

Summary
Although transgene-based reporter gene assays have been used to discover small molecules targeting expression of cancer-driving genes, the success is limited due to the fact that reporter gene expression regulated by incomplete cis-acting elements and foreign epigenetic environments does not faithfully reproduce chemical responses of endogenous genes. Here we present an IRES-based strategy for bicistronically co-expressing reporter genes with an endogenous gene in the native gene locus, yielding an in-situ reporter assay closely mimicking endogenous gene expression without disintegrating its function. This strategy combines the CRISPR-Cas9-mediated genome-editing tool with the recombinase-mediated cassette exchange technology, and allows for rapid development of orthogonal assays for excluding false hits generated from primary screens. We validated this strategy by developing a screening platform for identifying compounds targeting oncogenic eIF4E, and demonstrated that the novel reporter assays are powerful in searching for transcription-targeted lead compounds with high confidence.
Graphical Abstract
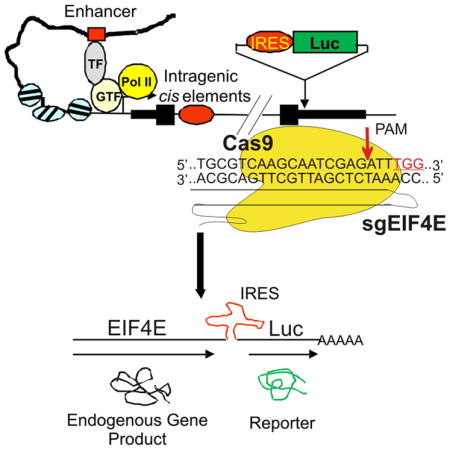
Introduction
Aberrant gene expression is a hallmark of cancer, and often drives growth, survival, and metastasis of malignant cells. Since cancer cells frequently develop dependency on altered expression of cancer-driving genes, it is generally believed that therapeutic agents capable of rectifying these abnormalities are promising in curing this deadly disease (Yan and Paul, 2013). The eukaryotic translation initiation factor 4E (eIF4E), for instance, is frequently overexpressed in human cancer and contributes to cancer development by selectively promoting translation of genes essential for cancer cell growth and survival (e.g., c-myc, VEGF, BCL-2) (Graff et al., 2008). As eIF4E hyperactivity is the convergence point of common oncogenic pathways, downregulation of EIF4E expression could be an ideal strategy for therapeutic intervention of cancer (Hsieh and Ruggero, 2010; Bitterman and Polunovsky, 2012). Indeed, an EIF4E-specific antisense oligonucleotide was shown to inhibit growth of a wide range of cancer cells and has entered clinical trials (Graff et al., 2007).
However, aberrant gene expression, which is often a consequence of dysregulated transcription, is traditionally considered as a “undruggable” target (Yan and Paul, 2013), mainly due to the lack of reliable high-throughput screening (HTS) assays that can be employed to search for small molecules regulatory for gene expression. Reporter assays whereby bioluminescent reporter genes (e.g., firefly luciferase, FLuc) are typically fused to a cloned promoter and stably integrated into a random genomic location provide a rapid, convenient, and cost-efficient means to monitor alternations in gene expression upon chemical treatments, and have been successfully used in high-throughput drug discovery (Rapisarda et al., 2002; Nakahara et al., 2007). Given that randomly-integrated reporters are often epigenetically silenced by flanking condensed chromatin (Yan and Boyd, 2006), we recently developed a technology that can integrate a reporter gene into a predefined permissive genomic location through homologous recombination mediated by the Flp recombinase (Yan et al., 2004; Nair et al., 2008). Despite these successes, current reporter assays are mainly based on cloned, transgenic promoters, and often unreliable in reproducing responses of endogenous genes to chemical treatments due to two major limitations. Firstly, cloned promoters often lack essential cis-acting elements far-removed from transcription start sites (TSS) (e.g. enhancers). While they may localize more than 40 kb apart from TSS, these distal cis-regulatory elements interact with proximal promoters through DNA looping, strongly influencing activities of the latter to drive gene expression (Pennacchio et al., 2013; Levine et al., 2014). Consequently, drug screens based on cloned promoters would miss a substantial number of small molecules regulatory for distal cis-elements. Secondly, transgenic promoters are integrated into genomic locations divergent from their native counterparts. Whereas a previously-“naked” (non-chromatinized) transgenic promoter is assembled into chromatin structurally similar to the integration site upon integration (Yan and Boyd, 2006), the “foreign” chromatin environment could alter the reporter gene expression in a manner atypical of the endogenous gene. Accordingly, screens based on transgenic promoters would yield high rates of false positives while agents targeting epigenetic mechanisms for regulating endogenous gene expression would be missed. Although recent studies attempting to insert reporter genes into sites immediate downstream of endogenous promoter (Lyssiotis et al., 2009) partly address these concerns, the close proximity of a large size of exogenous DNA to TSS results in not only disintegration of the endogenous gene, but also changes in transcription initiation and/or enhancer looping essential for gene transcription (Pennacchio et al., 2013).
To address these limitations, we employed emerging genome-editing tools to develop a novel reporter assay whereby a reporter gene was engineered into a genomic site immediately downstream of the coding region of an endogenous gene, and co-expressed bicistronically with the endogenous gene as a single transcript under the control of the native transcriptional regulatory machinery. We demonstrated that this bicistronic in-situ reporter assay is powerful in searching for transcription-targeted lead compounds for treating cancer.
