

Abstract
Background and Purpose
11β‐hydroxysteroid dehydrogenase type I (11β‐HSD1), a target for Type 2 diabetes mellitus, converts inactive glucocorticoids into bioactive forms, increasing tissue concentrations. We have compared the pharmacokinetic‐pharmacodynamic (PK/PD) relationship of target inhibition after acute and repeat administration of inhibitors of 11β‐HSD1 activity in human, rat and mouse adipose tissue (AT).
Experimental Approach
Studies included abdominally obese human volunteers, rats and mice. Two specific 11β‐HSD1 inhibitors (AZD8329 and COMPOUND‐20) were administered as single oral doses or repeat daily doses for 7–9 days. 11β‐HSD1 activity in AT was measured ex vivo by conversion of 3H‐cortisone to 3H‐cortisol.
Key Results
In human and rat AT, inhibition of 11β‐HSD1 activity was lost after repeat dosing of AZD8329, compared with acute administration. Similarly, in rat AT, there was loss of inhibition of 11β‐HSD1 activity after repeat dosing with COMPOUND‐20 with continuous drug cover, but effects were substantially reduced if a ‘drug holiday’ period was maintained daily. Inhibition of 11β‐HSD1 activity was not lost in mouse AT after continuous cover with COMPOUND‐20 for 7 days.
Conclusions and Implications
Human and rat AT, but not mouse AT, exhibited tachyphylaxis for inhibition of 11β‐HSD1 activity after repeat dosing. Translation of observed efficacy in murine disease models to human for 11β‐HSD1 inhibitors may be misleading. Investigators of the effects of 11β‐HSD1 inhibitors should confirm that desired levels of enzyme inhibition in AT can be maintained over time after repeat dosing and not rely on results following a single dose.
Abbreviations
- 11β‐HSD1
11β‐hydroxysteroid dehydrogenase type I
- PK/PD
pharmacokinetic‐pharmacodynamic
- AT
adipose tissue
- DIO
diet induced obese
- IHC
International Conference on Harmonisation
- GCP
Good Clinical Practice
- b.i.d.
twice daily
- u.i.d.
once daily
- HPMC
hydroxypropylmethylcellulose
- IC70
concentration that delivers 70% of the maximum effect
- IC90
concentration that delivers 90% of the maximum effect
- fu
fraction unbound
- Cmax
maximum achieved concentration
- Cmin
minimum or trough concentration
- E0
baseline
- Emax
maximum effect
- ANCOVA
analysis of covariance
Tables of Links
| TARGETS |
|---|
| Enzymes |
| 11β‐HSD1, 11β‐hydroxysteroid dehydrogenase type I |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Glucocorticoids have been implicated in the development of the metabolic syndrome, characterized by central obesity, Type 2 diabetes and cardiovascular disease (Wallerius et al., 2003). However, circulating levels of cortisol do not appear to be elevated in the metabolic syndrome (Gathercole and Stewart, 2010). 11β‐hydroxysteroid dehydrogenase type I (11β‐HSD1) is a ubiquitous enzyme, but quantitatively, mainly expressed in the liver and adipose tissue (AT), with a role to amplify the action of glucocorticoids by converting inactive 11‐ketometabolites to their bioactive forms thus increasing their local tissue concentrations. It is hypothesized that cortisol regeneration in tissues could be responsible for the clinical manifestations of the metabolic syndrome. Over the past decade, several pharmaceutical companies therefore have targeted 11β‐HSD1 selective inhibitors as a potential treatment for obese individuals with Type 2 diabetes.
Pre‐clinical studies with selective 11β‐HSD1 inhibitors have shown beneficial effects in diet‐induced obese (DIO) mice, with decreased body weight, food intake, fasting plasma glucose and insulin levels (Hermanowski‐Vosatka et al., 2005; Morgan and Tomlinson, 2010; Veniant et al., 2009; Wan et al., 2009; Wang et al., 2006).
In our view, published clinical studies with 11β‐HSD1 selective inhibitors have so far been unsuccessful in showing commercially attractive results in diabetic populations despite claiming substantial levels of 11β‐HSD1 inhibition in the liver and AT (Rosenstock et al., 2010; Shah et al., 2011; Feig et al., 2011). However, inhibition of 11β‐HSD1 activity in AT has mostly been demonstrated after acute administration (Hermanowski‐vosatka et al., 2005; Yeh et al., 2006; Webster et al., 2007; Hale et al., 2008; Gibbs et al., 2011). One pre‐clinical study has examined inhibition of 11β‐HSD1 activity in mouse AT after repeat dosing, but this was not compared with acute inhibition (Lloyd et al., 2009). Nevertheless, it is generally assumed that the inhibition of 11β‐HSD1 activity in AT achieved after acute dosing will be maintained with repeat dosing of 11β‐HSD1 inhibitors. This assumption is now challenged by our findings in clinical phase 1 studies suggesting that there is a loss of inhibition of 11β‐HSD1 activity in human AT after repeat dosing with 11β‐HSD1 inhibitors.
There is, however, a lack of published robust pharmacokinetic and pharmacodynamic (PK/PD) analysis of the effect of repeat administration of selective 11β‐HSD1 inhibitors in the inhibition of 11β‐HSD1 activity in AT. In the present study, we report a series of experiments where we have performed PK/PD analysis of the 11β‐HSD1 inhibition in human, rat and mouse tissue and have observed clear differences between species.
Methods
Human studies
The study was approved by the Ethics Committee National Research Ethics Service, St Thomas' Hospital, London, UK. The study was performed in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with the International Conference on Harmonisation (ICH)/Good Clinical Practice (GCP) and applicable regulatory requirements and the AstraZeneca policy on Bioethics. Informed consent was obtained from all subjects before initiation of any study related procedure. One single‐centre study included abdominally obese (waist >102 cm), otherwise, healthy males. AZD8329 25, 100, and 300 mg (n = 6 at each dose) or placebo (n = 8) was orally given (as a solution) twice daily (b.i.d.) for 9 days. Subcutaneous abdominal adipose tissue biopsies were performed by syringe aspiration under local anaesthesia. Clinical trial registration is available at ClinicalTrials.gov NCT01207089
Animal studies
All animal care and experimental procedures complied with the UK Home Office Animal Scientific Procedures Act 1986 and were approved by AnimalWelfare Ethical Review Committee (AWERB). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath and Kilkenny, 2010).
A total of 269 male Han Wistar rats (11‐ to 12‐week old, 260–330 g) (Harlan, UK) and 83 male C57B6Jax mice (11 to 12‐week old, 25–30 g) (AstraZeneca, UK) were used in these studies. Animals were housed under controlled temperature (21 ± 2 °C), humidity (45 ± 10%) and light (12‐h light, 12‐h dark cycle, lights on at 06.00 h), with ad libitum water and standard laboratory diet (Rat and Mouse No. 1 maintenance SQC expanded, Special Diet Services Ltd, UK). In all in vivo studies carried out, the animals were randomly allocated to treatment groups, adjusting for body weight to ensure that animals were comparable across groups.
