

Abstract
Composting is an appropriate management alternative for municipal solid waste; however, our knowledge about the microbial regulation of this process is still scare. We employed metaproteomics to elucidate the main biodegradation pathways in municipal solid waste composting system across the main phases in a large-scale composting plant. The investigation of microbial succession revealed that Bacillales, Actinobacteria and Saccharomyces increased significantly with respect to abundance in composting process. The key microbiologic population for cellulose degradation in different composting stages was different. Fungi were found to be the main producers of cellulase in earlier phase. However, the cellulolytic fungal communities were gradually replaced by a purely bacterial one in active phase, which did not support the concept that the thermophilic fungi are active through the thermophilic phase. The effective decomposition of cellulose required the synergy between bacteria and fungi in the curing phase.
Introduction
Composting is an aerobic process, during which organic waste is biologically degraded by microorganisms to humus-like material (Partanen et al., 2010). The products have a commercial value as soil conditioner or organic fertilizer (Negro et al., 1999). The typical composting batch could be divided into three phases: the mesophilic phase (10–42°C), the thermophilic phase (45–70°C) and the cooling and maturation phase (65–23°C) (Joséphine and Philippe, 2004; Bonito et al., 2010). Both bacteria and fungi play a key role in a typical composting process. Thus, a thorough understanding of microbial communities throughout the composting process is crucial for understanding the system and optimizing compost product quality (De Gannes et al., 2013a).
The information of microbial communities in composts has been examined by culture-independent deoxyribonucleic acid (DNA)-based methods. These investigations have focused on analysis of shifts in microbial community composition via amplified ribosomal DNA restriction analysis, single strand-conformation polymorphism, restriction fragment length polymorphism, denaturing gradient gel electrophoresis and nucleic acid microarrays (Peters et al., 2000; Dees and Ghiorse, 2001; Franke-Whittle et al., 2009; 2014; Baharuddin et al., 2010; Bonito et al., 2010; Partanen et al., 2010; Charbonneau et al., 2012; Tian et al., 2013). However, little is known about global gene expression of compost as relevant studies have not been performed.
Proteins reflect the actual functionality with respect to metabolic reactions and regulatory cascades, and give more direct information about microbial activity than functional genes and even the corresponding messenger RNAs (Wilmes and Bond, 2006). Furthermore, the use of proteins also bears the potential to reveal the identity of the active microorganisms via database analysis using the level of homology to other species (Benndorf et al., 2007). Thus, the presence of specific proteins in environmental samples is a potentially reliable indicator for microbial function (Benndorf et al., 2007). Wilmes and Bond (2004) proposed the term ‘metaproteomics’ for the large-scale characterization of the entire protein complement of environmental microbiota at a given point in time. By determining proteins which have been synthesized by microorganisms present at the time of sampling, metaproteomics enables the reconstruction of microbial processes and metabolic pathways that are central to the functioning of the ecosystem (Williams et al., 2012). Until now, several authors presented metaproteome data from different environmental systems, such as soil (Benndorf et al., 2007; Chourey et al., 2010), activated sludge (Wilmes and Bond, 2006), water (Benndorf et al., 2007; Lauro et al., 2011; Williams et al., 2012) and leaf litter (Schneider et al., 2012). Additionally, metaproteomics was used to define key catabolic players at contaminated sites to predict pollutant degradation networks in the environment (Guazzaroni et al., 2012). Metaproteomics has become an efficient tool to unravel and characterize metabolic networks as well as ecological interactions during complex environments.
For economical and capacity reasons, there is always a tendency to push the capacity limits and minimize the retention time at composting plants (Partanen et al., 2010). This may lead to unwanted anaerobic conditions and failure to attain the temperatures required for hygienization. To adjust these conditions at the large-scale composting plants, comprehensive information is needed regarding total biodiversity and metabolic processes. In the current study, we analysed the metaproteome from composting samples collected in different phases in a large-scale composting plant. By examining expressed proteins in the samples, we were able to infer the predominant metabolic processes performed by bacteria and fungi present in composting pile, and consider the significance of the differences between main composting phases.
Results and discussion
Physicochemical data for the composting samples
The composting pile achieved thermophilic temperature shortly after pile establishment. This temperature was maintained for 16 days, and gradually descended to ambient values thereafter. The pH values of the composting pile increased from acidic values in earlier phase to alkaline values on day 50 (Table 1). The highest water content level was seen in the active phase, with approximately 30% water content loss occurring afterwards, which might be explained by microbial heat generation causing enhanced desiccation. The total organic carbon (C) content reductions were found in composting pile, reflecting a notable mineralization of organic matter over time. The total nitrogen (N) percentage showed an increasing trend with composting duration, which is due to the concentration effect caused by carbon loss associated with mineralization of the organic matter (Shemekite et al., 2014). The C/N ratio decreased significantly over time, as C was lost in the form of CO2 through microbial respiration and N was recycled (Ryckeboer et al., 2003).
Table 1.
Physical and chemical properties of the samples
| LS1 | LS2 | LS3 | |
|---|---|---|---|
| Age (d) | 5 | 20 | 50 |
| Temperature (°C) | 60.3 | 62.4 | 47.0 |
| pH | 6.46 | 6.90 | 7.46 |
| Water content (%) | 58.80 | 53.02 | 34.84 |
| Organic C (%) | 63.71 | 52.29 | 47.17 |
| N (%) | 1.70 | 1.77 | 1.95 |
| C : N ratio | 37.48 | 29.54 | 24.19 |
Bacterial diversity represented in the composting metaproteome
A total of 640 proteins were detected (222 from the sample LS1, 220 from the sample LS2 and 198 from the sample LS3). Of the 640 proteins, 449 were with highest sequence identity to bacterial proteins and 191 fungal proteins. Within the bacterial subset, the majority had the best match (highest sequence identity) to proteins from members of the Gammaproteobacteria (181), followed by Bacilli (65), Alphaproteobacteria (41), Actinobacteria (37) and Betaproteobacteria (31) (Table 2). Within the Gammaproteobacteria, most of the matches had highest identity to proteins from the order Pseudomonadales (61) and Enterobacteriales (50). Within Pseudomonadales, the genus Azotobacter (5) can carry out the denitrification process (Szántó, 2009). The order Enterobacteriales includes the genus Salmonella, most of which are pathogens, and Escherichia, which also includes pathogenic strains (Sundberg et al., 2011). Within the Alphaproteobacteria, most of the proteins had the best match to members of three groups: order Rhizobiales (22), Rhodobacterales (7) and Rhodospirillales (6). The Rhizobium is the nitrogen-fixing bacteria; they take nitrogen from the air and convert it into ammonia, a form of nitrogen that plants can use. Within Rhodobacterales, some are denitrifying bacteria such as Paracoccus denitrificans (Baumann et al., 1996; Siddavattam et al., 2011). The order Rhodospirillales includes nitrogen-fixing bacteria such as Rhodobacter sphaeroides (Kontur et al., 2012). In the groups related to Betaproteobacteria, most of the matches had highest identity to proteins from the order Burkholderiales (17), Methylophilales (5), Neisseriales (4) and Nitrosomonadales (3). Cupriavidus and Bordetella were the most dominant genuses among the order Burkholderiales. Cupriavidus is adapted to several form of heavy metal stress (Nies, 1999; 2000). Bordetella is best known for species that are opportunistic human pathogens, but is also identified as a soil bacterium (Eriksson et al., 2003). The order Nitrosomonadales was found as nitrifying bacteria (Szántó, 2009). Pathogens such as Neiseria were also found among the Betaproteobacteria.
