

Abstract
Background and Purpose
Recent findings suggest the importance of inflammation in the pathogenesis of cerebral ischaemia and its potential as a therapeutic target. Cinnamaldehyde is a diterpene with a wide range of anti‐inflammatory effects thus may be advantageous in the treatment of cerebral ischaemia. The present study examined the potential therapeutic effects of cinnamaldehyde on cerebral ischaemia using a mouse model with permanent middle cerebral artery occlusion.
Experimental Approach
Male CD‐1 mice, which had the middle cerebral artery occluded, were treated (i.p.) with cinnamaldehyde. Neuroprotection by cinnamaldehyde was analysed by evaluating neurological deficit scores, brain oedema and infarct volume. Expressionsof signal transduction molecules and inflammatory mediators were measured by Western blotting, qRT‐PCR and immunohistochemical staining. Activation of NF‐κB was assessed by Western blotting, immunohistochemistry and immunofluorescence.
Key Results
Cinnamaldehyde reduced the neurological deficit scores, brain oedema and infarct volume. Cinnamaldehyde suppressed the activation of signal transduction molecules including toll‐like receptor 4, tumour necrosis receptor‐associated factor 6 and NF‐κB, attenuated the increased levels of TNF‐α, IL‐1β, CCL2 and endothelial‐leukocyte adhesion molecule‐1 and ultimately reduced leukocyte infiltration into the ischaemic brain areas after cerebral ischaemia.
Conclusions and Implications
Cinnamaldehyde protects against cerebral ischaemia injury by inhibiting inflammation, partly mediated by reducing the expression of toll‐like receptor 4, tumour necrosis receptor‐associated factor 6 and the nuclear translocation of NF‐κB. Our findings suggest that cinnamaldehyde may serve as a new candidate for further development as a treatment for stroke.
Abbreviations
- BBB
blood brain barrier
- ELAM‐1
endothelial leukocyte adhesion molecule‐1
- IHC
immunohistochemical staining
- IRAK
IL‐1 receptor‐associated kinase
- MAPKs
mitogen‐activated protein kinases
- MPO
myeloperoxidase
- pMCAO
permanent middle cerebral artery occlusion
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- rCBF
relative regional cerebral blood flow
- TRAF6
tumour necrosis factor receptor‐associated factor 6
- TTC
2,3,5‐triphenyltetrazolium chloride
Tables of Links
| TARGETS |
|---|
| Catalytic receptor a |
| TLR4 |
| Enzymes b |
| IRAK, IL‐1 receptor‐associated kinase |
| MAPK |
| MPO, myeloperoxidase |
| LIGANDS |
|---|
| CCL2 |
| Cinnamaldehyde |
| IL‐1β |
| TNF‐α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a bAlexander et al., 2013a, 2013b).
Introduction
Stroke is the third leading cause of death and the most frequent cause of permanent disability in adults worldwide (Donnan et al., 2008). Thrombolysis is the only currently effective and available stroke therapy, but it is limited to a small proportion of patients because of its narrow time window and risk of intracranial haemorrhage. This emphasizes the need for new therapeutic approaches to stroke treatment. Cerebral ischaemia results in a cascade of pathophysiological events including inflammation, oxidative stress, excitotoxicity and apoptosis (Moskowitz et al., 2010). Inflammatory response has been confirmed to play an important role in the pathogenesis of brain injury secondary to ischaemia (Danton and Dietrich, 2003; Kleinig and Vink, 2009; Dong et al., 2013). Many of the molecules involved in inflammatory response are potential therapeutic targets for stroke. Therefore, it is believed that pharmacological inhibition of the inflammatory response is one of the promising approaches for stroke therapy.
Activation of NF‐κB induced by the toll‐like receptors (TLRs) has been recognized as a key contributor to the pro‐inflammatory response (Medzhitov, 2001). Activation of TLRs triggers the downstream stimulation of NF‐κB and the induction of genes that encode inflammation‐associated molecules and cytokines (O'Neill, 2003; Akira and Takeda, 2004). Accumulating evidence suggests that TLRs and NF‐κB are important mediators in cerebral ischaemic inflammatory injury (Caso et al., 2007; Downes and Crack, 2010; Wang et al., 2011; Jiang et al., 2012). We have also shown that several TLRs, such as TLR2, TLR4, TLR5 and TLR9, played a pivotal role in the inflammatory response following cerebral ischaemia (Fan et al., 2009; Qiao et al., 2012; Zhang et al., 2013). Tumour necrosis factor receptor‐associated factor 6 (TRAF6) is a critical signalling adapter and acts downstream of the TLRs (Kobayashi et al., 2004). All of these results suggest that TLRs and the downstream signalling molecules such as TRAF6 and NF‐κB can be excellent therapeutic targets for ischaemic stroke.
Cinnamaldehyde (CA), the major constituent of the essential oil of Cassia bark, is a safe, widely used flavouring agent in food stuffs such as beverages, ice cream, sweets and chewing gum. Cinnamaldehyde has been reported to suppress TLR4‐dependent inflammation in vitro (Youn et al., 2008). Recent studies further suggested that cinnamaldehyde exerted a protective effect on myocardial ischaemia damage in vivo. Therefore, in the present study, we aimed to assess the potential anti‐inflammatory and neuroprotective effect of cinnamaldehyde in a mouse model of permanent middle cerebral artery occlusion (pMCAO) and whether this therapeutic benefit was associated with the activation of TLR4, TRAF6 and NF‐κB.