Results
Bicistronic co-expression of a reporter gene with an endogenous gene via IRES
Given the above-discussed limitations of reporter assays, we sought to develop a reliable reporter assay whereby the reporter gene expression would closely mimic endogenous gene expression under chemical treatments. Our strategy is to insert the FLuc gene led by an internal ribosome entry site (IRES) into a genomic site immediately downstream of the coding region of an endogenous gene (e.g., EIF4E) (Figure 1). We reasoned that IRES could allow expression of a single transcript comprised of the reporter gene and the endogenous gene under the control of the endogenous gene promoter, the distal cis-regulatory elements, and the native chromatin environment (Figure 1), thereby yielding a reliable screening assay faithfully reproducing responses of the endogenous gene to chemical treatments.
Figure 1. The schematic shows co-expression of a bicistronic in-situ reporter with an endogenous gene under the control of the native transcriptional regulatory machinery.
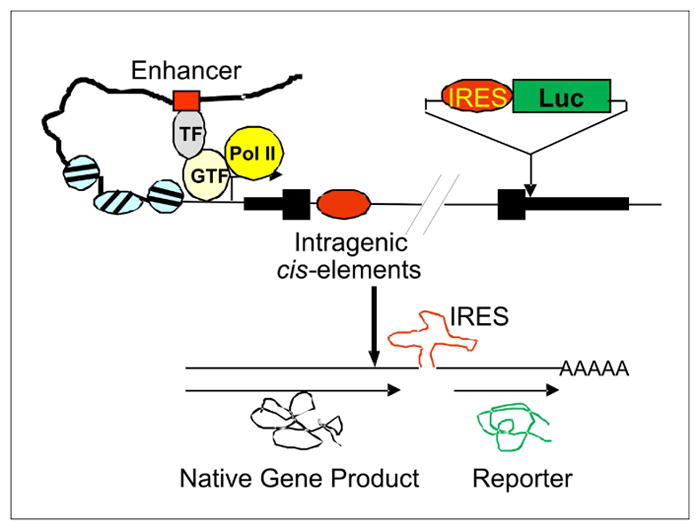
IRES, internal ribosome entry site; Luc, luciferase gene; TF, transcription factors; GTF, general transcription factors; PolII, RNA polymerase II.
We chose to knock the IRES-FLuc DNA into the native EIF4E gene locus given that eIF4E-targeted drugs are highly desirable for cancer treatments (Bitterman and Polunovsky, 2012). We took a two-step strategy to attain this goal. We first employed the emerging CRISPR-Cas9 genome-editing tool (Mali et al., 2013) to insert a fusion selection gene (tk-ble) into a site between the stop codon (TAA) and the polyadenylation sites of the EIF4E gene, and then replaced the selection gene with the FLuc gene through Flp-mediated cassette exchange (RMCE) (Baer and Bode, 2001) (Figure 2A). The tk-ble fusion gene confers Zeocin resistance and ganciclovir (GCV) toxicity and thus respectively allows for positive selection for EIF4E-targeting and negative selection for RMCE events. RMCE can mediate rapid insertions of other reporter genes into the same targeted locus thereby facilitating the development of orthogonal screening assays in our later experiments (see below).
Figure 2. Genome editing by CRISPR-Cas9 followed by RMCE generates cells harboring a reporter gene in the endogenous EIF4E locus.

(A) The schematic showing the two-step strategy for inserting the IRES-FLuc cassette into the EIF4E gene locus through CRISPR-Cas9 and RMCE. DSB, DNA double-strand break; LA, left homology arm; RA, right homology arm; PAM, protospacer adjacent motif; F and F3, wild-type and mutant FRT sites; sgEIF4E, EIF4E-specific sgRNA; pA, polyadenylation signal; TAA, the stop codon. (B) Identification of Zeocin-resistant clones carrying the tk-ble gene in the targeted EIF4E locus. Genomic DNAs from resistant clones were subjected to PCR using a primer pair RA-F and RA-R indicated in (B). Positive clones were expected to generate a ~1.5-kb fragment. (C) The tk-ble gene was replaced by the IRES-FLuc cassette in E8-FLuc clones confirmed by PCR. The primers are indicated in (B). The E8 clone carrying the tk-ble gene in the targeted EIF4E locus was chosen for RMCE. (D) Northern blotting confirmed co-expression of the FLuc gene and the endogenous EIF4E gene as single transcripts. Arrows indicate the fused, bicistronic transcripts. (E) Immunoblotting detected the eIF4E protein. No fusion of eIF4E to FLuc was found in F8-FLuc cells. (F) FLuc was highly expressed in recombinant cells. Clones were lysed for firefly luciferase activity assays. The FLuc expression level was comparable among clones. (G) E8-Fluc clones exhibited similar responses to chemical treatments. The indicated clones were treated with 2.5 μM of NSC607097 for 16 h for luciferase activity assays. See also Figure S1.
To insert the selection gene into the EIF4E gene locus, we constructed an editing vector that carries the tk-ble gene flanked by a wild-type (F) and a mutated (F3) FRT fragment (Schlake and Bode, 1994) (Figure S1A), and then cloned the EIF4E homology arms into the vector to generate a targeting vector pEIF4E-Target (Figure 2A). We transfected the targeting vector along with a single guided RNA (sgRNA), which specifically recognizes a region downstream of the EIF4E stop codon (Figure 2A), into human fibrosarcoma HT1080 cells. The sgRNA could guide the Cas9 nuclease to generate a double-strand break (DSB) thereby facilitating high-efficient integration of the tk-ble gene through homology-directed repair (Figure 2A) (Ran et al., 2013). Indeed, we identified 8 EIF4E-targeted clones from 62 Zeocin-resistant clones by PCR (Figure 2B). The targeting efficiency was 12.9%.