Experimental design
AZD8329 rat acute dose response
Results from three separate studies (n = 109) were used to derive the acute dose response. In all three studies, rats received an oral administration at 08.00 h of either hydroxypropylmethylcellulose (HPMC) vehicle (water containing 0.5% (w/v) HPMC and 0.1% w/v polysorbate 80 and meglumine) (n = 29) or AZD8329 at 0.3, 1, 3, 10, 30, 60, 120 or 250 mg · kg−1 (n = 10). At 1, 3 or 12‐h after this dose, animals were terminally anaesthetized by a rising concentration of CO2. Cardiac blood samples were taken into EDTA anticoagulant for plasma compound concentration measurement. Death was ensured by cervical dislocation. Epididymal adipose depots were dissected and stored on wet ice for up to 1h prior to ex vivo enzyme activity evaluation.
AZD8329 Rat single and repeat dosing study A
Rats (n = 58) were dosed orally once daily (u.i.d.) at 08.00 h for seven consecutive days with either vehicle (n = 8), 60 (n = 25) or 120 (n = 25) mg · kg−1 of AZD8329 in HPMC vehicle. Acute arm rats were given an oral administration of 60 or 120 mg · kg−1 AZD8329 at 08.00 h on day 7 only. On day 7, rats were killed at 3 or 12‐h after dosing, cardiac blood and AT samples were collected and stored as described above.
AZD8329 rat single and repeat dosing study B
Rats were dosed orally u.i.d. at 07.00 h for six consecutive days receiving either 120 mg · kg−1 AZD8329 (n = 16) or HPMC vehicle (n =24). In this experiment, a satellite group of animals was used to establish plasma compound concentrations on study day 6. Satellite animals (n = 6) were treated as the main study group on days 1–6 of the study. On study day 6, at 4, 7 and 11.5‐h after dosing, 300 μL blood samples were taken via 21‐gauge butterfly needles into EDTA coated tubes, centrifuged and the resultant plasma stored at −20 °C for compound concentration measurement. At 12‐h after dose, animals were killed by a rising concentration of CO2.
On day 7, vehicle‐treated study animals (during days 1–6) received a single oral dose of either vehicle (n = 8), 19 or 78 mg · kg−1 (n = 8) of AZD8329 to achieve plasma exposures of IC70 or IC90, respectively, based on the previously established acute PK/PD (Table 2). Study animals treated with AZD8329 (during days 1–6) received a single oral dose of either 9 or 71 mg · kg−1 (n = 8) AZD8329 to also achieve the same plasma exposures of IC70 or IC90 respectively. The doses given to the latter group were reduced to account for residual AZD8329 present in the blood from days 1–6. At 3‐h after dosing, the animals were killed and AT was dissected as described above.
Table 2.
PK/PD parameters for model fit for AZD8329 in the ex vivo 11β‐HSD1 activity assay in rat AT, after an acute and repeat oral administration
| Day | Parameter | Estimate | Units | %CV |
|---|---|---|---|---|
| Acute | E0 | 102 | – | 3.9 |
| IC50 | 0.10 | μM | 33 | |
| Emax | 10.0 | – | 55 | |
| Repeat | E0 | 104 | – | 5.6 |
| IC50 | 1.24 | μM | 33 | |
| Emax | 10.0 | – | FIXED |
COMPOUND‐20 rat single and repeat dosing study
Rats were dosed orally b.i.d. at 07.00 and 19.00 h for six consecutive days. They received either HPMC vehicle b.i.d. (acute and vehicle control groups) (n = 32), 20 mg · kg−1 (n = 24) COMPOUND‐20 at 07.00 h and HPMC vehicle at 19.00 h (u.i.d. group) or 60 mg · kg−1 (n = 24) COMPOUND‐20 b.i.d. These doses were selected to maintain free plasma concentrations of COMPOUND‐20 above the acute in vivo free IC50 (Goldberg et al., 2012) for the whole duration of the study (60 mg · kg−1 COMPOUND‐20 b.i.d.: ‘continuous cover’) or to provide a daily ‘drug holiday’ where free plasma concentration of COMPOUND‐20 was only maintained above the in vivo free IC50 for approximately 10 h in each 24‐h period (20 mg · kg−1 COMPOUND‐20 u.i.d.: ‘drug holiday’). Plasma compound concentrations were confirmed in a group of satellite animals on day 6. On day 7, animals treated with vehicle (during days 1–6) received a single oral dose of either vehicle (n = 8), 2.5 or 17 mg · kg−1 (n = 12); the 20 mg · kg−1 u.i.d.‐treated animals received a single oral dose of either 2.5 or 17 mg · kg−1 (n = 12) COMPOUND‐20 to achieve IC70 or IC90 respectively (based on acute PK/PD response, Goldberg et al., 2012). The animals treated with 60 mg · kg−1 b.i.d. (during days 1–6) received a reduced dose of either 1.8 or 16 mg · kg−1 (n = 12) COMPOUND‐20 to achieve the same IC70 or IC90 respectively. The doses given to the latter group were reduced to account for the remaining COMPOUND‐20 present in blood from days 1–6. Animals were killed at 1‐h after dosing and AT was dissected as described above.
COMPOUND‐20 mouse single and repeat dosing study
Mice (n = 83) were dosed orally b.i.d. at 07.00 and 19.00 h for six consecutive days. They received either HPMC vehicle bid (acute and vehicle control groups) (n = 31), 3 mg · kg−1 COMPOUND‐20 at 07.00 h and HPMC vehicle at 19.00 h (u.i.d. group) (n = 26) or 30 mg · kg−1 COMPOUND‐20 bid (n = 26). These doses were selected to maintain free plasma concentrations of COMPOUND‐20 above the mouse acute in vivo free IC50 (Goldberg et al., 2012) for the whole duration of the study (30 mg · kg−1 COMPOUND‐20 b.i.d.: continuous cover) or to provide a daily ‘drug holiday’ (3 mg kg−1 COMPOUND‐20 u.i.d.: drug holiday). Plasma compound concentrations were confirmed at Tmax in a group of satellite animals on day 6 (results not shown).
At the 19.00‐h dose on day 6, all animals received an oral dose of HPMC vehicle so no residual COMPOUND‐20 would be present in the blood at the time of the final dosing on day 7. In a previous experiment, we determined this short compound ‘washout’ for just a few hours did not affect the PK/PD response following 6 days of repeated dosing (results not shown). Moreover, because of the small doses required to achieve free plasma concentrations of COMPOUND‐20 in mice, equivalent to IC70 and IC90, this was deemed the best experimental design to ensure all animals had comparable free plasma concentrations of COMPOUND‐20 at termination of the study in the morning of day 7. On day 7, all animals received an oral dose of either vehicle (n = 5) 0.3 mg · kg−1 (n = 13, for acute, u.i.d. and b.i.d. groups, respectively) or 1 mg · kg−1 (n = 13, for acute, u.i.d. and b.i.d. groups, respectively) of COMPOUND‐20 to achieve IC70 or IC90, respectively, based on a previously determined acute PK/PD response (Goldberg et al., 2012). Animals were killed at 1‐h after dosing by a rising concentration of CO2. Cardiac blood and AT samples were collected and stored as described earlier.