Methods
Animal preparation and pMCAO
All animal care and experimental procedures were approved by the Animal Care and Management Committee of Second Hospital of Hebei Medical University (permit number HMUSHC‐130318). All studies involving animals were reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 230 male CD‐1 mice (25–30 g) purchased from the Vital River, Beijing, China, were kept on a 12 h light/12 h dark cycle with free access to food and water, and they were allowed to acclimatize the new surrounding for at least 3 days before using in experiments. Animals were anaesthetized by i.p. injection of 10% chloral hydrate (400 mg·kg−1; Yongda Chemical Reagent Co., Ltd., Tianjin, China). Body temperature was monitored and maintained at 36.5 to 37.5 °C. A standard model of intraluminal middle cerebral artery occlusion was used to make permanent focal ischaemia by intraluminal placement of a filament (Beijing Shadong BioTechnoiogiies Co., Ltd.), as described previously (Longa et al., 1989). Sham‐operated control mice received the same procedure except filament insertion. To confirm successful blockade of the artery, an optic fibre was glued to the skull (1 mm posterior and 3 mm lateral to bregma) and connected to a laser Doppler flowmeter (moorVMS‐LDF, Moor Instruments Ltd., Axminster, Devon, UK) for monitoring the relative regional cerebral blood flow (rCBF) in the core territory of the right middle cerebral artery. The rCBF, as measured by a flexible probe and laser Doppler flowmetry, decreased by 85–95% in this mouse model of pMCAO.
Experimental groups
Cinnamaldehyde (Aike Biotechnology Co, Chengdu, Sichuan, China) with purity of more than 98% was dissolved in saline including 0.2% dimethyl sulfoxide (DMSO). All the mice were randomly divided into seven groups using a random number table generated by the spss software 13.0 (SPSS Inc., Chicago, IL, USA) as described (Jiang et al., 2014): in preliminary experiments, sham groups: mice received sham operation with equal volume of normal sodium or 0.2% DMSO; MCAO groups: mice received pMCAO with equal volume of normal sodium or 0.2% DMSO and cinnamaldehyde groups: mice were treated i.p. with cinnamaldehyde at 25 (CA25), 50 (CA50) and 75 mg·kg−1 (CA75), immediately after cerebral ischaemia and then once daily thereafter. We found that neurological deficit scores, brain water content and infarct volume were comparable between mice treated with saline and mice treated with 0.2% DMSO in sham and MCAO groups (data not shown), so we could exclude the biological effect of vehicle. Sham group (mice receiving sham operation with 0.2% DMSO, n = 42), MCAO group (mice received MCAO with 0.2% DMSO, n = 54), CA25 group (n = 24), CA50 group (n = 54) and CA75 (n = 24) group were finally included for the subsequent experiments. At 24 and 72 h after cerebral ischaemia, mice were killed via rapid decapitation under deep anaesthesia, and samples were collected for further study. To further estimate the neuroprotective effect of cinnamaldehyde administered at different time‐points, one group of mice was administered with cinnamaldehyde (50 mg·kg−1) at 1 h before, 4 or 8 h after cerebral ischaemia (n = 6 per time‐point).
Neurological deficit scores
A neurological test was administered by the same examiner, unaware of the experimental groups, at 24 and 72 h (n = 6 per group per time‐point) after cerebral ischaemia following a modified scoring system based on that developed from Bederson (Bederson et al., 1986a) as follows: 0, normal motor function; 1, contralateral forelimb weakness and torso turning to the ipsilateral side when held by tail; 2, circling to affected side but normal posture at rest; 3, leaning to contralateral side at rest and 4, no spontaneous activity or barrel rolling. The higher the neurological deficit scores, the more severe impairment of motor function.
Brain water content
Brain water content was observed using the standard wet–dry method at 24 and 72 h after cerebral ischaemia (n = 6 per group per time‐point) (Hatashita et al., 1988). After dissecting free 3 mm of frontal pole, a coronal brain slice (approximately 2 mm thick) was cut, and the slice was divided into ipsilateral and contralateral hemispheres. The two hemispheres packaged respectively with tinfoil, wet weights measured and then dried in an oven at 100 °C for 24 h to provide the dry weights. Brain water content was then calculated as follows: (wet weight–dry weight)/(wet weight) × 100%.
Brain infarct volume
Infarct volume after cerebral ischaemia was determined by 2,3,5‐triphenyltetrazolium chloride (TTC) at 24 and 72 h after cerebral ischaemia (n = 6 per group per time‐point). Brain tissue was sliced into five coronal sections (2 mm thick), stained with a 2% solution of TTC at 37 °C for 20 min (Bederson et al., 1986b) and followed by fixation with 4% paraformaldehyde. Normal tissue was stained deep red, while the infarct area was stained pale grey. TTC‐stained sections were photographed, and the digital images were analysed using image analysis software (Image‐Pro Plus 5.1, Media Cybernetics, Inc, Bethesda, MD, USA) to calculate the infarct volume. To compensate for the effect of brain oedema, the percentage hemisphere lesion volume was calculated by the following formula (Tatlisumak et al., 1998): %HLV = {[total infarct volume – (volume of intact ipsilateral hemisphere–volume of intact contralateral hemisphere)]/contralateral hemisphere volume} × 100%.
