activating protein and tumour suppressor. J Cell Mol Med. 2007;11:1185–1207. doi: 10.1111/j.1582-4934.2007.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim TY, Vigil D, Der CJ, Juliano RL. Role of DLC-1, a tumor suppressor protein with RhoGAP activity, in regulation of the cytoskeleton and cell motility. Cancer Metastasis Rev. 2009;28:77–83. doi: 10.1007/s10555-008-9167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao YC, Shih YP, Lo SH. Mutations in the focal adhesion targeting region of deleted in liver cancer-1 attenuate their expression and function. Cancer Res. 2008;68:7718–7722. doi: 10.1158/0008-5472.CAN-08-2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narumiya S, Tanji M, Ishizaki T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009;28:65–76. doi: 10.1007/s10555-008-9170-7. [DOI] [PubMed] [Google Scholar]
- 9.Lo SH. Tensin. The international journal of biochemistry & cell biology. 2004;36:31–34. doi: 10.1016/s1357-2725(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 10.Lo SH, Janmey PA, Hartwig JH, Chen LB. Interactions of tensin with actin and identification of its three distinct actin-binding domains. The Journal of cell biology. 1994;125:1067–1075. doi: 10.1083/jcb.125.5.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui Y, Liao YC, Lo SH. Epidermal growth factor modulates tyrosine phosphorylation of a novel tensin family member, tensin3. Molecular cancer research: MCR. 2004;2:225–232. [PubMed] [Google Scholar]
- 12.Hafizi S, Alindri F, Karlsson R, Dahlback B. Interaction of Axl receptor tyrosine kinase with C1-TEN, a novel C1 domain-containing protein with homology to tensin. Biochem Biophys Res Commun. 2002;299:793–800. doi: 10.1016/s0006-291x(02)02718-3. [DOI] [PubMed] [Google Scholar]
- 13.Davis S, Lu ML, Lo SH, Lin S, Butler JA, Druker BJ, Roberts TM, An Q, Chen LB. Presence of an SH2 domain in the actin-binding protein tensin. Science. 1991;252:712–715. doi: 10.1126/science.1708917. [DOI] [PubMed] [Google Scholar]
- 14.Chen H, Duncan IC, Bozorgchami H, Lo SH. Tensin1 and a previously undocumented family member, tensin2, positively regulate cell migration. Proc Natl Acad Sci U S A. 2002;99:733–738. doi: 10.1073/pnas.022518699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H, Ishii A, Wong WK, Chen LB, Lo SH. Molecular characterization of human tensin. The Biochemical journal. 2000;351(Pt 2):403–411. [PMC free article] [PubMed] [Google Scholar]
- 16.Lo SH, Lo TB. Cten, a COOH-terminal tensin-like protein with prostate restricted expression, is down-regulated in prostate cancer. Cancer Res. 2002;62:4217–4221. [PubMed] [Google Scholar]
- 17.Liao YC, Si L, Devere RW, Lo White SH. The phosphotyrosine-independent interaction of DLC-1 and the SH2 domain of cten regulates focal adhesion localization and growth suppression activity of DLC-1. The Journal of cell biology. 2007;176:43–49. doi: 10.1083/jcb.200608015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lo SH, Yu QC, Degenstein L, Chen LB, Fuchs E. Progressive kidney degeneration in mice lacking tensin. The Journal of cell biology. 1997;136:1349–1361. doi: 10.1083/jcb.136.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katz M, Amit I, Citri A, Shay T, Carvalho S, Lavi S, Milanezi F, Lyass L, Amariglio N, Jacob-Hirsch J, Ben-Chetrit N, Tarcic G, Lindzen M, Avraham R, Liao YC, Trusk P, Lyass A, Rechavi G, Spector NL, Lo SH, Schmitt F, Bacus SS, Yarden Y. A reciprocal tensin-3-cten switch mediates EGF-driven mammary cell migration. Nat Cell Biol. 2007;9:961–969. doi: 10.1038/ncb1622. [DOI] [PubMed] [Google Scholar]
- 20.Heering J, Erlmann P, Olayioye MA. Simultaneous loss of the DLC1 and PTEN tumor suppressors enhances breast cancer cell migration. Exp Cell Res. 2009;315:2505–2514. doi: 10.1016/j.yexcr.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 21.Yam JW, Ko FC, Chan CY, Jin DY, Ng IO. Interaction of deleted in liver cancer 1 with tensin2 in caveolae and implications in tumor suppression. Cancer Res. 2006;66:8367–8372. doi: 10.1158/0008-5472.CAN-05-2850. [DOI] [PubMed] [Google Scholar]
- 22.Qian X, Li G, Asmussen HK, Asnaghi L, Vass WC, Braverman R, Yamada KM, Popescu NC, Papageorge AG, Lowy DR. Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and Rho-specific GTPase-activating protein activities. Proc Natl Acad Sci U S A. 2007;104:9012–9017. doi: 10.1073/pnas.0703033104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao X, Voss C, Zhao B, Kaneko T, Li SS. Differential regulation of the activity of deleted in liver cancer 1 (DLC1) by tensins controls cell migration and transformation. Proc Natl Acad Sci U S A. 2012;109:1455–1460. doi: 10.1073/pnas.1114368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tripathi BK, Qian X, Mertins P, Wang D, Papageorge AG, Carr SA, Lowy DR. CDK5 is a major regulator of the tumor suppressor DLC1. The Journal of cell biology. 2014;207:627–642. doi: 10.1083/jcb.201405105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ravi A, Kaushik S, Ravichandran A, Pan CQ, Low BC. Epidermal growth factor activates the Rho GTPase-activating protein (GAP) Deleted in Liver Cancer 1 via focal adhesion kinase and protein phosphatase 2A. J Biol Chem. 2015;290:4149–4162. doi: 10.1074/jbc.M114.616839. [DOI] [PMC free article] [PubMed] [Google Scholar]