Table 2.
Most abundant bacterial order/suborder for the predominant phyla
| Taxonomy | Protein number |
|---|---|
| Proteobacteria | |
| Gammaproteobacteria | 181 |
| Pseudomonadales | 61 |
| Enterobacteriales | 50 |
| Alphaproteobacteria | 41 |
| Rhizobiales | 22 |
| Rhodobacterales | 7 |
| Rhodospirillales | 6 |
| Betaproteobacteria | 31 |
| Burkholderiales | 17 |
| Methylophilales | 5 |
| Neisseriales | 4 |
| Nitrosomonadales | 3 |
| Deltaproteobacteria | 15 |
| Firmicutes | |
| Bacilli | 65 |
| Bacillales | 45 |
| Lactobacillales | 20 |
| Clostridia | 19 |
| Actinobacteria | 37 |
| Corynebacterineae | 19 |
| Streptosporangineae | 5 |
| Frankineae | 4 |
| Tenericutes | 13 |
| Cyanobacteria | 12 |
| Bacteroidetes | 8 |
| Spirochaetes | 7 |
| Thermotogae | 5 |
| Aquificae | 5 |
The bacteria found in the metaproteome, such as members of the Actinobacteria, Bacilli, Alphaproteobacteria, Betaproteobacteria and Gammaproteobacteria, were typical for municipal solid waste and lignocellulosic materials (bagasse, coffee and rice) compost. Our data are consistent with the phylogenetic diversity obtained by previous gene-based analyses of facility composting, which identified the relative abundance of Actinobacteria, Bacteroidetes, Firmicutes and Proteobacteria (Partanen et al., 2010; Sundberg et al., 2011; De Gannes et al., 2013b). Exception included Bacteroidetes, which were minor community constituents in our study.
Fungal diversity represented in the composting metaproteome
Matches to the fungi were mainly to Saccharomycetes (114), followed by Schizosaccharomycetes (25), Sordariomycetes (16), Eurotiomycetes (16) and Basidiomycota (9) (Table 3). Within the Saccharomycetes, nearly half (51) of the matches had highest identity to proteins from the genus Saccharomyces; other proteins had the best match to members of genus Candida (14), Eremothecium (14) and Kluyveromyces (11). Some of the yeasts genuses are pathogens, such as Candida, which are opportunistic human pathogens (Bonito et al., 2010), and Eremothecium (also known as Ashbya), which are plant pathogens (Ashby and Nowell, 1926). Within the Sordariomycetes, most of the proteins had the best match to members of genus Neurospora (8) and Chaetomium (3), which relate to the degradation of cellulose (Umikalsom et al., 1997; Phillips et al., 2011; Sygmund et al., 2012). Matches to the Eurotiomycetes were mainly to Aspergillus (13), which includes pathogenic strains (Dehghani et al., 2012). Basidiomycetes are known to produce powerful degradation enzymes (Bonito et al., 2010). In our study, Phanerochaete chrysosporium, which produced cellobiose dehydrogenase, was detected in earlier phase (Table 4).
Table 3.
Most abundant fungal genera for the predominant phyla
| Taxonomy | Protein number |
|---|---|
| Ascomycota | |
| Saccharomycetes | 114 |
| Saccharomyces | 51 |
| Candida | 14 |
| Eremothecium | 14 |
| Kluyveromyces | 11 |
| Yarrowia | 6 |
| Pichia | 4 |
| Schizosaccharomycetes | 25 |
| Schizosaccharomyces | 25 |
| Sordariomycetes | 16 |
| Neurospora | 8 |
| Chaetomium | 3 |
| Eurotiomycetes | 16 |
| Aspergillus | 13 |
| Basidiomycota | 9 |
| Microsporidia | 8 |
Table 4.
Proteins belonged to carbohydrate metabolic pathway identified in the composting metaproteome
| Protein name | Accession | Functional group | Species | Taxonomy |
|---|---|---|---|---|
| LS1 | ||||
| Glyceraldehyde-3-phosphate dehydrogenase | P0A1P0 | Glycolysis / Gluconeogenesis | Salmonella enterica | Gammaproteobacteria |
| Enolase | A8AY46 | Glycolysis / Gluconeogenesis | Streptococcus gordonii | Bacilli; Lactobacillales |
| Glyceraldehyde-3-phosphate dehydrogenase 2 | Q6FSM4 | Glycolysis / Gluconeogenesis | Candida glabrata | Saccharomycetes |
| Glyceraldehyde-3-phosphate dehydrogenase | Q00584 | Glycolysis / Gluconeogenesis | Claviceps purpurea | Sordariomycetes |
| Acetyl-coenzyme A synthetase | Q3IFM6 | Pyruvate metabolism | Pseudoalteromonas haloplanktis | Gammaproteobacteria |
| Acetate kinase | A4VSL6 | Pyruvate metabolism | Streptococcus suis | Bacilli; Lactobacillales |
| Aconitate hydratase 2 | Q9I2V5 | TCA | Pseudomonas aeruginosa | Gammaproteobacteria |
| Aconitate hydratase 1 | Q9I3F5 | TCA | Pseudomonas aeruginosa | Gammaproteobacteria |
| Isocitrate lyase | Q9I0K4 | TCA | Pseudomonas aeruginosa | Gammaproteobacteria |
| Malate synthase G | Q3K5N4 | TCA | Pseudomonas fluorescens | Gammaproteobacteria |
| Malate dehydrogenase | Q4FQU7 | TCA | Psychrobacter arcticus | Gammaproteobacteria |
| Isocitrate dehydrogenase [NADP] 2 | P41561 | TCA | Colwellia maris | Gammaproteobacteria |
| Malate dehydrogenase | A5UCQ1 | TCA | Haemophilus influenzae | Gammaproteobacteria |
| Citrate synthase | O33915 | TCA | Sinorhizobium meliloti | Alphaproteobacteria |
| Malate synthase G | Q2J0A5 | TCA | Rhodopseudomonas palustris | Alphaproteobacteria |
| Malate synthase G | C0RES0 | TCA | Brucella melitensis | Alphaproteobacteria |
| Malate dehydrogenase | Q2GCH6 | TCA | Neorickettsia sennetsu | Alphaproteobacteria |
| Malate synthase G | A1UGU7 | TCA | Mycobacterium sp. | Actinobacteria |
| Succinate dehydrogenase [ubiquinone] flavoprotein subunit, mitochondrial | Q00711 | TCA | Saccharomyces cerevisiae | Saccharomycetes |
| Citrate synthase, mitochondrial | P00890 | TCA | Saccharomyces cerevisiae | Saccharomycetes |
| Aconitate hydratase, mitochondrial | P19414 | TCA | Saccharomyces cerevisiae | Saccharomycetes |
| Malate dehydrogenase, cytoplasmic | P83778 | TCA | Candida albicans | Saccharomycetes |
| Putative exoglucanase type C | P46238 | Cellulose degradation | Fusarium oxysporum | Sordariomycetes |
| Exoglucanase 1 | P38676 | Cellulose degradation | Neurospora crassa | Sordariomycetes |
| Cellobiose dehydrogenase | Q01738 | Cellulose degradation | Phanerochaete chrysosporium | Basidiomycota |
| Mannose-6-phosphate isomerase | Q870Y1 | Amino sugar and nucleotide sugar metabolism | Neurospora crassa | Sordariomycetes |
