

Abstract
Secondary metabolite genes are often clustered together and situated in particular genomic regions, like the subtelomere, that can facilitate niche adaptation in fungi. Solanapyrones are toxic secondary metabolites produced by fungi occupying different ecological niches. Full-genome sequencing of the ascomycete Ascochyta rabiei revealed a solanapyrone biosynthesis gene cluster embedded in an AT-rich region proximal to a telomere end and surrounded by Tc1/Mariner-type transposable elements. The highly AT-rich environment of the solanapyrone cluster is likely the product of repeat-induced point mutations. Several secondary metabolism-related genes were found in the flanking regions of the solanapyrone cluster. Although the solanapyrone cluster appears to be resistant to repeat-induced point mutations, a P450 monooxygenase gene adjacent to the cluster has been degraded by such mutations. Among the six solanapyrone cluster genes (sol1 to sol6), sol4 encodes a novel type of Zn(II)2Cys6 zinc cluster transcription factor. Deletion of sol4 resulted in the complete loss of solanapyrone production but did not compromise growth, sporulation, or virulence. Gene expression studies with the sol4 deletion and sol4-overexpressing mutants delimited the boundaries of the solanapyrone gene cluster and revealed that sol4 is likely a specific regulator of solanapyrone biosynthesis and appears to be necessary and sufficient for induction of the solanapyrone cluster genes. Despite the dynamic surrounding genomic regions, the solanapyrone gene cluster has maintained its integrity, suggesting important roles of solanapyrones in fungal biology.
INTRODUCTION
The subtelomere, the region upstream of the telomere on a linear chromosome, is characterized by richness in repetitive sequences and frequent chromosomal rearrangement and lacks synteny among closely related species and even within the same species in fungi (1–4). Subtelomeric regions are also enriched with duplicate, translocated, and horizontally transferred genes and thought to be evolutionarily labile and important for niche adaptation (2, 5, 6). These plastic chromosomal regions often include contingency genes, such as genes involved in membrane transport (2, 6), nutrient assimilation (2, 7), anaerobiosis (8), and virulence (1, 9, 10). Secondary metabolite (SM) gene clusters also exhibit a subtelomeric bias (11–14).
In fungi, SMs play diverse roles in host and niche specialization, serving as virulence factors, antibiotics, and metal ion-chelating agents (15, 16). Solanapyrones are polyketide-derived SMs that were originally found in the plant-pathogenic fungi Alternaria solani and Ascochyta rabiei (17, 18), as well as in some other ascomycetous fungi occupying different ecological niches (19–21). The fact that solanapyrones are produced by distantly related species like Nigrospora (Sordariomycetes) but not by closely related species of Ascochyta or Alternaria (Dothideomycetes) suggests the acquisition of the entire gene cluster by horizontal gene transfer (HGT) events. Recently, the solanapyrone biosynthesis gene cluster was identified in Al. solani (22). However, a lack of genomic information on the solanapyrone gene cluster in other fungi has hindered further examination of the HGT hypothesis. A. rabiei is a host-specific parasite with a narrow host range, infecting cultivated chickpea (Cicer arietinum L.) and its wild relatives (23, 24). Among solanapyrones, solanapyrone A was proven to be the most toxic to chickpea, and it has been claimed to be a key virulence factor in A. rabiei (25, 26). However, a recent functional study showed that solanapyrones are not required for A. rabiei and Al. solani to cause disease in the host plants (27).
The genomes of filamentous fungi are larger and more complex than the genomes of prokaryotes because of the presence of AT-rich regions containing large noncoding regions and degenerated genes (i.e., pseudogenes). These AT-rich regions are also rich in transposable elements (TEs) and repetitive DNA sequences, which scatter along the genome and alternate with large GC-equilibrated regions in which most of the predicted genes reside (28). Only a few TEs are currently active. However, TEs are thought to have greatly impacted gene function and genome structure during genome evolution (29, 30). TEs can be broadly categorized into two major groups, retrotransposons and DNA transposons (31). Although the majority of TEs in the genomes of the fungal class Dothideomycetes are retrotransposons, members of the Tc1/Mariner superfamily of DNA transposons are frequently found in certain members of the fungal class (32–34). The Tc1/Mariner-type TEs are highly variable in size but are generally delimited by terminal inverted repeats and encode a single transposase (35). A large number of Tc1/Mariner-type TEs have been degenerated by extensive mutations, and a few functional copies can be found across the genome of Cochliobolus heterostrophus (33).
In most ascomycetous fungi, TEs are targets for repeat-induced point mutation (RIP), which involves the transitions of the dinucleotide CpA to TpA (and TpG to TpA for the complementary strand) in pairs of duplicated DNA sequences during the dikaryotic phase between meioses (36, 37). RIP is considered a genome surveillance mechanism that mutates repetitive sequences to limit the accumulation of TEs (38). Recent comparative genomic studies suggested that RIP also plays a significant role in the evolution of pathogenicity-related effector genes that populate repeat-rich regions (39, 40). The higher mutation rate associated with the RIP process could introduce new functional alleles with premature stop codons or nonsynonymous substitutions into the genes adjacent to repetitive sequences.
The solanapyrone gene cluster in Al. solani comprises six genes (sol1 to sol6) covering about 20 kb of the genome, and their roles were tentatively assigned to the proposed solanapyrone biosynthesis scheme (22). The sol1 gene encodes a polyketide synthase (PKS) that initiates the solanapyrone biosynthesis pathway. The sol2 gene (encoding an O-methyltransferase) and sol6 gene (encoding a cytochrome P450) are thought to modify functional groups in the α-pyrone ring, producing prosolanapyrone II, the immediate precursor of solanapyrone A (22). Recently, genetic evidence that the sol5 gene, encoding a Diels-Alderase, catalyzes the final step of solanapyrone biosynthesis both in A. rabiei and in Al. solani was obtained (27). The sol3 gene (encoding a dehydrogenase) was proposed to convert solanapyrone A to a less phytotoxic metabolite, solanapyrone B (22, 25). However, some studies showed that solanapyrone B is not detectable in culture filtrates of A. rabiei, and the role of the sol3 gene in solanapyrone metabolism remains to be elucidated (17, 27).
Boundaries of SM gene clusters can be defined through identification of their respective pathway-specific regulators that coordinate the biosynthesis of the SMs (41). The solanapyrone gene clusters in A. rabiei and Al. solani are known to contain a transcription factor (TF; encoded by sol4) similar to the fungus-specific Zn(II)2Cys6 zinc cluster transcription factor (C6 zinc cluster TF) family (22, 27). The C6 zinc cluster TF family represents one of the largest classes of transcriptional regulators in fungi, as exemplified by Gal4 from the yeast Saccharomyces cerevisiae (42). C6 zinc cluster TFs have been characterized to be regulators of primary and secondary metabolism and many different cellular processes (43, 44). This TF family contains a highly conserved N-terminal DNA-binding domain consisting of six cysteines in the pattern CX2CX6CX6CX2CX6C which complexes with two Zn(II) ions (45). Immediately following the DNA-binding domain is the coiled-coil region, a very short stretch of amino acids (15 residues in the case of Gal4) which provides an interface for homo- or heterodimerization (43, 46). Many fungus-specific C6 zinc cluster TFs have been reported to dimerize through the coiled-coil region and bind to inverted repeat sequences, like CGG triplets (44, 47). Another conserved feature of C6 zinc cluster TFs is the presence of a weak homology region located in the middle of the protein, termed the middle homology region (MHR) (46). The exact role of MHR is not well understood; however, it is thought to regulate the transcriptional activity of the C6 zinc cluster TFs upon cognate ligand binding (43).
During an investigation of the solanapyrone biosynthesis gene cluster and examination of its surrounding genomic regions in A. rabiei, we discovered a novel type pathway-specific regulator and the degeneracy of adjacent genomic regions. Here we describe the dynamic genomic environment surrounding the solanapyrone cluster and functionally characterize a novel type C6 zinc cluster transcription factor for the cluster genes. We speculate on the role of horizontal transfer of the solanapyrone gene cluster, the potential impacts of RIP on the genomic environment, and the regulation of solanapyrone biosynthesis.
MATERIALS AND METHODS
Fungal strains and whole-genome sequencing.
A. rabiei strains AR628 (ATCC 201622) and AR21 (ATCC 76502) were obtained from the worldwide A. rabiei collection maintained at the USDA Western Regional Plant Introduction Station. Genomic DNA (10 μg) of strain AR628 was sequenced on a PacBio RS platform (Pacific Biosciences, Menlo Park, CA) at the Washington State University Laboratory for Bioanalysis and Biotechnology. The raw data from 15 SMRT cells were assembled using the HGAP protocol of SMRT Analysis (v2.0.0) software (Pacific Biosciences).
Sliding-window analysis of RIP indices and GC content.
To determine how the nucleotide compositions of the 100-kb region from the telomeric repeats change as a function of the position relative to the telomere, the GC content and RIP indices in a window of 200 nucleotides were calculated. The window was slid in 50-bp increments (in the centromere-to-telomere direction), and values were recalculated for each position. This was reiterated until the right-hand edge of the window reached the 3′ end of the contig.
The GC content was measured as the percentage of G and C nucleotides in each window. RIP index I was calculated as the number of ApT residues/number of TpA residues, and RIP index II was calculated as (number of CpA residues + number of TpG residues)/(number of ApC residues + number of GpT residues) (37). These operations were performed automatically using a Perl script (RIPindex.pl; kindly provided by M. Farman, University of Kentucky). To analyze the GC content and RIP indices for transposable elements, the values were calculated in a window of 200 nucleotides with 20-bp increments in the 5′-to-3′ direction.
Generation of gene replacement constructs for sol4 deletion.
