

Phosphoproteomic and functional analysis of the developmental progression of Trypanosomes demonstrates that this transition shows bistability, with commitment to differentiation requiring new protein synthesis, and that the protein kinase NRK is a key regulator.
Abstract
The life cycle of Trypanosoma brucei involves developmental transitions that allow survival, proliferation, and transmission of these parasites. One of these, the differentiation of growth-arrested stumpy forms in the mammalian blood into insect-stage procyclic forms, can be induced synchronously in vitro with cis-aconitate. Here, we show that this transition is an irreversible bistable switch, and we map the point of commitment to differentiation after exposure to cis-aconitate. This irreversibility implies that positive feedback mechanisms operate to allow commitment (i.e., the establishment of “memory” of exposure to the differentiation signal). Using the reversible translational inhibitor cycloheximide, we show that this signal memory requires new protein synthesis. We further performed stable isotope labeling by amino acids in cell culture to analyze synchronized parasite populations, establishing the protein and phosphorylation profile of parasites pre- and postcommitment, thereby defining the “commitment proteome.” Functional interrogation of this data set identified Nek-related kinase as the first-discovered protein kinase controlling the initiation of differentiation to procyclic forms.
Introduction
To survive, proliferate, and cooperate, cells must make switch-like decisions, such as whether to express particular genes, to divide, or to differentiate. Bistability is an important property of many biological switches, which has been fundamental for understanding mitotic progression as well as the control of developmental transitions in both prokaryotic and eukaryotic organisms. Bistable switches are characterized by the lack of stable intermediate states between the initial and final states. That is, the transition from one state to the next is not a progressive response to a signal; rather, it is a digital change from one state to the next. Crucially, this change occurs at a specific stimulus threshold. By definition, and unlike ultrasensitive or sigmoidal responses to external signals, the “on” and “off” responses of bistable switches follow different trajectories, such that at a given level of signal, the cell state can be stably on or off, depending on the prior exposure to the signal. This is referred to as hysteresis (Tyson et al., 2003) and enables the clear-cut transition between states necessary for robust decision making (Fig. 1, A–C), such as progression through cell cycle checkpoints, as an adaptive response to a changing environment, or in cell differentiation (Xiong and Ferrell, 2003; Pomerening, 2008; Wang et al., 2009). It also enables decisive responses in the context of a noisy environment in which an extrinsic signal may be transient or fluctuate in levels after the initial stimulatory event (Losick and Desplan, 2008).
Figure 1.

Models for biological signal responses. (A–C) Schematic signal-response curves for cellular differentiation. Signal responses for cell differentiation can reflect an ultrasensitive, reversible response (A), a bistable reversible response (B), or an irreversible bistable response (C). The dashed lines represent unstable intermediates between the response profiles, with ascending and descending levels of signal. (D) Once a differentiation signal is received, the response can be stabilized using positive feedback loops operating at different levels in the signal-response pathway, generating bistability in the system. The solid arrows represent progression from signal reception to signal transduction (two components are shown) and thereafter to effector molecules that drive changes in gene expression and differentiation state. Dotted arrows represent possible sites of positive feedback driving the signal response.
Bistability requires positive feedback loops, which ensure separation and stability of the on and off states and whose activation results in commitment to transitions between states (Ferrell and Xiong, 2001; Tyson et al., 2003; Mitrophanov and Groisman, 2008). These are common in transcriptional and signaling networks; for example, they occur where transcription factors activate their own transcription or when protein kinases autophosphorylate. These responses can be sustained, such that removal of the initial signal does not reverse the response. They may also trigger downstream or overlapping feedback mechanisms that make a transition irreversible. Hence, bistable switches can operate at different and/or multiple levels (Fig. 1 D), but the consequence is that the cell progresses unidirectionally to its next state (Losick and Desplan, 2008).
Protozoan parasites are one group of organisms that progress through an irreversible series of cytological states in a highly regulated manner. Commonly, these parasites have complex life cycles whereby they transition between life in a mammalian host and life in an invertebrate vector (Matthews, 2011). This includes the major pathogens Plasmodium spp. (responsible for malaria; Aly et al., 2009) and kinetoplastid parasites responsible for several important tropical diseases including Chagas disease (Trypanosoma cruzi; Goldenberg and Avila, 2011), Leishmaniasis (Leishmania spp.; Zilberstein and Shapira, 1994), and African trypanosomiasis (Trypanosoma brucei; MacGregor et al., 2012). In the life cycle of each of these parasites, a developmental transition accompanies the progression of the parasite between the vertebrate and the invertebrate host and vice versa, and there are also further developmental transitions within each environment. Each of these is characterized by being precisely controlled, adaptive for the changed environment, and irreversible. They are also frequently linked to cell cycle progression (Turner, 1992; Matthews and Gull, 1994; Pollitt et al., 2011). Hence, the life cycle of important human pathogens provides exemplars of potentially bistable transitions in eukaryotic biology whose disruption could provide important targets to block virulence or transmission. However, the molecular processes that underlie the developmental transitions and their regulatory control are poorly characterized compared with the mitotic progression of eukaryotic cells, for example.
One differentiation step that is tractable both cytologically and in molecular terms is the development of African trypanosomes from their bloodstream form to the form found in the midgut of their tsetse fly vector. Trypanosomes proliferate in the bloodstream of mammals as morphologically “slender forms” before undergoing a density-dependent differentiation to G1 arrested “stumpy forms” (Vassella et al., 1997). Within experimental infections, uniform populations of stumpy forms can be purified and stimulated, in vitro, by exposure to cis-aconitate (CA) and reduced temperature to initiate development to the procyclic forms that normally colonize the tsetse midgut (Ziegelbauer et al., 1990). If initiated with a population enriched in stumpy forms, the differentiation is remarkably synchronous, allowing events at the single cell level to be interpreted from population-level analysis, with sufficient cell numbers being available from experimental infections to allow both cytological and molecular studies (Matthews and Gull, 1994). Such experiments have revealed that the CA signal is detected via a surface family of proteins (PAD proteins; Dean et al., 2009), and that this activates a signaling cascade whereby a “master” regulatory tyrosine phosphatase, TbPTP1, is inactivated (Szöőr et al., 2006) and its substrate, a serine threonine phosphatase TbPIP39, is activated. TbPIP39 is then trafficked to glycosomes, specialist organelles in trypanosomes that compartmentalize the glycolytic enzymes and other enzymatic activities (Szöőr et al., 2010). Differentiation can also be stimulated by pronase, acting independently of TbPIP39 signaling (Szöőr et al., 2013).
Beyond signaling events, the first cytological marker for cell differentiation to procyclic forms is the expression of the glutamic acid and proline (EP) procyclin surface protein coat (Roditi and Liniger, 2002). This occurs in the first 2 h and is followed by the loss of the bloodstream-form variable surface antigen (at 5–6 h; Matthews and Gull, 1994), and then repositioning of the mitochondrial genome, the kinetoplast, to midway between the cell posterior and cell nucleus (at 6–8 h; Matthews et al., 1995). Thereafter, the stumpy forms reenter their cell cycle, involving DNA synthesis, kinetoplast segregation, mitosis, and cytokinesis (12 h after differentiation is triggered; Matthews and Gull, 1994). The commitment to differentiation has been mapped using immunofluorescence for the expression of EP procyclin; however, the molecular basis of cell commitment and the extent to which this represents an irreversible bistable switch has not been investigated.
Here, we examined commitment to differentiation in synchronized populations in detail and investigated its underlying molecular regulation. We conclusively establish that trypanosome differentiation represents a bistable transition, with commitment occurring between 2 and 3 h after exposure to the differentiation signal. Moreover, we establish that the “memory” of exposure to the signal requires protein synthesis; on the basis of this, we detail the changes in the cellular proteome that occur at commitment in response to the differentiation signal. Concomitantly, we generated a high-resolution map of the changes in the phosphoproteome early during differentiation events, generating a “commitment proteome” that can provide a framework for dissecting the molecular events controlling differentiation. Finally, we identified the stumpy-enriched protein kinase Nek-related kinase (NRK) as a key regulator of the differentiation process.