We next employed the RMCE technology to generate recombinant cells co-expressing FLuc and the endogenous EIF4E gene as single transcripts. Towards this end, we co-transfected an EIF4E-targeted clone, E8, with a cassette-exchange vector carrying the IRES-FLuc cassette flanked by the F and F3 fragments (Figure S2B, and Figure 2A), and a Flp-expressing plasmid, for GCV selection. We found that the tk-ble gene was replaced by the IRES-FLuc DNA in almost all tested GCV-resistant clones evidenced by PCR amplification of a DNA fragment composed of the EIF4E and the IRES fragment (Figure 2C). Northern blotting assays detected 3 mRNA bands that were hybridized to both EIF4E and FLuc probes (Figure 2D, lanes 3 and 6, arrows), demonstrating that the FLuc gene was expressed as bicistronic transcripts fused to the EIF4E mRNA in the recombinant cells. The multiple double-hybridized bands might be generated from fusion of the FLuc mRNA to different EIF4E splice variants. Indeed, the EIF4E gene was transcribed as several mRNAs evidenced in lane 1, Figure 2D. On the contrary, immunoblotting assays showed that the FLuc gene was not expressed as a protein fused to eIF4E (Figure 2E, lane 3). Whereas these recombinant cells expressed a high level of FLuc (Figure 2F), their responses to a small molecule (NSC607097) capable of decreasing EIF4E expression (see below) were comparable. Therefore, we developed a method allowing for highly-efficient generation of recombinant cells expressing a reporter gene under the control of the authentic regulatory mechanism for endogenous EIF4E gene expression.
Bicistronic reporters yield screens with a decreased false-hit rate
We reasoned that reporter genes expressed under this condition would better mimic chemical responses of endogenous genes, and accordingly, chemical screening based on these recombinant cells would yield fewer false hits with improved success rates. To test this, we treated one of these recombinant cells (E8-FLuc) with ~4,800 small molecules from the NCI and a commercial (Chembridge) chemical library (Figure 3A). We were interested in identifying compounds that decrease EIF4E expression as they could be further developed into agents for treating cancer overexpressing eIF4E. The Z′-factor of the reporter assay equaled 0.67 (Figure S2A), indicating its suitability for HTS. As a comparison, we also generated cells carrying a FLuc gene driven by a cloned EIF4E promoter (−1512 ~ −1) in a defined, foreign genomic location (F55-pEIF4E-luc) (Figure 3A) using a previously-developed, Flp-based strategy (Figure S2B) (Nair et al., 2008), and subjected the cells to HTS similarly. We identified 11 hits from the E8-FLuc-based screen and 28 hits from the screen using the F55-pEIF4E-luc cells (Figure 3B, Table S1); 10 compounds were found to decrease the firefly luciferase activity in both assays (Figure 3C). We carried out qRT-PCR to validate these hits, and found 6 hits from the E8-FLuc screen were true positives that indeed decreased EIF4E expression in cancer cells (Figure 3D, and Figure S3A). These hits decreased the expression of the bicistronic FLuc reporter in a concentration-dependent manner (Figure S3B), and these effects were unlikely caused by cytotoxicity (Figure S3C). Similar effects were also observed in another independent clone (F89-FLuc) carrying the same bicistronic reporter (Figure S3B). Previously, these compounds were found to have distinct biological activities, including transcription inhibition (NSC607097 and NSC146109) (Chau et al., 2005; Wang and Yan, 2011), direct DNA (NSC71795) (Stiborova et al., 2001) or protein binding (NSC607097 and NSC255109) (Schulte and Neckers, 1998; Kahsai et al., 2006), metal chelation (NSC86372) (Burnett et al., 2003), and protein kinase inhibition (NSC56346) (Gschwendt et al., 1994) (Table S2). Interestingly, in one of our early studies, we demonstrated that NSC146109 inhibits transcription of another cancer-causing gene MDMX (Wang and Yan, 2011), suggesting that EI4E and MDMX might share a common mechanism for transcriptional regulation. Of note, it is unlikely that these compounds are pan-assay interference compounds (PAINS) (Baell and Holloway, 2010; Baell and Walters, 2014), as we did not identify these compounds, except NSC146109, as positive hits in our previous screen using a FLuc reporter driven by a cloned MDMX promoter (Wang et al., 2011). Indeed, we confirmed that one of these hits, NSC607097, decreased EIF4E expression in several other human cancer cells (Figure S3D), and that it was not a general transcription inhibitor as it did not alter the MDMX promoter activity (Figure S3E). Out of these compounds, 5 were also identified as positive in the F55-pEIF4E-luc-based screen (Figure 3E). However, NSC56346, one validated EIF4E inhibitor, was missed in the latter screen using the cloned, transgenic EIF4E promoter (Figure 3E), arguing for the notion that cis-regulatory elements modulated by some chemicals might be omitted from cloned promoters. Therefore, the false-positive rate of the screen based on the in-situ reporter was 45.4%, which was significantly lower than that (82.1%) of the F55-pEIF4E-luc-based screen (p=0.044, Fisher’s Exact test). Our results thus demonstrated that the screening assay based on the bicistronic in-situ reporter was more efficient.