Immunohistochemical staining (IHC)
Paraffin‐embedded sections were used to assess the expression of TLR4, TRAF6 and NF‐κB, according to standard histological procedures, at 24 and 72 h after cerebral ischaemia (n = 3 per group per time‐point). Brain tissues were fixed in 4% paraformaldehyde in phosphate‐buffered saline (PBS; 0.01 M, pH 7.4) over 24 h at 4 °C and then dehydrated in a graded series of alcohols and embedded in paraffin. Brain tissues were cut at 5 µm using a Leica® RM1850 rotary microtome (Leica Microsystem, IL, Hesja, Germany). Brain sections were incubated in 3% H2O2 to eliminate the endogenous peroxidase activity, and 3% normal goat serum, then incubated with rabbit polyclonal antibody of NF‐κB (1:100, Bioworld Biotechnology), TRAF6 (1:100, Bioworld Biotechnology) and TLR4 (1:50, Bioworld Biotechnology) in 0.01 M·L−1 PBS overnight at 4 °C. They were rinsed with PBS and incubated with secondary antibodies at 37 °C for 45 min. They were rinsed again with PBS and incubated with secondary biotinylated conjugates at 37 °C. Slices were developed with diaminobenzidine and counterstained with haematoxylin. The secondary antibodies, secondary biotinylated conjugates and diaminobenzidine from the streptavidin‐peroxidase kit (Zhongshan Biology Technology Company, Beijing, China) were used to visualize the signals. The immunoreactive cells were counted under a 400× light microscope in five visual fields of the ischaemic cortex region around the infarct core. The average number was used for statistical analysis and represented the immunopositive cells of that mouse.
Western blotting
Protein extraction was obtained from the cortex using a total protein extraction kit and nuclear‐cytosol extraction kit (Applygen Technologies Inc., Beijing, China) following the manufacturer's protocols at 24 and 72 h after cerebral ischaemia (n = 6 per group per time‐point). Total protein for TLR4, TRAF6 and nuclear protein for NF‐κB were prepared. Protein concentration of the supernatant was determined using a bicinchoninic acid protein assay reagent kit (Novagen, Madison, WI, USA) with bovine serum albumin as the standard. An equivalent amount of 50 µg total protein samples, as well as 30 µg nuclear protein samples, was subjected to electrophoresis on 10% sodium dodecyl sulfate–polyacrylamide gels for 45 min at 80 V followed by 100 min at 100 V and then transferred onto polyvinylidene difluoride membranes (Millipore Corporation, Billerica, MA, USA) for 2 h at 100 V. The membrane was blocked with 5% skimmed milk/PBS containing 0.1% Tween‐20 (TPBS) (10 mM Tris–HCl, 150 mM NaCL and 0.05% Tween‐20) for 2 h at room temperature then incubated with the corresponding primary antibodies (TLR4, 1:200, Bioworld Biotechnology; TRAF6, 1:500, Bioworld Biotechnology and NF‐κB, 1:500, Bioworld Biotechnology) at 4 °C overnight. GAPDH (1:1000, Bioworld Biotechnology) was used as an internal control. The following day, membranes were washed with TPBS (10 min × 3) each time and subsequently incubated in TPBS containing fluorescent labelling second antibodies (IRDye® 800‐conjugated goat anti‐rabbit IgG, 1:10 000 dilution, Rockland, Gilbertsville, PA, USA) for 1 h at room temperature. Membranes were then washed three times with TPBS (10 min × 3), and the relative density of bands was analysed on an Odyssey infrared scanner (LICOR Bioscience, Lincoln, NE, USA). Densitometric values were normalized with respect to GAPDH immunoreactivity to correct for any loading and transfer differences among samples.
Quantitative real‐time polymerase chain reaction (qRT‐PCR)
The qRT‐PCR was used to analyse the mRNA levels of TNF‐α, IL‐1β, CCL2, endothelial leukocyte adhesion molecule‐1 (ELAM‐1), TLR4, TRAF6 and NF‐κB at 24 and 72 h after cerebral ischaemia (n = 6 per group per time‐point). Mice were anaesthetized, and the brains were removed and frozen in liquid nitrogen. Total RNA was extracted from ischaemic cortex using the Trizol reagent (Invitrogen, Carlsbad, CA, USA) and was reverse‐transcribed into cDNA using revert aid first strand cDNA synthesis kit (Fermentas International Inc., Burlington, Canada) for qRT‐PCR (MX 3005P, USA) in the presence of a fluorescent dye (SYBR Green I, Cwbio). Absorbance was read at 260 and 280 nm using an UV spectrophotometer. Only RNA samples with an OD260/OD280 value > 1.8 were considered appropriate for use. The mRNA level was normalized to the GAPDH RNA and was calculated by the 2−ΔΔCt method. The primer sequences are as follows: TLR4 forward 5′‐GAA TGA GGA CTG GGT GAG AAA C‐3′, reverse 5′‐CTC AGC AAG GAC TTC TCC ACT T‐3′; TRAF6 forward 5′‐TGG ATT CTA CAC AGG CAG ACC‐3′, reverse 5′‐TCA AAG CGG GTA GAG ACT TCA‐3′; NF‐κB forward 5′‐GGT GGA GTT TGG GAA GGA TTT G‐3′, reverse 5′‐TTT TCT CCG AAG CTG AAC AAA CAC‐3′; GAPDH forward 5′‐TGA ACG GGA AGC TCA CTG G‐3′, reverse 5′‐GCT TCA CCA CCT TCT TGA TGT C‐3′; IL‐1β forward 5′‐TGA AAT GCC ACC TTT TGA CAG‐3′, reverse 5′‐CCA CAG CCA CAA TGA GTG ATA C‐3′; TNF‐α forward 5′‐GTC GTA GCA AAC CAC CAA GTG‐3′, reverse 5′‐CAG ATT TGT GTT GTG GTC CTT C‐3′; CCL2 forward 5′‐GTC GTA GCA AAC CAC CAA GTG‐3′, reverse 5′‐TGA GGT GGT TGT GGA AAA GGT AGT G‐3′; ELAM‐1 forward 5′‐GAA GCC TGA ACT GCT CCC ACC‐3′, reverse 5′‐GCA CTC CAC TCT CCA GAG GAC GTA‐3′.