The sol4 gene was deleted using the split-marker method (48), similar to that we used previously to delete the sol5 gene (27). DNA fragments of the gene replacement constructs overlapping within the hygromycin B phosphotransferase (hph) gene were amplified using a double-joint PCR method (49) with minor modifications. In the first-round PCR with genomic DNA of the AR628 strain, an 838-bp fragment upstream of the sol4 coding sequence was amplified with the primer-1/primer-2 primer pair, and a 1,094-bp fragment downstream of the sol4 coding sequence was amplified with the primer-3/primer-4 primer pair. A 1,372-bp hph cassette was amplified from pDWJ5 using the HYG-F/HYG-R primer pair. The primers primer-2 and primer-3 carried 27-bp sequence tails that overlapped the 5′ and 3′ ends of the hph cassette, respectively. In the second-round PCR, each sol4 flanking DNA fragment was fused to the hph cassette through PCR by overlap extension. The vectors cloned with the upstream flanking DNA construct and the downstream flanking DNA construct were used as the templates for the third-round PCR with the nested primer pairs primer-7/HY-R for the upstream flanking DNA construct and primer-8/YG-F for the downstream flanking DNA construct. The upstream and downstream split-marker constructs were used for the transformation of strains AR628 and AR21. The primers used for split-marker construction are listed in Table S1 in the supplemental material.
sol4 gene knockout transformation and confirmation of transformants.
Preparation of fungal protoplasts and transformation were conducted as previously described for the knocking out of the sol5 gene (27). Putative transformants were subcultured on V8 agar containing hygromycin B (200 μg/ml), and two rounds of single spore isolation were conducted to obtain homokaryotic transformants. The selected transformants were screened by a diagnostic PCR with the primer pair primer-11/primer-12 pair to amplify the full-length DNA sequence of the sol4 gene in the wild-type (WT) strains and the hph cassette in putative Δsol4 mutants (see Fig. S1 in the supplemental material). Homologous integration of the split-marker DNA constructs into the sol4 gene locus was verified by PCR with an upstream flanking primer, primer-1, and primer HYG-R.
Solanapyrone extraction and detection.
Liquid cultures of the WT strains (AR628 and AR21) and their Δsol4 mutants were established by inoculating 30 ml Czapek Dox medium supplemented with cations (50) in 250-ml Erlenmeyer flasks with 100 μl of conidial suspensions of the strains (1 × 107 spores ml−1), and the conidial suspensions were incubated at 20°C as a stationary culture. After 18 days, the cultures were filtered through four layers of Miracloth (Calbiochem, USA) in a vacuum to remove the mycelium. The mycelium in each flask was dried to determine its dry weight, and solanapyrones in the culture filtrates were extracted using Sep-Pak Vac 6 cc (1-g) tC18 cartridges (Waters Corp., Milford, MA, USA). Individual cartridges were eluted with 2 ml of methanol, and the eluents were subjected to liquid chromatography (LC)-mass spectrometry analysis. Chromatographic separation was achieved using an Acquity ultraperformance LC system as described elsewhere (27). The presence or absence of peaks corresponding to solanapyrones in the culture extracts of the WT and Δsol4 mutants was confirmed by comparing the retention times and mass and UV spectra of the solanapyrone A standard (kindly provided by Hideaki Oikawa, Hokkaido University) and purified solanapyrone C (27) in separate LC runs.
Growth measurement and virulence assays.
Agar plugs (diameter, 3 mm) containing actively growing mycelia on potato-dextrose agar (PDA) were transferred to fresh PDA, and the cultures were incubated for 12 h under a fluorescent light at 20 ± 2°C. For each strain, the colony diameters in the orthogonal direction in five replicate plates (diameter, 13.5 cm) were measured at 3-day intervals for up to 36 days. The virulence of the Δsol4 mutants was compared with that of their respective progenitors on two chickpea genotypes using the minidome technique as previously described (27).
Quantitative real-time RT-PCR analyses.
RNA was extracted from PDA cultures of the AR628 WT strain and the Δsol5 and Δsol4 mutants at 9 days after growth, using an RNeasy plant minikit (Qiagen) and adopting the method described previously (51). The Δsol5 mutant was from a previous study (27). On-column DNA digestion was performed using an RNase-free DNase set (Qiagen) to remove genomic DNA. First-strand cDNA was synthesized from total RNA using an iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's instructions.
The real-time reverse transcription-PCR (RT-PCR) conditions were those described previously (27). Quantification of transcript levels was performed by normalization by the comparative threshold cycle method (52). The transcript levels of the target genes were normalized against the Actin1 gene transcript levels, and the relative gene expressions levels were log2 transformed. The means from three biological replicates and the standard deviations of the relative expression values are presented. The primers used in the real-time RT-PCR analyses are listed in Table S1 in the supplemental material.
Generation of 4OE strains and RT-PCR analysis.
For generating sol4-overexpressing (4OE) strains, full-length DNA sequences of sol4 (1,741 bp) and its downstream untranslated region (276 bp) were amplified from the genomic DNA of strain AR628 with 5′ and 3′ primers tailed with BamHI and HindIII restriction sites, respectively. The resulting PCR product was partially digested with the BamHI restriction enzyme, as one BamHI site is present in the coding sequence, and subsequently digested with the HindIII enzyme. The larger DNA fragment (2,029 bp) was eluted and then ligated to the vector pHNU3PelA, carrying the hph gene (53). pHNU3PelA had been precut with the same restriction enzymes. The vector contains the pelA promoter from Aspergillus nidulans, which can be induced by polygalacturonic acid (PGA) and repressed by the glucose in the medium (53). The vector carrying the sol4 gene was transformed into the AR628 strain as described previously (54). Putative transformants were initially selected by their hygromycin resistance, and PCR analysis was conducted to verify integration of the plasmid by a single crossover at the native sol4 genomic locus (see Fig. S2 in the supplemental material), as described earlier (55). The resulting transformants carried two copies of the sol4 gene; one was driven by the inducible pelA promoter, and the other was driven by the native sol4 promoter.
Two sol4-overexpressing strains (4OE2-2 and 4OE6-2) were tested to see if sol4 overexpression activates other sol genes under PGA induction. AR628 or sol4-overexpressing strains were first grown in half-strength PDB for 2 days by shaking at 120 rpm. Mycelia were harvested, washed twice with distilled water, and transferred to CM broth (56) containing 1% PGA. The cultures were further incubated for 4 days, and total RNA was extracted from the mycelia after washing with distilled water two times. For RT-PCR analysis, 200 ng RNA was reverse transcribed and amplified using a OneStep RT-PCR kit (Qiagen). The sol genes (sol1 to sol6) were amplified for 30 to 32 cycles; Actin1 was amplified for 28 cycles (annealing temperature, 62°C). The experiment was repeated three times using different batches of RNA samples. The primer sets for RT-PCR analysis were designed to amplify flanking exons, including one intron. Inclusion of an intron in the amplified region facilitated distinguishing mRNA from pre-mRNA and/or possible genomic DNA contamination. The primer sequences used for plasmid constructions, verification of integration, and RT-PCR analysis are presented in Table S1 in the supplemental material.
Synteny block analysis.
Synteny blocks between the draft genomes of A. rabiei, Didymella exigua, and Leptosphaeria maculans were computed by use of the SyMAP synteny browser (57). The repeat-masked draft genome sequences of L. maculans (39) and D. exigua were obtained from the Joint Genome Institute (JGI) website (http://www.jgi.doe.gov/). Genomic sequences were first aligned using PROmer/MUMmer software (58). Raw anchors resulting from MUMmer were clustered into gene anchors. The minimum number of anchors for synteny block detection was set to 7 as a default value. The sequence of the A. rabiei contig (835 kb) harboring the solanapyrone gene cluster was pairwise compared with the sequences of L. maculans supercontig 1 and D. exigua scaffold 6.
Nucleotide sequence accession number.
The sequence of the subtelomeric region has been submitted to GenBank and may be found under accession number KR139658.
RESULTS
The solanapyrone biosynthesis gene cluster is situated in a subtelomeric region.
The genome of the AR628 strain of A. rabiei was sequenced to a coverage of 72 times, using the PacBio RS platform. The genome assembly had a total size of 40.89 Mb, scaffolded into 144 contigs, and had an N50 of 1.1 Mb. The solanapyrone biosynthesis gene cluster was found in a contig with a size of 835 kb. The contig had a tandem array of telomeric repeats [(TTAGGG)9] at the 3′ end and also included a telomere-linked helicase (TLH) gene within about 15 kb from the repeats (Fig. 1; Table 1). The TLH gene appears to be mutated by point mutations and indels and shows a partial sequence match with a full-length TLH gene. In addition, the GC-equilibrated region within approximately 10 kb from the repeats was filled with a mosaic of gene relics, most of which showed sequence similarity to the sequences of TLH genes in Pyrenophora teres f. teres (not shown).
FIG 1.

Solanapyrone biosynthesis gene cluster and its genomic environment. Sliding-window analysis of the nucleotide composition was conducted with the chromosome end harboring the genes of the solanapyrone biosynthesis gene cluster. The GC content (in percent; top, black line), RIP index I (number of ApT residues/number of TpA residues; bottom, blue line), and RIP index II [(number of CpA residues + number of TpG residues)/(number of ApC residues + number of GpT residues; bottom, red line] were calculated in a 200-bp window, which was slid in 50-bp increments across the terminal 100 kb from the telomere end. The x axis shows the distance from the left edge of the window. The (TTAGGG)9 telomere repeats were found in the rightmost region. A schematic diagram of the arrangement and orientation of open reading frames (O1 to O10; red arrows) and pseudogenes (P1 to P8; blue arrows) is shown in the middle. The shaded area indicates the region harboring the solanapyrone cluster genes (sol1 to sol6). The relative positions and details of the gene content in the 100-kb region are listed in Table 1. The horizontal solid line in the top panel shows the 50% GC mark, while the dashed line in the bottom panel shows a RIP index value of 1.
TABLE 1.