Figure 3. A drug screen using the bicistronic in-situ reporter assays identifies EIF4E inhibitors with a significantly reduced false positive rate.

(A) The schematic showing the screening strategy. A random E8-FLuc clone and a clone (F55-pEIF4E-luc) carrying a ~ 1.5 kb EIF4E promoter in a permissive genomic site were treated with library chemicals in microplates for luciferase activity assays. (B) The results of E8-FLuc-based screen (red) and F55-pEIF4E-luc-based screen (blue). Recombinant cells in 96-well plates were treated with ~ 4,800 compounds (2.5 μM) for 16 h, and then lysed for firefly luciferase activity assays. The relative luciferase activities were converted into logarithm values (binary logarithm, i.e., log2) and plotted for each compound. The cutoff values set for positives are ±0.5849, i.e., either decrease to at least 66.7% or increase to at least 150% of the DMSO group, and indicated by the dotted lines. Chemicals that decreased the luciferase activity due to cytotoxicity were identified by MTT assays, and excluded from further investigation in this study. These chemicals were not shown in this graph. (C) Venn Diagram showing the numbers of hits (i.e., decreasing the FLuc activity) from the screens using E8-FLuc or F55-pEIF4E-luc cells. (D) qRT-PCR validation of the hits from two screens. The dotted lines indicate the cutoff (<0.667) for positives. Error bars represent SD for three replicate measurements. (E) Venn Diagram showing the validated EIF4E inhibitors. See also Figure S2 and Figure S3.
Orthogonal assays readily developed through RMCE identifies false hits targeting luciferase
However, up to 45% of the hits from the new screening assay were still false positives. These artifacts might be caused by small molecules that interfere with the reaction catalyzed by FLuc, or affect the stability of the FLuc protein/mRNA (Feng et al., 2007; Auld et al., 2008). Indeed, a recent study suggests that more than 40% of positive hits from FLuc-based assays could be FLuc inhibitors, and many of these FLuc inhibitors counterintuitively increase FLuc activity (Thorne et al., 2010). An approach to identify these false positives is to counter-screen them using an orthogonal assay that measures the activity of a reporter distinct from FLuc (e.g., Renilla luciferase or RLuc) (Auld et al., 2008; Thorne et al., 2010). To test this strategy, we employed RMCE (Figure 4A) to generate recombinant cells carrying the RLuc gene in the same genomic location as the E8-FLuc cells by transfecting E8 cells with a new cassette-exchange construct carrying an IRES-RLuc cassette (Figure S2C). The new recombinant cells (E8-RLuc) verified by PCR (Figure 4B) responded to treatments with the validated EIF4E inhibitor NSC607097 in a way as same as E8-FLuc cells (Figure 4C). Interestingly, none of the tested compounds that increased the FLuc activity in the primary screen altered the RLuc activity in the new recombinant cells (Figure 4D). Moreover, whereas the 6 validated EIF4E inhibitors could decreased the RLuc activity as expected, 4 out of 5 false positives identified in the primary screen did not alter the RLuc activity (Figure 4D). The only false hit that was missed by the orthogonal screening assay, NSC321239, was found to directly inhibit both the FLuc and the RLuc activity (Figure 4E). Thus, an orthogonal screening assay using a RLuc reporter gene engineered to reside in the same genomic location as FLuc could identify and exclude a majority of false positives. The RMCE technology employed in our cell-engineering strategy allows for fast, convenient development of such an orthogonal screening assay. Overall, the screens based on our new reporter assays yielded a false-positive rate that was lower than 10%.
Figure 4. An orthogonal screening assay based on RLuc inserted in the same EIF4E locus identifies a majority of false hits.

(A) Diagram showing that RMCE mediates rapid insertion of the RLuc gene into the same genomic location as FLuc. (B) PCR results confirmed the replacement of the tk-ble gene with RLuc in E8-RLuc cells. (C) E8-RLuc cells responded to chemical treatments as same as E8-FLuc cells. Cells were treated with NSC607097 for 16 h for luciferase activity assays. (D) Effects of primary hits (including those compounds that increased the FLuc activity by at least 1 fold) on EIF4E mRNA level and reporter gene activity. The only false hit (NSC321239) that could not be identified by the orthogonal assay is indicated by the red arrow. (E) NSC321239 is an inhibitor of both FLuc and RLuc. Lysates from cells expressing FLuc or RLuc were incubated with 2.5 μM of NSC321239, or Pifithrin-α (a known FLuc inhibitor), on ice before assaying for FLuc or RLuc activity. Error bars represent SD for three replicate measurements.
Discussion
Targeting aberrant expression of cancer-driving genes using small molecules is a promising anti-cancer strategy (Yan and Paul, 2013), yet its success is limited due to lack of reliable HTS assays. In this report, we present a proof of principle for a versatile strategy that can be employed to engineer cultured cells and allow them to express reporter genes under control of the native mechanism for regulation of endogenous gene expression, thereby yielding screening assays closely mimicking responses of endogenous genes to chemical treatments. This strategy also allows for convenient insertions of different reporter genes into the same genomic locations, and therefore facilitates rapid development of orthogonal screening assays for identification of false positives generated from primary screens. We demonstrated that these bicistronic in-situ reporter assays are powerful in searching for therapeutic agents targeting abnormal gene expression in cancer with high confidence and improved success rates.