Results
Trypanosome differentiation is an irreversible bistable switch
To characterize the response of trypanosomes to CA, uniform populations of stumpy forms were exposed to this developmental signal and monitored for their EP procyclin expression by flow cytometry. This provides a sensitive and quantitative measure that can be compared with earlier studies using scoring by immunofluorescence assay (Matthews and Gull, 1997), EP procyclin 3′UTR-linked reporter assays (Sbicego et al., 1999), or cell growth after differentiation (Engstler and Boshart, 2004). We also stained the nuclei and kinetoplasts with DAPI to follow cell cycle reentry as a confirmation of differentiation (unpublished data). Fig. 2 A demonstrates that EP procyclin was detected on most cells in the population after 1–2 h of exposure to CA, consistent with earlier studies. Thereafter, we calibrated the sensitivity and response time of the parasites to CA to map the point of commitment and to test the bistability of the underlying developmental switch. First, the response of the parasites to varying levels of CA was analyzed by the incubating stumpy forms with a titration of CA from 10−4 mM to 6 mM CA. This demonstrated that 1 mM CA at 27°C was required for effective procyclin expression after 24 h (Fig. 2 B), matching the outgrowth of differentiated parasites at this temperature reported by Engstler and Boshart (2004) and EP-3′UTR–based reporter studies (Sbicego et al., 1999). Next, commitment to differentiation was analyzed by exposing stumpy forms to 6 mM CA for 1, 2, or 3 h, which were then washed free of CA and incubated for a further 24 h, whereupon EP procyclin expression was measured. Fig. 2 C demonstrates that although some cells (42%) expressed procyclin at 24 h after 1-h exposure to CA, procyclin expression was maintained at a high level for 24 h after 2- and 3-h exposure to CA (72% and 84%, respectively), positioning commitment at 2–3 h. The bimodal shapes of the EP expression profiles also indicated that cells either do or do not commit to differentiation, and that there is some noise in their decision, probably because of as-yet-unexplored differences between stumpy cells. Finally, hysteresis of the response was tested by exposing parasites to 6 mM CA for 3 h, after which cells were washed and returned to a titration of CA concentrations matching the earlier signal-response analysis (i.e., 10−4 to 6 mM CA) for 24 h (Fig. 2 D). In all cases, the cells remained procyclin positive, despite being maintained at a signal level below the threshold necessary to stimulate the initial response. This also showed that the level of EP expression was not dependent on the CA concentration, which strengthens the notion that differentiation is independent of the signal after commitment. Memory of prior exposure to a CA level above the triggering threshold defines this transition as an irreversible bistable switch, and the data from Fig. 2 define the signal-response curve (or one-parameter bifurcation diagram) of this developmental switch.
Figure 2.

Trypanosome differentiation is an irreversible bistable switch. (A) Time course of EP procyclin expression after trypanosome exposure to 6 mM CA. Procyclin expression was detectable within 1 h and maximum expression was achieved at 3 h. Some reduction in EP procyclin expression was routinely observed at 24 h after induction in all experiments. Quantitation of the EP procyclin expression at each time point is shown. (B) Signal sensitivity of differentiation. Parasites were exposed to a titration of CA at 27°C and monitored for the expression of EP procyclin at 24 h. Quantitation of the EP procyclin expression at 24 h is shown. (C) Commitment to differentiation. Parasites were exposed to 6 mM CA for 1, 2, or 3 h and then washed free of CA. The expression of EP procyclin was then monitored at 24 h. Cells maintained in CA throughout the 24 h of the experiment were also assayed. Quantitation of the EP procyclin expression at each time point is shown. (D) Differentiation is irreversible. Parasites were exposed to 6 mM CA for 3 h and then returned to medium containing varying levels of CA and assayed for EP procyclin expression. Quantitation of the EP procyclin expression at 24 h is shown.
Signal memory requires new protein synthesis
The demonstration that trypanosomes exhibited signal memory through irreversible commitment to differentiation after 2–3-h exposure to CA led us to investigate the molecular basis of this. Commitment should depend on the triggering of a positive feedback loop, which could involve posttranslational modifications (e.g., phosphorylation) and/or mRNA or protein synthesis. To test whether new protein synthesis was necessary for commitment, we used the reversible protein synthesis inhibitor cycloheximide (CHX). For this, cells were exposed to 6 mM CA in the presence of CHX to block protein synthesis during the commitment phase. We also incubated cells with CA without CHX as a commitment and differentiation control. From 1, 2, or 3 h after exposure to CA and CHX (or CA alone), cells were washed and maintained in the absence of both CA and CHX (Fig. 3 A). Fig. 3 B demonstrates that in the differentiation control (+CA and −CHX) cells exhibited ∼80% EP-positive cells after 3 h. As expected, cells incubated with CA and CHX did not express EP procyclin, confirming the efficacy of protein synthesis block (Fig. 3 C). However, the cells also did not go on to express EP procyclin when CA and CHX were washed out at 1, 2, or 3 h and then incubated for a further 24 h. This indicated that in the presence of CHX, the cells did not retain a “memory” of their prior exposure to the CA signal (i.e., any stable molecular change necessary for detection of the signal or commitment) and thus could not differentiate once protein synthesis was allowed to resume. To ensure that the effect of CHX was reversible and to rule out the possibility that CHX results in complete loss of capacity to differentiate (or irreparable cell damage), stumpy cells were incubated with both CA and CHX for 3 h, washed free of CHX, and then reincubated with CA and monitored for EP procyclin expression. Fig. 4 shows that cells induced EP procyclin expression between 1 and 2 h after washing free of CHX. Hence, protein synthesis inhibition by CHX was reversible; even after 3 h of exposure to CHX, cells remained capable of differentiation once the protein synthesis block was removed, as long as they were newly exposed to the differentiation signal CA. These experiments established that signal memory in the parasites was dependent on new protein synthesis (i.e., the parasites required newly made proteins to “remember” that they had been exposed to CA).
Figure 3.

The commitment to differentiation requires new protein synthesis. (A) Schematic of the experimental regimen. (B and C) Stumpy forms were incubated with 6 mM CA in the absence (B) or presence (C) of CHX. The expression of EP procyclin was measured at 0, 1, 2, 3, or 24 h. At each time point, some cells were washed free of CHX and incubated until 24 h, at which point their expression of EP procyclin was determined. Cells maintained for 24 h in CHX were swollen and immotile; here, detection of EP procyclin probably reflects nonspecific antibody trapping by dead cells or the expression of some EP procyclin as cells lose viability in CHX. Quantitation of the EP procyclin expression at each time point is shown.
Figure 4.

Protein synthesis “sets the clock” for differentiation. (A) Schematic of the experimental regimen. (B and C) Stumpy forms were exposed to 6 mM CA in the absence (B) or presence (C) of CHX. After 3 h, the cells were washed free of CHX and exposed to CA and then monitored for EP procyclic expression after 1, 2, 3, and 24 h. EP procyclin expression commenced at 1 h, confirming the removal of CHX. Quantitation of the EP procyclin expression at each time point is shown.
Protein expression and protein phosphorylation during commitment
These experiments established the importance of newly made proteins for commitment but did not preclude an additional contribution from a positive feedback loop involving protein phosphorylation/dephosphorylation events on existing or newly synthesized proteins. To explore this, we analyzed changes in the protein and phosphorylation profile of the “commitment proteome.” Specifically, a quantitative proteomic and phosphoproteomic analysis was performed on stumpy forms either before or during commitment in the presence of CA (Fig. 5). Because metabolic labeling of stumpy forms by stable heavy isotopes in vivo is not experimentally tractable, a “spike-in” stable isotope labeling by amino acids in cell culture (SILAC) approach was used, whereby a reference heavy isotope–labeled sample of cultured cells was used as an internal standard (Ishihama et al., 2005). To maximize the proteome coverage of the reference, equal amounts of monomorphic bloodstream forms and cultured procyclic forms were combined. The heavy isotope–labeled standard then was used to compare unlabeled ex vivo stumpy cells or the same stumpy cells incubated with or without CA for 1 or 3 h, in biological triplicate. For two of the replicates, the commitment profile of the cells was validated by removing an aliquot at 1 h and 3 h, washing the cells free of CA, and then assaying their EP procyclin expression at 24 h. This confirmed that each sample showed the expected commitment profile 1–3 h after exposure to CA (one example is shown in Fig. 5). The commitment to cell cycle reentry was also monitored by analyzing the flow cytometry profile of cells with DNA stained with DAPI (Fig. 5). This matched the expression of EP procyclin and confirmed differentiation at 1–3 h after exposure to CA.
Figure 5.

Schematic representation of the analysis of the commitment proteome and phosphoproteome. Proteomic workflow. Ex vivo stumpy cells were incubated at 37°C in the presence or absence of CA for 1 and 3 h. An aliquot of each sample was withdrawn, washed, and cultured in the absence of CA before analysis of EP procyclin expression and reentry into the cell cycle to confirm that commitment occurred on the expected timescale. The remainder of the each sample was mixed with an equal number of Arg6Lys4 heavy isotope–labeled cultured T. brucei cells, digested with trypsin, and peptides were subjected to proteomic or phosphoproteomic analysis. Experiments were conducted in biological triplicate. SCX, strong cation exchange.