| Endo-1,3(4)-beta-glucanase 1 | P53753 | Cell wall degradation | Saccharomyces cerevisiae | Saccharomycetes |
| LS2 | ||||
| Glyceraldehyde-3-phosphate dehydrogenase | P0A1P0 | Glycolysis / Gluconeogenesis | Salmonella enterica | Gammaproteobacteria |
| Glyceraldehyde-3-phosphate dehydrogenase | Q00584 | Glycolysis / Gluconeogenesis | Claviceps purpurea | Sordariomycetes |
| Phosphoenolpyruvate carboxykinase [GTP] | A8L175 | Glycolysis / Gluconeogenesis | Frankia sp. | Actinobacteria |
| Fructose-1,6-bisphosphatase class 1 | C0QTP7 | Glycolysis / Gluconeogenesis | Persephonella marina | Aquificae |
| Glyoxylate/hydroxypyruvate reductase B | Q0W9V5 | Pyruvate metabolism | Yersinia pestis | Gammaproteobacteria |
| Acetyl-coenzyme A synthetase | Q9F7R5 | Pyruvate metabolism | uncultured marine gamma proteobacterium | Gammaproteobacteria |
| Acetyl-coenzyme A synthetase 1 | Q9Z3R3 | Pyruvate metabolism | Sinorhizobium meliloti | Alphaproteobacteria |
| Pyruvate, phosphate dikinase | Q59754 | Pyruvate metabolism | Sinorhizobium meliloti | Alphaproteobacteria |
| Malate dehydrogenase | A5WGM2 | TCA | Psychrobacter sp. | Gammaproteobacteria |
| Citrate lyase subunit beta | P17725 | TCA | Klebsiella pneumoniae | Gammaproteobacteria |
| Probable malate : quinone oxidoreductase | B3QK65 | TCA | Rhodopseudomonas palustris | Alphaproteobacteria |
| Succinate dehydrogenase flavoprotein subunit | Q59661 | TCA | Paracoccus denitrificans | Alphaproteobacteria |
| Succinate dehydrogenase iron-sulfur subunit | Q1RGP3 | TCA | Rickettsia bellii | Alphaproteobacteria |
| Succinate dehydrogenase flavoprotein subunit | P08065 | TCA | Bacillus subtilis | Bacilli; Bacillales |
| Malate dehydrogenase | C1DB66 | TCA | Laribacter hongkongensis | Betaproteobacteria |
| Endoglucanase | P10475 | Cellulose degradation | Bacillus subtilis | Bacilli; Bacillales |
| Endoglucanase E-2 | P26222 | Cellulose degradation | Thermobifida fusca | Actinobacteria |
| Endo-1,4-beta-xylanase B | P26515 | Hemicellulose degradation | Streptomyces lividans | Actinobacteria |
| 1,4-alpha-glucan branching enzyme GlgB | B0TZI5 | Starch and sucrose metabolism | Francisella philomiragia | Gammaproteobacteria |
| Phosphoglucosamine mutase | Q5NNT4 | Amino sugar and nucleotide sugar metabolism | Zymomonas mobilis | Alphaproteobacteria |
| Phosphoglucosamine mutase | A8AWM5 | Amino sugar and nucleotide sugar metabolism | Streptococcus gordonii | Bacilli; Lactobacillales |
| Beta-N-acetylhexosaminidase | P49610 | Amino sugar and nucleotide sugar metabolism | Streptococcus pneumoniae | Bacilli; Lactobacillales |
| 2-keto-3-deoxy-L-rhamnonate aldolase | Q0TFJ7 | Fructose and mannose metabolism | Escherichia coli | Gammaproteobacteria |
| Xylose isomerase | P19148 | Fructose and mannose metabolism | Thermoanaerobacterium thermosulfurigenes | Clostridia |
| LS3 | ||||
| 6-phosphofructokinase | A8GLB0 | Glycolysis / Gluconeogenesis | Serratia proteamaculans | Gammaproteobacteria |
| Glyceraldehyde-3-phosphate dehydrogenase | P24750 | Glycolysis / Gluconeogenesis | Escherichia hermannii | Gammaproteobacteria |
| Glyceraldehyde-3-phosphate dehydrogenase | O32755 | Glycolysis / Gluconeogenesis | Lactobacillus delbrueckii | Bacilli; Lactobacillales |
| Hexokinase-2 | P50521 | Glycolysis / Gluconeogenesis | Schizosaccharomyces pombe | Schizosaccharomycetes |
| Triosephosphate isomerase | Q9HGY8 | Glycolysis / Gluconeogenesis | Aspergillus oryzae | Eurotiomycetes |
| Glyceraldehyde-3-phosphate dehydrogenase | Q00584 | Glycolysis / Gluconeogenesis | Claviceps purpurea | Sordariomycetes |
| Phosphoenolpyruvate carboxykinase [GTP] | A8L175 | Glycolysis / Gluconeogenesis | Frankia sp. | Actinobacteria |
| Aryl-phospho-beta-D-glucosidase BglC | P42403 | Glycolysis / Gluconeogenesis | Bacillus subtilis | Bacilli; Bacillales |
| Pyruvate dehydrogenase E1 component | Q59637 | Pyruvate metabolism | Pseudomonas aeruginosa | Gammaproteobacteria |
| Glyoxylate/hydroxypyruvate reductase B | Q0W9V5 | Pyruvate metabolism | Yersinia pestis | Gammaproteobacteria |
| Acetyl-coenzyme A synthetase | Q9F7R5 | Pyruvate metabolism | uncultured marine Gammaproteobacterium | Gammaproteobacteria |
| Acetyl-coenzyme A synthetase 1 | Q9Z3R3 | Pyruvate metabolism | Sinorhizobium meliloti | Alphaproteobacteria |
| Malate dehydrogenase | A5WGM2 | TCA | Psychrobacter sp. | Gammaproteobacteria |
| Citrate lyase subunit beta | P17725 | TCA | Klebsiella pneumoniae | Gammaproteobacteria |
| Succinate dehydrogenase flavoprotein subunit | Q59661 | TCA | Paracoccus denitrificans | Alphaproteobacteria |
| Malate dehydrogenase | B2JQD2 | TCA | Burkholderia phymatum | Betaproteobacteria |
| Succinate dehydrogenase flavoprotein subunit | P08065 | TCA | Bacillus subtilis | Bacilli; Bacillales |
| Malate synthase G | A4IN50 | TCA | Geobacillus thermodenitrificans | Bacilli; Bacillales |
| Endoglucanase | P10475 | Cellulose degradation | Bacillus subtilis | Bacilli; Bacillales |
| Endoglucanase E-2 | P26222 | Cellulose degradation | Thermobifida fusca | Actinobacteria |
| Probable beta-glucosidase I | A2R989 | Cellulose degradation | Aspergillus niger | Eurotiomycetes |
| Endo-1,4-beta-xylanase B | P26515 | Hemicellulose degradation | Streptomyces lividans | Actinobacteria |
| 1,4-alpha-glucan branching enzyme GlgB | A8GKV0 | Starch and sucrose metabolism | Serratia proteamaculans | Gammaproteobacteria |
| Phosphoglucosamine mutase | Q5NNT4 | Amino sugar and nucleotide sugar metabolism | Zymomonas mobilis | Alphaproteobacteria |
| 2-keto-3-deoxy-L-rhamnonate aldolase | Q31Z78 | Fructose and mannose metabolism | Shigella boydii | Gammaproteobacteria |
| Sensor protein ChvG | P72292 | Cell wall degradation | Sinorhizobium meliloti | Alphaproteobacteria |
Compared with bacteria, the diversity of fungi was lower, but also generally the species richness of fungi is not as high as that of bacteria. This finding was congruent with those of other investigators (Hultman et al., 2010; De Gannes et al., 2013a).