The solanapyrone biosynthesis gene cluster and flanking ORFs and pseudogenes in Ascochyta rabiei
| Genea | Relative positionb (5′- 3′) | Length (bp) | Predicted function | Closest match (GenBank accession no.) by blast analysis | IDc (%) | E value |
|---|---|---|---|---|---|---|
| P1 | 6,363–7,942 | 1,580 | Transposase | TLS1 in AM toxin region (AB525200) | 68 | 5e−92 |
| P2 | 9,046–9,783 | 738 | Ubiquitin C-terminal hydrolase | Ubiquitin hydrolase (XM_001259501) | 78 | 6e−166 |
| O1 | 12,384–10,712 | 1,673 | Major facilitator superfamily | Hypothetical protein (XP_008030938) | 71 | 0 |
| P3 | 13,063–14,396 | 1,334 | Aminotransferase | Hypothetical protein (XM_007837320) | 77 | 0 |
| O2 | 15,169–19,287 | 4,121 | Nonribosomal peptide synthetase | Hypothetical protein (XP_007835510) | 69 | 0 |
| O3 | 20,805–21,410 | 606 | Fe(II) dioxygenase | Hypothetical protein (XP_008030935) | 84 | 0 |
| O4 | 22,905–30,959 | 8,055 | Polyketide synthase | sol1, polyketide synthase (BAJ09789) | 97 | 0 |
| O5 | 32,264–33,753 | 1,490 | O-Methyltransferase | sol2, O-methyltransferase (BAJ09788) | 98 | 0 |
| P4 | 34,253–36,118 | 1,866 | Transposase | S. nodorum transposon Molly (AJ488502) | 69 | 8e−157 |
| P5 | 42,611–44,476 | 1,866 | Transposase | S. nodorum transposon Molly (AJ488502) | 70 | 0 |
| O6 | 47,917–47,009 | 909 | Dehydrogenase | sol3, dehydrogenase (BAJ09787) | 96 | 0 |
| O7 | 50,140–48,400 | 1,741 | Transcription factor | sol4, transcription factor (BAJ09786) | 96 | 0 |
| O8 | 52,989–51,240 | 1,750 | Diels-Alderase | sol5, oxidase/Diels-Alderase (BAJ09785) | 98 | 0 |
| O9 | 53,583–55,242 | 1,660 | P450 monooxygenase | sol6, P450 monooxygenase (BAJ09784) | 97 | 0 |
| O10 | 62,240–61,527 | 714 | P450 monooxygenase | Hypothetical protein (XP_007784573) | 75 | 4e−127 |
| P6 | 63,499–65,364 | 1,867 | Transposase | S. nodorum transposon Molly (AJ488502) | 68 | 2e−132 |
| P7 | 82,019–80,154 | 1,866 | Transposase | S. nodorum transposon Molly (AJ488502) | 68 | 4e−135 |
| P8 | 85,301–88,831 | 3,531 | Helicase | Telomere-linked helicase (XM_001942430) | 64 | 6e−164 |
The gene designations are according to those in Fig. 1. P and O, pseudogene and ORF, respectively.
Relative positions of genes found within a 100-kb region from the telomere end. Telomeric repeats [(TTAGGG)9] were positioned at residues 99,948 to 100,001.
ID, amino acid sequence identity. For the pseudogenes (P1 to P8), the percent sequence identity at the DNA level is presented.
The solanapyrone gene cluster was situated ≈45 kb from the telomere end (Fig. 1). The gene cluster is comprised of six genes (sol1 to sol6) with the same order and orientation of the solanapyrone gene cluster found in Al. solani (22), with the homologous genes showing a remarkably high level of amino acid sequence identity (96 to 98%; Table 1). However, unlike the solanapyrone gene cluster in Al. solani, the gene cluster in A. rabiei is interrupted by an AT-rich region (about 13 kb) between the sol2 and sol3 genes, separating the gene cluster into halves (Fig. 1).
We identified other putative SM-related genes in the flanking regions of the solanapyrone gene cluster. Three open reading frames (ORFs; ORF1, ORF2, and ORF3) found in the flanking region distal to the telomere showed homology to the membrane transporter, nonribosomal peptide synthetase, and dioxygenase, respectively (Fig. 1; Table 1). Also found were some pseudogenes with no apparent ORF; however, they showed significant hits with known protein families by blast analysis (>77% identity at the DNA sequence level), such as the ubiquitin carboxyl-terminal hydrolase and aminotransferase. Only a single ORF (ORF10) could be found within the region (about 45 kb) between the gene cluster and the telomere end. ORF10 encodes a P450 monooxygenase and includes only a partial P450 catalytic domain; thus, it is likely nonfunctional.
RIP has affected TEs and adjacent genes but not the solanapyrone gene cluster.
In the subtelomeric region (100 kb; GenBank accession number KR139658), transposable elements (TEs) are scattered along AT-rich regions (Fig. 1). The five TEs found in the genomic locus are all DNA transposons with characteristics of the Tc1/Mariner superfamily; four have a high level of DNA sequence similarity to the sequence of the Molly transposon from Stagonospora nodorum, and the sequence of the remaining TE is similar to that of the transposase-like gene found in the AM toxin gene cluster from Alternaria alternata (Table 1).
In fungi (especially filamentous ascomycetes), the presence of AT-rich regions is often indicative of the action of RIP, a defense mechanism that likely acts to protect the genome from invasive repeated sequences, such as those of viruses and TEs (38). To survey whether the AT-rich regions were caused by RIP, we conducted a sliding-window analysis to calculate two RIP indices (RIP indices I and II) across the 100-kb genomic locus. Regions with a RIP index I of >1 and a RIP index II of <1 are likely to have undergone RIP (37, 59). As indicated by both indices, there was a strong sign of RIP in the AT-rich regions within the solanapyrone gene cluster and between the cluster and the telomere (Fig. 1). However, the regions in which the solanapyrone cluster genes were located appeared to be unaffected by RIP.
The TEs located close to the solanapyrone cluster genes exhibited a high degree of structural conservation with the Molly transposon found in S. nodorum, with the elements having terminal inverted repeat (TIR) and transposase domains of nearly identical lengths (see Fig. S3 in the supplemental material). However, the overall GC content of the proximal TEs (39 to 41%) was lower than that of the Molly transposon (52%). RIP analyses indicated that the low GC content of the proximal TEs arose from RIP, suggesting that the TEs are degenerated copies of Molly transposons (Fig. 2A). Concomitantly, several premature stop codons in the coding regions of a putative transposase were identified in the degenerated TEs, and these may have been introduced directly by RIP (see Fig. S3 in the supplemental material). It is likely that the AT-rich regions found in the subtelomeric region resulted from a RIP that mutated these TEs and other repeated DNA sequences.
FIG 2.
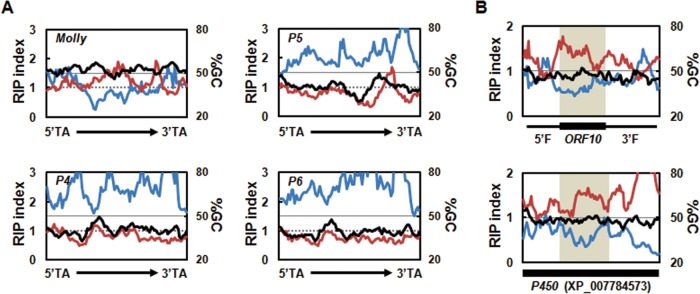
Degeneracy of transposable elements and ORF10 located near the solanapyrone gene cluster. (A) Sliding-window analyses of the nucleotide composition were conducted for the full-length functional Molly transposon from Stagonospora nodorum (1,866 bp; GenBank accession number AJ488502) and pseudogenes (P4 to P6) found in the subtelomeric region (P4 and P5, 1,866 bp; P6, 1,867 bp), including the 5′ and 3′ TA insertion sites, terminal inverted repeats, and putative transposase. Values for GC content (in percent; black line), RIP index I (number of ApT residues/number of TpA residues; blue line), and RIP index II [(number of CpA residues + number of TpG residues)/(number of ApC residues + number of GpT residues; red line] are shown. These values were calculated across a 200-bp window, which was slid in 20-bp increments across the full-length elements. Horizontal solid lines, the 50% GC mark; dashed lines, a RIP index value of 1. (B) Sliding-window analyses of the nucleotide composition of ORF10 plus its 5′ and 3′ flanking regions (2,029 bp in total) (top) and the homologous P450 gene (2,020 bp, GenBank accession number XP_007784573) (bottom). The black box below each graph indicates the position of the coding regions of the sequences. ORF10 with a 5′ flanking region (5′F) and a 3′ flanking region (3′F) and the corresponding homologous region in the P450 gene are shaded for comparison.
SM-related genes near TEs are often affected by RIP, and the degree of RIP on genes proximal to a repeat sequence was inversely proportional to the distance from the repeated sequence (32). ORF10, encoding a partial P450 enzyme, was embedded in the middle of AT-rich regions proximal to a degenerated TE (Fig. 1). This raised the possibility that the upstream and downstream sequences of ORF10 were affected and degenerated by RIP and otherwise encoded the rest of a functional P450 domain.
The deduced amino acid sequence of ORF10 was best matched (75% identity) with the gene for a full-length P450 enzyme from Coniosporium apollinis, which belongs to the class Eurotiomycetes. The DNA sequence similarities between the upstream and downstream regions of ORF10 and the corresponding regions of the orthologous P450 gene were lower, but the sequences still showed 52 and 54% identities at the DNA level, respectively, indicating that the flanking regions had diverged to a greater extent (see Fig. S4 in the supplemental material). This led us to question whether the sequence divergence might be the consequence of the RIP processes. Although it was not prominent in the RIP responses, there was an indication of the potential occurrence of RIP in upstream and downstream regions of ORF10, especially when the sequences of those regions were compared to the sequences of the corresponding regions of the functional P450 gene homolog (Fig. 2B). These results suggest that the abolished N and C termini of the otherwise functional P450 domain of ORF10 is affected by RIP to some extent.
The sol4 gene encodes a novel type Zn(II)2Cys6 TF.
The genomic environment around the solanapyrone gene cluster revealed that additional SM-related genes, such as genes for a nonribosomal peptide synthetase (ORF2) and dioxygenase (ORF3), were neighbored closely to the solanapyrone cluster. It is uncertain that these genes are involved in solanapyrone biosynthesis or coordinately regulated with other solanapyrone cluster genes. In the 100-kb subtelomeric region, we could not identify transcription factor (TF)-encoding genes other than the sol4 gene, which was previously predicted to encode a putative TF (22). However, the deduced amino acid sequence of the sol4 gene lacked a DNA-binding domain and contained only the middle homology region (MHR) commonly found in typical C6 zinc cluster TFs like yeast Gal4 (Fig. 3). The two best hits obtained by blast analysis using the protein sequence as a query were with hypothetical proteins in Podospora anserina and Aspergillus niger, with the sequences sharing 41 and 40% amino acid sequence identities, respectively. Unlike the Sol4 protein, those two hypothetical proteins contained the conserved DNA-binding domain and MHR, the hallmarks of the C6 zinc cluster TF family (see Fig. S5 in the supplemental material).