Proteomic data overview
The proteomic data set provides a snapshot of the complex changes that occur during signal perception and commitment events in the early stages of differentiation, as well as the changes in metabolism, protein coat, and reentry into the cell cycle. To enable quantitation of changes in both protein abundance and phosphorylation, each biological sample was split and subjected either to proteomic analysis (5%) or to enrichment of phosphopeptides (95%) before analysis (Fig. 5). Altogether, 324 liquid chromatography (LC) tandem mass spectrometry (MS/MS) runs identified a total of 5,911 protein groups containing 6,869 proteins, with 10,159 phosphorylation sites identified on 2,334 proteins (>0.75 localization probability) with a 1% false discovery rate. Quantitation of 5,285 protein groups containing 6,232 proteins was obtained using only data from the proteomic experiments, and 9,856 phosphorylation sites were quantified using only data from the enriched samples. Reproducibility was high, with biological replicates showing a Pearson correlation of 0.85–0.96 and 0.78–0.96 with the mean at the protein and phosphorylation site levels, respectively (Fig. 6, A and B; and Fig. S1). The Pearson correlation between different samples reflected their exposure to CA; untreated samples were most similar to each other and to t = 0 h, whereas upon CA treatment, over time, the correlation with t = 0 h decreased and the correlation with the procylic form increased (Fig. 6, C and D). That such patterns in correlation are discernible at even these early time points suggests that the changes observed are part of a defined program of events to retool the biology of the parasite in response to a perceived change in its environment.
Figure 6.

Summary of the changes in the phosphoproteome at each time point under each experimental regimen. (A) The protein SILAC H/L ratio of the individual biological replicates compared pairwise to the mean ratio (with Pearson correlation), after 3 h in the absence of CA (3 h − CA, top panel) or 3 h in the presence of CA (3 h + CA, bottom panel). (B) Pairwise comparison of phosphorylation site data, presented as described for A. (C) Pairwise comparison of the mean protein SILAC H/L ratio determined under each experimental regimen: 0 h – stumpy, t = 0 h; 1 h − CA, t = 1 h no CA; 3 h − CA, t = 3 h no CA; 1 h + CA, t = 1 h with CA; and 3 h + CA, t = 3 h with CA. The matrix is colored according to the Pearson correlation between each pair. (D) Pairwise comparison of phosphorylation site data under each experimental regimen, presented as described for C. Pcf, cultured procylic form cell.
The changes in the proteome and phosphoproteome in response to exposure to CA before (1 h) and after (3 h) commitment were quantified by calculating the difference between the treated and untreated samples at each time point, using the heavy isotope–labeled standard as an internal control (Tables S1, S2, and S3). Overall, the changes in phosphorylation sites were larger and occurred earlier than the changes in protein abundance. For example, at 1 h (precommitment), 356 phosphorylation sites (on 251 proteins) and 25 proteins changed by more than fourfold (log2 >2); at 3 h (postcommitment), 371 phosphorylation sites (on 257 proteins) and 36 proteins changed by more than fourfold (Fig. S2). Although a similar number of changes occurred at each time point, a largely distinct subset of proteins and phosphorylation sites changed after commitment at 3 h, with respect to before commitment at 1 h (Fig. 7), suggesting that there is a distinction between signal perception events occurring precommitment and events that occur once commitment has been reached. The observed changes were driven by changes in the treated samples, rather than recovery of the untreated samples (e.g., from cold shock; Fig. S3). The balance between phosphorylation and dephosphorylation events altered significantly before and after commitment, with events at 1 h dominated by dephosphorylation (72% > |log2 2|), whereas a balance between dephosphorylation (48% > |log2 2|) and phosphorylation was reached at 3 h.
Figure 7.
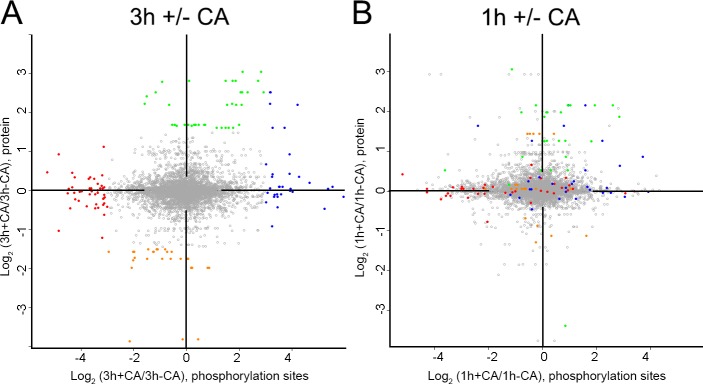
Distinct changes occur in the proteome before and after commitment. (A and B) Changes in protein and phosphorylation site abundance in cells treated with CA compared with untreated cells at time points after commitment (3 h; A) and before commitment (1 h; B). Coloring represents the greatest changes that occur at 3 h, defined as follows: blue, phosphorylation site up at 3 h + CA (log2 [3 h + CA/3 h − CCA], phosphorylation site ≥3.0); red, phosphorylation sites down 3 h + CA (log2 [3 h + CA/3 h − CCA], phosphorylation site less than −3.0); green, proteins up 3 h + CA (log2 [3 h + CA/3 h − CCA], protein ≥1.5); and orange, proteins down 3 h + CA (log2 [3 h + CA/3 h − CCA], protein less than −1.5).
Agreement with known biology
The changes in the proteome during differentiation from the stumpy form to the procyclic form reflect the parasite’s adaptation of its surface coat and metabolism for its new host environment, as well as reentry into the cell cycle. These changes are already evident in the proteomic data at only 3 h after exposure to CA (Table 1). Our data show significant enrichment of procyclic-specific surface proteins among proteins up-regulated more than fourfold (25%) compared with those changing less than fourfold (6.5%), in agreement with a recent study demonstrating that the T. brucei cell surface is enriched in stage-specific proteins (Shimogawa et al., 2015). We observed the expected up-regulation of MSP-B (Tb927.8.1640), linked to VSG release during differentiation (Gruszynski et al., 2006), and elevation of the procyclic-specific surface protein PSSA-2 (Tb927.10.11220; Fragoso et al., 2009), nucleoside transporter NT10 (Tb927.9.7470; Sanchez et al., 2004), and trans-sialidase (Tb927.7.6850; Engstler et al., 1992). Down-regulation was detected for the bloodstream form–specific surface protein ISG75 as well as BRCA2 (Tb927.1.640) linked to DNA repair and antigenic variation in the bloodstream (Trenaman et al., 2013). Developmental changes in metabolism included up-regulation of THT2A, gMDH, and sterol 24-C-methyltransferase, and down-regulation of the bloodstream-enriched PGK-B (Tb927.1.710; Blattner and Clayton, 1995). Reflecting the reinitiation of protein synthesis early during differentiation (Kabani et al., 2009), there was increased phosphorylation of eIF2β and eIF4ε. Interestingly, we also observed multiple changes in the phosphorylation site of the cytoskeleton-associated calpain-like proteases Tb927.1.2100 (log2 +3.30 to −1.51) and Tb927.11.1090 (log2 +4.25 to −1.80), the latter localizing to the flagella attachment zone and reported to be responsible for maintaining the bloodstream-form trypomastigote cell morphology (Hayes et al., 2014).
Table 1. Examples of changes observed in the differentiation proteome.
| TriTrypDB ID | Description | Log2 T3C/T3N protein | Log2 T3C/T3N phosphorylationa | Classification |
|---|---|---|---|---|
| Tb927.8.1640 | MSP-B, major surface protease | +3.67 | — | Surface |
| Tb927.7.6850 | TS, trans-sialidase | +1.68 | — | Surface |
| Tb927.10.11220 | PSSA-2, procyclic form surface protein | +1.45 | — | Surface |
| Tb927.1.2100 | Calpain-like cysteine peptidase | +0.36 | S81 +4.25; S366 +3.23; T365 +2.72; S95 +2.64 | Surface |
| Tb927.11.17870 | VSG, variant surface glycoprotein | −1.75 | — | Surface |
| Tb927.5.370 | ISG75, 75-kD invariant surface glycoprotein | −2.09 | — | Surface |
| Tb927.10.8530 | THT2A, glucose transporter | +3.05 | T526 +2.83 | Metabolism |
| Tb927.10.15410 | gMDH, glycosomal malate dehydrogenase | +2.43 | — | Metabolism |
| Tb927.10.6950 | Sterol 24-C-methyltransferase | +2.12 | — | Metabolism |
| Tb927.1.710 | PGKB, phosphoglycerate kinase B | −3.58 | — | Metabolism |
| Tb927.1.2600 | PUF9, pumilio RNA binding protein | −1.98 | Cell cycle | |
| Tb927.11.11770 | eIF4e, eukaryotic translation initiation factor 4e | +0.20 | T159 +2.89 | Cell cycle |
| Tb927.11.1820 | eIF2b, eukaryotic translation initiation factor 2b | +0.19 | S126 +2.57; T127 +2.57 | Cell cycle |
| Tb927.9.6100 | PIP39, PTP1-interacting protein | +2.50 | — | Signaling |
| Tb927.7.5940 | PAD2, protein associated with differentiation 2 | +0.05 | T577 -4.46; T587 −3.65 | Signaling |
| Tb927.7.5990 | PAD7, protein associated with differentiation 7 | −0.15 | T584 +7.01; S580 +3.44; S582 +2.71 | Signaling |
| Tb927.6.3490 | ZFP1, zinc finger protein 1 | — | S18 +2.81; S16 +2.56 | Signaling |
For proteins with multiple phosphorylation sites, only those with the largest changes are given; see Table S3 for full details. Missing values were either not observed or not quantified.