Myeloperoxidase (MPO) activity assay
Brain cortices were collected at 24 and 72 h after cerebral ischaemia (n = 6 per group per time‐point). The samples were rinsed, weighed and then homogenized in 19 volumes of 9 g·L−1 ice‐cold saline for 10 min using a Dounce tissue grinder (Kimble and Kontes, Vineland, NJ, USA). Supernatant homogenate was collected after centrifugation at 4000× g for 10 min at 4 °C. The kit (A003, Nanjing Jiancheng Bioengineering Institute, Nanjing, China) was selected for MPO activity measurement according to the manufacturer's instructions.
Immunofluorescence staining
Mice were anaesthetized by 10% chloral hydrate and transcardially perfused with saline quickly followed by 4% paraformaldehyde in PBS. Frozen coronal brain sections (30 µm thick) were permeabilized with 0.3% Triton X‐100 for 10 min, blocked with 10% normal donkey serum for half an hour at room temperature and then incubated with rabbit anti‐NF‐κB (1:100, Bioworld Biotechnology) and mouse anti‐Neun (1:500, Millipore Corporation) or mouse anti‐glial fibrillary acidic protein (1:500, Millipore Corporation) or goat anti‐MPO (1:100, Santa Cruz Biotechnology) overnight at 4 °C. Slices were washed with PBS on the second day and incubated with anti‐rabbit tetramethylrhodamine‐conjugated secondary antibody (1:200, Zhongshan Biology Technology Company, Beijing, China), anti‐mouse or anti‐goat FITC‐conjugated secondary antibody (1:600, Zhongshan Biology Technology Company) for 2 h. In order to identify the nucleus, brain sections were counterstained with Hoechst 33342 (10 µg·mL−1) for 15 min. To acquire colour images, a 20× laser scanning confocal microscope (Olympus FV10‐ASW, Tokyo, Japan) was used.
Data analysis
Results are expressed as means ± SEM, except for the neurological scores and mortality. Statistical analysis was performed by the spss software 13.0 software (SPSS Inc.). Statistical comparisons were performed by one‐way ANOVA followed by Student–Newman–Keuls tests for multiple comparisons. For neurological scores, data were analysed by the chi‐square (χ 2) method followed by the Mann–Whitney U test for comparisons between two groups. The mortality of mice after cerebral ischaemia was assessed with the chi‐square (χ 2) method. g*power 3.1.9.2 software (http://www.gpower.hhu.de/fileadmin/redaktion/Fakultaeten/Mathematisch‐Naturwissenschaftliche_Fakultaet/Psychologie/AAP/gpower/GPowerWin_3.1.9.2.zip) was used to calculate the sample size. Differences with P < 0.05 were considered statistically significant.
Results
Cinnamaldehyde reduced the neurological deficit scores, brain oedema and infarct volume after cerebral ischaemia
A total of 14 mice died before completion of the experiment and were excluded from the study: five mice (9.3%) in MCAO group, three mice (12.5%) in the CA25 group, four mice (7.4%) in the CA50 group and two mice in CA75 (8.3%). Post mortem examinations did not reveal the occurrence of intracerebral or subarachnoid haemorrhage in any of these animals, and no significant differences were found among the number of deaths of each group.
To investigate whether cinnamaldehyde could attenuate cerebral ischaemia injury, we determined the neurological deficit scores, brain water content and infarct volume at 24 and 72 h after cerebral ischaemia. Compared with MCAO group, there was a significant improvement in neurological deficit scores in the CA50 and CA75 groups (P < 0.05), but no significant changes was shown in the scores of the CA25 group (P > 0.05). The neurological deficit scores of each group at 24 h after cerebral ischaemia were shown in Table 1.
Table 1.
Effects of cinnamaldehyde on neurological deficit scores in mice
| Neurological deficit scores | No. of mice in MCAO group | No. of mice in the CA25 group | No. of mice in the CA50 group | No. of mice in the CA75 group |
|---|---|---|---|---|
| 1 | 0 | 0 | 0 | 3 |
| 2 | 0 | 1 | 3 | 3 |
| 3 | 4 | 4 | 3 | 0 |
| 4 | 2 | 1 | 0 | 0 |
| Median | 3 | 3 | 2.5* | 1.5* |
| 95%CI | 2.79–3.88 | 2.34–3.67 | 1.93–3.07 | 0.93–2.07 |
MCAO, middle cerebral artery occlusion; CA, cinnamaldehyde; CI, confidence interval.
The number of mice with each particular neurological scores in each group was shown in the table (n = 6). χ 2 = 55.5, d.f. = 16 and P < 0.001. *P < 0.05 versus MCAO group (Mann–Whitney U test).
Brain oedema in the ischaemic hemisphere of each group was shown in Figure 1A. Compared with MCAO group, there was a significant reduction in brain water content in the CA75 group (P < 0.05; n = 6 per time‐point), and in the CA50 group (P < 0.05; n = 6 per time‐point), but there was no significant reduction of brain oedema in the CA25 group (P > 0.05).
Figure 1.