FIG 3.
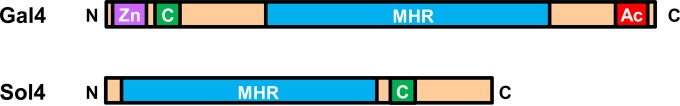
Structural organization of the typical C6 zinc cluster transcription factor (Gal4) and the Sol4 protein. C6 zinc cluster TFs consist of a highly conserved N-terminal zinc cluster DNA-binding domain consisting of six cysteines coordinating two zinc ions (Zn; purple), immediately followed by a coiled-coil region (C; green) that involves protein-protein interactions, a large C-terminal domain dubbed the MHR (blue) that regulates transcriptional activities, and an acidic activation domain (Ac; red) in the distal C-terminal end. Note that the Sol4 protein lacks the characteristic N-terminal zinc cluster DNA-binding domain of the C6 zinc cluster TF family but contains the MHR, followed by a coiled-coil region.
The sol4 gene is required for solanapyrone production but not for growth, sporulation, or virulence.
To examine if the sol4 gene encodes a bona fide TF and specifically regulates the solanapyrone cluster genes, we generated sol4 deletion (Δsol4) mutants from two wild-type (WT) strains (AR628 and AR21). Deletion of the sol4 gene resulted in the complete loss of solanapyrone production, whereas the WT strains produced solanapyrones under the same conditions (solanapyrone A, which eluted at 4.19 min; solanapyrone C, which eluted at 4.03 min) (Fig. 4A). The growth of the Δsol4 mutant in the culture was similar to that of the WT strain, as indicated by the dry weight of the mycelial mats (Fig. 4B).
FIG 4.
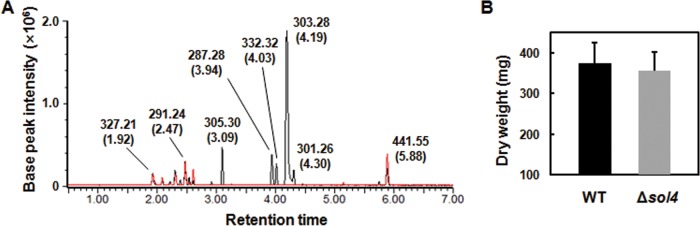
Lack of solanapyrone production in Δsol4 mutant culture. (A) Overlaid base peak ion chromatograms of equivalent quantities of culture extracts of the AR628 WT strain (black line) and its Δsol4 mutant (red line), indicated by the observed m/z value and retention time (in minutes, as indicated in parentheses) for each compound at the peak. Peaks corresponding to solanapyrones A and C (which eluted at 4.19 and 4.03 min, respectively, in the WT culture extract) are not found in the Δsol4 mutant. (B) Mean dry weight of the WT strain and the Δsol4 mutant. Mycelial mats were dried and weighed when the cultures were harvested and were analyzed for solanapyrone production 18 days after inoculation. Error bars are standard deviations (n = 28 flasks).
Deletion of the sol4 gene itself had no apparent effect on growth because both WT strains and the Δsol4 mutants initially grew at similar rates in the first 2 weeks (Fig. 5C). As solanapyrone accumulated in the WT cultures, the growth rate of the WT strains became lower and growth became inhibited, with the strains showing a restricted colony morphology, whereas the Δsol4 mutants continued to grow at the initial rate, showing uninhibited growth and an expansive colony morphology until the colony reached the edge of the agar plates. This difference in colony morphology late in culture was apparently due to the toxicity of solanapyrones (Fig. 5A) and was not due to the deletion of the sol4 gene itself (Fig. 5C). The toxicity of solanapyrone to mycelial growth could be seen by close examination of the hyphal tips at the colony margins only 3 weeks after incubation (Fig. 5B, right). No defects in the sporulation or conidial morphology of the Δsol4 mutants were observed, although the spore masses oozing out from the pycnidia differed in color between the WT strains and theΔsol4 mutants (Fig. 5B, left). Virulence assays showed that the Δsol4 mutants remained as virulent as their respective progenitors on two chickpea genotypes (see Fig. S7 in the supplemental material).
FIG 5.
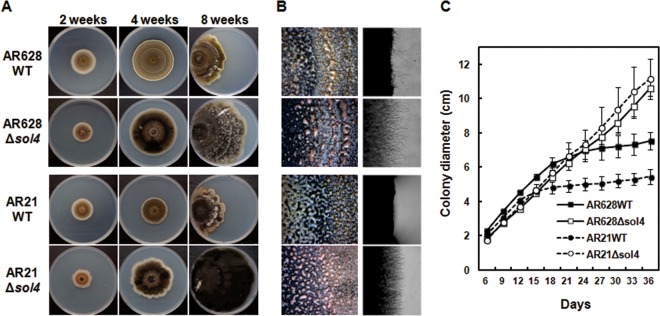
Colony morphology of WT and Δsol4 mutant strains. (A) Colony morphology of AR628 and AR21 WT strains and their corresponding Δsol4 mutants at 2, 4, and 8 weeks after incubation on PDA. For 8-week-old cultures, strains were grown from the left-hand edge of the plates. Note that the restricted colony size for the WT strains is due to the production and accumulation of solanapyrones. (B) (Left) Normal sporulation of both the WT strains and Δsol4 mutants at 3 weeks after incubation on PDA. The mass of spores oozing out from the pycnidia was visible on the colony surface of both strains. (Right) Inhibition of mycelial growth and the toxic effect of solanapyrone on mycelial growth are visible at the colony margins in the wild-type strain but not in the Δsol4 mutant strains. See panel A for the strains shown in the four panels in each column. (C) Growth curves of the WT strains and their corresponding Δsol4 mutants on PDA. Colony diameters were measured at 3-day intervals. Error bars are standard deviations (n = 5). The initial growth rates were similar among the WT and Δsol4 mutant strains in the first 2 weeks, but the growth rate became lower in the WT strain due to inhibition by solanapyrones.
The sol4 gene is a pathway-specific regulator of solanapyrone biosynthesis.
To investigate the effect of the sol4 gene in the control of solanapyrone cluster genes, the transcript levels of the sol genes (sol1 to sol6) were compared between the AR628 WT strain and its Δsol4 mutant. Additionally, the Δsol5 mutant, which lacks the enzyme required for the final step of solanapyrone biosynthesis, was included in the gene expression study because it was reported that the sol4 gene is autonomously overexpressed in the Δsol5 mutant, possibly due to a positive-feedback loop (i.e., the accumulation of intermediates triggers the entire metabolic pathway) (27). The DNase treatment in the RNA isolation step was effective in removing genomic DNA contamination because the Actin1 gene did not show the genomic DNA/pre-mRNA band (Fig. 6A). As expected, no sol4 transcripts were detected in the Δsol4 mutant, and deletion of the sol4 gene also significantly reduced the levels of expression of the other sol genes (Fig. 6A). Real-time RT-PCR analysis showed that the levels of expression of all the sol genes decreased in the Δsol4 mutant (Fig. 7). In particular, the expression level of the sol1 gene, encoding a polyketide synthase that initiates the solanapyrone biosynthesis pathway, was dramatically reduced in the Δsol4 mutant, showing an approximately 700-fold decrease compared to the level of expression in the WT strain. In contrast, the expression levels of sol1, sol2, sol4, and sol6 were significantly elevated in the Δsol5 mutant, presumably due to the inherently elevated level of sol4 gene expression (Fig. 7).
FIG 6.

Transcriptional activation of solanapyrone cluster genes by the sol4 gene. The expression levels of the solanapyrone cluster genes by the WT strain and the Δsol4 mutant grown on PDA (A) and in the WT strain and two independent sol4-overexpressing strains (4OE2-2 and 4OE6-2) grown in CM broth upon induction with polygalacturonic acid (B) were analyzed by RT-PCR. The expected band sizes of mRNA (lower bands) and pre-mRNA/genomic DNA (gDNA) (upper bands) are indicated (the numbers to the left and right of the gels are in base pairs). Similar results were obtained from three independent experiments. Lanes M, 100-bp DNA ladder. Since all the primer pairs were designed to amplify flanking exons, including one intron, the absence of the upper band in the Actin1 reference gene (A) indicates that there was no genomic DNA contamination.
FIG 7.

Pathway-specific control of the sol4 gene for solanapyrone cluster genes. The expression levels of solanapyrone cluster genes (sol1 to sol6), flanking open reading frames (ORF2, ORF3, and ORF10), and other secondary metabolite-related genes (PKS1 and PKS2) in the AR628 WT strain and its Δsol5 and Δsol4 mutants are shown. Total RNA was extracted from a 9-day-old colony grown on potato-dextrose agar. Quantitative RT-PCR was performed, and the levels of gene expression were normalized by the Actin1 expression levels in the respective samples. The relative gene expressions levels were log2 transformed. Error bars are standard deviations from three biological replicates. Asterisks, no transcripts were detected from the samples due to the specific deletion of the respective genes.
To check if the sol4 gene affects the expression of genes outside the solanapyrone cluster and to define the boundaries of the cluster, the transcript levels of the flanking ORFs (ORF2, ORF3, and ORF10) and two known polyketide synthase genes (PKS1 and PKS2) were compared between the WT strain and the Δsol4 and Δsol5 mutants. The genes outside the solanapyrone cluster remained unaffected by deletion of either the sol4 or the sol5 gene (Fig. 7). Student's t test showed that there was no difference in the transcript levels of the flanking ORFs, PKS1, and PKS2 between the strains. It is currently unknown whether the flanking ORFs and PKS2 are functional, but the PKS1 gene is known to be involved in melanin biosynthesis (54). We also attempted to compare, if the gene was transcribed, the transcript levels of one of the degenerated TEs (pseudogene 6) next to ORF10. No transcripts could be amplified from any of the tested strains, further supporting the fact that this element has been pseudogenized.
The sol4 gene alone is sufficient for induction of solanapyrone biosynthesis genes.
