

Abstract
Background and Purpose
Ischaemic heart disease can lead to serious, life‐threatening complications. Traditional therapies for ischaemia aim to increase oxygen delivery and reduce the myocardial ATP consumption by increasing the coronary perfusion and by suppressing cardiac contractility, heart rate or blood pressure. An adjunctive treatment option for ischaemia is to improve or optimize myocardial metabolism.
Experimental Approach
Metabolic suppression in the ischaemic heart is characterized by reduced levels of high‐energy molecules: ATP and NAD+. Because NAD+ is required for most metabolic processes that generate ATP, we hypothesized that restoration of NAD+ would be a prerequisite for ATP regeneration and examined the role of the major NAD+ anabolic and catabolic pathways in the bioenergetic restoration process following oxygen–glucose deprivation injury in a cardiomyocyte cell line (H9c2 cells).
Key Results
Salvage of NAD+ via nicotinamide phosphoribosyl transferase was essential for bioenergetic recovery in cardiomyocytes. Blockade of nicotinamide phosphoribosyl transferase prevented the restoration of the cellular ATP pool following oxygen–glucose deprivation injury by inhibiting both the aerobic and anaerobic metabolism in the cardiomyocytes. NAD+ consumption by PARP‐1 also undermined the recovery processes, and PARP inhibition significantly improved the metabolism and increased cellular ATP levels in cardiomyocytes.
Conclusions and Implications
We conclude that the NAD+ salvage pathway is essential for bioenergetic recovery in post‐hypoxic cardiomyocytes and PARP inhibition may represent a potential future therapeutic intervention in ischaemic heart disease.
Abbreviations
- CVD
cardiovascular disease
- FK866
(E)‐N‐[4‐(1‐benzoylpiperidin‐4‐yl)butyl]‐3‐(pyridin‐3‐yl)acrylamide
- JC‐1
5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethyl‐imidacarbocyanine iodide
- MitoSOX Red
MitoSOX™ Red mitochondrial superoxide indicator
- MTT
3‐(4,5‐dimethyl‐2‐thiazolyl)‐2,5‐diphenyl‐2H‐tetrazolium bromide
- NamPRT
nicotinamide phosphoribosyltransferase
- NMNAT
nicotinamide mononucleotide adenylyl transferase
- OGD
oxygen–glucose deprivation
- PJ34
N‐(6‐oxo‐5,6‐dihydrophenanthridin‐2‐yl)‐(N,N‐dimethylamino) acetamide hydrochloride
- RNF146
ring finger protein 146
Tables of Links
| TARGETS |
|---|
| Enzymes |
| PARP‐1 |
| LIGANDS |
|---|
| FK866, daporinad |
| NAD+ |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Ischaemic heart disease is a major cause of death and disability in developed countries. Cardiovascular disease (CVD) accounts for 31% of deaths, and coronary heart disease alone causes one of six deaths in the United States (Go et al., 2014; Go et al., 2014). Currently, CVD costs more than any other diagnostic group including cancer. The estimated cost of CVD was $315.4 billion in the United States in 2010 while the costs of all cancer and benign neoplasms was $201.5 billion (Go et al., 2014). Ischaemic heart disease is caused by reduced coronary blood supply to the heart that stops to provide enough oxygen and energy source for the myocardium. This leads to decreased oxygen consumption and to diminished ATP and NAD+ levels in the heart (Nunez et al., 1974; Cave et al., 2000; Schriewer et al., 2013). The imbalance between oxygen delivery and myocardial ATP demand causes energetic failure that may result in myocardial dysfunction or serious complications including angina pectoris, heart failure and acute myocardial infarction.
Revascularization is the most effective therapy to limit the infarct size and preserve the systolic function, but it involves secondary damage to the myocardium (reperfusion injury). Traditional pharmacological therapies use haemodynamic approaches to restore the balance between ATP formation and utilization: increasing the blood flow (oxygen delivery) via coronary vasodilation or decreasing the heart rate, arterial blood pressure and cardiac contractility to reduce the oxygen consumption (Stanley, 2001). Alternative (or adjunctive) treatments address the reperfusion injury and aim to optimize the energy metabolism in the ischaemic heart. Various pharmacological agents were tested to address the oxidative stress (antioxidants, PARP inhibitors) (Liaudet et al., 2001; Morrow et al., 2009), the mitochondrial permeability transition pore opening (cyclosporine A) (Hausenloy et al., 2002; Piot et al., 2008) and the metabolism (glucose–insulin–potassium, atorvastatin and glucagon‐like peptide exenatide) (Diaz et al., 1998; Bell and Yellon, 2003; Sack and Yellon, 2003; Lonborg et al., 2012) and even mechanical interventions (ischaemic post‐conditioning, remote ischaemic conditioning and therapeutic hypothermia) were used with success in myocardial infarction (Staat et al., 2005). Interestingly, these earlier approaches share a common mechanism of action: the activation of survival kinases, which were also designated as the reperfusion injury salvage kinase (RISK) pathway (Hausenloy and Yellon, 2004; Frohlich et al., 2013; Hausenloy and Yellon, 2013). Activation of survival kinases leads to inhibition of apoptotic effectors and also increases the translocation of glucose transporters to the cell membrane and improves glucose uptake (Sack and Yellon, 2003; Hausenloy and Yellon, 2004). However, the major limitation of ATP synthesis is not glucose deficiency in the ischaemic tissues but the reduced cellular NAD+ content (Nunez et al., 1974). Because CVD is a disease of the elderly, the basal tissue NAD+ content is also lower as a result of age‐related decrease in NAD+ biosynthesis enzymes (Braidy et al., 2011; Stein and Imai, 2012). Furthermore, NAD+ deficiency may be aggravated by PARP‐mediated NAD+ consumption in oxidative stress (Szabo et al., 2004; Molnar et al., 2006).
In mammalian cells, NAD+ is produced via two biosynthetic pathways: (1) the de novo (or kynurenine) pathway that uses tryptophan as substrate and (2) the salvage pathway that regenerates NAD+ from nicotinamide. The dominant route is the salvage pathway in which the rate‐limiting step is catalysed by nicotinamide phosphoribosyltransferase (NamPRT) (Chiarugi et al., 2012). Interestingly, there is also a secreted (extracellular) isoform of NamPRT (also called visfatin, pre‐B cell colony‐enhancing factor) that acts as a cytokine (Houtkooper et al., 2010). NAD+ has a short half‐life in the cells, and it is constantly recycled within a day; the estimated daily NAD+ synthesis is around 1 g∙kg−1 tissue weight (Chiarugi et al., 2012). Because NAD+ is compartmentalized in the cells, its distribution also needs to be considered in conditions of depletion. For instance, in cardiac myocytes, ~70% of the cellular NAD+ pool is in the mitochondria and cannot support glycolysis (Nikiforov et al., 2011; Stein and Imai, 2012). Because NAD+ is a prerequisite for energy production, rapid resynthesis of the cellular NAD+ content is expected to promote the recovery following hypoxia. Counterintuitively, inhibition of NAD+ biosynthesis by blocking NamPRT was protective in myocardial infarction (Montecucco et al., 2013). While this action was associated with blockage of the extracellular NamPRT and an anti‐inflammatory mechanism was suggested, it should be noted that the currently available NamPRT inhibitors block both the extracellular and the intracellular isoforms of the enzyme.
To investigate whether NamPRT played an essential role in the recovery following hypoxia, we studied the bioenergetic restoration in an in vitro model of ischaemia‐reperfusion injury, using a rat cardiomyocyte cell line (H9c2 cells). Because PARP is the most important NAD+ consumer in the cells, we also explored its role in the recovery process. We found that bioenergetic recovery was abolished by NamPRT inhibition but PARP inhibition significantly improved the cellular bioenergetics in the absence of NamPRT activity.
Methods
Cell culture
H9c2 rat cardiomyocytes were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in DMEM (Biochrom AG, Berlin, Germany) supplemented with 4 mM glutamine, 10% FBS (PAA Laboratories Inc, Westborough, MA), 100 IU∙mL−1 penicillin and 100 µg∙mL−1 streptomycin (Invitrogen, Carlsbad, CA) at 37°C in 10% CO2 atmosphere.