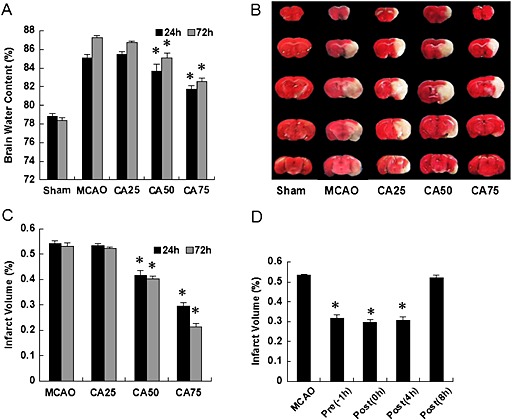
Effect of cinnamaldehyde on cerebral ischaemia injury. (A) Effect of cinnamaldehyde at different doses on brain oedema in ischaemic hemisphere. Sham group (mice received sham operation with 0.2% DMSO, n = 6 per time‐point), MCAO group (mice received MCAO with 0.2% DMSO, n = 6 per time‐point), the CA25 group (cinnamaldehyde of 25 mg·kg−1 given i.p. immediately after cerebral ischaemia, n = 6 per time‐point), the CA50 group (cinnamaldehyde of 50 mg·kg−1 given i.p. immediately after cerebral ischaemia, n = 6 per time‐point) and the CA75 group (cinnamaldehyde of 75 mg·kg−1 given i.p. immediately after cerebral ischaemia, n = 6 per time‐point). Results shown are the means ± SEM, n = 6 * P < 0.05, versus same time‐point MCAO group;one‐way ANOVA with Student–Newman–Keuls test. (B) Representative TTC‐stained sections at 24 h after cerebral ischaemia. Normal tissue was stained deep red, while the infarct area was stained pale grey. (C) Effect of cinnamaldehyde at different doses on infarct volume at 24 and 72 h after cerebral ischaemia. Results shown are the means ± SEM, n = 6 * P < 0.05, versus same time‐point MCAO group;one‐way ANOVA with Student–Newman–Keuls test. (D) Effect of cinnamaldehyde (50 mg·kg−1) administered at different time‐points on infarct volume at 24 h after cerebral ischaemia. Results shown are the means ± SEM, n = 6 * P < 0.05, versus same time‐point MCAO group;one‐way ANOVA with Student–Newman–Keuls test.
The infarct volume of each group at 24 h after cerebral ischaemia was shown in Figure 1B. In MCAO group, an extensive lesion was found in both striatum and cortex (Figure1C). In agreement with neurological deficit scores and brain water content, compared with MCAO group, cinnamaldehyde reduced the infarct volume in a dose‐dependent manner with a significant therapeutic effect in the CA75 group (P < 0.05) and the CA50 group (P < 0.05), but not in the CA25 group (Figure 1C).
Because these results demonstrated therapeutic effects at 50 and 75 mg·kg−1 cinnamaldehyde, we used the 50 mg·kg−1 dose in all subsequent studies.
To further estimate the neuroprotective effect of CA, one group of mice was treated with cinnamaldehyde (50 mg·kg−1) at different time‐points (1 h before and 4 or 8 h after cerebral ischaemia). The infarct volume of mice treated at 1 h before cerebral ischaemia, 0 h after and 4 h after cerebral ischaemia was significantly smaller than that in MCAO group at 24 h after cerebral ischaemia (P < 0.05 for all, Figure 1D). Similarly, the neurological deficit scores of mice treated at 1 h before cerebral ischaemia and at 0 or 4 h after cerebral ischaemia were significantly decreased compared with those in the MCAO group (P < 0.05 for all). However, compared with the MCAO group, there was no significant decrease in infarct volume and neurological deficit scores when cinnamaldehyde was administered at 8 h after cerebral ischaemia (P > 0.05).
Cinnamaldehyde reduced the mRNA levels of inflammatory mediators
The mRNA levels of the cytokines, TNF‐α and IL‐1β, the chemokine CCL2 and the adhesion molecules (ELAM‐1) were assayed. At 24 h after cerebral ischaemia, all the values of inflammatory mediators measured were significantly higher than that in the sham group (P < 0.05), and all these effects were partly reversed by treatment with cinnamaldehyde (Figures 2A–2D). At 72 h after cerebral ischaemia, the mRNA levels of TNF‐α, CCL2 and ELAM‐1 were higher than that in the sham group, and cinnamaldehyde reduced the mRNA levels of TNF‐α and CCL2. However, the levels of IL‐1β and ELAM‐1 in the CA50 group showed no significant difference from those of the MCAO group, at 72 h after cerebral ischaemia (Figures 2A–2D).
Figure 2.
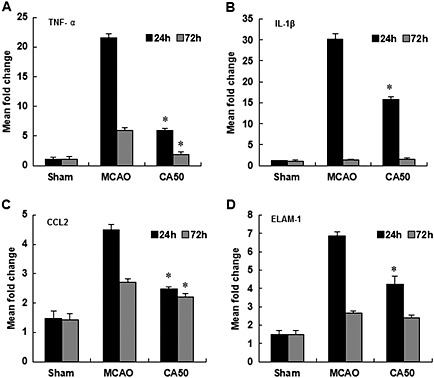
Effect of cinnamaldehyde on mRNA expression of TNF‐α, IL‐1β, CCL2 and ELAM‐1. The mRNA levels of inflammatory mediators TNF‐α (A), IL‐1β (B), CCL2 (C) and ELAM‐1 (D) in cerebral cortex were shown by bar graphs at 24 and 72 h after cerebral ischaemia. Results shown are the means ± SEM, n = 6 * P < 0.05, versus same time‐point MCAO group;one‐way ANOVA with Student–Newman–Keuls test.
Cinnamaldehyde reduced leukocyte infiltration
To determine whether cinnamaldehyde could reduce leukocyte infiltration into the ischaemic area, we quantified the number of MPO‐positive cells, MPO activity and MPO expression in the ischaemic cortex. The MPO‐positive cells of each group at 24 h after cerebral ischaemia are shown in Figure 3A. The number of MPO‐positive cells was significantly decreased in the CA50 group at 24 and 72 h after cerebral ischaemia (P < 0.05, Figure 3B). MPO activity is a good indicator of inflammation and neutrophil accumulation and can be quantified by the MPO activity assay (Barone et al., 1992; Jiang et al., 1995). MPO activity in the ischaemic cortex was also reduced in the CA50 group at 24 and 72 h after cerebral ischaemia (P < 0.05, Figure 3C). Western blotting analysis demonstrated that ischaemia caused an increase in MPO levels in the right cortex at 24 and 72 h after cerebral ischaemia, whereas cinnamaldehyde attenuated such increases (Figures 3D and 3E).