The upregulation of the cluster genes in the Δsol5 mutant may result directly from the elevated expression of the sol4 gene. To verify this, we generated sol4-overexpressing (4OE) strains carrying an additional copy of the sol4 gene from the WT strain but under the control of the pelA promoter. The second copy of the sol4 gene can be inducible when the 4OE strains are grown on polygalacturonic acid (PGA)-containing media, as PGA induces the pelA promoter. After induction with PGA, the expression levels of the cluster genes (sol1 to sol6) were compared between the WT strain and the 4OE strains. RT-PCR analysis showed that the sol4 gene was highly overexpressed in the 4OE strains and the expression levels of all of the cluster genes except for the sol3 gene were increased in the 4OE strains compared to the WT strain (Fig. 6B). The primer sets for the RT-PCR analysis were designed to amplify flanking exons, including one intron, in order to distinguish mRNA (lower bands) from pre-mRNA and/or possible genomic DNA contamination (upper bands) (Fig. 6). A similar trend was observed in the Δsol5 mutant, where the expression level of the sol3 gene was not significantly different from that in the WT strain (Fig. 7), suggesting a requirement for another factor for sol3 gene induction.
It is notable that pre-mRNA species of the sol1, sol2, and sol5 genes accumulated in the WT strain, while the degrees of pre-mRNA accumulation were reduced in the 4OE strains (Fig. 6). The elevated expression of the sol genes in 4OE strains is likely attributable to the more efficient processing of pre-mRNA to mRNA. In the same context, considerable amounts of pre-mRNA species of the sol genes were still present in the Δsol4 mutants compared to the amounts in the WT strain on PDA (Fig. 6A). The absence of the upper band for the Actin1 gene in all the RNA samples and the similar mRNA band intensities of the Actin1 and sol5 genes in the WT indicate that there was no genomic DNA contamination, and thus, the accumulation of the upper bands likely arose from less efficient pre-mRNA processing of the sol genes in the WT and Δsol4 mutants than in the 4OE strains (Fig. 6).
DISCUSSION
We found that the solanapyrone gene cluster is housed within the 80 kb from a telomere end in A. rabiei (Fig. 1). In fungi, conditionally dispensable chromosomes often accommodate genes involved in the production of SMs, host-selective toxins, and effectors that have a role in host range delineation and specialization (60–63). Synteny analyses of the A. rabiei draft genome data and data on the genomes of other fungal species within the order Pleosporales, available on the JGI website (64), showed that a large portion of the contig harboring the solanapyrone gene cluster is syntenic (mesosyntenic sensu stricto) to the largest scaffold of related species (the 3.38-Mb scaffold of Leptosphaeria maculans and the 4.03-Mb scaffold of Setosphaeria turcica), indicating that the solanapyrone gene cluster resides in one of the core chromosomes of A. rabiei. Accordingly, despite the high degree of karyotype variation observed in the A. rabiei strains (65), all strains examined so far are known to produce solanapyrones (26, 27, 66).
Synteny block analysis of the contig harboring the solanapyrone gene cluster with syntenic scaffolds of the two most related Dothideomycetes, Didymella exigua and L. maculans, whose genomes are available on the JGI website, showed that although GC-equilibrated regions of the contig share many synteny blocks with regions of the scaffolds of the two related species, the subtelomeric region (100 kb from telomeric repeats) harboring the solanapyrone gene cluster is unique to A. rabiei (Fig. 8). We also attempted to compare the genomic environments of the solanapyrone gene cluster between A. rabiei and Al. solani. Unfortunately, we could obtain only a short contig (20 kb) encompassing the sol1 and sol2 genes and an approximately 10-kb flanking region of the sol1 gene from the genome sequence data for Al. solani. The gene profile in the 10-kb flanking region of the Al. solani sol1 gene was different from that of A. rabiei, with the 10-kb flanking region of the Al. solani sol1 gene containing genes involved in nutrient assimilation and antibiotic resistance (see Fig. S6 in the supplemental material). Unlike the genes found in the flanking region of the A. rabiei sol1 gene, the genes found in the flanking region of the Al. solani sol1 gene were highly similar to those of members of the Dothideomycetes, such as Cochliobolus miyabeanus and S. turcica. Although a species-specific genomic region can be properly assessed only through a more extensive survey of closely related species like legume-infecting Ascochyta species, the subtelomeric region is likely a species-specific region at the end of a core chromosome in A. rabiei.
FIG 8.
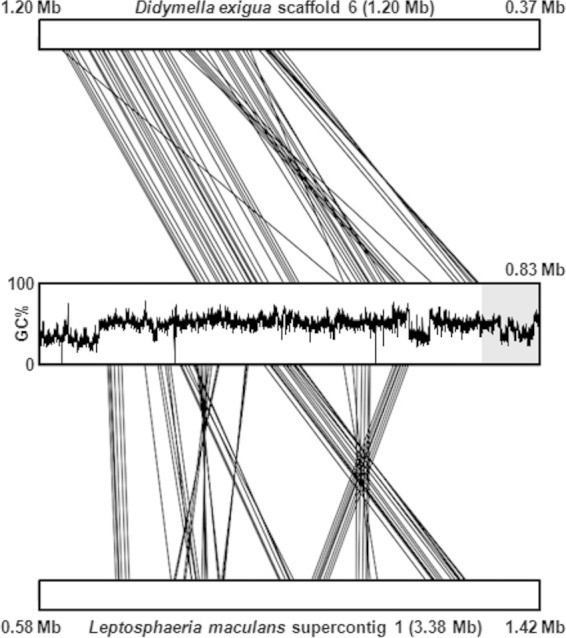
Synteny blocks shared between the A. rabiei contig harboring the solanapyrone biosynthesis gene cluster and scaffolds of related Dothideomycetes. The sequences of Didymella exigua scaffold 6 (top) and Leptosphaeria maculans supercontig 1 (bottom) were compared pairwise to the sequence of the contig of A. rabiei, and only the 835-kb regions showing synteny with the A. rabiei contig are presented. For the A. rabiei contig, the GC content across the entire contig (835 kb) is provided, and the subtelomeric region (100 kb) harboring the solanapyrone biosynthesis gene cluster is highlighted in a gray box. Note that L. maculans supercontig 1 showed mesosynteny with the A. rabiei contig, while the D. exigua is largely macrosyntenic to the A. rabiei contig, owing to its close taxonomic relationship with A. rabiei (teleomorph, Didymella rabiei).
The solanapyrone gene cluster resides in a species-specific genomic island.
The horizontal transfer of a gene (or an entire gene cluster) can be supported by circumstantial evidence, such as a patchy phyletic distribution of the gene within closely related species, a phylogenetic incongruence of the gene with the known species phylogeny, and variations in codon usage or GC content (67). Legume-associated Ascochyta fungi form a monophyletic group (68–70). However, PCR primers that can amplify the sol5 gene in both A. rabiei and Al. solani failed to amplify any DNA in other legume-infecting Ascochyta species (W. Kim and W. Chen, unpublished data). Furthermore, sol genes were not found in the draft genome data for closely related Ascochyta species infecting different legumes (J. Lichtenzveig, personal communication). Also, the exceptionally high amino acid sequence similarity (>96%) of the cluster genes between A. rabiei and Al. solani suggests their acquisition by an HGT event between the two species or their independent acquisition from a third organism.
The solanapyrone gene cluster in A. rabiei is situated in a subtelomeric region. AT-rich regions with a repetitive DNA sequence near the telomere are prone to undergo ectopic recombination at a rate much higher than that expected for homologous recombination (71, 72). Moreover, several copies of degenerated Tc1/Mariner-type TEs were found around the gene cluster (Fig. 1). Tc1/Mariner-type TEs are widely found in taxa as diverse as fungi, rotifers, insects, and mammals (73–75) and are known to be horizontally transferred between distant taxa in mammals or insects (76, 77). The finding that the solanapyrone cluster in A. rabiei is placed in such a dynamic genomic environment further supports the likelihood of HGT of the gene cluster to A. rabiei.
RIP may play a role in diversification of secondary metabolites.
RIP normally operates on repeated sequences, but it can leak to regions flanking the repeated sequence (32). There is increasing evidence that RIP leakage to adjacent DNA sequences and coding regions contributes to the diversification of pathogenicity-related effector genes (40, 78). Extensive genome sequencing of related fungal species revealed that SM gene clusters have been diversified likely due to differences in evolutionary trajectories among the species, resulting in rearrangement, pseudogenization, and/or loss of the gene constituents and in fragmentation of the SM gene cluster in separate genomic loci (79, 80).
ORF10, encoding a truncated P450 domain, is located proximally to a degenerated TE. Some putative homologs encoding full-length P450 enzymes were found from species in the classes Eurotiomycetes and Sordariomycetes but not in the Dothideomycetes, implying either its foreign origin or fast divergence. Given the taxonomic distance, the degree of sequence identity (75% at the amino acid sequence level) between ORF10 and the putative P450 gene homolog from Coniosporium apollinis was considerably high. Also, reciprocal best hits in the A. rabiei and C. apollinis genomes (Broad Institute, MA, USA) by blast analysis supported the orthology of the two genes. The upstream and downstream regions of ORF10 were mildly affected by RIP that may have originated from surrounding AT-rich regions containing repeated sequences and TEs. However, despite the discovery of extended homology and evidence for RIP, there were some indels and point mutations not relevant to RIP in the regions upstream and downstream of ORF10, whose sequences did not align very well with the sequence of the homologous P450 gene, suggesting that RIP is not the sole source of mutation acting on the genomic loci.
Solanapyrone analogs with chemical structures more complex than those of the solanapyrones produced by A. rabiei and Al. solani have been identified from distant fungal species (19–21). Considering the structural diversity of solanapyrone analogs, additional genes may be involved in their production in other solanapyrone-producing fungi. It is currently unknown whether ORF10 is part of the solanapyrone gene cluster. However, it is plausible that RIP-mediated degradation of biosynthetic genes for a metabolite could be one of the mechanisms used to achieve such a diverse chemical structure of SMs sharing the common backbone, analogous to the effector diversification processes in plant-pathogenic fungi that occurred during genome evolution. A future direction of study is to identify solanapyrone gene clusters and neighboring genes from other solanapyrone-producing fungi in order to compare gene contents, gene degeneracy, and metabolic outputs.