A summary table of all experiments presented in this paper is provided as Supporting Information.
Measurements made
Ex vivo measurement of 11β‐HSD1 activity in human adipose tissue
Activity of human 11β‐HSD1 activity in AT biopsies was measured as the conversion of 3H‐cortisone to 3H‐cortisol, following a previously reported ex vivo AT assays (Sjostrand et al., 2010). Approximately 100 mg of tissue was weighed and incubated, in triplicate, for 3 h in the presence of 3H‐cortisone (20 nM, 1 μCi · mL−1, Amersham Biosciences GE Healthcare, Buckinghamshire, UK) containing DMEM supplemented with 6 mM glucose (Sigma Chemical Co., St Louise, MO, USA), 10% FCS (Sigma) and 1% penicillin–streptomycin (PEST; Invitrogen Corporation, Paisley, UK) in 37 °C at 5% CO2. After AT incubations, medium samples were collected, frozen and stored in −20 °C before analysis of cortisone to cortisol conversion. An index of 11β‐HSD1 activity was measured using radiometric HPLC, and activity was expressed as percent conversion of 3H‐cortisone to 3H‐cortisol per hour per 100 mg AT for AT assays. Medium was then analysed for radio‐labelled cortisone and cortisol by LC after extraction with ethyl acetate. 3H‐labelled steroids were detected with flow scintillation detection. The results were presented as % conversion of 3H‐cortisone to 3H‐cortisol per 100 mg tissue during 3 h (area of 3H‐cortisol/[area of 3H‐cortisone + 3H‐cortisol]/tissue weight).
Ex vivo measurement of 11β‐HSD1 activity in rat and mouse adipose tissue
Activity of the 11β‐HSD1 enzyme in epididymal AT was measured as the conversion of 3H‐cortisone to 3H‐cortisol. Epididymal AT pieces were weighed (approximately 100–200 mg), cut into 2–3 mm3 pieces using scissors and incubated for 60 min (mouse AT) or 90 min (rat AT) in the presence of 3H‐cortisone (20 nM, 1 μCi · mL−1, specific activity 1.97 GBq/mmol, Perkin Elmer) containing DMEM Ham F12 media (Sigma‐Aldrich, Poole, UK) supplemented with 10% FCS and 1% penicillin–streptomycin (Sigma‐Aldrich, Poole, UK) in 37 °C at 5% CO2. After tissue incubations, media samples were collected for analysis of 3H‐cortisone to 3H‐cortisol conversion. Radio‐labelled steroids were extracted using ethyl acetate, samples were evaporated to dryness under nitrogen and re‐suspended in mobile phase for HPLC analysis adapted from Napolitano et al. (1998). Radioactivity was measured using a flow scintillation analyser. 11β‐HSD1 activity was expressed as % conversion of 3H‐cortisone to 3H‐cortisol per 100 mg tissue.
Measurement of drug levels in blood
Blood samples (50 μL) were protein‐precipitated with ice cold acetonitrile (Sigma‐Aldrich) containing a generic internal standard compound from the AstraZeneca compound library. The samples were then mixed and centrifuged at 4500 g for 10 mins. A sample of the supernatant (50 μL) was removed and diluted with 300 μL of water prior to injection (50 μL) onto the LC‐MS/MS system. Free plasma levels of compounds were calculated based on measured concentrations in blood and corrected using a constant free fraction (fu) for each compound and species. Free fraction values were generated in house in vitro using previously published methodology (Buttar et al., 2010). AZD8329 fu was 0.012 and 0.013 in man and rat respectively. COMPOUND‐20 fu was 0.55 and 0.60 in rat and mouse respectively. Samples from the clinical study were analysed at York Bioanalytical solutions, York, UK on behalf of AstraZeneca using a very similar method.
Data analysis
Pharmacokinetic and pharmacodynamic modelling of AZD8329 results
Phoenix™ WinNonlin® 6.2 Certara L.P. software (Certara USA Inc., Princeton, NJ, USA)was used for all PK/PD modelling.
Human
A direct response Emax‐type model was used in the PK/PD analysis of 11β‐HSD1 activity (expressed as % conversion of 3H‐cortisone to 3H‐cortisol per 100 mg tissue) versus free concentrations of compound in plasma. The mathematical equation used to fit the results was as follows:
Where E0 = % conversion per 100 mg of tissue of the placebo‐treated group, C = free compound concentration in plasma and IC50 = free compound concentration in plasma that delivers 50% of the max effect.
Rat
To generate the PK/PD response after acute administration, results from three separate studies in the rat were combined and normalized. Results from the sub‐acute study A was used for the repeat dosing PK/PD fit and was also normalized (each study was normalized to its respective vehicle‐treated controls to allow comparison across studies). A direct response Emax‐type model was used in the PK/PD analysis of 11β‐HSD1 activity (expressed as normalized conversion in AT) versus free concentrations of compound in plasma. The mathematical equation used to fit the results was as follows:
Where E0 = normalized conversion in AT for vehicle‐treated groups, Emax = minimum normalized conversion achievable (i.e. maximum inhibition), C = free compound concentration in plasma and IC50 = free compound concentration in plasma that delivers 50% of the max effect. The Emax parameter (max inhibition) for the repeat dosing fit was fixed to the one obtained after the acute PK/PD fit; this assumes that the same maximum inhibition can theoretically be achieved after repeat dosing compared with acute administration if concentrations of compound in plasma could reach high enough. As we have observed a rightward shift in the PK/PD after repeat dosing, unfortunately, we could not achieve high enough plasma exposure with AZD8329 in the rat to confirm this assumption. This modelling approach was consistent with the analysis of the human data where it was also assumed that the same maximum inhibition could be achieved after acute or repeat administration.
Statistical analysis
Human study
Results (excluding the placebo group) were analysed on a log scale using anova adjusting for dose, subject within dose and study day (1 or 9). The analysis also included an exploration of the relevance of free plasma concentrations of compound to ex vivo response and whether the difference between study days was significantly associated with the dose group (an interaction between dose and study day). Neither was statistically significant and so these terms were removed from the model. Study day 9 (sub‐acute) was compared with study day 1 (acute) using a fold change of the geometric means (within subject) and a 95% confidence interval calculated. A statistical test was also performed to establish the statistical significance of the fold change using a two‐sided 5% t‐test, based on the within‐subject standard deviation.