Figure 3.
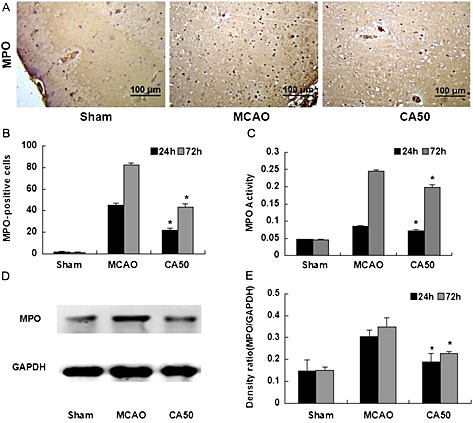
Effect of cinnamaldehyde on MPO expression and activity. (A) MPO staining in cerebral cortex in different groups at 24 h after cerebral ischaemia (200 × magnification). (B) Quantification of MPO‐positive cells in cerebral cortex in different groups at 24 and 72 h after cerebral ischaemia. Results shown are the means ± SEM, n = 6 * P < 0.05, versus same time‐point MCAO group;one‐way ANOVA with Student–Newman–Keuls test. (C) Bar graph of MPO activity in cerebral cortex in different groups at 24 and 72 h after cerebral ischaemia. Results shown are the means ± SEM, n = 6 * P < 0.05, versus same time‐point MCAO group;one‐way ANOVA with Student–Newman–Keuls test. (D) Western blotting analysis of MPO in cerebral cortex in different groups at 24 h after cerebral ischaemia. (E) Quantification of protein level of MPO in different groups at 24 and 72 h after cerebral ischaemia. Results shown are the means ± SEM, n = 6 * P < 0.05, versus same time‐point MCAO group;one‐way ANOVA with Student–Newman–Keuls test.
Cinnamaldehyde suppressed the expression of TLR4, TRAF6 and NF‐κB
The localization of TLR4, TRAF6 and NF‐κB was identified by immunohistochemistry at 24 and 72 h after cerebral ischaemia, and the quantification was performed by manually counting the number of immunoreactive cells separately stained for TLR4, TRAF6 and NF‐κB. The immunohistochemical staining of TLR4, TRAF6 and NF‐κB of each group at 24 h after cerebral ischaemia was shown in Figures 4A–4C. Few cells were stained for TLR4, TRAF6 and NF‐κB in the cortex in sham group. Compared with the sham group, the number of cells positive for TLR4, TRAF6 and NF‐κB in the MCAO group was significantly increased at 24 and 72 h after cerebral ischaemia (P < 0.05); in these cells, NF‐κB was located in both the cytoplasm and the nucleus. As expected, compared with the MCAO group, the number of cells positive for TLR4, TRAF6 and NF‐κB in the CA50 group was significantly decreased at 24 and 72 h after cerebral ischaemia (P < 0.05). Moreover, the number of nuclei positive for NF‐κB was also reduced and many of the cells labelled by NF‐κB were stained only in the cytoplasm (Figures 4D–4F).
Figure 4.
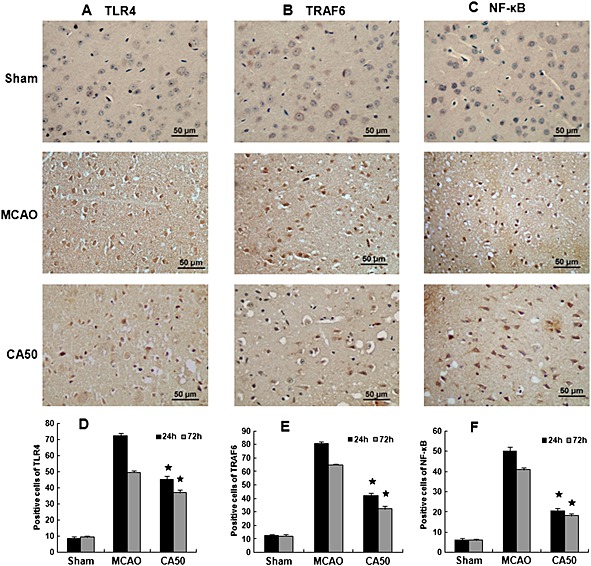
Immunohistochemical staining for TLR4, TRAF6 and NF‐ κB in the cerebral cortex. (A)–(C) Immunohistochemical staining for TLR4, TRAF6 and NF‐κB in different groups at 24 h after cerebral ischaemia (400 × magnification). Results shown are the means ± SEM, n = 3 * P < 0.05, versus same time‐point MCAO group;one‐way ANOVA with Student–Newman–Keuls test. (D)–(F) Quantification of the number of immunoreactive cells (TLR4, TRAF6 and NF‐κB) in different groups at 24 and 72 h after cerebral ischaemia. Results shown are the means ± SEM, n = 3 * P < 0.05, versus same time‐point MCAO group;one‐way ANOVA with Student–Newman–Keuls test.