The data set also captured changes involved in signal perception and commitment to differentiation; for instance, up-regulation of TbPIP39 and dephosphorylation of PAD2 were observed, as was increased phosphorylation of PAD7 and ZFP1. Although knowledge of the differentiation signaling pathway is incomplete, PAD proteins (Dean et al., 2009), the tyrosine phosphatase TbPTP1 (Szöőr et al., 2006), and the glycosomal protein TbPIP39 (Szöőr et al., 2010) are known to be involved in the process. Unfortunately, we were not able to observe the phosphotyrosine sites PIP39-pY278 or NOPP44/46-pY181 (both substrates of TbPTP1; Chou et al., 2010; Szöőr et al., 2010) because the flanking sequence meant that neither produced phosphopeptides detectable by LC mass spectrometry (MS; Table S1), although both were observed at the protein level. We also did not observe widespread changes in phosphotyrosine abundance or in glycosomal proteins (Güther et al., 2014) upon treatment; these changes may occur at later time points. Other signaling molecules with roles in differentiation, such as ZFK, MAPK5, TOR4, and RDK2, were observed but did not change significantly in abundance upon CA stimulation, suggesting that they are not dynamically regulated at the time points studied. A significant subset of the proteins identified as displaying “loss of function in differentiation only” from genome-wide RNAi analysis were observed to undergo substantial changes, including many hypothetical proteins (Alsford et al., 2011). Likewise, a subset of the potential RNA binding proteins identified by an artificial RNA-tethering screen (Erben et al., 2014) underwent substantial changes. Given the post-transcriptional control of trypanosome gene expression (Clayton and Shapira, 2007), such modulation of RNA binding proteins is likely to play a role in early differentiation, perhaps analogous to the role of transcription factors in differentiation in other eukaryotes.
Molecules regulated during commitment and molecules that regulate differentiation
Having derived a comprehensive picture of the phosphoproteome during commitment, we sought to identify molecules implicated in its regulation. First, we explored the proteome and phosphoproteome data for molecules that exhibited changes in abundance and phosphorylation early during differentiation. Of these, one obviously regulated molecule was a predicted RNA binding protein, ZC3H22 (Tb927.7.2680). Hence, pleomorphic trypanosomes were generated where ZC3H22 was silenced by RNAi and its effects on stumpy formation or differentiation were analyzed. Fig. S4 shows that effective depletion of ZC3H22 was achieved in bloodstream forms, but that the generation of stumpy forms proceeded normally, precluding a role for this molecule during that developmental transition. When analyzed during differentiation to procyclic forms, EP procyclin was expressed on the expected timescale and the efficiency of differentiation was unchanged whether RNAi was induced or not (Fig. S5 A). However, when RNAi was induced in three independently derived RNAi lines in procyclic forms, cell growth was inhibited (Fig. S5 B). This suggests that ZC3H22 is required for procyclic form viability or fitness and is activated during differentiation to fulfill this function. The previously determined RNAi target sequencing (RIT-seq) profile for ZC3H22 (1-1-1-0, where 1 denotes no growth phenotype and 0 denotes a significant growth phenotype in day 3 bloodstream forms, day 6 bloodstream forms, differentiating cells, and procyclic forms, respectively) was consistent with this interpretation (Alsford et al., 2011).
As an alternative route to identify molecules contributing to the regulation of differentiation, we explored proteins that were present only in the light fraction (and were hence enriched in stumpy forms with respect to the spiked heavy-labeled slender or procyclic form samples) and with a predicted role in signaling events (Table S3). This identified three molecules: MPK10 (Tb927.8.3770), Rac serine threonine kinase (Tb927.6.2250), and NRK, representing the NRKA (Tb927.4.5390) and NRKB (Tb927.8.6930) genes, which are 99.5% identical (Gale and Parsons, 1993). These were then analyzed for their published RIT-seq profile (Alsford et al., 2011), with MAPK4 having evidence for a role in procyclic forms (1-1-1-0), whereas Rac serine threonine kinase demonstrated no growth phenotype in any type analyzed (1-1-1-1). In contrast, NRK exhibited a specific differentiation defect by RIT-seq analysis (1-1-0-1) and was previously shown to be specifically expressed in T. brucei TREU667 stumpy forms at the protein, but not RNA, level (Gale and Parsons, 1993; Gale et al., 1994). Confirming this, an antibody recognizing NRK demonstrated enriched expression of the proteins (Fig. 8 A) in AnTat1.1 stumpy forms (Fig. 8 B). To explore the role of NRK in differentiation, pleomorphic RNAi lines were generated targeting NRKA/NRKB and the resulting cell lines were analyzed for effective protein depletion and growth in vitro and in vivo. In vitro, the NRK signal was strongly reduced after 48-h induction (Fig. 8 C), but this did not result in any obvious growth defect (not depicted). Thereafter, two independent RNAi lines were analyzed in vivo. Fig. 8 D shows that the induced samples generated a slightly reduced parasitemia but that each generated uniform populations of stumpy forms after 6 d, demonstrating the NRK was not required for the production of this life cycle stage. Once generated, the stumpy forms were induced to differentiate to procyclic forms either by exposure to 6 mM CA or by incubation with pronase for 10 min. These two stimuli promote differentiation to procyclic forms, with CA operating through the action of TbPTP1 on the glycosomally directed phosphatase TbPIP39, whereas pronase operates independently of TbPIP39 (Szöőr et al., 2013). In each case, differentiation of RNAi-induced or RNAi-uninduced cells was monitored after exposure to each trigger for the expression of EP procyclin at 0, 3, 6, and 24 h. Fig. 9 A demonstrates that the RNAi for NRK was effective in stumpy forms with low levels of protein in the induced sample. Upon exposure to the differentiation signals, uninduced cells responded to either CA or pronase very effectively, expressing procyclin at 3 h and beyond (Fig. 9 B), with the expression of NRK decreasing at 3 h and 6 h with respect to 0 h, consistent with the decreased expression of NRK in procyclic forms (Fig. 9 A). The cells also reentered into a proliferative cell cycle after 24 h (data not depicted). In contrast, the NRK-depleted cells showed reduced procyclin expression in response to CA at 3 h (mean 15.2% vs. 68.4%) or 6 h (mean 20.8% vs. 72.4%). After pronase treatment, EP procyclin expression was also reduced in the induced sample at 6 h, with respect to the RNAi-uninduced samples (mean 39.3% vs. 66.7%; Fig. 9 B).
Figure 8.

Depletion of NRK does not affect growth or differentiation of bloodstream-form trypanosomes. (A) Schematic representation of NRKA/NRKB. The two encoded proteins are 97% identical and hence indistinguishable by RNAi-based functional analysis. (B) Protein expression profile of NRKA/NRKB. Western blot of proteins from slender (Sl), stumpy (St), and procyclic (Pc) forms reveals that NRK is expressed in stumpy forms. α-Tubulin is used as a loading control. (C) RNAi against NRKA/NRKB. Two independent slender-form AnTat 1.1 lines were induced for RNAi and the expression of NRK monitored by Western blotting. The protein was depleted after 48-h induction in each case. An antibody to EF1α provided the loading control. (D) NRK RNAi does not result in a growth defect of parasites grown in vivo. When grown in vivo the induced parasites developed from slender to intermediate and stumpy forms with equivalent kinetics to parental and uninduced cell lines. Two replicate independent cell lines were analyzed in parallel, both with and without doxycycline (Dox) induction (each analyzed once, with n = 250 cells at each time point in each replicate). White boxes represent slender cells, gray boxes represent intermediate cells, and black boxes represent stumpy cells.
Figure 9.