Rat and mouse studies
A two‐stage procedure was adopted: (i) analysis of the vehicle and treated groups in a one‐way anova to compare each treatment group with control and then (ii) the vehicle group was excluded and the treatment groups re‐analysed to examine differences in treatment regimens. This analysis was a two‐factor analysis of covariance (ANCOVA) including the factors regimen (acute, u.i.d. or b.i.d.) and dose (IC70 or IC90) and included the exposure as a covariate. As part of the analysis, a test for an interaction (dose vs. regimen) was performed. If this test indicated that there was an interaction, the regimen effects were reported separately for each dose. If not, then the doses were combined to compare regimens. A multiple comparison adjustment was incorporated into the pairwise comparisons (Sidak's adjustment). The confidence intervals are 95% (also adjusted using Sidak's) so statistical significance was inferred if the 95% confidence interval did not span zero. All tests were performed using a two‐sided 5% test.
Materials
COMPOUND‐20 (Goldberg et al., 2012) and AZD8329 (Scott et al., 2012) are two selective, competitive and reversible 11β‐HSD1 inhibitors with full pharmacological characterization and compound structures previously published. These compounds were synthesized by AstraZeneca (Manchester, UK). Relevant primary pharmacological information is shown below:
| Compound | Human 11β‐HSD1 IC50 | Rat 11β‐HSD1 IC50 | Mouse 11β‐HSD1 IC50 |
|---|---|---|---|
| AZD8329 | 0.009 (μM) | 0.086 (μM) | 6.1 (μM) |
| COMPOUND‐20 | 0.028 (μM) | 0.271 (μM) | 0.027 (μM) |
Results
AZD8329
A PK/PD analysis of the 11β‐HSD1 activity in samples of AT and the free plasma concentration of AZD8329 at the time of tissue biopsy in the volunteers was performed. The resulting PK/PD fit and parameters are shown in Figure 1 and Table 1.
Figure 1.
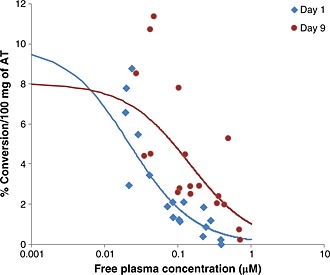
Comparison of the PK/PD relationship between ex vivo activity of the human 11β‐HSD1 enzyme in sub‐cutaneous AT biopsies (measured as the conversion of 3H‐cortisone to 3H‐cortisol) and free plasma concentrations of AZD8329 both after acute and repeat oral administration (n = 6 per dose level). A direct response (Emax) model was used to fit the PK/PD results. The observed results for day 1 and for day 9 are shown with the corresponding lines representing the model fit for each set of observations.
Table 1.
PK/PD parameters for model fit for AZD8329 in the ex vivo human adipose tissue 11β‐HSD1 activity assay after acute and repeat oral administration in humans
| Day | Parameter | Estimate | Units | %CV |
|---|---|---|---|---|
| Day 1 | IC50 | 0.022 | μM | 23 |
| E0 | 9.9 | %conv per 100 mg | 9.2 | |
| Day 9 | IC50 | 0.144 | μM | 37 |
| E0 | 8.0 | %conv per 100 mg | 13 |
The statistical comparison of day 9 and day 1 revealed that there was a significantly lower level of %conversion per 100 mg AT on day 1 than on day 9 (p = 0.001) in subjects treated with AZD8329. There was a trend for this difference to increase with increasing dose, but this did not reach statistical significance. Overall (pooling across dose groups), the %conversion per 100 mg AT on day 1 (1.77%) was 0.56‐fold of the level on day 9 (3.18%) indicating a lower level of inhibition on the 11β‐HSD1 activity in AT achieved by AZD8329 on repeat dosing (the adjusted 95% confidence interval for the fold chance was 0.41 to 0.76)
AZD8329 was also evaluated for inhibition of ex vivo 11β‐HSD1 activity in rat AT . Meta‐analysis of the results from multiple rat studies (acute dose response and study A in the methods and Supporting Information table) was used to generate the PK/PD response after acute administration of AZD8329 (Figure 2). The inhibition of 11β‐HSD1 activity in rat AT after repeat dosing of AZD8329 was evaluated after 7 days of dosing at either 60 or 120 mg · kg−1 u.i.d. The average free plasma concentrations of AZD8329 did not fall under 0.20 μM (Cmin of the 60 mg · kg−1 group), and based upon the acute PK/PD relationship (Table 2), all rats that received repeated doses for 7 days had free plasma concentrations of AZD8329 over the rat IC50 required to substantially inhibit 11β‐HSD1 in AT after an acute administration (i.e. they had ‘continuous cover’). The PK/PD parameters for the model fit after acute and repeat oral dosing are shown in Table 2.
Figure 2.
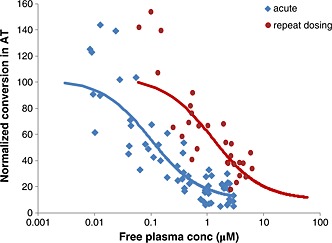
Meta‐analysis of multiple rat experiments to compare the PK/PD relationship between ex vivo activity of rat 11β‐HSD1 enzyme in epididymal AT (measured as conversion of 3H‐cortisone to 3H‐cortisol, normalized to vehicle treated rats) and free plasma concentrations of AZD8329 after acute and repeat oral administration. A direct response (Emax) model was used to fit the PK/PD data. The observed results for acute (n = 109) and repeat dosing for 7 days (n = 58) are shown with the corresponding lines representing the model fit for each set of observations.
The statistical comparison of acute and repeat dosing shows that there was a significantly lower level of %conversion per 100 mg AT after acute dosing compared with repeat dosing (p < 0.001). After acute dosing, the %conversion per 100 mg AT was 32.9% compared with 75.9% after repeat dosing. This represents a difference of 43% with a 95% confidence range (adjusted for using Sidak's) of (34.4, 51.6).
A second study with AZD8329 in the rat was conducted (rat study B in the methods and Supporting Information table) to further evaluate the differences in the inhibition of 11β‐HSD1 activity in rat AT between acute and repeated administration. The experiment was designed to enable the statistical comparison of the inhibition achieved after acute and repeat dosing. A ‘continuous cover’ dose (days 1–6) was selected to maintain plasma free drug levels above the in vivo IC50 (0.10 μM) based on acute PK/PD for the full duration of the study. Free plasma concentrations of AZD8329 during days 1–6 were measured in a group of satellite rats, and free concentrations were maintained between 1.4 μM (at Cmax) and 0.20 μM (at Cmin), which confirmed ‘continuous cover’ was achieved. On day 7, two dose levels (IC70 and IC90 based on acute PK/PD) were selected to test the inhibition of 11β‐HSD1 activity in AT. Acute and repeat dosing of AZD8329 significantly inhibited 11β‐HSD1 activity in rat AT compared with vehicle controls (Figure 3A). The PK/PD graph clearly showed two separate populations for the acute and repeat dosing (Figure 3B). There was a statistically significant difference between the two dosing regimens (Figure 3C). AZD8329 was not investigated in mice because of its low potency against mouse 11β‐HSD1 enzyme.