Oxygen–glucose deprivation injury
Oxygen–glucose deprivation (OGD) injury was conducted as previously described (Szabo et al., 2011; Szoleczky et al., 2012; Gero et al., 2014) with slight modifications. H9c2 cells (10 000 per well) were plated on 96‐well plates and cultured for 3 days prior to the induction of OGD. Cells transfected with siRNAs were subjected to OGD 48 h post‐transfection. Prior to hypoxia induction, the culture medium was replaced with glucose‐free DMEM. Then the plates were placed in gas‐tight incubation chambers (Billups‐Rothenberg Inc., Del Mar, CA), and the chamber atmosphere was replaced by flushing with 95% N2/5% CO2 mixture at 30 L∙min−1 flow rate for 10 min. Hypoxia was maintained for 8 or 10 h (as indicated) by incubation at 37°C. All assay plates subjected to hypoxia included wells exposed to glucose‐free medium (OGD) and medium containing 4.5 g∙L−1 glucose (hypoxia). Cells were simultaneously exposed to glucose‐free medium (glucose deprivation) or maintained in glucose‐containing medium (CTL) under normoxia. Following the hypoxia, glucose and serum concentration was restored by supplementing the culture medium with glucose and FBS, and the cells were incubated for 16 h at 37°C (recovery). In select cases, cells were treated with an inhibitor of NamPRT, FK866 (10 μM; (E)‐N‐[4‐(1‐benzoylpiperidin‐4‐yl)butyl]‐3‐(pyridin‐3‐yl)acrylamide, NIMH F‐901, Research Triangle Institute, Research Triangle Park, NC) and/or the PARP inhibitor PJ34 (3 μM; N‐(6‐oxo‐5,6‐dihydrophenanthridin‐2‐yl)‐(N,N‐dimethylamino) acetamide hydrochloride, Sigma‐Aldrich, St. Louis, MO) immediately following the hypoxic period. Measurement of cell viability, LDH release, ATP and NAD+ content, mitochondrial potential and oxidant production was carried out either immediately following the hypoxic period or at the end of the 16‐h‐long recovery period. Cellular viability was measured with MTT 3‐(4,5‐dimethyl‐2‐thiazolyl)‐2,5‐diphenyl‐2H‐tetrazolium bromide and by LDH release, as previously described (Gero et al., 2007). A calibration curve was created by measuring the MTT converting capacity of serial dilutions of H9c2 cells to calculate the number of viable cells using Gen5 data reduction software (Biotek, Winooski, VT). Viability values are shown as percent of control mean cell count.
Oxidant‐induced injury
Oxidant injury was carried out as previously described (Gero et al., 2007; Gero et al., 2014). H9c2 cells (10 000 per well) were plated on 96‐well plates and grown for 3 days. The cells were treated with H2O2 in fresh culture medium and incubated for 3 or 24 h. Cell viability was measured by the MTT and LDH assays (Gero et al., 2007). Viability values are shown as percent of vehicle‐treated control mean cell count. Cells transfected with siRNAs were subjected to H2O2 injury 48 h post‐transfection. To test the oxidant sensitivity of the cells exposed to hypoxia and/or glucose deprivation, the cells were treated with H2O2 in fresh culture medium immediately following the hypoxia and incubated for 3 h prior to viability measurements. To assay the role of NamPRT, cells were pretreated with FK866 (10 μM) for 1 h prior to the addition of H2O2.
ATP and NAD+ assays
ATP concentration was determined by the commercially available CellTiter‐Glo® Luminescent Cell Viability Assay (Promega, Madion, WI) as previously described (Gero et al., 2014). An ATP calibration curve was prepared by measuring the luminescent signal generated by serial dilutions of ATP. The ATP content of each well was calculated by Gen5 data reduction software (Biotek, Winooski, VT), and values are shown as percent of control mean ATP content.
The cellular NAD+ content was measured using an alcohol dehydrogenase‐based assay (Queval and Noctor, 2007; Modis et al., 2012). NAD+ extraction was performed by precipitating the cell samples in 0.2 M hydrochloric acid, then the lysates were centrifuged at 16 000× g for 15 min. The cleared supernatant (200 μL) was heated to 100°C for 3 min and neutralized by adding 20 μL 0.4 M NaH2PO4 and 160 μL 0.2 M NaOH. The NAD+ reaction mix was prepared by diluting MTT (250 μM), N‐methylphenazonium methyl sulfate (1 mM) and alcohol dehydrogenase (7.5 U∙mL−1) in reaction buffer (100 mM HEPES, 2 mM EDTA, 10 mM nicotinamide, pH 7.5). The sample or NAD+ calibration standard (20 μL) was added to 160 μL reaction mixture and the reaction started by addition of the substrate (ethanol, 7.5%). The reaction was monitored kinetically at 570 nm for 1 h. NAD+ concentration was calculated using a calibration curve generated from the maximal velocity values of simultaneously measured NAD+ dilution series. NAD+ content is shown as percent of control mean NAD+ values.
Mitochondrial potential and superoxide production
Mitochondrial potential was measured with JC‐1 (Sigma‐Aldrich, St. Louis, MO) fluorescent probe. The cells were loaded with the dye by exposing them to JC‐1 stain solution (containing 10 μM JC‐1 and 0.6 mM β‐cyclodextrin (Sigma‐Aldrich, St. Louis, MO)) for 30 min. Subsequently, the cells were washed in PBS, and the red (Ex/Em: 485/528 nm) and green (Ex/Em: 530/590 nm) fluorescence were measured on a microplate reader. The mitochondrial potential is expressed as the relative ratio of the mitochondrial J‐aggregates (red fluorescence) and the cytoplasmic monomer form of the dye (green fluorescence).
Mitochondrial reactive oxygen species (ROS) production was measured using the mitochondrial superoxide sensor MitoSOX™ Red (Invitrogen, Carlsbad, CA) as previously described (Gero et al., 2013).
siRNA‐mediated gene silencing and gene expression assays
H9c2 cells (10 000 per well) were plated on 96‐well plates; the following day, the cells were transfected with Silencer Select PARP‐1, NMNAT1, NMNAT3 or negative control #1 siRNA (1 pmol per well, assay IDs: s62054, s151624, s221491 and ID: 4390844, Life Technologies, Carlsbad, CA) using Lipofectamine 2000 transfection reagent. The knockdown efficiency was evaluated by realtime PCR and by Western blotting 24 and 48 h post‐transfection for PARP‐1 and by realtime PCR for NMNAT1 and NMNAT3 siRNAs 48 h following the transfection. RNA was isolated using a commercial RNA purification kit (SV total RNA isolation kit, Promega, Madison, WI) and reverse transcribed using High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) as previously described (Gero et al., 2013; Gero et al., 2013). PARP‐1, NMNAT1, NMNAT3 and NamPRT expressions were measured with Taqman assays (assay IDs: Rn00565018_m1, Rn01516826_m1, Rn01403958_m1 and Rn00822043_m1, Life Technologies, Carlsbad, CA) using TaqMan® Rodent GAPDH Control Reagents (Life Technologies, Carlsbad, CA) for normalization. Relative expression values are shown as percent of control mean expression.
Western blotting was performed as previously described using antibodies against PARP‐1 (Cell Signaling, Beverly, MA) and anti‐ring finger protein 146 (RNF146) (Abnova, Walnut, CA) (Gero et al., 2014). Blots were detected on a CCD camera‐based detection system (GBox, Syngene USA, Frederick, MD) with enhanced chemiluminescent substrate, and signals were quantitated using Genetools analysis software (Syngene USA, Frederick, MD). Normalized expression values are shown as percent of control mean expression.
Bioenergetic profiling by extracellular flux analysis
An XF24 Analyzer (Seahorse Biosciences, Billerica, MA) was used to measure cellular bioenergetics following hypoxia in H9c2 cells (Gero et al., 2013; Gero et al., 2013). It measures oxygen and proton concentrations in real time via specific fluorescent dyes and calculates oxygen consumption rate (expressed in pmol min‐1) and extracellular acidification rate (expressed in pH change per minute) that represent measures of the aerobic and anaerobic metabolism.