NRK RNAi prevents differentiation of stumpy trypanosomes to procyclic forms. (A) Western blot of stumpy forms induced or not to deplete NRK by RNAi during growth in vivo. Western blots are shown of parasites harvested from blood and analyzed before or 3 h after exposure to 6 mM CA or incubation with pronase (Pr), each representing an effective differentiation trigger. Western blots demonstrate the level of NRK, the differentiation regulator TbPIP39, this being elevated upon differentiation, and EIFα, used as a loading control. The overall level of protein in pronase-treated cells is less because of death of some parasites under this regimen. Samples from the parental AnTat1.1 90:13 line are shown, as well as two independent NRK RNAi lines, induced or not. (B) Expression of EP procyclin after 0, 3, 6, and 24 h after exposure to CA or pronase. Control cells and the uninduced RNAi lines differentiate effectively in response to both CA and pronase. RNAi-mediated depletion of NRK limits the efficiency of differentiation in response to both CA and pronase, this being consistent between two independent RNAi lines. Dox, doxycycline.
We conclude that NRK depletion inhibits differentiation to procyclic forms regardless of whether it is stimulated by CA or pronase. Because TbPIP39 is not required for differentiation in response to pronase, this places NRK action below where the CA and pronase signaling pathways have converged or on both pathways.
Discussion
The commitment of cells to a differentiation program is fundamental in eukaryotic development. Trypanosomes represent an interesting model to examine the cell biology of such unidirectional developmental events, these being among the most evolutionarily divergent eukaryotes (Sogin et al., 1986; Walker et al., 2011). Moreover, the regulatory processes underlying their life cycle control are likely to be conserved in a broad range of protozoan pathogens, in which progressive differentiation steps characterize their transition between different hosts, or in complex host-vector interactions. Here, we have exploited the synchronous and tractable in vitro differentiation of stumpy form parasites to tsetse midgut procyclic forms to examine parasite cell commitment from several complementary perspectives. First, we examined the reversibility of their cell commitment processes, formally demonstrating that it represents an irreversible bistable switch and further revealing that signal memory during commitment requires protein synthesis. Second, we examined the molecular events accompanying commitment, providing a detailed map of the parasite’s commitment proteome and phosphoproteome. Finally, we used this information, triangulated with prior knowledge, to identify a key regulatory kinase controlling the initiation of the differentiation program.
Initially, we reexamined earlier studies that used relatively subjective immunofluorescence detection of the EP procyclin surface antigen to map the commitment point during the synchronous differentiation from stumpy to procyclic forms (Matthews and Gull, 1997; Sbicego et al., 1999). Our quantitative analysis placed irreversible commitment to differentiation at 2–3 h after exposure to CA, such that the cells maintain the expression of procyclin (already expressed at commitment) but also go on to reenter a proliferative cell cycle (DNA synthesis having been mapped to occur at 8–10 h; Matthews and Gull, 1994). These studies also demonstrated that commitment (at 27°C; at 20°C and 37°C, cells commit on the same timescale; unpublished data) required exposure to at least 1 mM CA and that once this signal was present for 2–3 h, the cells retained memory of the signal exposure even when reincubated at levels of CA unable to drive differentiation ab initio. If this signal memory were dependent only on posttranslational modifications, we predicted that cells exposed to CA in the presence of CHX would progress into the differentiation program once the inhibitor was removed. This was not the case; parasites treated with CHX did not respond to CA unless the inhibitor was washed out and CA was added, after which they differentiated on a timescale consistent with de novo exposure to the signal. This demonstrated that new protein synthesis is required for the commitment to differentiation and sets the clock for the initiation of the process. Although phosphorylation events could also contribute, these studies establish that CA exposure does not generate a stable signal involving only posttranslational mechanisms, or an effector pathway requiring RNA alone.
To explore the molecular events accompanying the early steps in differentiation, we analyzed the commitment proteome and phosphoproteome. Previous studies have compared the protein and phosphorylation profile of different life cycle stages (Nett et al., 2009; Gunasekera et al., 2012; Urbaniak et al., 2013) but have not compared cells pre- and postcommitment in a synchronous differentiation model with accompanying biological validation of the cell fate of each sample. Providing reassurance on the quality of the data set, there was good correspondence between individual, independently performed, biological replicates, and key changes observed early in differentiation agreed well with prior characterization of this transition. The temporal analysis at 0 h, 1 h (precommitment), and 3 h (postcommitment) also confirmed the importance of new protein synthesis to commitment, such that few changes occurred in protein abundance at 1 h, with most changes occurring only at 3 h; this is in contrast to protein phosphorylation, which was already changed dramatically at 1 h. Although most changes in phosphorylation did not simply correspond to changes in protein abundance, one exception was ZC3H22, a predicted RNA binding protein strongly induced during differentiation with a concomitant multisite phosphorylation profile. This protein, however, appears to be unrelated to the developmental process itself, instead being expressed and required for the viability of procyclic forms. Other changes in protein abundance or phosphorylation await individual analysis and comparison with data sets analyzing the cell cycle of bloodstream and procyclic forms. This will provide an invaluable framework to distinguish differentiation-specific changes from the events of cell cycle reentry only.
As an approach to the identification of regulatory molecules that might control early steps in the differentiation program, we selected molecules on the basis of stumpy-enriched expression, signaling function, and RIT-seq phenotype. This focused our attention on the NRKA/NRKB protein kinases. These molecules have serine threonine kinase activity (Gale and Parsons, 1993; Gale et al., 1994) and are members of the expanded NEK kinase family in African trypanosomes (Jones et al., 2014). They have also been shown to be developmentally regulated at the translational level (Gale et al., 1994). RNAi against these molecules demonstrated that inducible depletion of NRK inhibited the response of the parasites to CA. Interestingly, the parasites were also less able to respond to an alternative differentiation trigger, pronase. This signals effective differentiation but operates independently of TbPIP39, a glycosomally directed phosphatase important in the CA-mediated differentiation pathway (Szöőr et al., 2010). The NRK-depleted parasites also differentiated less efficiently when exposed to BZ3, an inhibitor of the tyrosine phosphatase TbPTP1 that acts upstream of, and dephosphorylates, TbPIP39 (Szöőr et al., 2006; unpublished data). Consequently, NRK most likely operates downstream of where the CA and pronase pathways converge to drive differentiation, or it acts on both branches. Hence, NRK is identified as the first protein kinase necessary for trypanosome development to procyclic forms and its conservation and synteny, where known, in all other kinetoplastids, including Crithidia fasciculata and Bodo saltans, implies an important role throughout the order.
Combined, our studies represent a comprehensive cytological and molecular dissection of cell differentiation in an early diverging eukaryotic pathogen. Similar processes are fundamental in the life cycles of all protozoan pathogens, including related kinetoplastids (T. cruzi and Leishmania parasites) and those with an enormous impact on human health, such as malaria. Each of these parasites undergoes proliferation and then cell cycle arrest before reentry into their cell cycle, with concomitant differentiation to the next life cycle stage. In each case, the developmental cycles are also unidirectional and involve irreversible commitment steps. This regulatory framework ensures that the necessary conditions are met for successful progression to the next developmental step in the parasite’s life cycle and avoids lethal indecision in a changing environment.
Materials and methods
Trypanosomes
T. brucei AnTat1.1 parasites were used for all experiments. Transgenic parasites were T. brucei AnTat1.1 90:13 cells, which express the T7 RNA polymerase and tetracycline repressor protein (Engstler and Boshart, 2004). RNA constructs were based on the pALC 14 plasmid (Pusnik et al., 2007). Transfection and cell maintenance in HMI-9 medium containing 20% FCS (Hirumi and Hirumi, 1989) was performed as previously described (Szöőr et al., 2013).
In vivo growth involved i.p. injection of ∼105 parasites. At day 6 after infection, parasites were >80% stumpy by morphology, and trypanosomes were harvested and purified from blood by DEAE chromatography (Lanham and Godfrey, 1970). The purified parasites were incubated in HMI-9 medium at 37°C for 1.5 h at a concentration of 2 × 106/ml and were differentiation stimulated using an appropriate concentration of CA. For isolation of samples for flow cytometry, 3 ml of culture was concentrated at 3,000 rpm in a clinical centrifuge, washed briefly with PBS, and then centrifuged in a microfuge at 2,500 rpm. The cell pellet was resuspended in FACS fix solution (2% formaldehyde and 0.05% glutaraldehyde in PBS) and stored at 4°C. For commitment experiments, samples at time points throughout differentiation were harvested by centrifugation at 2,000 rpm in a clinical centrifuge before resuspension in HMI-9 20% FCS without CA. For protein synthesis inhibition experiments, cells were incubated with 0.1 mM CHX.