Figure 3.
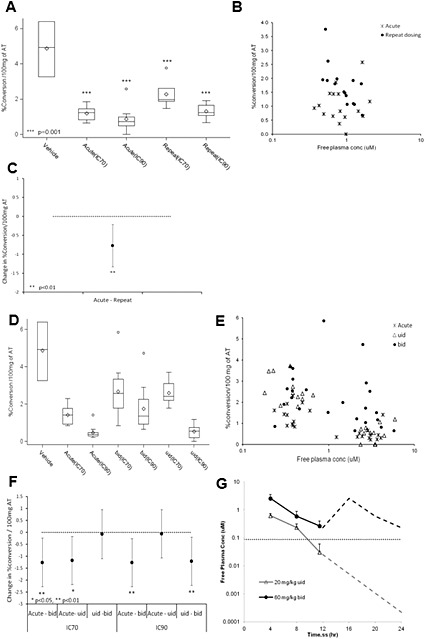
Rat study B with AZD8329: (A) Box and whisker plot of ex vivo activity of rat 11β‐HSD1 enzyme in epididymal AT (measured conversion of 3H‐cortisone to 3H‐cortisol) after acute and repeat oral administration, per treatment group of AZD8329 (n = 12), comparison of the vehicle (n = 8) versus the treated groups (box and whisker plot shows the median (central line), the upper and lower quartiles (top and bottom of the box) and the range of the data (whiskers). If there are data points more than 1.5× the interquartile range (IQR) above the upper quartile (or below the lower quartile), then these points are singled out and presented separately. *** P < 0.001, significantly different from vehicle. (B) Comparison of the PK/PD relationship between rat 11β‐HSD1 enzyme activity and free plasma concentrations of AZD8329 after acute and repeated (7 days) oral administration in rats.. (C) Treatment groups statistical comparison of the ex vivo activity of the rat 11β‐HSD1 enzyme in epididymal AT. The confidence intervals are 95% (with Sidak's multiple comparison adjustment) so statistical significance was inferred if the 95% confidence interval did not span zero. **P< ,0.01. Rat experiment with COMPOUND‐20: (D) Box and whisker plot of ex vivo activity of rat 11β‐HSD1 enzyme in epididymal AT (measured conversion of 3H‐cortisone to 3H‐cortisol) after acute and repeat oral administration (u.i.d. and b.i.d.), per treatment group of COMPOUND‐20 (n = 12), comparison of the vehicle (n = 8) versus the treated groups. (E) Comparison of the PK/PD relationship between rat 11β‐HSD1 enzyme activity and free plasma concentrations of COMPOUND‐20 after acute and repeat (b.i.d. or u.i.d. for 7 days) oral administration in rats. (F) Treatment groups statistical comparison of the ex vivo activity of the rat 11β‐HSD1 enzyme in epididymal AT. The confidence intervals are 95% (with Sidak's multiple comparison adjustment). *P < 0.05, **P < 0.01. (G) Free plasma concentrations (+SD) of COMPOUND‐20 in rats following a daily dose of COMPOUND‐20 at either 20 mg · kg−1 u.i.d. or 60 mg · kg−1 b.i.d.. Horizontal dotted line indicates ‘efficacious’ concentrations based on acute IC50 for the inhibition of rat 11β‐HSD1 activity in the adipose tissue. Results shown are measured plasma concentrations of COMPOUND‐20 for the first 12 h on day 6. The second 12 h are simulated based on results from the first 12 h.
COMPOUND‐20
COMPOUND‐20 is a potent selective 11β‐HSD1 inhibitor in mouse, rat and human but from a different chemical series than AZD8329. We have previously reported that an acute dose of COMPOUND‐20 inhibited 11β‐HSD1 activity in rat AT in a exposure‐dependent manner, with a resulting in vivo free IC50 of 0.087 μM (Goldberg et al., 2012)
A comparison of the effects of COMPOUND‐20 in 11β‐HSD1 activity in rat AT after both acute and repeat dosing was carried out. Doses of 60 mg · kg−1 b.i.d. and 20 mg · kg−1 u.i.d. were selected (days 1–6). Pharmacokinetic analysis of a satellite group of rats demonstrated that 60 mg · kg−1 b.i.d. maintained free plasma concentrations of COMPOUND‐20 in excess of the acute in vivo free IC50 (Figure 3G). Rats dosed with 20 mg · kg−1 u.i.d. had plasma concentrations of COMPOUND‐20 that delivered free compound levels in plasma above the acute free in vivo IC50 for approximately 10 h each day, providing a ‘drug holiday’ in each 24‐h period (Figure 3G).
On day 7, two dose levels (IC70 and IC90 based on acute PK/PD) were selected to test the inhibition of 11β‐HSD1 activity in AT. Acute and repeat dosing of COMPOUND‐20 significantly inhibited 11β‐HSD1 activity in rat AT compared with vehicle controls (Figure 3D). A comparison of the PK/PD relationship between rat 11β‐HSD1 enzyme activity and free plasma concentrations of COMPOUND‐20 after acute and repeat oral administration and their statistical comparison are shown in Figure 3E and F respectively.
A single dose of COMPOUND‐20 inhibited 11β‐HSD1 activity in mouse AT in a plasma exposure dependent manner with an in vivo free IC50 of 0.030 μM (Goldberg et al., 2012). In the repeat dosing study, mice dosed with 30 mg · kg−1 b.i.d. had continuous cover (above the acute in vivo free IC50) and mice dosed with 3 mg · kg−1 u.i.d. had approximately 10‐h cover (‘drug holiday’) (results not shown). Acute and repeat dosing of COMPOUND‐20 significantly inhibited 11β‐HSD1 activity in mouse AT compared with vehicle controls (Figure 4A). The PK/PD graph did not show two separate populations for the acute and repeat dosing (Figure 4B). There was a small statistically significant difference (P < 0.05) between the two repeat dosing regimens with the b.i.d. ‘continuous cover’ regimen apparently leading to greater inhibition than the u.i.d. ‘drug holiday’ regimen (Figure 4C).
Figure 4.