H9c2 cells transfected with PARP‐1 or CTL siRNA were exposed to 8‐h‐long hypoxia or OGD. Subsequently, glucose concentration and oxygen tension were normalized, and the cells were treated with FK866 (10 μM) or vehicle and incubated for 16 h before analysing the cellular metabolism. For all bioenergetic measurements, the culture medium was changed to unbuffered DMEM (pH 7.4) containing 5 mM glucose, 2 mM L‐glutamine and 1 mM sodium pyruvate. After determining the basal oxygen consumption rate and extracellular acidification rate values, oligomycin, carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone and antimycin A were injected (1 µg∙mL−1, 0.3 μM and 2 µg∙mL−1, respectively) to determine the oxygen consumption linked to ATP production, the non‐ATP‐linked oxygen consumption (proton leak), the maximal respiration capacity and the non‐mitochondrial oxygen consumption. Simultaneous recording of the proton production was used to calculate the capacity of the cells to compensate via anaerobic metabolism.
Data analysis
Data are shown as means ± SEM. One‐way anova was used to detect differences between groups or unpaired two‐tailed Student's t‐test to compare two groups. Post hoc comparisons were made using Tukey's test. A value of p < 0.05 was considered statistically significant. Non‐linear regression curves were fitted to raw viability and LDH activity values with Prism 4 software to calculate 50% reduction or increase values respectively. All statistical calculations were performed using Prism 4 analysis software (GraphPad Software, Inc., La Jolla, CA).
Results
OGD induces reversible injury that sensitizes the cells to oxidant injury
In ischaemia, the loss of blood flow decreases the oxygen and energy source (glucose) supply and results in cellular energy depletion (diminished ATP pool). To investigate the bioenergetic recovery processes from this state, we developed an in vitro model of reversible injury by exposing H9c2 cardiomyocytes to OGD. We found that 8‐h‐long OGD resulted in no change of cellular viability if the oxygen and energy source supplies were normalized subsequently (Figure 1A and B), while longer OGD periods clearly induced cell death (Figure 1A and B) (Szabo et al., 2011). The 8‐h‐long OGD severely diminished the cellular ATP and NAD+ pools that were completely restored during the 16‐h incubation with normalized oxygen and glucose levels (Figure 1C and D). Glucose deprivation or hypoxia alone did not decrease the ATP pool (Figure 1C); thus, the energy source reserve in the cells was sufficient to produce ATP via aerobic metabolism and the hypoxic cells could switch to anaerobic metabolism and produce adequate amount of ATP using glucose for energy source. Interestingly, glucose deprivation alone also caused a slight decrease in NAD+ content, but it was not comparable to the effect of OGD (Figure 1D).
Figure 1.
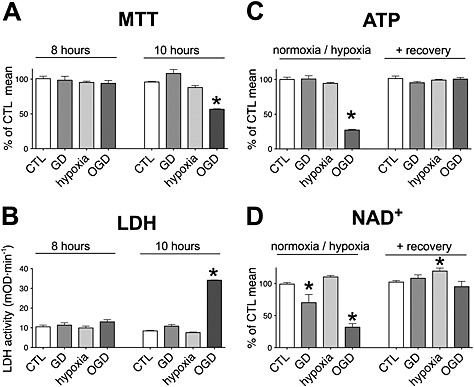
OGD induces reversible energy depletion. A, B: H9c2 cells were exposed to glucose deprivation (GD), hypoxia or OGD or maintained in glucose‐containing culture medium at normoxia (CTL) for 8 or 10 h. The cellular viability was determined by the MTT assay (A), and LDH release (B) was measured in the supernatant following a 16‐h‐long recovery period. 8‐h‐long OGD did not affect the cellular viability, while 10‐h‐long exposure induced significant decrease in viability. C, D: H9c2 cells were exposed to GD, hypoxia or OGD for 8 h followed by a 16‐h‐long recovery period. The cellular ATP and NAD+ contents were measured at the end of the 8‐h‐long hypoxic period and following the recovery. OGD induces significant decrease in cellular ATP and NAD+ contents that are completely restored during the recovery period. n = 4, *P < 0.05 compared with CTL.
PARP‐1 is the major NAD+ consumer in oxidative stress, and PARP inhibition (by genetic or pharmacological means) is protective in various heart disease models, including myocardial infarction (Pieper et al., 2000; Liaudet et al., 2001). We found that PARP‐1 expression was decreased following severe ischaemic injury in the heart and the removal of PARP‐1 was regulated by RNF146, an E3 ubiquitin ligase (Gero et al., 2014). RNF146 directly interacts with PARP‐1, and by capturing PARP‐1, it can modulate the cell fate in oxidative stress. High levels of RNF146 protect against cellular injury, while decreased RNF146 levels augment the injury.
To test whether similar changes occur in this model, we measured the expression of PARP‐1 and RNF146. We found that significant changes occur in the expression of PARP‐1 and RNF146 at the mRNA level following hypoxia, but these mostly return to normal by the end of the recovery period (Figure 2A and B). At the protein level, RNF146 expression is significantly suppressed following OGD, but it is normalized by the end of the recovery period (Figure 2C–F). On the other hand, PARP‐1 level becomes significantly elevated in the cells exposed to hypoxia alone, which can be the result of increased PARP‐1 and decreased RNF146 expression during hypoxia, as detected at the mRNA level. It should be noted that changes at the mRNA level may not be directly translated to the protein levels in case of these proteins, because the degradation of PARP‐1 and RNF146 can occur very rapidly (Gero et al., 2014).
Figure 2.
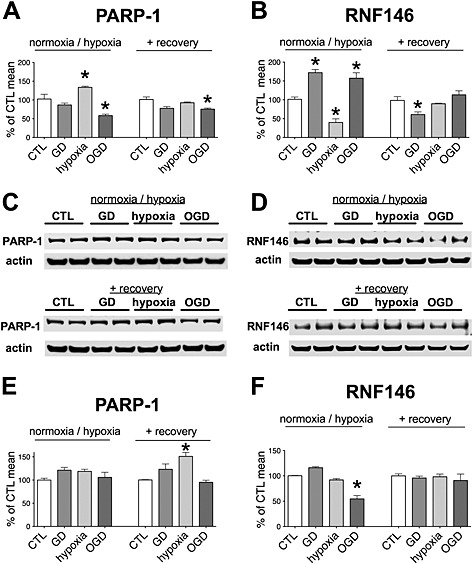
PARP‐1 and RNF146 expression in hypoxia‐reoxygenation injury. H9c2 cells were exposed to glucose deprivation (GD), hypoxia or OGD or maintained in glucose‐containing culture medium at normoxia (CTL) for 8 h followed by a 16‐h‐long recovery period. A, B: PARP‐1 (A) and RNF146 (B) mRNA level expression was measured by Taqman assays at the end of the hypoxic period and following the recovery. Relative expression levels were normalized to GAPDH expression. Hypoxia induces significant increase in PARP‐1 mRNA level and a decrease in RNF146 expression, while GD and OGD increases the expression of RNF146. C–F: PARP‐1 (C and E) and RNF146 (D and F) protein expression was measured by Western blotting at the end of the hypoxic period and following the recovery using actin normalization. Representative blots (C and D) and densitometric analysis results (E and F) are shown. Hypoxia increases the expression of PARP‐1 at the end of the recovery period. OGD induces a reversible decrease in RNF146 expression. n = 4, *P < 0.05 compared with CTL.