Flow cytometry analysis
Stored fixed cell samples in 2% formaldehyde and 0.05% glutaraldehyde were diluted in PBS, centrifuged at 2,500 rpm in a clinical centrifuge, and then resuspended in 500 µl PBS 2% BSA. Samples were stored for 30 min at room temperature to block. Thereafter, the tubes were filled with PBS, centrifuged, and resuspended in 200 µl EP procyclin antibody diluted 1:500 in PBS 2% BSA. After incubation for >1 h, samples were diluted in PBS, centrifuged, and washed once with PBS before being resuspended in a 1:1,000 dilution of α-mouse FITC secondary antibody and incubated for 30 min at room temperature. The samples were then washed twice in PBS and resuspended in PBS DAPI (20 ng/ml). Flow cytometry was performed using an LSRII flow cytometer (Becton Dickinson) and data were analyzed using FlowJo X 10.0.7r2 (Treestar Inc.)
Western blotting was performed as described in Szöőr et al. (2013) using anti-TbPIP39 (Szöőr et al., 2010) diluted 1:200, anti-TbNRK (Gale et al., 1994) diluted 1:100, or anti-EF1α diluted 1:5,000.
SILAC labeling of cultured cells
SILAC labeling of bloodstream and procyclic forms of T. brucei was performed, using media containing l-arginine U-13C6 and l-lysine 4,4,5,5-2H4 (R6K4; Cambridge Isotope Labs; Urbaniak et al., 2012, 2013). Monomorphic bloodstream-form MITat 1.2 cells (Wirtz et al., 1999) were diluted 10,000-fold into HMI11-SILAC plus R6K4 containing 2.5 µg/ml G418 at 37°C in a 5% CO2 incubator. Bloodstream-form cells were harvested after 3-d growth at ∼2 × 106 cells/ml. Procyclic form T. brucei clone 29.13.6 cells were washed with SDM-79-SILAC and then grown at 28°C without CO2 in SDM-79-SILAC plus R6K4 in the presence of 15 µg/ml G418 and 50 µg/ml hygromycin. Procyclic form cells were passaged every 2 d to enlarge the culture and to reach seven to eight cell divisions under labeling conditions, and they were then harvested at ∼2 × 107 cells/ml. Both cell types were harvested by centrifugation and hypotonically lysed at 109 cells/ml for 5 min on ice in the presence of protease inhibitors (0.1 µM 1-chloro-3-tosylamido-7-amino-2-heptone, 1 mM benzamidine, 1 mM phenyl-methyl sulfonyl fluoride, 1 µg/ml leupeptin, and 1 µg/ml aprotinin) and Phosphatase Inhibitor Mixture II (Calbiochem).
To produce the heavy-SILAC standard for use in the spike-in SILAC experiments, an equal volume of R6K4-labeled bloodstream and R6K4-labeled procyclic lysates were mixed, aliquoted, snap frozen, and stored at −80°C. To minimize variability, a single batch of the heavy-SILAC standard was used for the entire study.
Comparative proteomic and phosphoproteomic analysis
Ex vivo stumpy cells (5 × 108 cells) were maintained in HMI-9 and at 37°C for 1.5 h before initiation of the experiment to reduce any inconsistency resulting from their isolation and purification from blood, for example. The procedure was performed in triplicate, using distinct infections to generate the starting stumpy form population. For analysis, each sample was supplemented with an equal cell number of the R6K4-labeled standard, the mixture was solubilized with SDS, and tryptic peptides generated by an adaptation of the filter-aided sample preparation procedure (Wiśniewski et al., 2009; Urbaniak et al., 2013). Because phosphorylation occurs at low abundance and is difficult to detect by MS, the peptides were split between a proteomic (5%) and phosphoproteomic (95%) workflow. In the proteomic workflow, peptides were fractionated into six fractions by strong cation exchange chromatography before analysis by LC-MS/MS as described previously (Urbaniak et al., 2012). In the phosphoproteomic workflow, phosphopeptides were enriched and fractionated into six fractions by strong cation exchange (Beausoleil et al., 2004) before further enrichment by TiO2 in the presence of 5% TFA and 1 M glycolic acid (Thingholm et al., 2006; Jensen and Larsen, 2007), as described previously (Urbaniak et al., 2013).
MS data acquisition
LC-MS/MS was performed by the FingerPrints Proteomic Facility at the University of Dundee. LC was performed on a fully automated Ultimate U3000 Nano LC System (Dionex) fitted with a 1 × 5 mm PepMap C18 trap column and a 75 µm × 15 cm reverse-phase PepMap C18 nanocolumn (LC Packings; Dionex). Samples were loaded in 0.1% formic acid (buffer A) and separated using a binary gradient consisting of buffer A (0.1% formic acid) and buffer B (90% MeCN and 0.08% formic acid). Peptides were eluted with a linear gradient from 5% to 40% buffer B over 65 min. The HPLC system was coupled to an LTQ Orbitrap Velos Pro mass spectrometer (Thermo Fisher Scientific) equipped with a Proxeon nanospray ion source. For phosphoproteomic analysis, the mass spectrometer was operated in data-dependent mode to perform a survey scan over a range of 335–1,800 m/z in the Orbitrap analyzer (R = 60,000), with each MS scan triggering 15 MS2 acquisitions of the 15 most intense ions using multistage activation on the neutral loss of 98 and 49 Th in the LTQ ion trap (Schroeder et al., 2004). For proteomic analysis, the mass spectrometer was operated in data-dependent mode, with each MS scan triggering 15 MS2 acquisitions of the 15 most intense ions in the LTQ ion trap. The Orbitrap mass analyzer was internally calibrated on the fly using the lock mass of polydimethylcyclosiloxane at m/z 445.120025.
MS data processing
Data were processed using MaxQuant (version 1.5.2.8; Cox and Mann, 2008), which incorporates the Andromeda search engine (Cox et al., 2011). Proteins were identified by searching a protein sequence database containing T. brucei 927 annotated proteins (version 9.0, 11,568 protein sequences; TriTrypDB; Aslett et al., 2010), supplemented with the VSG221 sequence and frequently observed contaminants (porcine trypsin, bovine serum albumin, and mammalian keratins). Search parameters specified an MS tolerance of 6 ppm, an MS/MS tolerance at 0.5 D, and full trypsin specificity, allowing for up to two missed cleavages. Carbamidomethylation of cysteine was set as a fixed modification and oxidation of methionines, N-terminal protein acetylation, and N-pyroglutamate were allowed as variable modifications. Phosphoproteomic analysis included phosphorylation of serine, threonine, and tyrosine as additional variable modifications. Peptides were required to be at least seven amino acids in length and to have a MaxQuant score >5, with false discovery rates of 0.01 calculated at the levels of peptides, proteins, and modification sites based on the number of hits against the reversed sequence database. SILAC ratios were calculated where at least one peptide could be uniquely mapped to a given protein group, and they required a minimum of two SILAC pairs. To account for any errors in the counting of the number of cells mixed, the distribution of SILAC ratios was normalized within MaxQuant at the peptide level so that the median of log2 ratios was zero, as described by Cox and Mann (2008).
Before statistical analysis, the outputs from MaxQuant were filtered to remove known contaminants and reverse sequences, and phosphorylation sites with a MaxQuant localization probability <0.75 were discarded. SILAC ratios for phosphorylation sites were calculated using only data from the phosphoproteomic experiments, and SILAC ratios for proteins were calculated using only data from the proteomic experiments. Because the heavy SILAC–labeled cells (HSTD) were derived from a single batch, they constitute an invariable internal standard that can be used to compare different experimental regimen. Thus, the measured H/L ratios (HSTD/LA, HSTD/LB, etc.) were used to calculate the changes in abundance between individual ex vivo samples (i.e., log2 LA/LB = log2 HSTD/LB – log2 HSTD/LA).
Data were visualized using Perseus 1.5.1.6 (http://www.perseus-framework.org) and further information on the identified proteins was obtained from TriTrypDB (Aslett et al., 2010). To make our data accessible to the scientific community, we uploaded our study to TriTrypDB and deposited the Thermo RAW files and search engine output into the ProteomeXchange consortium via the Pride partner repository with the data set identifier PXD002165, enabling researchers to access the data presented here.
Online supplemental material
Fig. S1 shows reproducibility of the proteomic analysis. Fig. S2 shows changes in proteome and phosphoproteome in response to CA treatment. Fig. S3 shows that observed changes in the proteome are driven by changes induced by CA treatment. Fig. S4 shows in vivo growth of ZC3H22 RNAi lines and Northern blot of the ZC3H22 transcript after RNAi. Fig. S5 shows that ZC3H22 is required for procyclic form viability but not differentiation. Table S1 shows that TbPIP39 and NOPP44/46 phosphorylation sites are unsuitable of LC-MS. Table S2 shows quantitative proteomic analysis of commitment to differentiation. Table S3 shows quantitative phosphoproteomic analysis of commitment to differentiation. Table S4 shows phosphorylation sites observed only in the “light” experimental samples analysis. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201506114/DC1.