Microbial community differs between sampling times
The protein assignments illustrated that specific taxa of bacteria and fungi had different temporal abundances (Fig. 1). The Bacilli, Gammaproteobacteria and Alphaproteobacteria were further divided into the orders in order to study the phylogenetic differences between the three samples (Fig. 1A). The largest increase in the proportion of plant spectra was observed in Bacillales (4.8 to 14.1%) in the active phase, followed by Actinomycetales (6.2 to 11.3%). The proportion of Pseudomonadales sharply decreased in the active phase from 29.7% to 4.3%. As the composts transitioned into the curing phase, the proportion of order Rhizobiales decreased from 6.2% to 2.1%.
Figure 1.
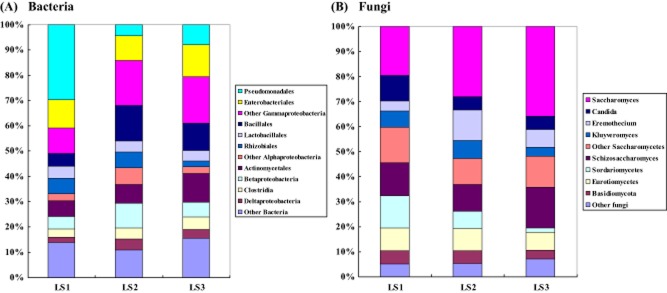
Phylogenetic assignment of (A) bacterial and (B) fungal proteins detected in the three samples. Relative abundances were calculated from the sum of SpCns found for each group at the respective sampling sites. (A) The sum is 717 565 and 443 respectively; (B) the sum is 315 225 and 242 respectively.
Bacteria may be present throughout the composting process as active or dormant cells, or as spores. Only their numbers and level of activity change during the composting process (Gentleman et al., 2004). Microorganisms belonging to the Bacillales and Actinobacteria, known to be critical for an efficient composting process, were sufficiently abundant in the waste to inoculate the subsequent composting process. However, the numbers were found to be lower than those of the Lactobacillales in most large-scale reactors, because the oxygen supply may be restricted (Partanen et al., 2010; Watanabe et al., 2010). To improve this situation, the active phase of the composting process should be steered towards conditions that favour thermotolerant microbes such as Bacillales and Actinobacteria. In this study, the main adjustment was turning the compost pile mechanically according to a schedule. The Bacillales and Actinobacteria groups became dominant as composting progressed, and there was no increase in Lactobacillales, suggesting that improving the internal aeration of the composting mass by mechanized turning was an efficient way to improve the performance of the composting plant.
The sharp decrease of the order Pseudomonadales could be explained by the effects of high temperature on Gram-negative bacteria (Dees and Ghiorse, 2001). On the other hand, as previously reported, the type of indigenous microorganisms such as actinomycetes and fungi is critical for the suppression of pathogens growth in compost (Kim et al., 2011). This might be another reason why the order Pseudomonadales decreased.
Since proteins representing the Saccharomycetes were by far the largest group, this class was further divided into genuses in order to study the phylogenetic differences (Fig. 1B). The largest increase in the proportion was observed in Saccharomyces (19.5% to 35.7%), followed by Schizosaccharomyces (13.0% to 16.1%). The smallest decrease was observed in Kluyveromyces (6.5% to 3.6%), followed by Candida (10.4% to 5.4%) and Sordariomycetes (13.0% to 1.8%). The proportion of Eremothecium rose to 12.3% in the active phase and decreased to 7.1% in the curing phase.
A high proportion of ascomycetous yeasts, included order Saccharomycetes and Schizosaccharomycetes, were detected at all examined phases. Yeasts being able to grow at low pH may promote a reduction in the acidity and an increase in the growth of thermophilic bacteria (Choi and Park, 1998). Previous studies have reported on the presence of yeasts during the early phases of composting (Bonito et al., 2010; Hultman et al., 2010). Although plant and human pathogens were found at the start of active phase, few pathogenic species were recovered from samples representing the curing phase of composting suggesting that the composting process is effective in the removal of fungal pathogens.
Carbohydrate metabolism in composting process
The most prevalent bacterial and fungal proteins in the metaproteome were components of carbohydrate metabolic enzymes (77 proteins; 12.0% of the total metaproteome), translation proteins (74; 11.6%), ribosomal subunits (60; 9.4%) and transport proteins (42; 6.6%). Deoxyribonucleic acid replication and repair proteins (41), amino acid metabolic enzymes (36), protein chaperones (36), transcription proteins (36) and outer membrane proteins (32) were also detected in the metaproteome (Supplementary Table S1 and S2).
Composting can be defined as an aerobic process of decomposition of organic matter. The active phase of composting involves the degradation of easily degradable compounds such as carbohydrates, amino acids, proteins and lipids. In the current metaproteome, the carbohydrate metabolic enzymes comprised a higher proportion, suggesting that the carbohydrate metabolism was the principal metabolic pathway in composting process. These enzymes were involved in different carbohydrate metabolic pathways (Fig. 2). The largest amounts (29 of the 77 total) of carbohydrate metabolic enzymes were involved in the citrate cycle (TCA cycle, Krebs cycle), including malate dehydrogenase (9), succinate dehydrogenase (6) and malate synthase (5), which were much more abundant than the enzymes were essential for glycolysis/gluconeogenesis (16), pyruvate metabolism (10), cellulose degradation (8) and amino sugar and nucleotide sugar metabolism (5) (Table 4).
Figure 2.