The solanapyrone gene cluster is controlled by a novel type of transcriptional regulator.
We identified the sol4 gene to be a pathway-specific regulator of solanapyrone biosynthesis. This unusual type of transcriptional regulator containing only MHR specifically regulates the solanapyrone cluster genes. Although several SM-related genes were found adjacent to the solanapyrone gene cluster, deletion of sol4 did not affect their expression (Fig. 7). The sharp demarcation of transcriptional control by sol4 also helped us to delimit the borders of the solanapyrone gene cluster in A. rabiei.
As far as we are aware, there are no reports on the regulatory roles of this type of regulator in biological processes. Notably, in the gene cluster of cercosporin (a polyketide produced by Cercospora spp.), three putative TF-encoding genes were found. One encodes a conventional C6 zinc cluster TF (CTB8) that is known to coordinately regulate genes in that cluster, but the other two are truncated versions of the C6 zinc cluster TF that have an unknown function and that lack the conserved DNA-binding domain (81). We could not identify any other TF-encoding genes around the solanapyrone gene cluster. However, given the fact that gene expression was not completely shut down by sol4 deletion and that the Sol4 protein contains a coiled-coil region and an MHR through which it interacts with other proteins and regulates transcriptional activity, it is conceivable that the sol4 gene activates the gene cluster in conjunction with yet unknown TFs.
C6 zinc cluster TFs are known to bind to an inverted repeat sequence like CGG(N6–11)CCG with some variations in the triplet sequence (42, 44). Using the MEME motif discovery tool (82), consensus inverted repeat sequences such as CCG(N8)CGG or GCC(N10)GGC were found upstream of the coding regions of the sol genes (see Table S2 in the supplemental material), implying that there is a C6 zinc cluster TF for the regulation of solanapyrone biosynthesis. C6 zinc cluster TFs usually repress the target genes under noninducing conditions, as is well illustrated in galactose catabolism by Gal4 in yeast (83). Therefore, Sol4 may have a role in derepression of the cluster genes by interacting with an as-yet-unknown C6 zinc cluster TF and act as a transcriptional coactivator. Identification of the specific partners interacting with the Sol4 protein will shed light on a novel regulatory mechanism for SM biosynthesis.
Solanapyrones likely exert their functions in the ecology of A. rabiei.
Solanapyrone has been claimed to be a virulence factor, as it exhibits strong phytotoxicity (25, 26). However, Δsol4 mutants generated from two WT strains (AR628 and AR21) of different pathotypes showed virulence levels similar to those of their respective WT progenitors on susceptible chickpea cultivars (see Fig. S7 in the supplemental material), supporting the previous finding with sol5 deletion mutants that solanapyrones are not required for virulence (27). Why, then, do all the field isolates of the fungus produce the auxiliary metabolite which may impose a metabolic cost to the organism? The universal production of solanapyrones among A. rabiei strains suggested that it may play an important role in the biology of the pathogen other than parasitism. In fact, solanapyrones A and C were recently shown to have selective antibiotic activity against Gram-positive bacteria, such as Bacillus subtilis, one of the common soil bacteria (84). We also observed that solanapyrone A inhibits the growth of a variety of fungal species as well as A. rabiei itself (Kim and Chen, unpublished). This antifungal activity and autotoxicity explain the restricted growth phenotype of the WT strains and the expansive growth of the Δsol4 mutants (Fig. 5).
The genomic environment of the solanapyrone gene cluster is marked by abundance in TEs and SM-related genes, many of which underwent RIP-mediated degradation and subsequent pseudogenization. Nevertheless, the gene cluster has remained intact in such a mutation hot spot region. Alternatively, it may be that, if the HGT hypothesis is valid, the gene cluster was recently transferred to the AT-rich region. It is difficult to conclude which is the case; however, either case implies the importance in maintaining the capacity for A. rabiei to produce solanapyrones; otherwise, the gene cluster would have been pseudogenized or if it had been horizontally transferred it would not have been fixed in the existing population of A. rabiei. Therefore, solanapyrones may confer a fitness advantage to the organism by acting as antibiotic agents. We are further examining the ecological roles of solanapyrones in the biology of the pathogen.
Supplementary Material
ACKNOWLEDGMENTS
The research reported in this publication was supported in part by the USDA Cool Season Food Legume Research Program and by the USA Dry Pea and Lentil Council. Mass spectrometric analysis was performed on an instrument acquired through a major research instrumentation grant (DBI-1229749) from the National Science Foundation to David R. Gang. The genome sequence data of D. exigua (CBS 183.55) were produced by the U.S. Department of Energy Joint Genome Institute (http://www.jgi.doe.gov/) in collaboration with the user community.
We are grateful to B. G. Turgeon and D. Wu (Cornell University, USA) for kindly providing pHNU3PelA, M. Farman (University of Kentucky, USA) for sharing a Perl script used in the RIP index analyses, H. Oikawa (Hokkaido University, Japan) for providing the solanapyrone A standard, and three anonymous reviewers for constructive suggestions and comments that improved the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00084-15.
REFERENCES
- 1.Barry JD, Ginger ML, Burton P, McCulloch R. 2003. Why are parasite contingency genes often associated with telomeres? Int J Parasitol 33:29–45. doi: 10.1016/S0020-7519(02)00247-3. [DOI] [PubMed] [Google Scholar]
- 2.Fairhead C, Dujon B. 2006. Structure of Kluyveromyces lactis subtelomeres: duplications and gene content. FEMS Yeast Res 6:428–441. doi: 10.1111/j.1567-1364.2006.00033.x. [DOI] [PubMed] [Google Scholar]
- 3.Fedorova ND, Khaldi N, Joardar VS, Maiti R, Amedeo P, Anderson MJ, Crabtree J, Silva JC, Badger JH, Albarraq A, Angiuoli S, Bussey H, Bowyer P, Cotty PJ, Dyer PS, Egan A, Galens K, Fraser-Liggett CM, Haas BJ, Inman JM, Kent R, Lemieux S, Malavazi I, Orvis J, Roemer T, Ronning CM, Sundaram JP, Sutton G, Turner G, Venter JC, White OR, Whitty BR, Youngman P, Wolfe KH, Goldman GH, Wortman JR, Jiang B, Denning DW, Nierman WC. 2008. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet 4:e1000046. doi: 10.1371/journal.pgen.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galagan JE, Calvo SE, Cuomo C, Ma L-J, Wortman JR, Batzoglou S, Lee S-I, Basturkmen M, Spevak CC, Clutterbuck J, Kapitonov V, Jurka J, Scazzocchio C, Farman M, Butler J, Purcell S, Harris S, Braus GH, Draht O, Busch S, D'Enfert C, Bouchier C, Goldman GH, Bell-Pedersen D, Griffiths-Jones S, Doonan JH, Yu J, Vienken K, Pain A, Freitag M, Selker EU, Archer DB, Penalva MA, Oakley BR, Momany M, Tanaka T, Kumagai T, Asai K, Machida M, Nierman WC, Denning DW, Caddick M, Hynes M, Paoletti M, Fischer R, Miller B, Dyer P, Sachs MS, Osmani SA, Birren BW. 2005. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438:1105–1115. doi: 10.1038/nature04341. [DOI] [PubMed] [Google Scholar]
- 5.Berriman M, Hall N, Sheader K, Bringaud F, Tiwari B, Isobe T, Bowman S, Corton C, Clark L, Cross GAM, Hoek M, Zanders T, Berberof M, Borst P, Rudenko G. 2002. The architecture of variant surface glycoprotein gene expression sites in Trypanosoma brucei. Mol Biochem Parasitol 122:131–140. doi: 10.1016/S0166-6851(02)00092-0. [DOI] [PubMed] [Google Scholar]
- 6.Coelho MA, Gonçalves C, Sampaio JP, Gonçalves P. 2013. Extensive intra-kingdom horizontal gene transfer converging on a fungal fructose transporter gene. PLoS Genet 9:e1003587. doi: 10.1371/journal.pgen.1003587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naumov GI, Naumova ES, Louis EJ. 1995. Genetic mapping of the alpha-galactosidase MEL gene family on right and left telomeres of Saccharomyces cerevisiae. Yeast 11:481–483. doi: 10.1002/yea.320110512. [DOI] [PubMed] [Google Scholar]
- 8.Rachidi N, Martinez M-J, Barre P, Blondin B. 2000. Saccharomyces cerevisiae PAU genes are induced by anaerobiosis. Mol Microbiol 35:1421–1430. [DOI] [PubMed] [Google Scholar]
- 9.Verstrepen KJ, Jansen A, Lewitter F, Fink GR. 2005. Intragenic tandem repeats generate functional variability. Nat Genet 37:986–990. doi: 10.1038/ng1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hernandez-Rivas R, Mattei D, Sterkers Y, Peterson DS, Wellems TE, Scherf A. 1997. Expressed var genes are found in Plasmodium falciparum subtelomeric regions. Mol Cell Biol 17:604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonagh A, Fedorova ND, Crabtree J, Yu Y, Kim S, Chen D, Loss O, Cairns T, Goldman G, Armstrong-James D, Haynes K, Haas H, Schrettl M, May G, Nierman WC, Bignell E. 2008. Sub-telomere directed gene expression during initiation of invasive aspergillosis. PLoS Pathog 4:e1000154. doi: 10.1371/journal.ppat.1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer JM, Keller NP. 2010. Secondary metabolism in fungi: does chromosomal location matter? Curr Opin Microbiol 13:431–436. doi: 10.1016/j.mib.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrin RM, Fedorova ND, Bok JW, Cramer RA, Wortman JR, Kim HS, Nierman WC, Keller NP. 2007. Transcriptional regulation of chemical diversity in Aspergillus fumigatus by LaeA. PLoS Pathog 3:e50. doi: 10.1371/journal.ppat.0030050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maiya S, Grundmann A, Li X, Li S-M, Turner G. 2007. Identification of a hybrid PKS/NRPS required for pseurotin A biosynthesis in the human pathogen Aspergillus fumigatus. Chembiochem 8:1736–1743. doi: 10.1002/cbic.200700202. [DOI] [PubMed] [Google Scholar]