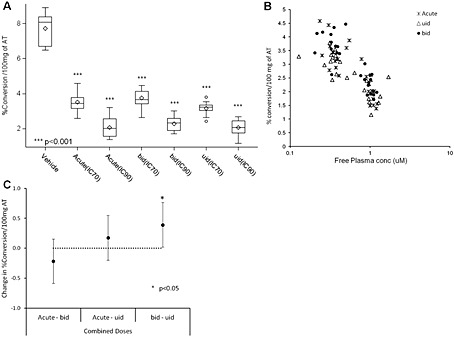
Mouse experiment with COMPOUND‐20: (A) Box and whisker plot of ex vivo activity of mouse 11β‐HSD1 enzyme in epididymal AT (measured conversion of 3H‐cortisone to 3H‐cortisol) after acute and repeat oral administration, per treatment group of COMPOUND‐20 (n = 13), comparison of the vehicle (n = 5) versus the treated groups (see legend of Figure 3 for description of box and whiskers plot). ***P < 0.001, significantly different from vehicle . (B) Comparison of the PK/PD relationship between mouse 11β‐HSD1 enzyme activity and free plasma concentrations of COMPOUND‐20 after acute and repeat (b.i.d. or u.i.d. for 7 days) oral administration in mice. (C) Treatment groups statistical comparison of the ex vivo activity of the mouse 11β‐HSD1 enzyme in epididymal AT. The confidence intervals are 95% (with Sidak's multiple comparison adjustment) so statistical significance was inferred if the 95% confidence interval did not span zero. *P < 0.05.
Discussion and conclusions
Pre‐clinical studies with selective 11β‐HSD1 inhibitors have shown efficacy in DIO mice, with decreased body weight, food intake, fasting plasma glucose and insulin levels (Hermanowski‐Vosatka et al., 2005; Morgan and Tomlinson, 2010; Veniant et al., 2009; Wan et al., 2009; Wang et al., 2006). There are few studies reported on administration of 11β‐HSD1 inhibitors to obese rats. However, clinical studies with 11β‐HSD1 selective inhibitors have so far been unsuccessful in showing similar magnitudes of efficacy in patients with diabetes, despite claims of substantial levels of 11β‐HSD1 inhibition in the liver and AT (Rosenstock et al., 2010; Shah et al., 2011; Feig et al., 2011). We compared the inhibition of 11β‐HSD1 in AT from two rodent species (rat and mouse) with the inhibition of 11β‐HSD1 in human AT. Species specificity is found with 11β‐HSD1 inhibitors, so, in the present study, we used two potent inhibitors for human and rat (AZ8329) and rat and mouse (COMPOUND‐20). AZD8329 has been trialled in human clinical studies, but as AZD8329 is a weak inhibitor of mouse 11β‐HSD1, we used COMPOUND‐20 to establish whether AT from mice and rats displays the same behaviour regarding dynamics of enzyme inhibition. To date, COMPOUND‐20 has not been tested in humans.
Published data regarding inhibition of 11β‐HSD1 activity in AT have mostly been reported after acute administration. However, it is assumed that inhibition of 11β‐HSD1 activity in AT is equally achieved after acute or repeat dosing of 11β‐HSD1 inhibitors, but this has not been documented. This assumption is now challenged by our findings in phase I human clinical studies suggesting that there is a loss of inhibition of 11β‐HSD1 activity in human AT after repeat dosing of 11β‐HSD1 inhibitors.
Pharmacokinetic and pharmacodynamic analysis of the effect of repeat dosing (for 9 days) of AZD8329 on 11β‐HSD1 activity in human AT has clearly shown a rightward shift in the PK/PD relationship after repeat dosing compared with acute administration (Figure 1). This tachyphylaxis has been quantified as around sevenfold based on the different in vivo free IC50 obtained when fitting the results (Table 1). Whether this tachyphylaxis in human AT will continue to exist after longer treatment periods is currently unknown.
Two separate experiments to assess the inhibition of 11β‐HSD1 activity by AZD8329 in rat AT after repeat dosing (for 7 days) were carried out. In both experiments, continuous cover (based on acute PK/PD results) was maintained for the duration of the experiment. A rightward shift in the PK/PD response of AZD8329 against inhibition of 11β‐HSD1 activity in rat AT was also observed following repeat dosing compared with acute administration (Figure 2). This tachyphylaxis was quantified as around 10‐fold based on the different in vivo free IC50 obtained when fitting the results (Table 2), which is consistent with the observations in man. Once again, whether this tachyphylaxis in rat AT would persist after longer treatment periods is currently unknown. The human and rat studies differ in duration by two days due to the varying experimental designs used for rodent and human investigations; however, these studies would both be classified as sub‐acute studies and the 2‐day difference should not affect interpretations.
In addition, COMPOUND‐20 also showed lower level of inhibition of 11β‐HSD1 activity in rat AT after 7 days of continuous cover compared with acute administration, despite achieving the same free compound plasma concentrations (Figure 3). Interestingly, this loss of inhibition of 11β‐HSD1 activity after 7 days was reduced if a ‘drug holiday’ period was achieved everyday (i.e. plasma compound concentrations allowed to fall below active levels, based upon acute PK/PD relationship). A ‘drug holiday’ group was introduced into the experimental design as we hypothesized that continuous inhibition of 11β‐HSD1 activity might cause accumulation of substrate (11‐dehydrocorticosterone) in adipose tissue, which might compete with the 11β‐HSD1 inhibitors. Importantly, pre‐clinical efficacy studies in mice with 11β‐HSD1 inhibitors have influenced experimental designs to have sustained maximal inhibition of 11β‐HSD1 for sustained efficacy. This suggests that a ‘drug holiday’ period (in our experiment around 14 h per day) was sufficient to allow rat AT 11β‐HSD1 to diminish the tachyphylaxis to compound‐mediated inhibition. However, further work to determine the optimum duration and degree of 11β‐HSD1 inhibition for the ‘drug holiday’ would need to be undertaken to evaluate if this approach would deliver significant efficacy in rodents and humans.
No studies have examined whether mouse AT exhibits tachyphylaxis to 11β‐HSD1 inhibition. AZD8329 could not be investigated in mice due to its poor potency against mouse 11β‐HSD1 enzyme, of which COMPOUND‐20 is a potent inhibitor. Surprisingly, repeat oral dosing of COMPOUND‐20 in either a ‘continuous cover’ or ‘drug holiday’ regimen did not show tachyphylaxis in the PK/PD of the inhibition of 11β‐HSD1 activity in mouse AT when compared with acute administration. Repeat dosing of a second potent mouse inhibitor, COMPOUND‐26 (Goldberg et al., 2012) did not exhibit tachyphylaxis in mouse AT compared with acute dosing (results not shown). Unfortunately, a combination of weaker potency against 11β‐HSD1 enzyme and poor PK/PD properties prevented us from testing COMPOUND‐26 in the rat.