The increased amount of PARP‐1 protein, and decreased amounts of RNF146 and NAD+ (the substrate of PARP‐1) may all predispose to oxidative stress‐induced injury; thus, we tested whether hypoxia, glucose deprivation or OGD (that itself does not induce irreversible injury) increases the sensitivity of the cells to oxidants. To test the role of PARP‐1 in the oxidant‐induced damage, we utilized siRNA‐mediated PARP‐1 silencing (Figure S1). We found that cells subjected to glucose deprivation, hypoxia or OGD showed decreased tolerance to oxidants, but PARP‐1 silencing had some protective effects in all cases (Figure 3). Cells exposed to OGD showed the weakest tolerance to oxidants, but cells subjected to glucose deprivation or hypoxia were also more sensitive than the cells maintained at normoxia. Glucose deprivation and OGD narrowed the concentration range of oxidants that was tolerated by the cells that might be the result of diminished NAD+ content of the cells. Hypoxia alone also sensitized the cells to oxidants, but it did not narrow the concentration range of oxidants that the cells survived.
Figure 3.
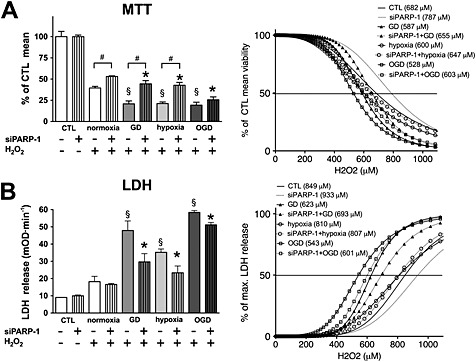
Oxygen and glucose deprivation increases the sensitivity to oxidants. H9c2 cells were transfected with PARP‐1 (siPARP‐1) or CTL siRNA, and 48 h later, the cells were exposed to glucose deprivation (GD), hypoxia or OGD for 8 h or maintained in glucose‐containing culture medium at normoxia (CTL). Subsequently, the cells were exposed to H2O2 (0–1100 μM) for 3 h, and cellular viability was determined by the MTT assay (A) and LDH release (B) was measured in the supernatant. Non‐linear curve‐fitting was applied to the raw data, and the concentration of H2O2 that caused 50% reduction in viability (A) or 50% increase in LDH release (B) was determined. Raw data measured at 700 μM H2O2 are shown (left panels), and curve‐fitting results with the concentrations that cause 50% reduction in MTT conversion or 50% increase in LDH release are shown (right panels). H2O2 induced significant reduction in cellular viability and induced significant increase in LDH release in all groups at 700 μM concentration (not labelled on the graph). Hypoxia, GD and OGD augment the H2O2‐induced injury, and PARP‐1 silencing provides protection. GD and OGD result in narrower range of H2O2 tolerance (steeper curves). n = 4, #P < 0.05 PARP‐1 silenced cells compared with respective CTL siRNA treated cells, §P < 0.05 CTL siRNA‐transfected, H2O2‐treated cells exposed to GD, hypoxia or OGD compared with CTL siRNA‐transfected cells maintained at normoxia, *P < 0.05 PARP‐1 siRNA‐transfected, H2O2‐treated cells exposed to GD, hypoxia or OGD compared with PARP‐1 siRNA‐transfected cells maintained at normoxia.
We tested whether the resistance to oxidants mediated by PARP‐1 silencing was associated with higher cellular ATP or NAD+ levels following hypoxia, but no difference was found between the PARP‐1 silenced and control cells (Figure 4A–D). Similarly, no difference was detectable in the cellular ATP and NAD+ levels between the PARP‐1 silenced and control cells following the recovery period. Glucose deprivation and OGD resulted in a recoverable increase in the mitochondrial potential following the 8‐h‐long exposure, and PARP‐1 silencing resulted in no change (Figure 4E and F). A slight mitochondrial dysfunction was measurable following the recovery period in the glucose‐deprived and oxygen–glucose‐deprived cells, as an increase in the mitochondrial superoxide production that was beneficially affected by PARP‐1 silencing (Figure 4G and H).
Figure 4.
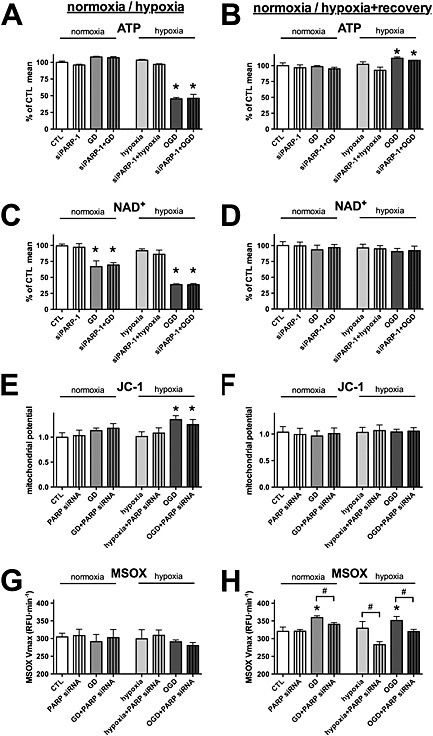
PARP‐1 silencing decreases the mitochondrial oxidant production following hypoxia. A–H: H9c2 cells were transfected with PARP‐1 (siPARP‐1) or CTL siRNA, and 48 h later, the cells were exposed to glucose deprivation (GD), hypoxia or OGD for 8 h. Control cells (CTL) were maintained in glucose‐containing culture medium at normoxia. Subsequently, the cells were subjected to a 16‐h‐long recovery. A–D: The cellular ATP (A and B) and NAD+ (C and D) concentrations were determined both at the end of the hypoxia (A and C) and following the recovery (B and D). E, F: The mitochondrial potential measured by JC‐1 at the end of the hypoxia (E) and following the recovery (F). G, H: Mitochondrial superoxide production was measured using MitoSOX Red (MSOX) staining at the end of the hypoxia (G) and following the recovery (H). PARP‐1 silencing does not affect the cellular ATP recovery or the mitochondrial potential following hypoxia but decreases the mitochondrial oxidant production. n = 3; *P < 0.05 compared with CTL, #P < 0.05 PARP‐1 silenced cells compared with respective CTL siRNA treated cells.
Overall, we conclude that both hypoxia and glucose deprivation sensitize the cells to oxidative stress and, when the two are combined, dysfunction and serious injury can be caused by levels of oxidative stress that scarcely cause damage in intact cells. Diminished cellular energy reserve (ATP and NAD+ pools) is responsible in large part for the increased oxidant sensitivity.
NamPRT is necessary for bioenergetic recovery following OGD
Because OGD not only decreased the cellular ATP content but also resulted in a severe decrease in the NAD+ level and both the ATP and NAD+ levels returned to normal during the recovery period, we hypothesized that NAD+ resynthesis was necessary for the ATP recovery. As mentioned in the introduction, NAD+ synthesis can occur via two major pathways: (1) the de novo synthesis and (2) the salvage pathway (Houtkooper et al., 2010). We blocked the NAD+ salvage with the NamPRT inhibitor FK866 during the recovery period following hypoxia, glucose deprivation or OGD (Hasmann and Schemainda, 2003). NamPRT inhibition completely blocked the recovery of the cellular ATP content following OGD exposure and also resulted in significantly lower ATP content following glucose deprivation and hypoxia (Figure 5A and B), and it blocked the normalization of the mitochondrial potential and induced mitochondrial ROS production (Figure 5C–F). To test whether NAD+ consumption by PARP‐1 affected the recovery, we blocked PARP activity with PJ34 during the recovery. Consistent with previous findings showing that silencing of PARP‐1 did not affect ATP regeneration, PARP inhibition by itself had no effect on ATP recovery (Figure 5A and B). However, inhibition of PARP significantly increased the ATP level in glucose‐deprived and OGD‐exposed cells if NAD+ salvage was blocked, showing that PARP‐1 was responsible for a significant basal NAD+ consumption in the cells. PARP inhibition also restored the mitochondrial potential and reduced the mitochondrial ROS production (Figure 5C–F).
Figure 5.