- 15.Michael JP, Pattenden G. 1993. Marine metabolites and metal ion chelation: the facts and the fantasies. Angew Chem Int Ed Engl 32:1–23. doi: 10.1002/anie.199300013. [DOI] [Google Scholar]
- 16.Turgeon BG, Bushley KE. 2010. Secondary metabolism, p 376–395. In Borkovich KA, Ebbole DJ (ed), Cellular and molecular biology of filamentous fungi. ASM Press, Washington, DC. [Google Scholar]
- 17.Alam SS, Bilton JN, Slawin AMZ, Williams DJ, Sheppard RN, Strange RN. 1989. Chickpea blight: production of the phytotoxins solanapyrones A and C by Ascochyta rabiei. Phytochemistry 28:2627–2630. doi: 10.1016/S0031-9422(00)98054-3. [DOI] [Google Scholar]
- 18.Ichihara A, Tazaki H, Sakamura S. 1983. Solanapyrones A, B and C, phytotoxic metabolites from the fungus Alternaria solani. Tetrahedron Lett 24:5373–5376. doi: 10.1016/S0040-4039(00)87872-7. [DOI] [Google Scholar]
- 19.Schmidt LE, Gloer JB, Wicklow DT. 2007. Solanapyrone analogues from a Hawaiian fungicolous fungus. J Nat Prod 70:1317–1320. doi: 10.1021/np070251m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu SH, Chen YW, Shao SC, Wang LD, Yu Y, Li ZY, Yang LY, Li SL, Huang R. 2009. Two new solanapyrone analogues from the endophytic fungus Nigrospora sp. YB-141 of Azadirachta indica. Chem Biodivers 6:79–85. doi: 10.1002/cbdv.200700421. [DOI] [PubMed] [Google Scholar]
- 21.Trisuwan K, Rukachaisirikul V, Sukpondma Y, Preedanon S, Phongpaichit S, Sakayaroj J. 2009. Pyrone derivatives from the marine-derived fungus Nigrospora sp. PSU-F18. Phytochemistry 70:554–557. doi: 10.1016/j.phytochem.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 22.Kasahara K, Miyamoto T, Fujimoto T, Oguri H, Tokiwano T, Oikawa H, Ebizuka Y, Fujii I. 2010. Solanapyrone synthase, a possible Diels-Alderase and iterative type I polyketide synthase encoded in a biosynthetic gene cluster from Alternaria solani. Chembiochem 11:1245–1252. doi: 10.1002/cbic.201000173. [DOI] [PubMed] [Google Scholar]
- 23.Chen W, Coyne CJ, Peever TL, Muehlbauer FJ. 2004. Characterization of chickpea differentials for pathogenicity assay of ascochyta blight and identification of chickpea accessions resistant to Didymella rabiei. Plant Pathol 53:759–769. doi: 10.1111/j.1365-3059.2004.01103.x. [DOI] [Google Scholar]
- 24.Frenkel O, Peever TL, Chilvers MI, Ozkilinc H, Can C, Abbo S, Shtienberg D, Sherman A. 2010. Ecological genetic divergence of the fungal pathogen Didymella rabiei on sympatric wild and domesticated Cicer spp. (chickpea). Appl Environ Microbiol 76:30–39. doi: 10.1128/AEM.01181-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamid K, Strange RN. 2000. Phytotoxicity of solanapyrones A and B produced by the chickpea pathogen Ascochyta rabiei (Pass.) Labr. and the apparent metabolism of solanapyrone A by chickpea tissues. Physiol Mol Plant Pathol 56:235–244. doi: 10.1006/pmpp.2000.0272. [DOI] [Google Scholar]
- 26.Kaur S. 1995. Phytotoxicity of solanapyrones produced by the fungus Ascochyta rabiei and their possible role in blight of chickpea (Cicer arietinum). Plant Sci 109:23–29. doi: 10.1016/0168-9452(95)04144-J. [DOI] [Google Scholar]
- 27.Kim W, Park C-M, Park J-J, Akamatsu HO, Peever TL, Xian M, Gang DR, Vandemark G, Chen W. 2015. Functional analyses of the Diels-Alderase gene sol5 of Ascochyta rabiei and Alternaria solani indicate that the solanapyrone phytotoxins are not required for pathogenicity. Mol Plant Microbe Interact 28:482–496. doi: 10.1094/MPMI-08-14-0234-R. [DOI] [PubMed] [Google Scholar]
- 28.Eyre-Walker A, Hurst LD. 2001. The evolution of isochores. Nat Rev Genet 2:549–555. doi: 10.1038/35080577. [DOI] [PubMed] [Google Scholar]
- 29.Ayarpadikannan S, Kim H-S. 2014. The impact of transposable elements in genome evolution and genetic instability and their implications in various diseases. Genomics Inform 12:98–104. doi: 10.5808/GI.2014.12.3.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kazazian HH. 2004. Mobile elements: drivers of genome evolution. Science 303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 31.Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH. 2007. A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 32.Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, Barry KW, Condon BJ, Copeland AC, Dhillon B, Glaser F, Hesse CN, Kosti I, LaButti K, Lindquist EA, Lucas S, Salamov AA, Bradshaw RE, Ciuffetti L, Hamelin RC, Kema GHJ, Lawrence C, Scott JA, Spatafora JW, Turgeon BG, de Wit PJGM, Zhong S, Goodwin SB, Grigoriev IV. 2012. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog 8:e1003037. doi: 10.1371/journal.ppat.1003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santana M, Silva J, Mizubuti E, Araujo E, Condon B, Turgeon B, Queiroz M. 2014. Characterization and potential evolutionary impact of transposable elements in the genome of Cochliobolus heterostrophus. BMC Genomics 15:536. doi: 10.1186/1471-2164-15-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manning V, Pandelova I, Dhillon B, Wilhelm L, Goodwim S, Berlin A, Figueroa M, Freitag M, Hane J, Henrissat B, Holman W, Kodira C, Martin J, Oliver R, Robbertse B, Schackwitz W, Schwartz D, Spatafora J, Turgeon B, Yandava C, Young S, Zhou S, Zeng Q, Grigoriev I, Ma L, Ciuffetti L. 2013. Comparative genomics of a plant-pathogenic fungus, Pyrenophora tritici-repentis, reveals transduplication and the impact of repeat elements on pathogenicity and population divergence. G3 (Bethesda) 3:41–63. doi: 10.1534/g3.112.004044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marini M, Zanforlin T, Santos P, Barros R, Guerra A, Puccia R, Felipe M, Brigido M, Soares C, Ruiz J, Silveira J, Cisalpino P. 2010. Identification and characterization of Tc1/mariner-like DNA transposons in genomes of the pathogenic fungi of the Paracoccidioides species complex. BMC Genomics 11:130. doi: 10.1186/1471-2164-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Selker EU, Cambareri EB, Jensen BC, Haack KR. 1987. Rearrangement of duplicated DNA in specialized cells of Neurospora. Cell 51:741–752. doi: 10.1016/0092-8674(87)90097-3. [DOI] [PubMed] [Google Scholar]
- 37.Margolin BS, Garrett-Engele PW, Stevens JN, Fritz DY, Garrett-Engele C, Metzenberg RL, Selker EU. 1998. A methylated Neurospora 5S rRNA pseudogene contains a transposable element inactivated by repeat-induced point mutation. Genetics 149:1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galagan JE, Selker EU. 2004. RIP: the evolutionary cost of genome defense. Trends Genet 20:417–423. doi: 10.1016/j.tig.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 39.Rouxel T, Grandaubert J, Hane JK, Hoede C, van de Wouw AP, Couloux A, Dominguez V, Anthouard V, Bally P, Bourras S, Cozijnsen AJ, Ciuffetti LM, Degrave A, Dilmaghani A, Duret L, Fudal I, Goodwin SB, Gout L, Glaser N, Linglin J, Kema GHJ, Lapalu N, Lawrence CB, May K, Meyer M, Ollivier B, Poulain J, Schoch CL, Simon A, Spatafora JW, Stachowiak A, Turgeon BG, Tyler BM, Vincent D, Weissenbach J, Amselem J, Quesneville H, Oliver RP, Wincker P, Balesdent M-H, Howlett BJ. 2011. Effector diversification within compartments of the Leptosphaeria maculans genome affected by repeat-induced point mutations. Nat Commun 2:202. doi: 10.1038/ncomms1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rep M, Kistler HC. 2010. The genomic organization of plant pathogenicity in Fusarium species. Curr Opin Plant Biol 13:420–426. doi: 10.1016/j.pbi.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 41.Keller NP, Hohn TM. 1997. Metabolic pathway gene clusters in filamentous fungi. Fungal Genet Biol 21:17–29. doi: 10.1006/fgbi.1997.0970. [DOI] [PubMed] [Google Scholar]
- 42.Todd RB, Andrianopoulos A. 1997. Evolution of a fungal regulatory gene family: the Zn(II)2Cys6 binuclear cluster DNA binding motif. Fungal Genet Biol 21:388–405. doi: 10.1006/fgbi.1997.0993. [DOI] [PubMed] [Google Scholar]
- 43.Näär AM, Thakur JK. 2009. Nuclear receptor-like transcription factors in fungi. Genes Dev 23:419–432. doi: 10.1101/gad.1743009. [DOI] [PubMed] [Google Scholar]
- 44.MacPherson S, Larochelle M, Turcotte B. 2006. A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol Mol Biol Rev 70:583–604. doi: 10.1128/MMBR.00015-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan T, Coleman JE. 1990. GAL4 transcription factor is not a “zinc finger” but forms a Zn(II)2Cys6 binuclear cluster. Proc Natl Acad Sci U S A 87:2077–2081. doi: 10.1073/pnas.87.6.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schjerling P, Holmberg S. 1996. Comparative amino acid sequence analysis of the C6 zinc cluster family of transcriptional regulators. Nucleic Acids Res 24:4599–4607. doi: 10.1093/nar/24.23.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reece R, Ptashne M. 1993. Determinants of binding-site specificity among yeast C6 zinc cluster proteins. Science 261:909–911. doi: 10.1126/science.8346441. [DOI] [PubMed] [Google Scholar]