Why is inhibition of 11β‐HSD1 enzyme in mice different from that in humans and rats? The loss of inhibition of 11β‐HSD1 activity in AT with repeat dosing of AZD8329 and COMPOUND‐20 in rat and human cannot be easily explained based on published results. As the loss of inhibition of 11β‐HSD1 activity is first reported in the present studies, it needs further investigation. The 11β‐HSD1 inhibitors were present at sufficient levels in the blood to cause inhibition, and we have evidence showing that concentrations of AZD8329 in ATs were enough to cover 11β‐HSD1 inhibition (Supporting Information Figure 1), therefore, a reduced penetration of 11β‐HSD1 inhibitors into adipose tissue on repeat dosing can be ruled out. 11β‐HSD1 is a bidirectional enzyme (reductase or dehydrogenase) that is located in the intra‐luminal compartment of the endoplasmic reticulum and catalyses the equilibrium between active and inactive glucocorticoids. The 11β‐HSD1 inhibitors used in the present study mainly inhibited only the reductase activity. AT 11β‐HSD1 activity is regulated by pentose phosphate pathway flux (McCormick et al., 2006; McCormick et al., 2008) and by the supply of glucose‐6‐phosphate, which has been described as a modulator of 11β‐HSD1 inhibitors (Balazs et al., 2009). A high concentration ratio of pyridine nucleotides (predominately NADPH/NADP) drives 11β‐HSD1 in the reductase direction, rather than the dehydrogenase direction (Dzyakanchuk et al., 2009; Balazs et al., 2009). Consequently, glucose metabolism in mouse AT is higher (than rat/human AT) and the increased pentose phosphate flux might overcome 11β‐HSD1 inhibition. The present study raises important questions about whether there are differences in regulation of the redox potential in mouse versus rat and human ATs, as potential differences in the capacity of human AT to generate NADPH have already been noted (Lee et al., 2008). There was no change in expression of 11β‐HSD1 in rat or human AT after repeat dosing with AZD8329 compared with the respective vehicle‐treated group (results not shown). Human AT biopsies collected from AZD8329 clinical trial were run in an ex vivo assay but with high concentrations of AZD8329 added in vitro (about 25× acute IC50). Under these conditions, complete inhibition of human AT 11β‐HSD1 activity was achieved (results not shown). This indicates that complete enzyme inhibition is possible, even after 9‐day treatment, but much higher compound concentrations were required.
In our view, published clinical studies with 11β‐HSD1 selective inhibitors (INCB‐13739 and MK‐0916) have, so far, been unsuccessful in demonstrating commercially attractive results in patients with Type 2 diabetes despite achieving plasma concentrations that delivered marked 11β‐HSD1 inhibition as measured after single administration (Rosenstock et al., 2010; Shah et al., 2011; Feig et al., 2011). However, the degree of inhibition of 11β‐HSD1 activity in AT in the phase IIB studies has not been reported. Based on our findings, it is possible that full inhibition of 11β‐HSD1 activity in AT was not achieved in the phase IIB clinical studies due to tachyphylaxis.
Finally, in the present study, human and rat AT exhibited tachyphylaxis in the inhibition of 11β‐HSD1 activity after repeat dosing but this was not demonstrated in mice. It is hypothesized that inhibition of 11β‐HSD1 in AT is important for significant delivery of efficacy. Therefore, translation of efficacy observed in disease models from mouse to human for 11β‐HSD1 inhibitors might be misleading. Investigators of 11β‐HSD1 inhibitors should confirm that the desired level of 11β‐HSD1 enzyme inhibition in AT can be maintained for the duration of the study and not rely on the results following a single dose.
Conflicts of interest
All authors were all employed by AstraZeneca at the time the studies were carried out and analysed.
Author contributions
All authors below contributed to drafting the work or revising it critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
In addition, each individual author made specific contributions as follows:
P M G substantially contributed to the conception and design of the work, and analysis and interpretation of data; A G substantially contributed to the conception and design of the work, and acquisition and interpretation of data; J deS substantially contributed to the acquisition, analysis and interpretation of data; P C substantially contributed to the conception and design of the work, and analysis and interpretation of data; J S substantially contributed to the acquisition and analysis of data; C S substantially contributed to the acquisition and analysis of data; M S substantially contributed to the conception and design of the work, and interpretation of data; J W E substantially contributed to the conception and design of the work, and interpretation of data; C N substantially contributed to the conception and design of the work, and analysis and interpretation of data; B L substantially contributed to the conception and design of the work and interpretation of data.
Supporting information
Table S1 Summary table of experiments presentedinthispaper.
Figure S1 Comparison of the PK/PD relationship between rat 11β‐HSD1 enzyme activity and adipose tissue concentrations of AZD8329 after acute and repeated oral administration in rats; blue diamonds are observed results after acute administration and red dots are the observed results after 7 days of repeat dosing.
Supporting info item
Acknowledgements
The authors wish to thank the entire 11β‐HSD1 team at AstraZeneca R&D for valuable support. Special thanks to Martin Wild, Mark Denn, Pam Tyers, Elizabeth Pilling, Elizabeth Kelly, Fred Goldberg and AS & W for their help and support to carry out this work.
Morentin Gutierrez, P. , Gyte, A. , deSchoolmeester, J. , Ceuppens, P. , Swales, J. , Stacey, C. , Eriksson, J. W. , Sjöstrand, M. , Nilsson, C. , and Leighton, B. (2015) Continuous inhibition of 11β‐hydroxysteroid dehydrogenase type I in adipose tissue leads to tachyphylaxis in humans and rats but not in mice. British Journal of Pharmacology, 172: 4806–4816. doi: 10.1111/bph.13251.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balazs Z, Nashev LG, Chandsawangbhuwana C, Baker ME, Odermatt A (2009). Hexose‐6‐phosphate dehydrogenase modulates the effect of inhibitors and alternative substrates of 11beta‐hydroxysteroid dehydrogenase 1. Mol Cell Endocrinol 301: 117–122. [DOI] [PubMed] [Google Scholar]
- Buttar D, Colclough N, Gerhardt S, MacFaul PA, Phillips SD, Plowright A et al. (2010). A combined spectroscopic and crystallographic approach to probing drug‐human serum albumin interactions. Bioorg Med Chem 18: 7486–7496. [DOI] [PubMed] [Google Scholar]