Previously, a drug-screening platform based on a FLuc gene knocked into the genomic site immediately downstream of the Nanog promoter was developed for identifying small molecules that can increase Nanog expression (Lyssiotis et al., 2009). Although this knock-in reporter assay was successful in identifying compounds that functionally replace KLF4 to induce pluripotent stem cells, its development utilizes embryonic stem cells and requires production of genetically-engineered mice (Lyssiotis et al., 2009). Moreover, the platform has limitations in that (1) the endogenous Nanog gene was disrupted, and that (2) it is highly likely that the exogenous DNA (i.e., FLuc-pA) proximal to TSS influences transcription initiation and/or the interplay between the promoter and distal cis-acting elements essential for gene expression (Levine et al., 2014). A recent reporter system wherein a luciferase gene was inserted into the CCND1 exon 1 by a zinc finger nuclease (ZFN) has the same limitations, although the reporter gene was expected to be co-expressed with CCND1 through translational skipping (Samsonov et al., 2013). On the contrary, knocking reporter genes into 3′-untranslated regions (3′-UTRs), which are often distant from TSS, maintains the integrity of endogenous genes thereby minimizing disturbances of their expression by genome editing. Indeed, we found that the insertion of IRES-FLuc into the EIF4E locus did not alter eIF4E expression in the recombinant cells (Figure 2E). Therefore, bicistronic co-expression of reporter genes appears to be more advantageous over currently-available assays for drug screening.
We chose IRES rather than 2A peptides for reporter co-expression as the latter, although smaller in size, add extra residues to endogenous proteins while the 2A peptide sequences need to be seamlessly fused with the EIF4E and FLuc coding sequence (de Felipe et al., 2006). Although IRES-dependent translation of reporter genes might not be as efficient as that of endogenous genes (Martin et al., 2006), bioluminescent reporter assays are highly sensitive and thus would allow for readily measuring alterations in reporter gene expression upon chemical treatments. Indeed, the luminescence reading reached to 5 digits in our assays (Figure 2F).
Genome editing including inserting exogenous DNA into endogenous gene loci in cultured cells can be achieved through homologous recombination mediated by recombinant adenoassociated viruses (rAAV) or artificial gene-specific nucleases (e.g., ZFN and TALEN) (Khan et al., 2011; Urnov et al., 2010; Bogdanove and Voytas, 2011). However, these approaches are either laborious (e.g, ZFN and TALEN), or low efficient (e.g., rAAV). Because of its simplicity in use and high efficiency in gene targeting, the CRISPR-Cas9 technology has quickly emerged as a powerful tool for genome editing (Mali et al., 2013; Ran et al., 2013). Indeed, we readily obtained 8 EIF4E-targeted clones in a single targeting experiment. In contrast, using a rAAV-based approach, we only identified 1 positive clone from 95 Zeocin-resistant clones (data not shown). Of note, the editing vector that we developed contains the backbone of a rAAV vector (see Experimental Procedures) and thus can also be packed into viral particles for rAAV-mediated targeting. It is worth noting that the CRISPR-Cas9 technology currently suffers from a limitation that the Cas9 nuclease may generate double-strand breaks (DSBs) at multiple unintended genomic sites owing to short recognition sequences of guided RNAs, which can lead to off-target editing. Whereas small insertions or deletions (indels) are produced by non-homologous end joining at DSBs (Ran et al., 2013), however, homologous recombination mediated by the EIF4E homology arms could efficiently prevent insertions of the selection gene (tk-ble) into off-target sites. Indeed, while off-target integration events could be readily identified and excluded by PCR screening, our recombinant cells expressed the FLuc gene as single transcripts fused with EIF4E (Fig 2D), indicating that the reporter gene was only inserted into the intended genomic site. Probable generation of indels in other genomic sites can interfere with gene functions, but might be least problematic for HTS studies as the possibility that indels alter pathways regulatory for expression of the genes of interest is remote.
We also employed RMCE to develop our in-situ reporter assays. This technology utilizes the yeast Flp recombinase to mediate cassette exchange through homologous recombination between two FRT pairs (Schlake and Bode, 1994; Baer and Bode, 2001), resulting in rapid insertion of any reporter genes into a FRT-flanked genomic site (Figure 2A). It is therefore possible to readily re-engineer recombinant cells by replacing one reporter gene with another, generating orthogonal assays that are critical to the success of reporter-based drug screening (Cheng and Inglese, 2012). RMCE also allows for convenient generation of reporter cells carrying improved reporters, such as codon-optimized, destabilized, or secreted luciferases, and thus provides maximal flexibility for HTS applications. As RMCE excises the selection gene from the target site (Figure 2A), it is also possible to engineer cells for multiple times to insert two or more reporter genes into distinct gene loci via different FRT combinations (Baer and Bode, 2001), and accordingly multiplex reporter-based HTS assays for high-effective drug discovery.
During the preparation of this manuscript, a TALEN-based genome-editing strategy was reported to generate HTS assays wherein a luciferase gene was engineered into the 3′-UTR and co-expressed with the endogenous PMP22 gene through a 2A peptide (Inglese et al., 2014). Interestingly, this most recent research also identified a compound that was escaped from a previous screen based on a randomly-integrated reporter (Inglease et al., 2014). Therefore, HTS assays built on co-expressed in-situ reporters appear to be more reliable for searching for small molecules targeting transcription.