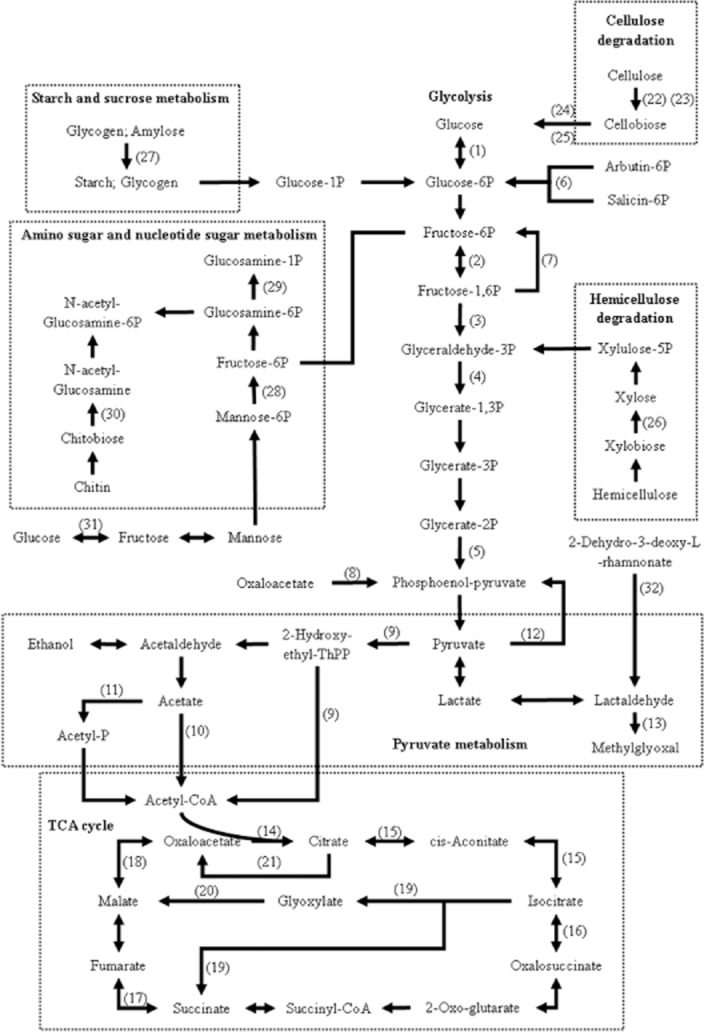
Depiction of the carbohydrate metabolic characteristics of microbial communities inferred from the metaproteome. Proteins shown are: (1) Hexokinase; (2) 6-phosphofructokinase; (3) Triosephosphate isomerase; (4) Glyceraldehyde-3-phosphate dehydrogenase; (5) Enolase; (6) Aryl-phospho-beta-D-glucosidase; (7) Fructose-1,6-bisphosphatase; (8) Phosphoenolpyruvate carboxykinase; (9) Pyruvate dehydrogenase; (10) Acetyl-coenzyme A synthetase; (11) Acetate kinase; (12) Pyruvate, phosphate dikinase; (13) Glyoxylate/hydroxypyruvate reductase; (14) Citrate synthase; (15) Aconitate hydratase; (16) Isocitrate dehydrogenase; (17) Succinate dehydrogenase; (18) Malate dehydrogenase; (19) Isocitrate lyase; (20) Malate synthase; (21) Citrate lyase; (22) Exoglucanase; (23) Endoglucanase; (24) Cellobiose dehydrogenase; (25) Beta-glucosidase; (26) Endo-1,4-beta-xylanase B; (27)1,4-alpha-glucan branching enzyme GlgB; (28) Mannose-6-phosphate isomerase; (29) Phosphoglucosamine mutase; (30) Beta-N-acetylhexosaminidase; (31) 2-keto-3-deoxy-L-rhamnonate aldolase; (32) Xylose isomerase.
The identified enzymes related to carbohydrate metabolism were mainly affiliated to Gammaproteobacteria (25), Alphaproteobacteria (16), Bacilli (11), Actinobacteria (7), Saccharomycetes (6) and Sordariomycetes (6) members (Table 4). The proportion of Saccharomyces proteins increased significantly in composting process, but no proteins involved in carbohydrate metabolism were detected except the earlier phase.
Key cellulolytic community in different composting stages is different
Ten cellulase and hemicellulase were identified in composting samples (Table 4). All three cellulase in the sample LS1 matched to fungi, included Fusarium oxysporum, Neurospora crassa and Phanerochaete chrysosporium, whereas all cellulase and hemicellulase in the sample LS2 matched to bacteria, such as Bacillus subtilis and Thermobifida fusca.
The most common complex carbohydrate available in the composting substrates is cellulose. A key function of bacteria and fungi during composting is to produce cellulolytic enzymes (Hubbe et al., 2010). In the earlier phase, cellulolytic fungal communities specifically targeted the breakdown of the cellulose. However, the cellulolytic fungal communities were replaced by a purely bacterial one in the active phase. As the composts transitioned into the curing phase, cellulolytic fungal communities were recovered and the mixed community of bacteria and fungi enhance the decomposition of cellulose. Previously, it has been shown that thermophilic fungi are active through the thermophilic phase (Tchobanoglous et al., 1993). Our results revealed that the cellulolytic fungal communities played a vital role in earlier phase, after which they gradually disappeared and cellulolytic bacterial communities gradually played a leading role. This suggested that the key microbiologic population for cellulose degradation in different composting stages was different.
Cellulose is a polymer of glucose, which is digested by a variety of enzymes. Cellulose itself requires three types of enzymes for its decomposition: Endoglucanases hydrolyse accessible intramolecular β-1,4-glucosidic bonds of cellulose chains randomly to produce new chain ends; exoglucanases or cellobiohydrolases processively cleave cellulose chains at the ends to release soluble cellobiose or glucose; and β-glucosidases hydrolyse cellobiose to glucose in order to eliminate cellobiose inhibition (Percival Zhang et al., 2006). In the current study, all three types of cellulases were detected in the metaproteome, suggesting the effective breakdown of cellulose in the composting process. Interestingly, exoglucanases and β-glucosidases were only produced by fungi, whereas endoglucanases were only produced by bacteria. Beta-glucosidase is a limited enzyme in cellulose decomposition, and it was only found in the curing phase. This indicated that the curing phase may be the key phase, in which mixed communities of bacteria and fungi worked together to digested cellulose.
Nitrogen flows in composting process
In composting processes, the nitrogen flows can be best described through ammonia (NH3) dynamics, which encompasses the production, volatilization, conversion and assimilation of ammonia (Szántó, 2009). Proteins that could be assigned to a function in the production of ammonia were identified in the metaproteome, including proteases and urease (Table 5). This suggested that the protein and urea were the source of ammonia in the municipal solid waste.
Table 5.