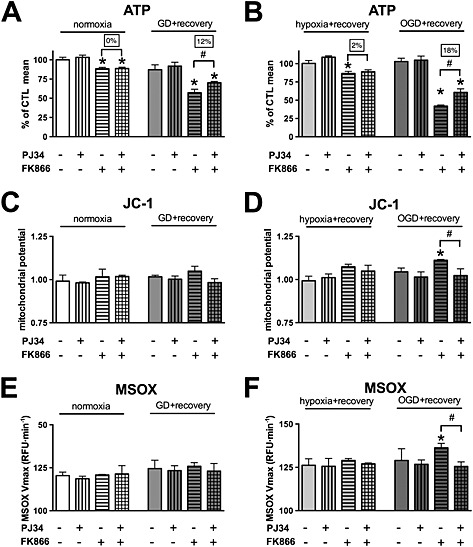
NamPRT is necessary for regeneration of the cellular ATP pool following OGD. A–F: H9c2 cells were exposed to glucose deprivation (GD), hypoxia or OGD or maintained in glucose‐containing culture medium at normoxia (CTL) for 8 h. Subsequently, the cells were treated with NamPRT inhibitor FK866 (10 μM), PARP inhibitor PJ34 (3 μM) or vehicle and were subjected to 16‐h recovery. A, B: The cellular ATP concentration was measured. C, D: The mitochondrial potential determined by JC‐1. E, F: Mitochondrial ROS production was determined by measuring the oxidation of the MitoSOX (MSOX) superoxide sensor. NamPRT inhibition blocks the regeneration of the cellular ATP pool following OGD. PARP inhibition improves the ATP recovery following NamPRT inhibition, restores the mitochondrial potential and reduces the ROS production. n = 3; *P < 0.05 compared with CTL, #P < 0.05 compared with respective PARP inhibitor treated cells.
These data suggest that NAD+ synthesis from nicotinamide (the salvage pathway) plays a dominant role in NAD+ biosynthesis in cells following hypoxia. To test whether its dominant function is restricted to post‐hypoxic recovery, we also exposed H9c2 cells to oxidant injury and measured the viability changes after 3 and 24 h. We found that NamPRT inhibition caused little difference 3 h following oxidant exposure, but it significantly augmented cell damage at 24 h (Figure 6), suggesting that NamPRT was involved in the cellular recovery from oxidative stress.
Figure 6.
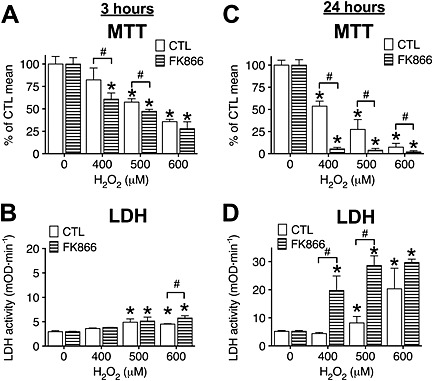
NamPRT is necessary for recovery following oxidant‐induced injury. A–D: H9c2 cells were treated with NamPRT inhibitor FK866 (10 μM, 1 h) or vehicle (CTL) and exposed to H2O2 (400, 500 or 600 μM) for 3 (A and B) or 24 h (C and D). Cellular viability was determined by the MTT assay (A and C), and the LDH release (B and D) was measured in the supernatant. NamPRT inhibition augments cellular oxidant‐induced cellular injury. n = 3; *P < 0.05 compared with cells not exposed to H2O2, #P < 0.05 compared with respective CTL cells.
To further investigate the role of NamPRT in cellular bioenergetics in post‐hypoxic cells, we conducted extracellular flux analysis. Oxidative phosphorylation was slightly diminished in cells prior exposed to OGD, but the anaerobic metabolism was not affected (Figure 7A–D). NamPRT inhibition did not affect the basal respiration or the anaerobic metabolism in cells exposed to hypoxia only but strongly decreased both respiration and acid production in cells pre‐exposed to OGD, confirming that NamPRT is necessary for bioenergetic recovery following OGD that caused cellular NAD+ and ATP decrease. We used PARP‐1 silencing to simultaneously test whether NAD+ consumption by PARP‐1 affected the cellular metabolism. Consistent with our results of PARP inhibition (Figure 5), PARP‐1 silencing significantly improved both the aerobic and anaerobic metabolism in the cells exposed to OGD, when resynthesis of NAD+ was blocked (Figure 7A–D).
Figure 7.
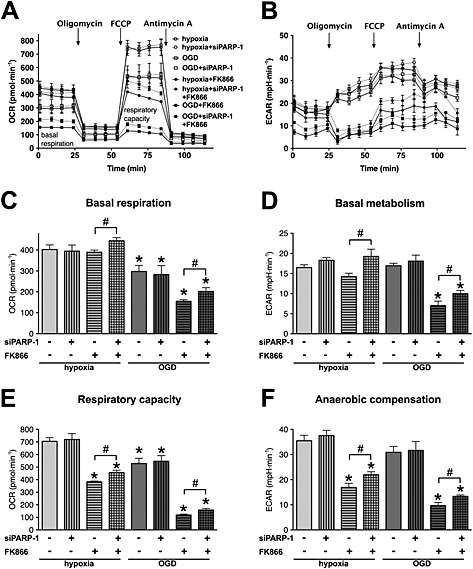
NamPRT is necessary for bioenergetic recovery following OGD. A–F: H9c2 cells were transfected with PARP‐1 (siPARP‐1) or CTL siRNA, and 48 h later, the cells were exposed to hypoxia or OGD for 8 h. Subsequently the cells were treated with NamPRT inhibitor FK866 (10 μM) or vehicle and were subjected to 16‐h‐long recovery. A, B: Metabolic profile of the cells was studied by sequential injections of oligomycin (1 µg∙mL−1), carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (0.3 μM) and antimycin A (2 µg∙mL−1) and by measurement of (A) the cellular oxygen consumption rate (OCR) and (B) the extracellular acidification rate using extracellular flux analysis. C, D: Basal oxygen consumption (C) and acid production of the respective basal metabolism (D) are shown. E, F: Total respiratory capacity (E) was determined following the addition of carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone, and respective acid production via anaerobic compensation (F) is shown. NamPRT inhibition severely blocks the recovery of the respiratory capacity and prevents the anaerobic metabolic compensation. n = 3, *P < 0.05 compared with hypoxia control, #P < 0.05 PARP‐1 silenced cells compared with respective CTL siRNA treated cells.
Metabolic profiling also revealed diminished respiratory capacity in cells pre‐exposed to OGD and the respiratory capacity was greatly reduced by NamPRT inhibition (Figure 7E). Importantly, anaerobic metabolism (glycolysis) showed an even higher degree of blockage in both post‐hypoxic and post‐OGD cells, when oxidative phosphorylation and NAD+ resynthesis were inhibited (by oligomycin and FK866, respectively) (Figure 7F), showing that the lack of NAD+ seriously inhibited glycolysis. PARP‐1 silencing significantly improved the respiratory capacity and anaerobic compensation, when NAD+ salvage was inhibited (Figure 7E and F). These results confirmed consumption of NAD+ by PARP‐1.
The next step of NAD+ biosynthesis is catalysed by nicotinamide mononucleotide adenylyl transferases (NMNATs). There are three NMNAT isoforms in mammals: the (predominantly) nuclear NMNAT1 and the mitochondrial NMNAT3 are ubiquitous, while NMNAT2 is localized to the Golgi and the cytoplasm but its expression is restricted to the brain (Berger et al., 2005; Nikiforov et al., 2011). To selectively test whether NMNAT1 or NMNAT3 is essential for the recovery from oxidant‐ induced or OGD‐induced injury, we used siRNA‐mediated gene silencing. NMNAT1 silencing induced a slight decrease in the basal ATP levels of the cells, but glucose deprivation and hypoxia did not result in any further decrease in the cellular ATP level, and neither the OGD‐induced decrease nor the recovery was affected by the decreased NMNAT1 expression (Figure 8A–F). Similarly, reduced NMNAT3 level resulted in a slight decrease in the ATP content in the hypoxia injury, but it neither affected the OGD‐induced decrease nor the recovery of the cellular ATP content (Figure 9A and D). NMNAT3 silencing did not affect the cellular viability in the OGD injury (Figure 9A–F), and unlike NMNAT1 silencing (Figure 8B), the reduced NMNAT3 expression did not result in decreased tricarboxylic acid cycle activity (as measured by the MTT assay) following hypoxia (Figure 9B). We also tested the effect of NMNAT silencing in oxidant‐induced injury and found that neither NMNAT1 nor NMNAT3 silencing affected the cell survival or the recovery following the oxidant exposure (Figure S2 and S3).