Significance
The success of transcription-targeted therapy is hindered by lack of reliable reporter gene assays for high-throughput drug screening. Traditional assays do not faithfully reproduce chemical responses of endogenous genes as they are based on cloned, transgenic promoters, which often contain incomplete cis-acting elements and are also affected by flanking epigenetic factors. The novel genome-editing strategy presented herein provides a rapid and efficient means to generate recombinant cells carrying reporter genes bicistronically co-expressed with endogenous genes under control of native transcriptional regulatory machineries, yielding a powerful drug-screening platform for discovering lead compounds targeting aberrant gene transcription in cancer and other human diseases.
Experimental Procedures
Vector Construction
Standard molecular cloning methods were used for vector construction. To construct the Editing Vector (pAAV-F-TKZeo-F3), we first PCR amplified the tk-ble fusion gene from pORF9-HSV-tk::Shble (InvivoGen), and then cloned it to pAAV-TK-Acceptor (Kim et al., 2008) after excising the loxP-Neo-loxP cassette from the latter plasmid. Oligonucleotides containing the wild-type FRT site and the mutated F3-FRT site (Schlake and Bode, 1994) were then inserted into the plasmid at the SpeI/BamH I site and the SacII/AvrII site, respectively. To construct the EIF4E-targeting vector, the left and right homology arms were amplified by PCR and respectively cloned into the AscI/Spe I site and the EcoRI/AvrII site of the Editing Vector. To construct the Cassette-exchange Vector (pF-luc2pA-F3), the IRES fragment was amplified from p414(Liao et al., 2011) by PCR, cloned into pGL4.10 (Promega) (SacI/XhoI sites), followed by addition of the F and F3 fragments to sites flanking the luc2 gene (SpeI/SacI and BamHI/SalI sites, respectively). The luc2 gene in this plasmid was then replaced with a RLuc gene amplified from pRL-TK (Promega), generating the Cassette-exchange Vector for RLuc (pF-RLucpA-F3). Table S3 lists the sequences of PCR primers used for cloning or other applications described below.
CRISPR-Cas9-based Genome Editing
The CRISPR-Cas9 genome editing tool developed by the Zhang laboratory (Ran et al., 2013) was employed to insert the tk-ble gene into the EIF4E locus. We first identified a sgRNA specifically-targeted region (5′-gcgtcaagcaatcgagatt-3′) immediate downstream of the stop codon of the EIF4E gene, synthesized and ligated oligonucleotides containing this sequence to the pSpCa9(BB)-2A-Puro plasmid (Ran et al., 2013). HT1080 cells (9×105) in a 60-mm dish were transfected with 2 μg of the sgRNA-expressing plasmid and 4 μg of the EIF4E targeting vector using Lipofectamine 2000 (Invitrogen), and then trypsinized and re-plated into three 100-mm dishes 2 days later. After selected with 0.5 μg/ml of puromycin for 3 days, transfected cells were cultured in Zeocin-containing (175 μg/ml) medium until single clones grew up. These resistant clones were plated in 96-well plates, and lysed in 50 μl of a buffer containing 10 mM Tris-HCl, pH 7.5, 10 mM EDTA, 10 mM NaCl, 0.5% SDS and 1 mg/ml proteinase K at 37°C overnight. Genomic DNAs were then precipitated by adding 100 μl of cold NaCl/ethanol mixture (0.075M NaCl), washed with 75% ethanol, air-dried, and dissolved in 35 μl of TE buffer for PCR screening for targeting events using the primers listed in Table S3.
Flp-mediated Cassette Exchange
E8 (HT1080) cells in a 60-mm dish were transfected with 4 μg of pCAGGS-Flpe (Gene Bridges) and 2 μg of pF-luc2pA-F3, or pF-RLucpA-F3, for 2 days, and then re-plated into 100-mm dishes at a density of 5×104 per dish and cultured in a medium containing 7.5 μg/ml of GCV for 2~3 weeks. Resistant clones were expanded, and lysed for luciferase activity assays. Genomic DNAs were also prepared as described above and subjected to PCR to confirm the replacement of the tk-ble gene.
Northern Blotting and qRT-PCR
Total RNA was prepared using the Trizol Reagent (Life Technologies). For Northern blotting, denatured RNA samples were resolved in 1% agarose, transferred to Hybond N+ nylon membranes, and then hybridized with [32P]-labeled EIF4E fragment amplified by PCR. After extensive washes, the membrane was wrapped with plastic wrap, and subjected to autography. The EIF4E probe was then stripped by incubating the membrane with boiled 0.1% of SDS for 1 h, and re-hybridized with [32P]-labeled FLuc gene fragment for detection of FLuc mRNA. For qRT-PCR, total RNAs (1 μg) were reverse transcribed using the DyNAmo cDNA synthesis Kit. 1 μl of cDNA were then subjected to quantitative real-time PCR assay in a total of 20 μl using the SYBR Green PCR reagent (Qiagen) and the StepOne Plus Real-time PCR system as described previously (Yan and Boyd, 2006). The EIF4E mRNA level was normalized to the GAPDH mRNA level.