Proteins belonged to nitrogen metabolism identified in the composting metaproteome
| Protein name | Accession | Functional group | Species | Taxonomy |
|---|---|---|---|---|
| Lon protease 2 | P36774 | Protein degradation | Myxococcus xanthus | Deltaproteobacteria |
| Lon protease homolog 2, peroxisomal | Q6CWS4 | Protein degradation | Kluyveromyces lactis | Saccharomycetes |
| Probable M18 family aminopeptidase 2 | A4VJG1 | Protein degradation | Pseudomonas stutzeri | Gammaproteobacteria |
| Probable cytosol aminopeptidase | Q0SHL0 | Protein degradation | Rhodococcus jostii | Actinobacteria |
| ATP-dependent zinc metalloprotease FtsH | Q03Z46 | Protein degradation | Leuconostoc mesenteroides | Bacilli; Lactobacillales |
| ATP-dependent Clp protease ATP-binding subunit ClpX | B2JGL6 | Protein degradation | Burkholderia phymatum | Betaproteobacteria |
| ATP-dependent protease ATPase subunit HslU | Q73NE3 | Protein degradation | Treponema denticola | Spirochaetes |
| Urease subunit alpha | B1JR71 | Urea degradation | Yersinia pseudotuberculosis | Gammaproteobacteria |
| [Protein-PII] uridylyltransferase | Q3J5H6 | Nitrogen fixation | Rhodobacter sphaeroides | Alphaproteobacteria |
| Nitrous-oxide reductase | P19573 | Denitrification | Pseudomonas stutzeri | Gammaproteobacteria |
Nitrogen can be lost as NO3-, NO2-, N2 or N2O through the process nitrification–denitrification (Szántó, 2009). Only one denitrification enzyme (nitrous-oxide reductase) produced by Pseudomonas stutzeri was detected in the metaproteome (Table 5). This indicated that denitrification enzymes production of other denitrifying bacteria was not obvious, and the Pseudomonas might be the key player in denitrification pathway in composting process.
The high abundance of translation proteins (11.6%) and amino acid metabolic enzymes (5.6%) could be attributed to nitrogen assimilation in composting process. Microbial biomass such as bacteria, viruses and fungi require nitrogen for their cell matter, which they gain from ammonium through assimilation into the cell tissue (Szántó, 2009). Assimilation is the immobilization of the ammoniacal nitrogen by the consecutive amino acid and protein synthesis. Another nitrogen assimilation route is the process of nitrogen fixation, in which N2 gas is converted into the biologically useful forms. In the current study, nitrogen fixation enzyme was only produced by Rhodobacter sphaeroides (Table 5).
Conclusions
Our metaproteomic analysis provided insight into microbial succession and in the activity of certain phylogenetic groups in a large-scale composting plant. The investigation of microbial succession revealed that Bacillales, Actinobacteria and Saccharomyces increased significantly with respect to abundance in composting process. The Gammaproteobacteria were the single largest group accounting for 40.3% of the total bacterial proteins.
The carbohydrate metabolism was the principal metabolic pathway in composting process. Cellulose and hemicellulose were the main carbon sources. Three types of cellulase essential for cellulose degradation were detected. Fungi were found to be the main producers of cellulase in earlier phase. Only bacterial, but no fungal, cellulolytic enzymes were detected at the end of the active phase, a finding that strongly supported our conclusion that the key microbiologic population for cellulose degradation in different composting stages was different, which did not support the concept that the thermophilic fungi are active through the thermophilic phase. In the curing phase, mixed community of bacteria and fungi enhance decomposition of cellulose, suggesting that the curing phase may be a rate-limiting phase in the cellulose decomposition process.
Though some denitrifying bacteria such Azotobacter vinelandii, Azotobacter chroococcum and Paracoccus denitrificans were detected in the samples, only Pseudomonas stutzeri produced denitrification enzyme. This indicated that denitrification enzymes production of other denitrifying bacteria was not obvious, and the Pseudomonas might be the key player in denitrification pathway in composting process.
We are fully aware that the metaproteomics-based approach suffers from certain limitations such as the protein extraction efficiency and data analysis. For complex environmental samples, it has been estimated that « 1% of the total metaproteome can be resolved using current method (Wilmes and Bond, 2006; Leary et al., 2013). Database selection has a significant impact in metaproteomics, and provides critical indications for improving depth and reliability of metaproteomic results (Tanca et al., 2013). Nevertheless, we believe that the metaproteome analysis provides a deep insight into molecular details of the composting process. Thus, further proteome analyses will help to elucidate the main metabolic pathways occurring at the microbial level in different composting systems.
Experimental procedures
Sampling
Samples were collected from Asuwei Composting Plant in Beijing, China. More details of this composting plant and how composting was carried out has been described elsewhere (He et al., 2011). The entire composting process was divided into the active and curing phases. The active phase was carried out for 20 days during which the pile was turned every 2 days by forklift, and the average temperature was maintained at around 60°C. The curing phase took 30 days to complete, and the pile was turned mechanically every 7 days. The temperature began to decrease in 20 days and reached a constant level with ambient temperature after 50 days. Triplicate samples were collected at different points from the top to the bottom of the composting plies after 5, 20 and 50 days. At different sampling-times, triplicate samples were mixed, and the composite sample was divided into two parts. Metaproteome analyses were performed in duplicate.
Physicochemical analyses
The pH was measured with a pH meter (SevenEasy S20, Mettler Toledo, Shanghai, China) and a standard electrode by mixing the samples with deionized water at a weight/volume ratio of 1:10 (Laos et al., 2002). Water content was determined gravimetrically by drying the sample at 105°C for 6 h. Total carbon was evaluated by dry combustion at 550°C (Navarro et al., 1990), and total nitrogen by the Kjeldahl digestion analysis. Information on the sampling and the physical-chemical properties at the sampling dates is illustrated in Table 1.
Metaproteomics
Protein extraction from 5 g fresh composting samples followed the method of Keiblinger and colleagues (2012) using 20 ml extraction buffer [1: 1 (v: v) SDS–phenol buffer: 50 mM Tris, 1% SDS pH 7.5 + phenol (pH 8.0)].
Extracted proteins were separated by one-dimensional SDS-PAGE. Before electrophoresis, the protein pellet was dissolved in appropriate volumes sample buffer for SDS-PAGE and incubated 20 min at 100°C. After centrifugation, the supernatant (5 μl) was loaded on SDS gel (5% stacking gel, 10% separating gel) using a Mini-PROTEAN Tetra system (Bio-Rad, Beijing, China). After electrophoresis, gel was stained with Coomassie brilliant blue R250.
For identification of proteins from composting samples, each lane of the SDS-PAGE was cut into seven slices. The gel slices were further cut into 1 mm3 gel pieces and subjected to immediate in-gel tryptic digestion (Promega, Madison, WI, USA). Digested peptides were separated by nano-LC (Ultimate 3000, Dionex, Sunnyvale, CA, USA; column: Venusil XBP-C18, 3.0 μm, 150 × 0.075 mm, Agela Technologies, Wilmington, DE, USA; eluent: 0.1% formic acid, 0% to 60% acetonitrile) and analysed by MS/MS (Q Exactive, Thermo Scientific, Pittsburgh PA, USA) as described earlier (Williams et al., 2012).
Database searches were carried out with MS/MS ion search (MASCOT, http://www.matrixscience.com) against a non-redundant protein database, SwissProt2013 xyzzy (539 616 sequences; 191 569 459 residues). The following search parameters were applied: (i) trypsin was chosen as protein-digesting enzyme and one missed cleavages were tolerated, (ii) carbamidomethyl-cysteine was chosen as fixed modification and (iii) Gln- > pyro-Glu (N-term Q) and oxidation (M) were chosen as variable modification. Searches were performed with a peptide mass tolerance of ± 15 ppm and a fragment mass tolerance of ± 20 mmu. Mascot searches with a false discovery rate > 5% were rejected. Protein matches were only accepted if they were identified by a minimum of one unique peptide. All proteins were manually annotated with the aid of blastp, and the protein hit that showed the highest sequence identity was recorded, including the organism name (Supplementary Table S1 and S2). Higher protein abundance is represented by a higher number of MS/MS spectra acquired from peptides of the respective protein. Thus, protein abundances were calculated based on the normalized spectral counts (SpCn; Piersma et al., 2010). An important consideration with spectrum counting and similar approaches is the fact that small proteins tend to have fewer peptides identified per protein compared with large proteins (Florens et al., 2006). All phylogenetic group abundances presented in the metaproteome are based on SpCns.