Supplementary Material
Acknowledgments
We are grateful to Julie Young for technical support for in vivo experiments. We thank Bryan Jensen and Marilyn Parsons for providing antibody to NRK and Stephen M. Beverley and the Genome Institute, Washington University School of Medicine, for Crithidia fasciculata genome data accessed via TriTrypDB.
M.R. Domingo Sananes received a fellowship from the Centre for Immunity, Infection, and Evolution at the University of Edinburgh, supported by a Wellcome Trust Strategic Award (WT095831). Work in K. Matthews’ laboratory is supported by a Wellcome Trust Senior Investigator Award (WT103740). M. Urbaniak is supported by the Biotechnology and Biological Sciences Research Council (BB/M009556/1). M. Ferguson is supported by a Wellcome Trust Senior Investigator Award (WT101842). MS was performed in the Fingerprints Proteomics Facility at the University of Dundee, supported by Wellcome Trust Strategic Award (WT097945).
The authors declare no competing financial interests.
Footnotes
Abbreviations used in this paper:
- CA
- cis-aconitate
- CHX
- cycloheximide
- EP
- glutamic acid and proline
- LC
- liquid chromatography
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- NRK
- Nek-related kinase
- RIT-seq
- RNAi target sequencing
- SILAC
- stable isotope labeling by amino acids in cell culture
References
- Alsford S., Turner D.J., Obado S.O., Sanchez-Flores A., Glover L., Berriman M., Hertz-Fowler C., and Horn D.. 2011. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 21:915–924. 10.1101/gr.115089.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aly A.S., Vaughan A.M., and Kappe S.H.. 2009. Malaria parasite development in the mosquito and infection of the mammalian host. Annu. Rev. Microbiol. 63:195–221. 10.1146/annurev.micro.091208.073403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslett M., Aurrecoechea C., Berriman M., Brestelli J., Brunk B.P., Carrington M., Depledge D.P., Fischer S., Gajria B., Gao X., et al. 2010. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 38(Database):D457–D462. 10.1093/nar/gkp851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil S.A., Jedrychowski M., Schwartz D., Elias J.E., Villén J., Li J., Cohn M.A., Cantley L.C., and Gygi S.P.. 2004. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. USA. 101:12130–12135. 10.1073/pnas.0404720101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner J., and Clayton C.E.. 1995. The 3′-untranslated regions from the Trypanosoma brucei phosphoglycerate kinase-encoding genes mediate developmental regulation. Gene. 162:153–156. 10.1016/0378-1119(95)00366-E [DOI] [PubMed] [Google Scholar]
- Chou S., Jensen B.C., Parsons M., Alber T., and Grundner C.. 2010. The Trypanosoma brucei life cycle switch TbPTP1 is structurally conserved and dephosphorylates the nucleolar protein NOPP44/46. J. Biol. Chem. 285:22075–22081. 10.1074/jbc.M110.108860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton C., and Shapira M.. 2007. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol. Biochem. Parasitol. 156:93–101. 10.1016/j.molbiopara.2007.07.007 [DOI] [PubMed] [Google Scholar]
- Cox J., and Mann M.. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26:1367–1372. 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., and Mann M.. 2011. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10:1794–1805. 10.1021/pr101065j [DOI] [PubMed] [Google Scholar]
- Dean S., Marchetti R., Kirk K., and Matthews K.R.. 2009. A surface transporter family conveys the trypanosome differentiation signal. Nature. 459:213–217. 10.1038/nature07997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstler M., and Boshart M.. 2004. Cold shock and regulation of surface protein trafficking convey sensitization to inducers of stage differentiation in Trypanosoma brucei. Genes Dev. 18:2798–2811. 10.1101/gad.323404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstler M., Reuter G., and Schauer R.. 1992. Purification and characterization of a novel sialidase found in procyclic culture forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 54:21–30. 10.1016/0166-6851(92)90091-W [DOI] [PubMed] [Google Scholar]
- Erben E.D., Fadda A., Lueong S., Hoheisel J.D., and Clayton C.. 2014. A genome-wide tethering screen reveals novel potential post-transcriptional regulators in Trypanosoma brucei. PLoS Pathog. 10:e1004178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell J.E., and Xiong W.. 2001. Bistability in cell signaling: how to make continuous processes discontinuous, and reversible processes irreversible. Chaos. 11:227–236. 10.1063/1.1349894 [DOI] [PubMed] [Google Scholar]
- Fragoso C.M., Schumann Burkard G., Oberle M., Renggli C.K., Hilzinger K., and Roditi I.. 2009. PSSA-2, a membrane-spanning phosphoprotein of Trypanosoma brucei, is required for efficient maturation of infection. PLoS ONE. 4:e7074 10.1371/journal.pone.0007074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale M. Jr, and Parsons M.. 1993. A Trypanosoma brucei gene family encoding protein kinases with catalytic domains structurally related to Nek1 and NIMA. Mol. Biochem. Parasitol. 59:111–121. 10.1016/0166-6851(93)90012-M [DOI] [PubMed] [Google Scholar]
- Gale M. Jr, Carter V., and Parsons M.. 1994. Translational control mediates the developmental regulation of the Trypanosoma brucei Nrk protein kinase. J. Biol. Chem. 269:31659–31665. [PubMed] [Google Scholar]
- Goldenberg S., and Avila A.R.. 2011. Aspects of Trypanosoma cruzi stage differentiation. Adv. Parasitol. 75:285–305. 10.1016/B978-0-12-385863-4.00013-7 [DOI] [PubMed] [Google Scholar]
- Gruszynski A.E., van Deursen F.J., Albareda M.C., Best A., Chaudhary K., Cliffe L.J., del Rio L., Dunn J.D., Ellis L., Evans K.J., et al. 2006. Regulation of surface coat exchange by differentiating African trypanosomes. Mol. Biochem. Parasitol. 147:211–223. 10.1016/j.molbiopara.2006.02.013 [DOI] [PubMed] [Google Scholar]
- Gunasekera K., Wüthrich D., Braga-Lagache S., Heller M., and Ochsenreiter T.. 2012. Proteome remodelling during development from blood to insect-form Trypanosoma brucei quantified by SILAC and mass spectrometry. BMC Genomics. 13:556 10.1186/1471-2164-13-556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güther M.L., Urbaniak M.D., Tavendale A., Prescott A., and Ferguson M.A.. 2014. High-confidence glycosome proteome for procyclic form Trypanosoma brucei by epitope-tag organelle enrichment and SILAC proteomics. J. Proteome Res. 13:2796–2806. 10.1021/pr401209w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes P., Varga V., Olego-Fernandez S., Sunter J., Ginger M.L., and Gull K.. 2014. Modulation of a cytoskeletal calpain-like protein induces major transitions in trypanosome morphology. J. Cell Biol. 206:377–384. 10.1083/jcb.201312067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirumi H., and Hirumi K.. 1989. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 75:985–989. 10.2307/3282883 [DOI] [PubMed] [Google Scholar]
- Ishihama Y., Sato T., Tabata T., Miyamoto N., Sagane K., Nagasu T., and Oda Y.. 2005. Quantitative mouse brain proteomics using culture-derived isotope tags as internal standards. Nat. Biotechnol. 23:617–621. 10.1038/nbt1086 [DOI] [PubMed] [Google Scholar]
- Jensen S.S., and Larsen M.R.. 2007. Evaluation of the impact of some experimental procedures on different phosphopeptide enrichment techniques. Rapid Commun. Mass Spectrom. 21:3635–3645. 10.1002/rcm.3254 [DOI] [PubMed] [Google Scholar]
- Jones N.G., Thomas E.B., Brown E., Dickens N.J., Hammarton T.C., and Mottram J.C.. 2014. Regulators of Trypanosoma brucei cell cycle progression and differentiation identified using a kinome-wide RNAi screen. PLoS Pathog. 10:e1003886 10.1371/journal.ppat.1003886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabani S., Fenn K., Ross A., Ivens A., Smith T.K., Ghazal P., and Matthews K.. 2009. Genome-wide expression profiling of in vivo-derived bloodstream parasite stages and dynamic analysis of mRNA alterations during synchronous differentiation in Trypanosoma brucei. BMC Genomics. 10:427 10.1186/1471-2164-10-427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanham S.M., and Godfrey D.G.. 1970. Isolation of salivarian trypanosomes from man and other mammals using DEAE-cellulose. Exp. Parasitol. 28:521–534. 10.1016/0014-4894(70)90120-7 [DOI] [PubMed] [Google Scholar]