Figure 8.
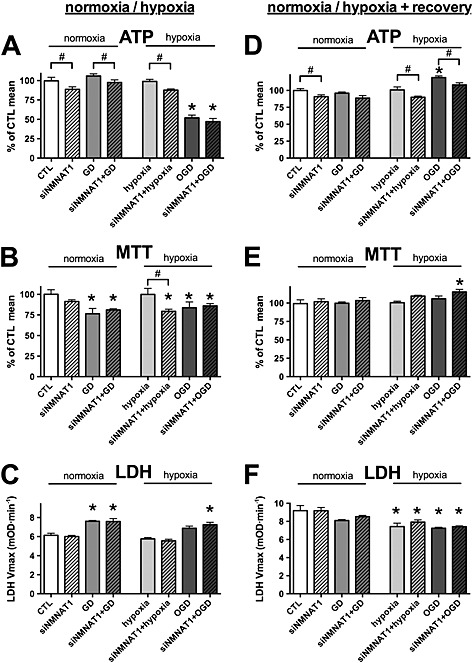
NMNAT1 silencing does not affect bioenergetic recovery following OGD. A–C: H9c2 cells were transfected with NMNAT1 siRNA (siNMNAT1) or CTL siRNA, and 48 h later, the cells were exposed to glucose deprivation (GD), hypoxia or OGD or maintained in glucose‐containing culture medium at normoxia (CTL) for 8 h. D–F: Following the 8‐h‐long GD, hypoxic injury or OGD, glucose concentration and oxygen tension were normalized, and the cells were subjected to 16‐h‐long recovery. A, D: The cellular ATP content was measured at the end of the hypoxic period and following the recovery period. B, E: The cellular viability was measured by the MTT assay. C, F: LDH release was determined in the supernatant. NMNAT1 siRNA decreases the cellular ATP content but does not inhibit regeneration of the cellular ATP following OGD. n = 3, *P < 0.05 compared with normoxia CTL, #P < 0.05 NMNAT1 silenced cells compared with respective CTL siRNA treated cells.
Figure 9.
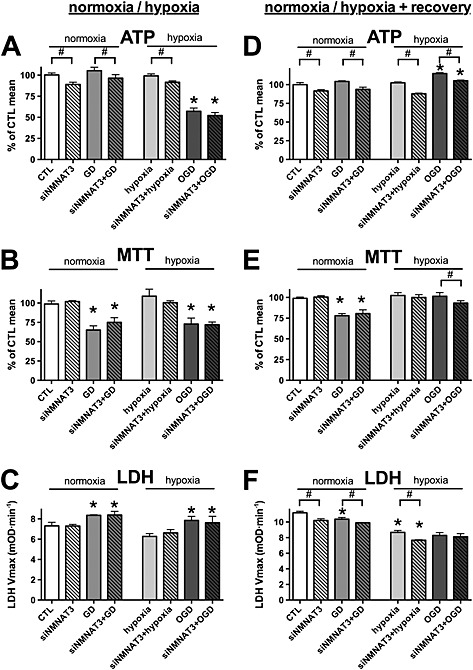
NMNAT3 silencing does not affect bioenergetic recovery following OGD. A–C: H9c2 cells were transfected with NMNAT3 siRNA (siNMNAT3) or CTL siRNA, and 48 h later, the cells were exposed to glucose deprivation (GD), hypoxia or OGD or maintained in glucose‐containing culture medium at normoxia (CTL) for 8 h. D–F: Following the 8‐h‐long GD, hypoxic injury or OGD, glucose concentration and oxygen tension were normalized, and the cells were subjected to 16‐h‐long recovery. A, D: The cellular ATP content was measured at the end of the hypoxic period and following the recovery period. B, E: The cellular viability was measured by the MTT assay. C, F: LDH release was determined in the supernatant. NMNAT3 siRNA decreases the cellular ATP content but does not inhibit regeneration of the cellular ATP following OGD. n = 3, *P < 0.05 compared with normoxia CTL, #P < 0.05 NMNAT3 silenced cells compared with respective CTL siRNA treated cells.
Taken together, these data confirmed that NAD+ salvage by NamPRT played a significant role in bioenergetic recovery of post‐hypoxic/post‐OGD cardiomyocytes, and that the suppressed NAD+ pool following OGD inhibited both aerobic and anaerobic metabolism in these cells. On the other hand, NMNAT1 and NMNAT3 did not play essential roles in the bioenergetic recovery following OGD, or alternatively, they may compensate for the loss of each other in this process. PARP‐1 consumed significant amounts of NAD+ in the post‐hypoxic cells, thus affecting the cellular metabolism. The basal activity of this enzyme could delay the bioenergetic recovery from the suppressed state caused by restricted oxygen and glucose supply.
Discussion
The main findings and conclusions of the present study are the following: (1) post‐hypoxic cells show increased sensitivity to oxidants due to diminished energy pools; (2) ATP regeneration is NAD+‐dependent in hypoxia‐reoxygenation injury; (3) NAD+ salvage by NamPRT is required for NAD+ and ATP regeneration; and (4) PARP‐1 is a significant NAD+ consumer in post‐hypoxic cells, and PARP inhibitors can be beneficial in ischaemia, even in the absence of acute complications.
ATP is the central coenzyme in the cells that functions as universal energy currency to transfer chemical energy. ATP is generated by catabolism of organic compounds via a series of redox reactions that involve the transfer of electrons from organic donor molecules to acceptor molecules (oxygen) to generate carbon dioxide and water. During these redox reactions, energy is ‘collected’ and stored mostly by reducing NAD+ to NADH in the cells. This energy is released from NADH to generate ATP using the proton gradient across the inner mitochondrial membrane as energy carrier in oxidative phosphorylation. Under basal conditions, fatty acids serve as major fuel substrates for energy production in the heart, but there is a metabolic switch in the ischaemic heart that leads to increased glucose utilization and glycolysis (Diaz et al., 1998; Stanley, 2001), similar to the fuel substrate switch that occurs in chronic hypoxia (induced by high altitude) (Essop, 2007). Glycolysis is also preferred to fatty acid utilization during the reperfusion because it is more oxygen‐efficient (Lopaschuk and Stanley, 1997; Essop, 2007; Lopaschuk et al., 2010). ATP is mostly generated via oxidative phosphorylation under normal conditions in the cells, with only a small portion of ATP provided by glycolysis. However, in hypoxia, the shortage of oxygen supply may completely stop mitochondrial respiration, and glycolysis will become the dominant ATP generation process.
Glycolysis is supplemented by lactic acid fermentation which helps regenerate the NAD+ molecules utilized through glycolysis. However, NAD+ consumed by the tricarboxylic acid cycle and by beta oxidation of fatty acids cannot be recycled in the absence of oxidative phosphorylation. Apart from the metabolic redox reactions catalysed by dehydrogenases, NAD+ is used in ADP–ribosylation reactions by PARPs and in deacetylation reactions by sirtuins in the cells (Hassa et al., 2006; Hirschey, 2011). These reactions produce nicotinamide that can be re‐used for NAD+ synthesis via the salvage pathway, but this pathway is an energy‐requiring (endothermic) process that itself consumes ATP. Thus, in the ischaemic heart, NAD+ depletion is a result of several processes: (1) reduced NAD+ regeneration from NADH, (2) increased NAD+ consumption by PARP activated by oxidative stress and (3) decreased NAD+ re‐synthesis from nicotinamide as a result of low metabolic output of the cells. The consequence of NAD+ insufficiency is the blockage of all processes in the cells that require NAD+ as coenzyme (Hassa et al., 2006; Hirschey, 2011). In the ischaemic heart, the metabolic blockade and energy depletion are well documented, and they constitute a risk that the cells cannot fulfil the energy requirements of heart function (Nunez et al., 1974; Stanley, 2001). Thus, the NAD+ metabolome is a rational therapeutic target in ischaemic heart disease.