Chemical library Screening
This was carried out as described elsewhere (Nair et al., 2008). Briefly, cells (1.2×104/well in 100 μl medium) in 96-well plates were treated with 2.5 μM of chemicals (0.5 μl) from the NCI Diversity-Set, the NCI Natural Products set, and a commercial (Chembridge) chemical library, for 16 h, and then washed with PBS using the Aquamax Plate Washer (Molecular Devices). In each plate, the non-response/negative control DMSO (0.5 μl) was added into wells A1, C1, E1, G1, B12, D12, F12, while cells in wells B1, D1, F1, H1, A12, C12, E12 and G12 were treated with the positive control actinomycin D (1 μM) or NSC607097 (2.5 μM). After the cells werProde lysed in 80 μl of Lysis buffer (1% Triton X-100, 25 mM Gly-Gly, pH 7.8, 15 mM MgSO4, and 4 mM EDTA) at room temperature for 20 min, 10 μl of cell lysates were dispended into white 96-well plates and then mixed with 50 μl of luciferase substrates (Promega) for luminescence reading using the SpectraMax L luminometer (Molecular Devices). The average luminescence intensity of DMSO-treated wells in each plate was used to calculate the relative reporter activity (relative luminescence) for each well treated with tested chemicals.
Supplementary Material
Highlights.
Reporter assays are unreliable due to ectopic integration and lack of enhancers;
The CRISPR-Cas9 system is employed to develop bicistronic in-situ reporter assays;
Orthogonal assays can be developed via recombinase-mediated cassette exchange;
Bicistronic reporter assays are powerful in drug discovery with high confidence.
Acknowledgments
This work was supported by NIH/NCI grants R01CA164006 and R01CA139107 to C.Y., and R01CA118450 to S.Y.S. We thank Dr. Todd Waldman for providing pAAV-TK-Acceptor, Dr. Feng Zhang for the pSpCa9(BB)-2A-Puro plasmid.
Footnotes
Author contributions
C.Y. conceived and designed the experiments, carried out vector construction and cell engineering, and wrote the manuscript. L.L. performed a part of plasmid/cell engineering, carried out chemical screening, and validated hits with the help of H.D. and X.C. S-Y.S. cloned the EIF4E promoter. GL helped in the design of the IRES strategy. H.D., X.C. and S-Y.S. commented on assay development and edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Auld DS, Southall NT, Jadhav A, Johnson RL, Diller DJ, Simeonov A, Austin CP, Inglease J. Characterization of chemical libraries for luciferase inhibitory activity. J Med Chem. 2008;51:2372–2386. doi: 10.1021/jm701302v. [DOI] [PubMed] [Google Scholar]
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Baell J, Walters MA. Chemical con artists foil drug discovery. Nature. 2014;513:481–483. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- Baer A, Bode J. Coping with kinetic and thermodynamic barriers: RMCE, an efficient strategy for the targeted integration of transgenes. Curr Opin Biotech. 2001;12:473–480. doi: 10.1016/s0958-1669(00)00248-2. [DOI] [PubMed] [Google Scholar]
- Bitterman PB, Polunovsky VA. Attacking a nexus of the oncogenic circuitry by reversing aberrant eIF4F-mediated translation. Mol Cancer Ther. 2012;11:1051–1061. doi: 10.1158/1535-7163.MCT-11-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanove AJ, Voytas DF. TAL effectors: Customizable proteins for DNA targeting. Science. 2011;333:1843–1846. doi: 10.1126/science.1204094. [DOI] [PubMed] [Google Scholar]
- Burnett JC, Schmidt JJ, Stafford RG, Panchal RG, Nguyen TL, Hermone AR, Vennerstorm JL, McGrath CF, Lane DJ, Sausville EA, Zaharevitz DW, Gussio R, Bavari S. Novel small molecule inhibitors of botulinum neurotoxin A metalloprotease activity. Biochem Biophys Res Comm. 2003;310:84–83. doi: 10.1016/j.bbrc.2003.08.112. [DOI] [PubMed] [Google Scholar]
- Chau NM, Rogers P, Aherne W, Carroll V, Collins I, McDonald E, Workman P, Ashcroft M. Identification of novel small molecule inhibitors of hypoxia-inducible factor-1 that differentially block hypoxia-inducible factor-1 activity and hypoxia-inducible factor-1alpha induction in response to hypoxic stress and growth factors. Cancer Res. 2005;65:4918–4928. doi: 10.1158/0008-5472.CAN-04-4453. [DOI] [PubMed] [Google Scholar]
- Cheng KC, Inglese J. A coincidence reporter-gene system for high-throughput screening. Nat Methods. 2012;9:937. doi: 10.1038/nmeth.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Felipe P, Luke GA, Hughes LE, Gani D, Halpin C, Ryan MD. E unum pluribus: Mutliple proteins from a self-processing polyprotein. Trend Biotechnol. 2006;24:68–75. doi: 10.1016/j.tibtech.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Feng BY, Simeonov A, Jadhav A, Bagaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J Med Chem. 2007;50:2385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- Graff J, Konicek B, Carter J, Marcusson E. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res. 2008;68:631–634. doi: 10.1158/0008-5472.CAN-07-5635. [DOI] [PubMed] [Google Scholar]