- 48.Catlett NL, Lee BN, Yoder OC, Turgeon BG. 2003. Split-marker recombination for efficient targeted deletion of fungal genes. Fungal Genet Newsl 50:9–11. [Google Scholar]
- 49.Yu J-H, Hamari Z, Han K-H, Seo J-A, Reyes-Domínguez Y, Scazzocchio C. 2004. Double-joint PCR: a PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet Biol 41:973–981. doi: 10.1016/j.fgb.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 50.Chen YM, Strange RN. 1991. Synthesis of the solanapyrone phytotoxins by Ascochyta rabiei in response to metal cations and development of a defined medium for toxin production. Plant Pathol 40:401–407. doi: 10.1111/j.1365-3059.1991.tb02397.x. [DOI] [Google Scholar]
- 51.Schumann U, Smith N, Wang M-B. 2013. A fast and efficient method for preparation of high-quality RNA from fungal mycelia. BMC Res Notes 6:71. doi: 10.1186/1756-0500-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 53.Yang G, Turgeon BG, Yoder OC. 1994. Toxin-deficient mutants from a toxin-sensitive transformant of Cochliobolus heterostrophus. Genetics 137:751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akamatsu HO, Chilvers MI, Stewart JE, Peever TL. 2010. Identification and function of a polyketide synthase gene responsible for 1,8-dihydroxynaphthalene-melanin pigment biosynthesis in Ascochyta rabiei. Curr Genet 56:349–360. doi: 10.1007/s00294-010-0306-2. [DOI] [PubMed] [Google Scholar]
- 55.Wu D, Oide S, Zhang N, Choi MY, Turgeon BG. 2012. ChLae1 and ChVel1 regulate T-toxin production, virulence, oxidative stress response, and development of the maize pathogen Cochliobolus heterostrophus. PLoS Pathog 8:e1002542. doi: 10.1371/journal.ppat.1002542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leach J, Lang BR, Yoder OC. 1982. Methods for selection of mutants and in vitro culture of Cochliobolus heterostrophus. J Gen Microbiol 128:1719–1729. [Google Scholar]
- 57.Soderlund C, Bomhoff M, Nelson WM. 2011. SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res 39:e68. doi: 10.1093/nar/gkr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu C, Kim Y-S, Smith KM, Li W, Hood HM, Staben C, Selker EU, Sachs MS, Farman ML. 2009. Characterization of chromosome ends in the filamentous fungus Neurospora crassa. Genetics 181:1129–1145. doi: 10.1534/genetics.107.084392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hatta R, Ito K, Hosaki Y, Tanaka T, Tanaka A, Yamamoto M, Akimitsu K, Tsuge T. 2002. A conditionally dispensable chromosome controls host-specific pathogenicity in the fungal plant pathogen Alternaria alternata. Genetics 161:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balesdent M-H, Fudal I, Ollivier B, Bally P, Grandaubert J, Eber F, Chèvre A-M, Leflon M, Rouxel T. 2013. The dispensable chromosome of Leptosphaeria maculans shelters an effector gene conferring avirulence towards Brassica rapa. New Phytol 198:887–898. doi: 10.1111/nph.12178. [DOI] [PubMed] [Google Scholar]
- 62.Ma L, van der Does H, Borkovich K, Coleman J, Daboussi M, Di Pietro A, Dufresne M, Freitag M, Grabherr M, Henrissat B, Houterman P, Kang S, Shim W, Woloshulk C, Xie X, Hu J, Antoniw J, Baker S, Bluhm B, Breakspear A, Brown D, Butchko R, Chapman S, Coulson R, Coutinho P, Danchin E, Diener A, Gale L, Gardiner D, Goff S. 2010. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium oxysporum. Nature 464:367–373. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mehrabi R, Bahkali AH, Abd-Elsalam KA, Moslem M, Ben M, Barek S, Gohari AM, Jashni MK, Stergiopoulos I, Kema GHJ, de Wit, PJGM . 2011. Horizontal gene and chromosome transfer in plant pathogenic fungi affecting host range. FEMS Microbiol Rev 35:542–554. doi: 10.1111/j.1574-6976.2010.00263.x. [DOI] [PubMed] [Google Scholar]
- 64.Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, Shabalov I. 2014. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res 42:D699–D704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Akamatsu HO, Chilvers MI, Kaiser WJ, Peever TL. 2012. Karyotype polymorphism and chromosomal rearrangement in populations of the phytopathogenic fungus, Ascochyta rabiei. Fungal Biol 116:1119–1133. doi: 10.1016/j.funbio.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 66.Höhl B, Weidemann C, Höhl U, Barz W. 1991. Isolation of solanapyrones A, B and C from culture filtrates and spore germination fluids of Ascochyta rabiei and aspects of phytotoxin action. J Phytopathol 132:193–206. doi: 10.1111/j.1439-0434.1991.tb00112.x. [DOI] [Google Scholar]
- 67.Fitzpatrick DA. 2012. Horizontal gene transfer in fungi. FEMS Microbiol Lett 329:1–8. doi: 10.1111/j.1574-6968.2011.02465.x. [DOI] [PubMed] [Google Scholar]
- 68.Barve MP, Arie T, Salimath SS, Muehlbauer FJ, Peever TL. 2003. Cloning and characterization of the mating type (MAT) locus from Ascochyta rabiei (teleomorph: Didymella rabiei) and a MAT phylogeny of legume-associated Ascochyta spp. Fungal Genet Biol 39:151–167. doi: 10.1016/S1087-1845(03)00015-X. [DOI] [PubMed] [Google Scholar]
- 69.Peever TL, Barve MP, Stone LJ, Kaiser WJ. 2007. Evolutionary relationships among Ascochyta species infecting wild and cultivated hosts in the legume tribes Cicereae and Vicieae. Mycologia 99:59–77. doi: 10.3852/mycologia.99.1.59. [DOI] [PubMed] [Google Scholar]
- 70.Chilvers MI, Rogers JD, Dugan FM, Stewart JE, Chen W, Peever TL. 2009. Didymella pisi sp. nov., the teleomorph of Ascochyta pisi. Mycol Res 113:391–400. doi: 10.1016/j.mycres.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 71.Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ, Thon M, Kulkarni R, Xu J-R, Pan H, Read ND, Lee Y-H, Carbone I, Brown D, Oh YY, Donofrio N, Jeong JS, Soanes DM, Djonovic S, Kolomiets E, Rehmeyer C, Li W, Harding M, Kim S, Lebrun M-H, Bohnert H, Coughlan S, Butler J, Calvo S, Ma L-J, Nicol R, Purcell S, Nusbaum C, Galagan JE, Birren BW. 2005. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434:980–986. doi: 10.1038/nature03449. [DOI] [PubMed] [Google Scholar]
- 72.Freitas-Junior LH, Bottius E, Pirrit LA, Deitsch KW, Scheidig C, Guinet F, Nehrbass U, Wellems TE, Scherf A. 2000. Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of P. falciparum. Nature 407:1018–1022. doi: 10.1038/35039531. [DOI] [PubMed] [Google Scholar]
- 73.Arkhipova IR, Meselson M. 2005. Diverse DNA transposons in rotifers of the class Bdelloidea. Proc Natl Acad Sci U S A 102:11781–11786. doi: 10.1073/pnas.0505333102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Plasterk RHA, Izsvák Z, Ivics Z. 1999. Resident aliens: the Tc1/mariner superfamily of transposable elements. Trends Genet 15:326–332. doi: 10.1016/S0168-9525(99)01777-1. [DOI] [PubMed] [Google Scholar]
- 75.Robertson HM. 1993. The mariner transposable element is widespread in insects. Nature 362:241–245. doi: 10.1038/362241a0. [DOI] [PubMed] [Google Scholar]
- 76.Robertson HM, Soto-Adames FN, Walden KKO, Avancini RMP, Lampe DJ. 1998. The mariner transposons of animals: horizontally jumping genes, p 268–284. In Syvanen M, Kado C (ed), Horizontal gene transfer. Chapman and Hall, London, United Kingdom. [Google Scholar]
- 77.Lampe DJ, Witherspoon DJ, Soto-Adames FN, Robertson HM. 2003. Recent horizontal transfer of mellifera subfamily mariner transposons into insect lineages representing four different orders shows that selection acts only during horizontal transfer. Mol Biol Evol 20:554–562. doi: 10.1093/molbev/msg069. [DOI] [PubMed] [Google Scholar]
- 78.Van de Wouw AP, Cozijnsen AJ, Hane JK, Brunner PC, McDonald BA, Oliver RP, Howlett BJ. 2010. Evolution of linked avirulence effectors in Leptosphaeria maculans is affected by genomic environment and exposure to resistance genes in host plants. PLoS Pathog 6:e1001180. doi: 10.1371/journal.ppat.1001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bradshaw RE, Slot JC, Moore GG, Chettri P, de Wit PJGM, Ehrlich KC, Ganley ARD, Olson MA, Rokas A, Carbone I, Cox MP. 2013. Fragmentation of an aflatoxin-like gene cluster in a forest pathogen. New Phytol 198:525–535. doi: 10.1111/nph.12161. [DOI] [PubMed] [Google Scholar]
- 80.Campbell MA, Staats M, Van Kan JAL, Rokas A, Slot JC. 2013. Repeated loss of an anciently horizontally transferred gene cluster in Botrytis. Mycologia 105:1126–1134. doi: 10.3852/12-390. [DOI] [PubMed] [Google Scholar]
- 81.Chen H, Lee M-H, Daub ME, Chung K-R. 2007. Molecular analysis of the cercosporin biosynthetic gene cluster in Cercospora nicotianae. Mol Microbiol 64:755–770. doi: 10.1111/j.1365-2958.2007.05689.x. [DOI] [PubMed] [Google Scholar]
- 82.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME suite: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Traven A, Jelicic B, Sopta M. 2006. Yeast Gal4: a transcriptional paradigm revisited. EMBO Rep 7:496–499. doi: 10.1038/sj.embor.7400679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang X-Z, Luo X-H, Xiao J, Zhai M-M, Yuan Y, Zhu Y, Crews P, Yuan C-S, Wu Q-X. 2014. Pyrone derivatives from the endophytic fungus Alternaria tenuissima SP-07 of Chinese herbal medicine Salvia przewalskii. Fitoterapia 99:184–190. doi: 10.1016/j.fitote.2014.09.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.