There are two options to increase the cellular NAD+ content: (1) NAD+ generation may be increased and (2) NAD+ consumption can be decreased. Therapeutic approaches for intervention tried to improve NAD+ biosynthesis by providing more nicotinamide (substrate) to NamPRT or by avoiding this step of the synthesis and supplementing with nicotinamide mononucleotide. Both approaches have been effective in ischaemia‐reperfusion injury of the heart (Sukhodub et al., 2010; Yamamoto et al., 2014).
Pharmacological interventions to decrease NAD+ catabolism have focused on PARP inhibition (Jagtap and Szabo, 2005). Unlike the energy‐producing metabolic steps, PARP function can be lost for longer periods without severe adverse effects (Piskunova et al., 2008). There are multiple PARP isoforms in the cells with various functions, but regarding NAD+ consumption and cellular bioenergetics, PARP‐1 is the most important, because it shows the highest expression level and it can use the most NAD+ (Schreiber et al., 2006). In preclinical studies, PARP inhibitors exerted a dramatic protective effect in myocardial reperfusion injury, and genetic ablation of PARP‐1 confirmed a similar effect (Zingarelli et al., 1997; Pieper et al., 2000; Liaudet et al., 2001). Beneficial effects of PARP inhibition has also been shown in various heart disease models including cardioplegic arrest, cardiopulmonary bypass and heart failure models and in age‐induced cardiac dysfunction (Pacher et al., 2004; Szabo et al., 2004; Szabo et al., 2004; Pacher et al., 2006). However, faith was lost in PARP inhibitors when it turned out that the cardioprotection seen with drug pretreatment was greatly reduced if PARP inhibitors were administered following the reperfusion (Morrow et al., 2009; Roesner et al., 2010). After the initial safety study of a single injection of the drug with 10 myocardial infarction patients per dose group, PARP inhibitor therapy was never tested in adequately powered cardiovascular clinical trials, but these inhibitors are now being tested in various cancer trials, which will provide data regarding their long‐term safety (Morrow et al., 2009; Garber, 2013). Because the compounds were shown to effectively block the enzyme in humans, we can expect that the beneficial effects of PARP inhibitors will be fully exploited in ischaemic heart disease in the coming years. We recently reported that PARP‐1 leaves the nucleus in myocardial ischaemia‐reperfusion injury and directly interacts with the ubiquitin E3 ligase protein RNF146 (Gero et al., 2014). Overexpression of RNF146 can provide protection against oxidant‐induced injury in cardiomyocytes, suggesting a novel option for reducing PARP‐1 activity by capturing PARP‐1 either with RNF146 or with a similar binding agent. Because this strategy is specific for PARP‐1, it may represent a valuable alternative to non‐specific inhibition of PARP.
In conclusion, metabolic therapy is a promising direction to supplement the current therapeutic options in ischaemic heart disease, and the NAD+ metabolome may be a prime target in this approach. Our results showed that NAD+ biosynthesis was crucial in bioenergetic recovery and blockade of NAD+ consumption by PARP‐1 had beneficial effects in post‐hypoxic cardiomyocytes. These are in line with previous data implying that both the NAD+ anabolic and catabolic processes can be exploited for therapeutic interventions in the ischaemic heart.
Author contributions
DG and CS designed the experiments, DG performed the experiments, and DG and CS analysed the data and wrote the paper.
Conflict of interest
The authors declare that they have no competing interests as defined by British Journal of Pharmacology or other interests that might be perceived to influence the results and discussion reported in this paper.
Supporting information
Figure S1 PARP‐1 silencing in H9c2 cells
Figure S2 NMNAT1 silencing cause enhanced sensitivity to oxidant‐induced injury. AG
Figure S3 NMNAT3 silencing does not affect the oxidant‐induced injury. A‐G
Supporting info item
Acknowledgements
This work was supported by the National Institutes of Health (to C. Szabo) (2P50GM060338).
Gero, D. , and Szabo, C. (2015) Salvage of nicotinamide adenine dinucleotide plays a critical role in the bioenergetic recovery of post‐hypoxic cardiomyocytes. British Journal of Pharmacology, 172: 4817–4832. doi: 10.1111/bph.13252.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RM, Yellon DM (2003). Atorvastatin, administered at the onset of reperfusion, and independent of lipid lowering, protects the myocardium by up‐regulating a pro‐survival pathway. J Am Coll Cardiol 41: 508–515. [DOI] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, Ziegler M (2005). Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 280: 36334–36341. [DOI] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Mansour H, Chan‐Ling T, Poljak A, Grant R (2011). Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One 6: e19194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave AC, Ingwall JS, Friedrich J, Liao R, Saupe KW, Apstein CS et al. (2000). ATP synthesis during low‐flow ischemia: influence of increased glycolytic substrate. Circulation 101: 2090–2096. [DOI] [PubMed] [Google Scholar]
- Chiarugi A, Dolle C, Felici R, Ziegler M (2012). The NAD metabolome – a key determinant of cancer cell biology. Nat Rev Cancer 12: 741–752. [DOI] [PubMed] [Google Scholar]
- Diaz R, Paolasso EA, Piegas LS, Tajer CD, Moreno MG, Corvalan R et al. (1998). Metabolic modulation of acute myocardial infarction. The ECLA (Estudios Cardiologicos Latinoamerica) Collaborative Group. Circulation 98: 2227–2234. [DOI] [PubMed] [Google Scholar]
- Essop MF (2007). Cardiac metabolic adaptations in response to chronic hypoxia. J Physiol 584: 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich GM, Meier P, White SK, Yellon DM, Hausenloy DJ (2013). Myocardial reperfusion injury: looking beyond primary PCI. Eur Heart J 34: 1714–1722. [DOI] [PubMed] [Google Scholar]
- Garber K (2013). PARP inhibitors bounce back. Nat Rev Drug Discov 12: 725–727. [DOI] [PubMed] [Google Scholar]
- Gero D, Modis K, Nagy N, Szoleczky P, Toth ZD, Dorman G et al. (2007). Oxidant‐induced cardiomyocyte injury: identification of the cytoprotective effect of a dopamine 1 receptor agonist using a cell‐based high‐throughput assay. Int J Mol Med 20: 749–761. [PubMed] [Google Scholar]
- Gero D, Szoleczky P, Chatzianastasiou A, Papapetropoulos A, Szabo C (2014). Modulation of Poly(ADP‐Ribose) Polymerase‐1 (PARP‐1) Mediated Oxidative Cell Injury by Ring Finger Protein 146 (RNF146) in Cardiac Myocytes. Mol Med . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gero D, Szoleczky P, Modis K, Pribis JP, Al‐Abed Y, Yang H et al. (2013. a). Identification of pharmacological modulators of HMGB1‐induced inflammatory response by cell‐based screening. PLoS One 8: e65994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gero D, Szoleczky P, Suzuki K, Modis K, Olah G, Coletta C et al. (2013. b). Cell‐based screening identifies paroxetine as an inhibitor of diabetic endothelial dysfunction. Diabetes 62: 953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ et al. (2014. a). Executive summary: heart disease and stroke statistics – 2014 update: a report from the American Heart Association. Circulation 129: 399–410. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ et al. (2014. b). Heart disease and stroke statistics – 2014 update: a report from the American Heart Association. Circulation 129: e28–e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasmann M, Schemainda I (2003). FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res 63: 7436–7442. [PubMed] [Google Scholar]
- Hassa PO, Haenni SS, Elser M, Hottiger MO (2006). Nuclear ADP‐ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev 70: 789–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM (2002). Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc Res 55: 534–543. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM (2013). Myocardial ischemia‐reperfusion injury: a neglected therapeutic target. J Clin Invest 123: 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM (2004). New directions for protecting the heart against ischaemia‐reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)‐pathway. Cardiovasc Res 61: 448–460. [DOI] [PubMed] [Google Scholar]