Conflict of interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Table S1. Detailed identifications of bacterial proteins extracted from samples.
Table S2. Detailed identifications of fungal proteins extracted from samples.
References
- Ashby SF. Nowell W. The fungi of stigmatomycosis. Ann Botany. 1926;7:69–84. [Google Scholar]
- Baharuddin AS, AbdRazak MN, Lim SH, Ahmad MN, Abd-Aziz S, AbdulRahman NA, et al. Isolation and characterization of thermophilic cellulase-producing bacteria from empty fruit bunches-palm oil mill effluent compost. Am J Appl Sci. 2010;7:56–62. [Google Scholar]
- Baumann B, Snozzi M, Zehnder AJ. Van Der Meer JR. Dynamics of denitrification activity of Paracoccus denitrificans in continuous culture during aerobic-anaerobic changes. J Bacteriol. 1996;178:4367–4374. doi: 10.1128/jb.178.15.4367-4374.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benndorf D, Balcke UG, Harms H. von Bergen M. Functional metaproteome analysis of protein extracts from contaminated soil and groundwater. ISME J. 2007;1:224–234. doi: 10.1038/ismej.2007.39. [DOI] [PubMed] [Google Scholar]
- Bonito G, Isikhuemhen SO. Vilgalys R. Identification of fungi associated with municipal compost using DNA-based techniques. Bioresour Technol. 2010;101:1021–1027. doi: 10.1016/j.biortech.2009.08.109. [DOI] [PubMed] [Google Scholar]
- Charbonneau MD, Meddeb-Mouelhi F, Boissinot M, Sirois M. Beauregard M. Identification of thermophilic bacterial strains producing thermotolerant hydrolytic enzymes from manure compost. Indian J Microbiol. 2012;52:41–47. doi: 10.1007/s12088-011-0156-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi MH. Park YH. The influence of yeast on thermophilic composting of food waste. Lett Appl Microbio. 1998;26:175–178. doi: 10.1046/j.1472-765x.1998.00307.x. [DOI] [PubMed] [Google Scholar]
- Chourey K, Jansson J, VerBerkmoes N, Shah M, Chavarria KL, Tom LM, et al. Direct cellular lysis/protein extraction protocol for soil metaproteomics. J Proteome Res. 2010;9:6615–6622. doi: 10.1021/pr100787q. [DOI] [PubMed] [Google Scholar]
- De Gannes V, Eudoxie G. Hickey WJ. Insights into fungal communities in composts revealed by 454-pyrosequencing: implications for human health and safety. Front Microbiol. 2013a;4:164. doi: 10.3389/fmicb.2013.00164. , and. doi: 10.3389/fmicb.2013.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gannes V, Eudoxie G. Hickey WJ. Prokaryotic successions and diversity in composts as revealed by 454-pyrosequencing. Bioresour Technol. 2013b;133:573–580. doi: 10.1016/j.biortech.2013.01.138. [DOI] [PubMed] [Google Scholar]
- Dees PM. Ghiorse WC. Microbial diversity in hot synthetic compost as revealed by PCR-amplified rRNA sequences from cultivated isolates and extracted DNA. FEMS Microbiol Ecol. 2001;35:207–216. doi: 10.1111/j.1574-6941.2001.tb00805.x. [DOI] [PubMed] [Google Scholar]
- Dehghani R, Asadi MA, Charkhloo E, Mostafaie G, Saffari M, Mousavi GA. Pourbabaei M. Identification of fungal communities in producing compost by windrow method. J Environ Prot. 2012;3:61–77. [Google Scholar]
- Eriksson M, Sodersten E, Yu Z, Dalhammar G. Mohn W. Degradation of polycyclic aromatic hydrocarbons at low temperature under aerobic and nitrate-reducing conditions in enrichment cultures from northern soils. Appl Environ Microbiol. 2003;69:275–284. doi: 10.1128/AEM.69.1.275-284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florens L, Carozza MJ, Swanson SK, Fournier M, Coleman MK, Workman JL. Washburn MP. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods. 2006;40:303–311. doi: 10.1016/j.ymeth.2006.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke-Whittle IH, Knapp BA, Fuchs J, Kaufmann R. Insam H. Application of COMPOCHIP microarray to investigate the bacterial communities of different composts. Microb Ecol. 2009;57:510–521. doi: 10.1007/s00248-008-9435-2. [DOI] [PubMed] [Google Scholar]
- Franke-Whittle IH, Confalonieri A, Insam H, Schlegelmilch M. Körner I. Changes in the microbial communities during co-composting of digestates. Waste Manag. 2014;34:632–641. doi: 10.1016/j.wasman.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guazzaroni ME, Herbst FA, Lores I, Tamames J, Peláez AI, López-Cortés N, et al. Metaproteogenomic insights beyond bacterial response to naphthalene exposure and bio-stimulation. ISME J. 2012;7:122–136. doi: 10.1038/ismej.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Xi B, Wei Z, Guo X, Li M, An D. Liu H. Spectroscopic characterization of water extractable organic matter during composting of municipal solid waste. Chemosphere. 2011;82:541–548. doi: 10.1016/j.chemosphere.2010.10.057. [DOI] [PubMed] [Google Scholar]
- Hubbe MA, Nazhad M. Sánchez C. Composting as a way to convert cellulosic biomass and organic waste into high-value soil amendments: a review. Bioresources. 2010;5:2808–2854. [Google Scholar]
- Hultman J, Vasara T, Partanen P, Kurola J, Kontro MH, Paulin L, et al. Determination of fungal succession during municipal solid waste composting using a cloning-based analysis. J Appl Microbio. 2010;108:472–487. doi: 10.1111/j.1365-2672.2009.04439.x. [DOI] [PubMed] [Google Scholar]
- Joséphine P. Philippe G. Environmental impacts of farm-scale composting practices. Water Air Soil Poll. 2004;153:45–68. [Google Scholar]
- Keiblinger KM, Wilhartitz IC, Schneider T, Roschitzki B, Schmid E, Eberl L, et al. Soil metaproteomics – comparative evaluation of protein extraction protocols. Soil Biol Biochem. 2012;54:14–24. doi: 10.1016/j.soilbio.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Miller CM, Shepherd MW, Liu X. Jiang X. Impact of indigenous microorganisms on Escherichia coli O157:H7 growth in cured compost. Bioresour Technolo. 2011;102:9619–9625. doi: 10.1016/j.biortech.2011.07.055. [DOI] [PubMed] [Google Scholar]
- Kontur WS, Schackwitz WS, Ivanova N, Martin J, Labutti K, Deshpande S, et al. Revised sequence and annotation of the Rhodobacter sphaeroides 2.4.1 genome. J Bacteriol. 2012;194:7016–7017. doi: 10.1128/JB.01214-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laos F, Mazzarino MJ, Walter I, Roselli L, Satti P. Moyano S. Composting of fish offal and biosolids in northwestern Patagonia. Bioresour Technol. 2002;81:179–186. doi: 10.1016/s0960-8524(01)00150-x. [DOI] [PubMed] [Google Scholar]
- Lauro FM, DeMaere MZ, Yau S, Brown MV, Ng C, Wilkins D, et al. An integrative study of a meromictic lake ecosystem in Antarctica. ISME J. 2011;5:879–895. doi: 10.1038/ismej.2010.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary DH, Hervey WJ, IV, Deschamps JR, Kusterbeck AW. Vora GJ. Which metaproteome? The impact of protein extraction bias on metaproteomic analyses. Mol Cell Probes. 2013;27:193–199. doi: 10.1016/j.mcp.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Navarro AF, Cegarra J, Roig A. Bernal P. 1990. , and Análisis de residuos urbanos, agrícolas, ganaderos y forestales: relación materia orgánicacarbono orgánico. III Cong Internac de Química de la ANQUE. Residuos sólidos y líquidos: su mejor destino. Fondo Editorial ANQUE, Madrid, pp. 447–456.