Then, we further analysed the protein expressions of total TLR4, TRAF6 and nuclear NF‐κB with Western blotting. Figures 5A–5C showed the protein levels of total TLR4, TRAF6 and nuclear/cytosolic NF‐κB in different groups at 24 h after cerebral ischaemia. In agreement with the immunohistochemical results, Western blotting also showed a significant decrease of total protein of TLR4 and TRAF6, and nuclear NF‐κB in the CA50 group at 24 and 72 h after cerebral ischaemia (P < 0.05, Figures 5D–5F). Similarly, qRT‐PCR assays showed that the expression of TLR4, TRAF6 and NF‐κB was decreased in the CA50 group at 24 and 72 h after cerebral ischaemia (P < 0.05, Figures 5G–5I).
Figure 5.
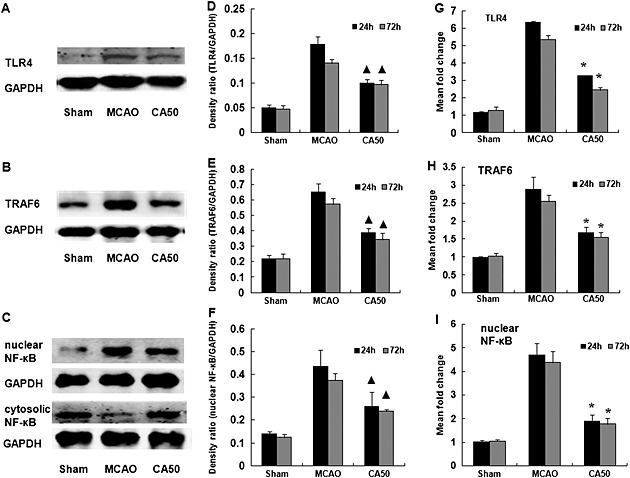
Effect of cinnamaldehyde on protein and mRNA expression of TLR4, TRAF6 and NF‐κB in the cerebral cortex. (A)–(C) Western blotting analysis of total TLR4, TRAF6 and nuclear/cytosolic NF‐κB in cerebral cortex in different groups at 24 h after cerebral ischaemia. (D)–(F) Quantification of protein level of total TLR4, TRAF6 and nuclear NF‐κB in different groups at 24 and 72 h after cerebral ischaemia. Results shown are the means ± SEM, n = 3. ▲ P < 0.05, versus same time‐point MCAO group;one‐way ANOVA with Student–Newman–Keuls test. (G)–(I) The mRNA levels of TLR4, TRAF6 and NF‐κB in cerebral cortex were shown by bar graphs at 24 and 72 h after cerebral ischaemia. Results shown are the means ± SEM, n = 6 * P < 0.05, versus same time‐point MCAO group;one‐way ANOVA with Student–Newman–Keuls test.
The location and translocation of NF‐κB in neurons, astrocytes and neutrophils after cerebral ischaemia
We examined the expressions of NF‐κB in neurons, astrocytes and neutrophils. In the sham group, NF‐κB was detectable in few neurons, almost no in astrocytes and neutrophils, and most cells labelled by NF‐κB were stained only in cytoplasm. In the MCAO group, the expression of NF‐κB was mainly in the neurons, few in the astrocytes and neutrophils and most of neuron NF‐κB was translocated into the nucleus, whereas cinnamaldehyde treatment attenuated the NF‐κB nuclear translocation (Figure 6).
Figure 6.
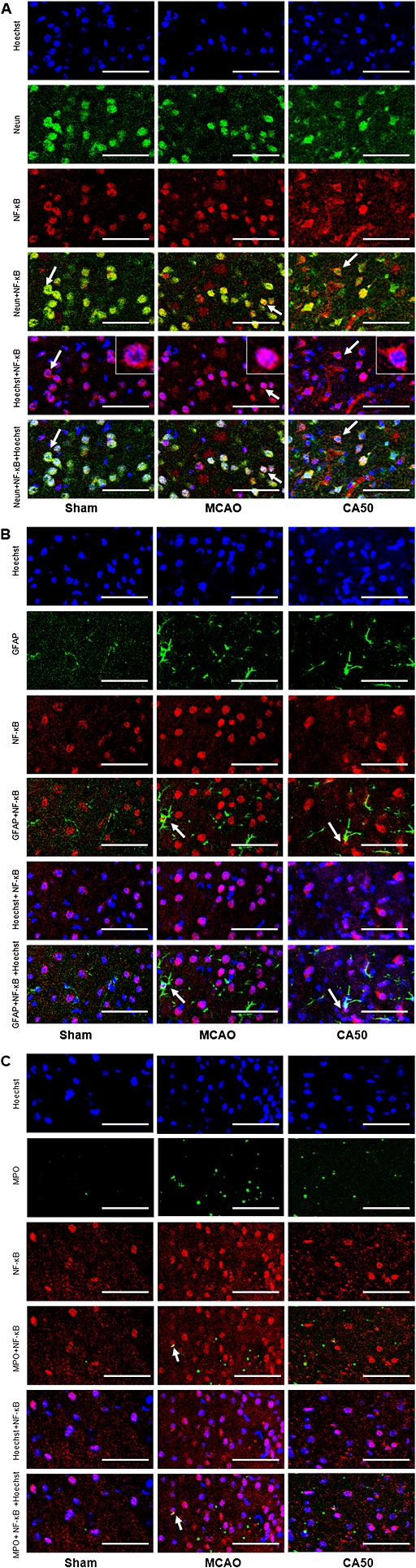
Immunofluorescence staining of NF‐κB in neurons, astrocytes and neutrophils in different groups at 24 h after cerebral ischaemia. When NF‐κB entered the nucleus, the combined figure would give rise to the purple colour. Scale bar = 100 µm. (A) Brain sections were triple‐stained with anti‐NF‐κB (red), anti‐Neun (green) and Hoechst 33342 (blue) to mark NF‐κB, neurons and nucleus. Arrows indicate the location of NF‐κB in neurons. (B) Brain sections were triple‐stained with anti‐NF‐κB (red), anti‐GFAP (green) and Hoechst (blue) to mark NF‐κB, astrocytes and nucleus. Arrows indicate the location of NF‐κB in astrocytes. (C) Brain sections were triple‐stained with anti‐NF‐κB (red), anti‐MPO (green) and Hoechst (blue) to mark NF‐κB, neutrophils and nucleus. Arrows indicate the location of NF‐κB in neutrophils.