- Dzyakanchuk AA, Balazs Z, Nashev LG, Amrein KE, Odermatt A (2009). 11beta‐Hydroxysteroid dehydrogenase 1 reductase activity is dependent on a high ratio of NADPH/NADP(+) and is stimulated by extracellular glucose. Mol Cell Endocrinol 301: 137–141. [DOI] [PubMed] [Google Scholar]
- Feig PU, Shah S, Hermanowski‐Vosatka A, Plotkin D, Springer MS, Donahue S et al. (2011). Effects of an 11beta‐hydroxysteroid dehydrogenase type 1 inhibitor, MK‐0916, in patients with type 2 diabetes mellitus and metabolic syndrome. Diabetes Obes Metab 13: 498–504. [DOI] [PubMed] [Google Scholar]
- Gathercole LL, Stewart PM (2010). Targeting the pre‐receptor metabolism of cortisol as a novel therapy in obesity and diabetes. J Steroid Biochem Mol Biol 122: 21–27. [DOI] [PubMed] [Google Scholar]
- Gibbs JP, Emery MG, McCaffery I, Smith B, Gibbs MA, Akrami A et al. (2011). Population pharmacokinetic/pharmacodynamic model of subcutaneous adipose 11beta‐hydroxysteroid dehydrogenase type 1 (11beta‐HSD1) activity after oral administration of AMG 221, a selective 11beta‐HSD1 inhibitor. J Clin Pharmacol 51: 830–841. [DOI] [PubMed] [Google Scholar]
- Goldberg FW, Leach AG, Scott JS, Snelson WL, Groombridge SD, Donald CS et al. (2012). Free‐Wilson and structural approaches to co‐optimizing human and rodent isoform potency for 11B‐Hydoxysteroid Dehydrogenase type 1 (11B‐HSD1) inhibitors. J Med Chem 55: 10652–10661. [DOI] [PubMed] [Google Scholar]
- Hale C, Veniant M, Wang Z, Chen M, McCormick J, Cupples R et al. (2008). Structural characterization and pharmacodynamic effects of an orally active 11beta‐hydroxysteroid dehydrogenase type 1 inhibitor. Chem Biol Drug Des 71: 36–44. [DOI] [PubMed] [Google Scholar]
- Hermanowski‐Vosatka A, Balkovec JM, Cheng K, Chen HY, Hernandez M, Koo GC et al. (2005). 11beta‐HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J Exp Med 202: 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Fried SK, Mundt SS, Wang Y, Sullivan S, Stefanni A et al. (2008). Depot‐specific regulation of the conversion of cortisone to cortisol in human adipose tissue. Obesity (Silver Spring, Md) 16: 1178–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd DJ, Helmering J, Cordover D, Bowsman M, Chen M, Hale C et al. (2009). Antidiabetic effects of 11beta‐HSD1 inhibition in a mouse model of combined diabetes, dyslipidaemia and atherosclerosis. Diabetes Obes Metab 11: 688–699. [DOI] [PubMed] [Google Scholar]
- McCormick KL, Wang X, Mick GJ (2006). Evidence that the 11 beta‐hydroxysteroid dehydrogenase (11 beta‐HSD1) is regulated by pentose pathway flux. Studies in rat adipocytes and microsomes. J Biol Chem 281: 341–347. [DOI] [PubMed] [Google Scholar]
- McCormick KL, Wang X, Mick GJ (2008). Modification of microsomal 11beta‐HSD1 activity by cytosolic compounds: glutathione and hexose phosphoesters. J Steroid Biochem Mol Biol 111: 18–23. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan SA, Tomlinson JW (2010). 11beta‐Hydroxysteroid Dehydrogenase Type 1 Inhibitors for the Treatment of Type 2 Diabetes. Expert Opin Investig Drugs 19: 1067–1076. [DOI] [PubMed] [Google Scholar]
- Napolitano A, Voice MW, Edwards CR, Seckl JR, Chapman KE (1998). 11Beta‐hydroxysteroid dehydrogenase 1 in adipocytes: expression is differentiation‐dependent and hormonally regulated. J Steroid Biochem Mol Biol 64: 251–260. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ; NC‐IUPHAR. (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstock J, Banarer S, Fonseca VA, Inzucchi SE, Sun W, Yao W et al. (2010). The 11‐beta‐hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care 33: 1516–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Hermanowski‐Vosatka A, Gibson K, Ruck RA, Jia G, Zhang J et al. (2011). Efficacy and safety of the selective 11beta‐HSD‐1 inhibitors MK‐0736 and MK‐0916 in overweight and obese patients with hypertension. Journal of the American Society of Hypertension: JASH 5: 166–176. [DOI] [PubMed] [Google Scholar]
- Scott JS, deSchoolmeester J, Kilgour E, Mayers RM, Packer MJ, Hargreaves D et al. (2012). Novel acidic 11B‐Hydoxysteroid dehydogenase type 1 (11B‐HSD1) inhibitor with reduced acyl glucoronide liability: the discovery of 4‐[4‐(2‐Adamantylcarbamoyl)‐5‐tert‐butyl‐pyrazol‐1‐yl]benzoic acid (AZD8329). J Med Chem 55: 10136–0147. [DOI] [PubMed] [Google Scholar]
- Sjostrand M, Jansson PA, Palming J, de Schoolmeester J, Gill D, Rees A et al. (2010). Repeated measurements of 11beta‐HSD‐1 activity in subcutaneous adipose tissue from lean, abdominally obese, and type 2 diabetes subjects–no change following a mixed meal. Hormone and metabolic research = Hormon‐ und Stoffwechselforschung = Hormones et metabolisme 42: 798–802. [DOI] [PubMed] [Google Scholar]
- Veniant MM, Hale C, Komorowski R, Chen MM, St Jean DJ, Fotsch C et al. (2009). Time of the day for 11beta‐HSD1 inhibition plays a role in improving glucose homeostasis in DIO mice. Diabetes Obes Metab 11: 109–117. [DOI] [PubMed] [Google Scholar]
- Wallerius S, Rosmond R, Ljung T, Holm G, Bjorntorp P (2003). Rise in morning saliva cortisol is associated with abdominal obesity in men: a preliminary report. J Endocrinol Invest 26: 616–619. [DOI] [PubMed] [Google Scholar]
- Wan ZK, Chenail E, Xiang J, Li HQ, Ipek M, Bard J et al. (2009). Efficacious 11beta‐hydroxysteroid dehydrogenase type I inhibitors in the diet‐induced obesity mouse model. J Med Chem 52: 5449–5461. [DOI] [PubMed] [Google Scholar]
- Wang SJ, Birtles S, de Schoolmeester J, Swales J, Moody G, Hislop D et al. (2006). Inhibition of 11beta‐hydroxysteroid dehydrogenase type 1 reduces food intake and weight gain but maintains energy expenditure in diet‐induced obese mice. Diabetologia 49: 1333–1337. [DOI] [PubMed] [Google Scholar]
- Webster SP, Ward P, Binnie M, Craigie E, McConnell KM, Sooy K et al. (2007). Discovery and biological evaluation of adamantyl amide 11beta‐HSD1 inhibitors. Bioorg Med Chem Lett 17: 2838–2843. [DOI] [PubMed] [Google Scholar]
- Yeh VS, Patel JR, Yong H, Kurukulasuriya R, Fung S, Monzon K et al. (2006). Synthesis and biological evaluation of heterocycle containing adamantane 11beta‐HSD1 inhibitors. Bioorg Med Chem Lett 16: 5414–5419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Summary table of experiments presentedinthispaper.
Figure S1 Comparison of the PK/PD relationship between rat 11β‐HSD1 enzyme activity and adipose tissue concentrations of AZD8329 after acute and repeated oral administration in rats; blue diamonds are observed results after acute administration and red dots are the observed results after 7 days of repeat dosing.
Supporting info item