- Graff J, Konicek B, Vincent T, Lynch R, Monteith D, Weir S, Schwier P, Capen A, Goode R, Dowless M, Chen Y, Zhang H, Sissons S, Cox K, McNulty A, Parsons S, Wang T, Sams L, Geeganage S, Douglass L, Neubauer B, Dean N, Blanchard K, Shou J, Stancato L, Carter J, Marcusson E. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J Clin Invest. 2007;117:2638–2648. doi: 10.1172/JCI32044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- Hsieh AC, Ruggero D. Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin Cancer Res. 2010;16:4914–4920. doi: 10.1158/1078-0432.CCR-10-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese J, Dranchak P, Moran JJ, Jang SW, Srinivasan R, Santiago Y, Zhang L, Guha R, Martinez N, MacArthur R, Cost GJ, Svaren J. Genome editing-enabled HTS assays expand drug target pathways for Charcot-Marie-Tooth disease. AMC Chem Biol. 2014;9:2594–2602. doi: 10.1021/cb5005492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahsai AW, Zhu S, Wardrop DJ, Lane WS, Fenteany G. Quinocarmycin analog DX-52-1 inhibits cell migration and targets radixin, disrupting interactions of radixin with actin and CD44. Chem Biol. 2006;13:973–983. doi: 10.1016/j.chembiol.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Khan IF, Hirata RK, Russell DW. AAV-mediated gene targeting methods for human cells. Nat Protocol. 2011;6:482–501. doi: 10.1038/nprot.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Bonifant C, BunZ F, Lane WS, Waldman T. Epitope tagging of endogenous genes in diverse human cell lines. Nucleic Acid Res. 2008;36:e127. doi: 10.1093/nar/gkn566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Cattoglio C, Tjian R. Looping back to leap forward: Transcription enters a new era. Cell. 2014;157:13–25. doi: 10.1016/j.cell.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G, Ma X, Liu G. An RNA-zipcode-independent mechanism that localizes Dia1 mRNA to the perinuclear ER through interactions between Dia1 nascent peptide and Rho-GTP. J Cell Science. 2011;124:589–599. doi: 10.1242/jcs.072421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyssiotis CA, Foreman RK, Staerk J, Garcia M, Mathur D, Markoulaki S, Hanna J, Lairson LL, Charette BD, Bouchez LC, Bollong M, Kunick C, Brinker A, Cho CY, Schultz PG, Jaenisch R. Reprogramming of murine fibroblasts to induced pluripotent stem cells with chemical complementation of Klf4. Proc Natl Acad Sci USA. 2009;106:8912–8917. doi: 10.1073/pnas.0903860106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10:957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P, Albagli O, Poggi MC, Boulukos KE, Pognonec P. Development of a new bicistronic retroviral vector with strong IRES activity. BMC Biotechnol. 2006;6:4. doi: 10.1186/1472-6750-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair R, Avila H, Ma X, Wang Z, Lennartz M, Darnay BG, Boyd DD, Yan C. A novel high-throughput screening system identifies a small molecule repressive for matrix metalloproteinase-9 expression. Mol Pharmacol. 2008;73:919–929. doi: 10.1124/mol.107.042606. [DOI] [PubMed] [Google Scholar]
- Nakahara T, Takeuchi M, Kinoyama I, Minematsu T, Shirasuna K, Matsuhisa A, Kita A, Tominaga F, Yamanaka K, Kudoh M, Sasamata M. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67:8014–8021. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Bickmore W, Dean A, Nobrega MA, Bejerano G. Enhancers: five essential questions. Nat Rev Genet. 2013;14:288–295. doi: 10.1038/nrg3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapisarda A, Uranchimeg B, Scudiero DA, Selby M, Sausville EA, Shoemaker RH, Melillo G. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002;62:4316–4324. [PubMed] [Google Scholar]
- Samsonov A, Zenser N, Zhang F, Zhang H, Fetter J, Malkov D. Tagging of genomic STAT3 and STAT1 with fluorescent proteins and insertion of a luciferase reporter in the cycline D1 gene provides a modified A549 cell line to screen for selective STAT3 inhibitors. PLoS ONE. 2013;8:e68391. doi: 10.1371/journal.pone.0068391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlake T, Bode J. Use of mutated FLP recognition target (FRT) sites for the exchange of expression cassettes at defined chromosomal loci. Biochemistry. 1994;33:12746–12751. doi: 10.1021/bi00209a003. [DOI] [PubMed] [Google Scholar]
- Schulte TW, Neckers LM. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binding to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharmacol. 1998;42:273–279. doi: 10.1007/s002800050817. [DOI] [PubMed] [Google Scholar]
- Stiborova M, Bieler CA, Wiessler M, Frei E. The anticancer agent ellipticine on activation by cytochrome P450 forms covalent DNA adducts. Biochem Pharmacol. 2001;62:1675–1684. doi: 10.1016/s0006-2952(01)00806-1. [DOI] [PubMed] [Google Scholar]
- Thorne N, Inglese J, Auld DS. Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol. 2010;17:646–657. doi: 10.1016/j.chembiol.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc fingr nucleases. Nat Rev Genet. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- Wang H, Ma X, Ren S, Buolamwini J, Yan C. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol Cancer Ther. 2011;10:69–79. doi: 10.1158/1535-7163.MCT-10-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yan C. A small-molecule p53 activator induces apoptosis through inhibiting MDMX expression in breast cancer cells. Neoplasia. 2011;13:611–619. doi: 10.1593/neo.11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Boyd DD. Histone H3 acetylation and H3 K4 methylation define distinct chromatin regions permissive for transgene expression. Mol Cell Biol. 2006;26:6357–6371. doi: 10.1128/MCB.00311-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Paul HJ. Drugging the undruggable: Transcription therapy for cancer. Biochim Biophys Acta. 2013;1835:76–85. doi: 10.1016/j.bbcan.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Wang H, Aggarwal B, Boyd DD. A novel homologous recombination system to study 92 kDa type IV collagenase transcription demonstrates that the NF-κB motif drives the transition from a repressed to an activated state of gene expression. FASEB J. 2004;18:540–541. doi: 10.1096/fj.03-0960fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.