- Hirschey MD (2011). Old enzymes, new tricks: sirtuins are NAD(+)‐dependent de‐acylases. Cell Metab 14: 718–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Canto C, Wanders RJ, Auwerx J (2010). The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 31: 194–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagtap P, Szabo C (2005). Poly(ADP‐ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov 4: 421–440. [DOI] [PubMed] [Google Scholar]
- Liaudet L, Szabo E, Timashpolsky L, Virag L, Cziraki A, Szabo C (2001). Suppression of poly (ADP‐ribose) polymerase activation by 3‐aminobenzamide in a rat model of myocardial infarction: long‐term morphological and functional consequences. Br J Pharmacol 133: 1424–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonborg J, Kelbaek H, Vejlstrup N, Botker HE, Kim WY, Holmvang L et al. (2012). Exenatide reduces final infarct size in patients with ST‐segment‐elevation myocardial infarction and short‐duration of ischemia. Circ Cardiovasc Interv 5: 288–295. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD, Stanley WC (1997). Glucose metabolism in the ischemic heart. Circulation 95: 313–315. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC (2010). Myocardial fatty acid metabolism in health and disease. Physiol Rev 90: 207–258. [DOI] [PubMed] [Google Scholar]
- Modis K, Gero D, Erdelyi K, Szoleczky P, DeWitt D, Szabo C (2012). Cellular bioenergetics is regulated by PARP1 under resting conditions and during oxidative stress. Biochem Pharmacol 83: 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar A, Toth A, Bagi Z, Papp Z, Edes I, Vaszily M et al. (2006). Activation of the poly(ADP‐ribose) polymerase pathway in human heart failure. Mol Med 12: 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecucco F, Bauer I, Braunersreuther V, Bruzzone S, Akhmedov A, Luscher TF et al. (2013). Inhibition of nicotinamide phosphoribosyltransferase reduces neutrophil‐mediated injury in myocardial infarction. Antioxid Redox Signal 18: 630–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow DA, Brickman CM, Murphy SA, Baran K, Krakover R, Dauerman H et al. (2009). A randomized, placebo‐controlled trial to evaluate the tolerability, safety, pharmacokinetics, and pharmacodynamics of a potent inhibitor of poly(ADP‐ribose) polymerase (INO‐1001) in patients with ST‐elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of the TIMI 37 trial. J Thromb Thrombolysis 27: 359–364. [DOI] [PubMed] [Google Scholar]
- Nikiforov A, Dolle C, Niere M, Ziegler M (2011). Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J Biol Chem 286: 21767–21778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez R, Calva E, Briones E, Lopez‐Soriano F (1974). Nicotinamide coenzymes in heart and coronary blood during myocardial infarction. Am J Physiol 226: 73–76. [DOI] [PubMed] [Google Scholar]
- Pacher P, Liaudet L, Mabley JG, Cziraki A, Hasko G, Szabo C (2006). Beneficial effects of a novel ultrapotent poly(ADP‐ribose) polymerase inhibitor in murine models of heart failure. Int J Mol Med 17: 369–375. [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Vaslin A, Benko R, Mabley JG, Liaudet L, Hasko G et al. (2004). A new, potent poly(ADP‐ribose) polymerase inhibitor improves cardiac and vascular dysfunction associated with advanced aging. J Pharmacol Exp Ther 311: 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ; NC‐IUPHAR. (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098‐1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper AA, Walles T, Wei G, Clements EE, Verma A, Snyder SH et al. (2000). Myocardial postischemic injury is reduced by polyADPripose polymerase‐1 gene disruption. Mol Med 6: 271–282. [PMC free article] [PubMed] [Google Scholar]
- Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N et al. (2008). Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 359: 473–481. [DOI] [PubMed] [Google Scholar]
- Piskunova TS, Yurova MN, Ovsyannikov AI, Semenchenko AV, Zabezhinski MA, Popovich IG et al. (2008). Deficiency in poly(ADP‐ribose) polymerase‐1 (PARP‐1) accelerates aging and spontaneous carcinogenesis in mice. Curr Gerontol Geriatr Res754190 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queval G, Noctor G (2007). A plate reader method for the measurement of NAD, NADP, glutathione, and ascorbate in tissue extracts: application to redox profiling during Arabidopsis rosette development. Anal Biochem 363: 58–69. [DOI] [PubMed] [Google Scholar]
- Roesner JP, Mersmann J, Bergt S, Bohnenberg K, Barthuber C, Szabo C et al. (2010). Therapeutic injection of PARP inhibitor INO‐1001 preserves cardiac function in porcine myocardial ischemia and reperfusion without reducing infarct size. Shock 33: 507–512. [DOI] [PubMed] [Google Scholar]
- Sack MN, Yellon DM (2003). Insulin therapy as an adjunct to reperfusion after acute coronary ischemia: a proposed direct myocardial cell survival effect independent of metabolic modulation. J Am Coll Cardiol 41: 1404–1407. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Ame JC, de Murcia G (2006). Poly(ADP‐ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7: 517–528. [DOI] [PubMed] [Google Scholar]
- Schriewer JM, Peek CB, Bass J, Schumacker PT (2013). ROS‐mediated PARP activity undermines mitochondrial function after permeability transition pore opening during myocardial ischemia‐reperfusion. J Am Heart Assoc 2: e000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L'Huillier I et al. (2005). Postconditioning the human heart. Circulation 112: 2143–2148. [DOI] [PubMed] [Google Scholar]
- Stanley WC (2001). Cardiac energetics during ischaemia and the rationale for metabolic interventions. Coron Artery Dis 12: S3–7. [PubMed] [Google Scholar]
- Stein LR, Imai S (2012). The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab 23: 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhodub A, Du Q, Jovanovic S, Jovanovic A (2010). Nicotinamide‐rich diet protects the heart against ischaemia‐reperfusion in mice: a crucial role for cardiac SUR2A. Pharmacol Res 61: 564–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, Liaudet L, Hagl S, Szabo C (2004. a). Poly(ADP‐ribose) polymerase activation in the reperfused myocardium. Cardiovasc Res 61: 471–480. [DOI] [PubMed] [Google Scholar]
- Szabo G, Soos P, Bahrle S, Zsengeller Z, Flechtenmacher C, Hagl S et al. (2004. b). Role of poly(ADP‐ribose) polymerase activation in the pathogenesis of cardiopulmonary dysfunction in a canine model of cardiopulmonary bypass. Eur J Cardiothorac Surg 25: 825–832. [DOI] [PubMed] [Google Scholar]
- Szabo G, Soos P, Mandera S, Heger U, Flechtenmacher C, Bahrle S et al. (2004. c). INO‐1001 a novel poly(ADP‐ribose) polymerase (PARP) inhibitor improves cardiac and pulmonary function after crystalloid cardioplegia and extracorporal circulation. Shock 21: 426–432. [DOI] [PubMed] [Google Scholar]
- Szabo G, Veres G, Radovits T, Gero D, Modis K, Miesel‐Groschel C et al. (2011). Cardioprotective effects of hydrogen sulfide. Nitric Oxide 25: 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szoleczky P, Modis K, Nagy N, Dori Toth Z, DeWitt D, Szabo C et al. (2012). Identification of agents that reduce renal hypoxia‐reoxygenation injury using cell‐based screening: purine nucleosides are alternative energy sources in LLC‐PK1 cells during hypoxia. Arch Biochem Biophys 517: 53–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Byun J, Zhai P, Ikeda Y, Oka S, Sadoshima J (2014). Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS One 9: e98972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingarelli B, Cuzzocrea S, Zsengeller Z, Salzman AL, Szabo C (1997). Protection against myocardial ischemia and reperfusion injury by 3‐aminobenzamide, an inhibitor of poly (ADP‐ribose) synthetase. Cardiovasc Res 36: 205–215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 PARP‐1 silencing in H9c2 cells
Figure S2 NMNAT1 silencing cause enhanced sensitivity to oxidant‐induced injury. AG
Figure S3 NMNAT3 silencing does not affect the oxidant‐induced injury. A‐G
Supporting info item