- Negro MJ, Solano ML, Ciria P. Carrasco J. Composting of sweet sorghum bagasse with other wastes. Bioresour Technol. 1999;67:89–92. [Google Scholar]
- Nies DH. Microbial heavy-metal resistance. Appl Microbiol Biotechnol. 1999;51:730–750. doi: 10.1007/s002530051457. [DOI] [PubMed] [Google Scholar]
- Nies DH. Heavy metal resistant bacteria as extremophiles: molecular physiology and biotechnological use of Ralstonia sp. CH34. Extremophiles. 2000;4:77–82. doi: 10.1007/s007920050140. [DOI] [PubMed] [Google Scholar]
- Partanen P, Hultman J, Paulin L, Auvinen P. Romantschuk M. Bacterial diversity at different stages of the composting process. BMC Microbiol. 2010;10:94. doi: 10.1186/1471-2180-10-94. , and. doi: 10.1186/1471-2180-10-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival Zhang YH, Himmel ME. Mielenz JR. Outlook for cellulase improvement: screening and selection strategies. Biotechnol Adv. 2006;24:452–481. doi: 10.1016/j.biotechadv.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Peters S, Koschinsky S, Schwieger F. Tebbe CC. Succession of microbial communities during hot composting as detected by PCR-single-strand-conformation polymorphism-based genetic profiles of small-subunit rRNA genes. Appl Environ Microbiol. 2000;66:930–936. doi: 10.1128/aem.66.3.930-936.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CM, Iavarone AT. Marletta MA. Quantitative proteomic approach for cellulose degradation by Neurospora crassa. J Proteome Res. 2011;10:4177–4185. doi: 10.1021/pr200329b. [DOI] [PubMed] [Google Scholar]
- Piersma SR, Fiedler U, Span S, Lingnau A, Pham TV, Hoffmann S, et al. Workflow comparison for label-free, quantitative secretome proteomics for cancer biomarker discovery: method evaluation, differential analysis, and verification in serum. J Proteome Res. 2010;9:1913–1922. doi: 10.1021/pr901072h. [DOI] [PubMed] [Google Scholar]
- Ryckeboer J, Mergaert J, Vaes K, Klammer S, De Clercq D, Coosemans J, et al. A survey of bacteria and fungi occurring during composting and self-heating processes. Ann Microbiol. 2003;53:349–410. [Google Scholar]
- Schneider T, Keiblinger KM, Schmid E, Sterflinger-Gleixner K, Ellersdorfer G, Roschitzki B, et al. Who is who in litter decomposition? Metaproteomics reveals major microbial players and their biogeochemical functions. ISME J. 2012;6:1749–1762. doi: 10.1038/ismej.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemekite F, Gómez-Brandón M, Franke-Whittle IH, Praehauser B, Insam H. Assefa F. Coffee husk composting: an investigation of the process using molecular and non-molecular tools. Waste Manag. 2014;34:642–652. doi: 10.1016/j.wasman.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddavattam D, Karegoudar TB, Mudde SK, Kumar N, Baddam R, Avasthi TS. Ahmed N. Genome of a novel isolate of Paracoccus denitrificans capable of degrading N,N-dimethylformamide. J Bacteriol. 2011;193:5598–5599. doi: 10.1128/JB.05667-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg C, Franke-Whittle IH, Kauppi S, Yu D, Romantschuk M, Insam H. Jönsson H. Characterisation of source-separated household waste intended for composting. Bioresour. 2011;102:2859–2867. doi: 10.1016/j.biortech.2010.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sygmund C, Kracher D, Scheiblbrandner S, Zahma K, Felice AK, Harreither W, et al. Characterization of the two Neurospora crassa cellobiose dehydrogenases and their connection to oxidative cellulose degradation. Appl Environ Microbiol. 2012;78:6161–6171. doi: 10.1128/AEM.01503-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szántó G. NH3. Wageningen, the Netherlands: Wageningen University; 2009. Dynamics in Composting – Assessment of the Integration of Composting in Manure Management Chains. [Google Scholar]
- Tanca A, Palomba A, Deligios M, Cubeddu T, Fraumene C, Biosa G, et al. Evaluating the impact of different sequence databases on metaproteome analysis: insights from a lab-assembled microbial mixture. PLoS ONE. 2013;8:e82981. doi: 10.1371/journal.pone.0082981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchobanoglous G, Theisen H. Vigil S. Integrated Solid Waste Management: Engineering Principles and Management Issues. New York, United States: McGraw Hill; 1993. [Google Scholar]
- Tian W, Sun Q, Xu DB, Zhang ZH, Chen D, Li CY, et al. Succession of bacterial communities during composting process as detected by 16S rRNA clone libraries analysis. Int Biodeter Biodegr. 2013;78:58–66. [Google Scholar]
- Umikalsom MS, Ariff AB, Shamsuddin ZH, Tong CC, Hassan MA. Karim MIA. Production of cellulose by a wild strain of Chaetomium globosum using delignified oil palm empty-fruit-bunch fibre as substrate. Appl Microbiol Biotechnol. 1997;47:590–595. [Google Scholar]
- Watanabe K, Nagao N, Toda T. Kurosawa N. Sustainable biotechnology: sources of renewable energy. London, England: Springer; 2010. [Google Scholar]
- Williams TJ, Long E, Evans F, DeMaere MZ, Lauro FM, Raftery MJ, et al. A metaproteomic assessment of winter and summer bacterioplankton from Antarctic Peninsula coastal surface waters. ISME J. 2012;6:1883–1900. doi: 10.1038/ismej.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmes P. Bond PL. The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ Microbiol. 2004;6:911–920. doi: 10.1111/j.1462-2920.2004.00687.x. [DOI] [PubMed] [Google Scholar]
- Wilmes P. Bond PL. Metaproteomics: studying functional gene expression in microbial ecosystems. Trends Microbiol. 2006;14:92–97. doi: 10.1016/j.tim.2005.12.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Detailed identifications of bacterial proteins extracted from samples.
Table S2. Detailed identifications of fungal proteins extracted from samples.