- Losick R., and Desplan C.. 2008. Stochasticity and cell fate. Science. 320:65–68. 10.1126/science.1147888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGregor P., Szöőr B., Savill N.J., and Matthews K.R.. 2012. Trypanosomal immune evasion, chronicity and transmission: an elegant balancing act. Nat. Rev. Microbiol. 10:431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews K.R. 2011. Controlling and coordinating development in vector-transmitted parasites. Science. 331:1149–1153. 10.1126/science.1198077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews K.R., and Gull K.. 1994. Evidence for an interplay between cell cycle progression and the initiation of differentiation between life cycle forms of African trypanosomes. J. Cell Biol. 125:1147–1156. 10.1083/jcb.125.5.1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews K.R., and Gull K.. 1997. Commitment to differentiation and cell cycle re-entry are coincident but separable events in the transformation of African trypanosomes from their bloodstream to their insect form. J. Cell Sci. 110:2609–2618. [DOI] [PubMed] [Google Scholar]
- Matthews K.R., Sherwin T., and Gull K.. 1995. Mitochondrial genome repositioning during the differentiation of the African trypanosome between life cycle forms is microtubule mediated. J. Cell Sci. 108:2231–2239. [DOI] [PubMed] [Google Scholar]
- Mitrophanov A.Y., and Groisman E.A.. 2008. Positive feedback in cellular control systems. BioEssays. 30:542–555. 10.1002/bies.20769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nett I.R., Martin D.M., Miranda-Saavedra D., Lamont D., Barber J.D., Mehlert A., and Ferguson M.A.. 2009. The phosphoproteome of bloodstream form Trypanosoma brucei, causative agent of African sleeping sickness. Mol. Cell. Proteomics. 8:1527–1538. 10.1074/mcp.M800556-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollitt L.C., MacGregor P., Matthews K., and Reece S.E.. 2011. Malaria and trypanosome transmission: different parasites, same rules?. Trends Parasitol. 27:197–203. 10.1016/j.pt.2011.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerening J.R. 2008. Uncovering mechanisms of bistability in biological systems. Curr. Opin. Biotechnol. 19:381–388. 10.1016/j.copbio.2008.06.009 [DOI] [PubMed] [Google Scholar]
- Pusnik M., Small I., Read L.K., Fabbro T., and Schneider A.. 2007. Pentatricopeptide repeat proteins in Trypanosoma brucei function in mitochondrial ribosomes. Mol. Cell. Biol. 27:6876–6888. 10.1128/MCB.00708-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roditi I., and Liniger M.. 2002. Dressed for success: the surface coats of insect-borne protozoan parasites. Trends Microbiol. 10:128–134. 10.1016/S0966-842X(02)02309-0 [DOI] [PubMed] [Google Scholar]
- Sanchez M.A., Drutman S., van Ampting M., Matthews K., and Landfear S.M.. 2004. A novel purine nucleoside transporter whose expression is up-regulated in the short stumpy form of the Trypanosoma brucei life cycle. Mol. Biochem. Parasitol. 136:265–272. 10.1016/j.molbiopara.2004.04.009 [DOI] [PubMed] [Google Scholar]
- Sbicego S., Vassella E., Kurath U., Blum B., and Roditi I.. 1999. The use of transgenic Trypanosoma brucei to identify compounds inducing the differentiation of bloodstream forms to procyclic forms. Mol. Biochem. Parasitol. 104:311–322. 10.1016/S0166-6851(99)00157-7 [DOI] [PubMed] [Google Scholar]
- Schroeder M.J., Shabanowitz J., Schwartz J.C., Hunt D.F., and Coon J.J.. 2004. A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal. Chem. 76:3590–3598. 10.1021/ac0497104 [DOI] [PubMed] [Google Scholar]
- Shimogawa M.M., Saada E.A., Vashisht A.A., Barshop W.D., Wohlschlegel J.A., and Hill K.L.. 2015. Cell surface proteomics provides insight into stage-specific remodeling of the host-parasite interface in Trypanosoma brucei. Mol. Cell. Proteomics. 14:1977–1988. 10.1074/mcp.M114.045146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin M.L., Elwood H.J., and Gunderson J.H.. 1986. Evolutionary diversity of eukaryotic small-subunit rRNA genes. Proc. Natl. Acad. Sci. USA. 83:1383–1387. 10.1073/pnas.83.5.1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szöőr B., Wilson J., McElhinney H., Tabernero L., and Matthews K.R.. 2006. Protein tyrosine phosphatase TbPTP1: a molecular switch controlling life cycle differentiation in trypanosomes. J. Cell Biol. 175:293–303. 10.1083/jcb.200605090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szöőr B., Ruberto I., Burchmore R., and Matthews K.R.. 2010. A novel phosphatase cascade regulates differentiation in Trypanosoma brucei via a glycosomal signaling pathway. Genes Dev. 24:1306–1316. 10.1101/gad.570310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szöőr B., Dyer N.A., Ruberto I., Acosta-Serrano A., and Matthews K.R.. 2013. Independent pathways can transduce the life-cycle differentiation signal in Trypanosoma brucei. PLoS Pathog. 9:e1003689 10.1371/journal.ppat.1003689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thingholm T.E., Jørgensen T.J., Jensen O.N., and Larsen M.R.. 2006. Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nat. Protoc. 1:1929–1935. 10.1038/nprot.2006.185 [DOI] [PubMed] [Google Scholar]
- Trenaman A., Hartley C., Prorocic M., Passos-Silva D.G., van den Hoek M., Nechyporuk-Zloy V., Machado C.R., and McCulloch R.. 2013. Trypanosoma brucei BRCA2 acts in a life cycle-specific genome stability process and dictates BRC repeat number-dependent RAD51 subnuclear dynamics. Nucleic Acids Res. 41:943–960. 10.1093/nar/gks1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner C.M.R. 1992. Cell cycle co-ordination in trypanosomes. Parasitol. Today (Regul. Ed.). 8:3–4. 10.1016/0169-4758(92)90295-D [DOI] [PubMed] [Google Scholar]
- Tyson J.J., Chen K.C., and Novak B.. 2003. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 15:221–231. 10.1016/S0955-0674(03)00017-6 [DOI] [PubMed] [Google Scholar]
- Urbaniak M.D., Guther M.L., and Ferguson M.A.. 2012. Comparative SILAC proteomic analysis of Trypanosoma brucei bloodstream and procyclic lifecycle stages. PLoS ONE. 7:e36619 10.1371/journal.pone.0036619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbaniak M.D., Martin D.M., and Ferguson M.A.. 2013. Global quantitative SILAC phosphoproteomics reveals differential phosphorylation is widespread between the procyclic and bloodstream form lifecycle stages of Trypanosoma brucei. J. Proteome Res. 12:2233–2244. 10.1021/pr400086y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassella E., Reuner B., Yutzy B., and Boshart M.. 1997. Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J. Cell Sci. 110:2661–2671. [DOI] [PubMed] [Google Scholar]
- Walker G., Dorrell R.G., Schlacht A., and Dacks J.B.. 2011. Eukaryotic systematics: a user’s guide for cell biologists and parasitologists. Parasitology. 138:1638–1663. 10.1017/S0031182010001708 [DOI] [PubMed] [Google Scholar]
- Wang L., Walker B.L., Iannaccone S., Bhatt D., Kennedy P.J., and Tse W.T.. 2009. Bistable switches control memory and plasticity in cellular differentiation. Proc. Natl. Acad. Sci. USA. 106:6638–6643. 10.1073/pnas.0806137106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz E., Leal S., Ochatt C., and Cross G.A.. 1999. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99:89–101. 10.1016/S0166-6851(99)00002-X [DOI] [PubMed] [Google Scholar]
- Wiśniewski J.R., Zougman A., Nagaraj N., and Mann M.. 2009. Universal sample preparation method for proteome analysis. Nat. Methods. 6:359–362. 10.1038/nmeth.1322 [DOI] [PubMed] [Google Scholar]
- Xiong W., and Ferrell J.E. Jr. 2003. A positive-feedback-based bistable ‘memory module’ that governs a cell fate decision. Nature. 426:460–465. 10.1038/nature02089 [DOI] [PubMed] [Google Scholar]
- Ziegelbauer K., Quinten M., Schwarz H., Pearson T.W., and Overath P.. 1990. Synchronous differentiation of Trypanosoma brucei from bloodstream to procyclic forms in vitro. Eur. J. Biochem. 192:373–378. 10.1111/j.1432-1033.1990.tb19237.x [DOI] [PubMed] [Google Scholar]
- Zilberstein D., and Shapira M.. 1994. The role of pH and temperature in the development of Leishmania parasites. Annu. Rev. Microbiol. 48:449–470. 10.1146/annurev.mi.48.100194.002313 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.