Discussion and conclusions
In this study, we tested the hypothesis that (1) cinnamaldehyde protected against brain injury in a mouse model of pMCAO by reducing neurological deficit scores, brain oedema and infarct volume; (2) cinnamaldehyde protected the brain from ischaemic injury when administered 1 h before, immediately or 4 h after cerebral ischaemia; (3) cinnamaldehyde suppressed inflammatory response following cerebral ischaemia by reducing the inflammatory mediators release and leukocyte infiltration and (4) the underlying mechanism of its neuroprotection may partly involve the down‐regulation of the TLR4/TRAF6/NF‐κB signalling pathway.
Cinnamaldehyde is a diterpene found in considerable quantities in the stem bark of Cinnamomum cassia (He et al., 2005) and exhibits anti‐inflammatory action in vitro (Reddy et al., 2004; Youn et al., 2008; Ho et al., 2013). Cinnamaldehyde also protected against cardiac ischaemia injury by inhibiting inflammation in vivo (Hwa et al., 2012) and a recent study showed that cinnamaldehyde provided neuroprotection in inflammation‐mediated neurodegenerative diseases (Pyo et al., 2013). All of these findings strongly suggested that cinnamaldehyde may be a therapeutic candidate for inflammation‐mediated cerebral ischaemia injury. In our study, we have demonstrated that cinnamaldehyde significantly reduced neurological deficit scores, brain oedema and infarct volume after cerebral ischaemia, in our mouse model. Nevertheless, we are aware that the effect of cinnamaldehyde on cerebral ischaemia should be further studied in a larger number of animals and in other higher‐order species, with different animal models of cerebral ischaemia.
Several studies have revealed that a therapeutic window of approximately 6 h exists between the onset of ischaemia and irreversible neuronal death (Williams et al., 2004; Xu et al., 2006). Neuroprotective agents are aimed at salvaging or delaying the infarction of the still‐viable ischaemic penumbra. Therefore, it is desirable that neuroprotective interventions should be attempted before a stroke occurs or very soon afterward. In our study, we found that cinnamaldehyde had neuroprotective effects when administered, not only immediately after cerebral ischaemia, but also 1 h before ischaemia or 4 h after the onset of ischaemia. Cinnamaldehyde administered 8 h after ischaemia offered no significant protection, suggesting that early treatment was important for a beneficial outcome. Although we have presented a considerable amount of data showing the efficacy of cinnamaldehyde, more detailed studies of dose‐response relationships, pharmacokinetics, safety and tolerability are still needed before this compound could be tested in a larger sample. Moreover, other routes of administration, more detailed time‐courses and the long‐term effects of cinnamaldehyde on cerebral ischaemia still need further study.
The inflammatory response plays an important role in the secondary injury following cerebral ischaemia. Brain ischaemic injury is undoubtedly associated with the expression of inflammatory mediators such as inflammatory cytokines, chemokine and adhesion molecules (Zheng and Yenari, 2004). Inflammatory cytokines or chemokines form immediately after the onset of cerebral ischaemia, stimulate the expression of adhesion molecules on leukocytes and endothelial cells and cause the adherence and extravasation of leukocytes into brain parenchyma (Barone and Feuerstein, 1999). From previous studies of cerebral ischaemia, it appears that TNF‐α, IL‐1β, CCL2 and ELAM‐1 are well established molecules, highly relevant the inflammatory response and appear to exacerbate cerebral ischaemic injury (Wang et al., 1995; Barone et al., 1997; Hughes et al., 2002; Mulcahy et al., 2003). In our study, a certain number of factors related to inflammatory response, such as inflammatory cytokines (IL‐1β and TNF‐α), chemokines (CCL2) and adhesion molecules (ELAM‐1), were induced by ischaemia and inhibited by cinnamaldehyde, indicating that the neuroprotective efficacy of cinnamaldehyde could be attributed, at least in part, to its anti‐inflammatory activities. This is in agreement with previous reports of the anti‐inflammatory effects of cinnamaldehyde, in vitro and in vivo (Ho et al., 2013; Pyo et al., 2013).
Inflammation in stroke has been traditionally identified, in histopathological terms, as neutrophil infiltration (Weston et al., 2007). MPO is the most abundant component in azurophilic granules of neutrophils and has been used extensively as a marker for quantifying neutrophil infiltration (Rausch et al., 1978; Weston et al., 2006). MPO is also a key finflammatory enzyme mainly secreted by activated neutrophils in the ischaemic tissues and can generate highly reactive oxygen species to cause additional damage in cerebral ischaemia (Breckwoldt et al., 2008). Recent studies indicated that cinnamaldehyde inhibited N‐formyl‐methionyl‐leucyl‐phenylalanine‐induced inflammatory infiltration of neutrophils in vitro (Lee et al., 2009). By using immunohistochemistry, Western blotting and the biochemical assay, we observed that MPO activity and expression increased over time from 24 to 72 h after MCAO, in the ischaemic area, similar to earlier findings (Weston et al., 2007; Breckwoldt et al., 2008) and that cinnamaldehyde significantly reduced acute neutrophil infiltration, indicating that this compound did modify the acute inflammatory processes of neutrophil recruitment, following cerebral ischaemia.
The TLRs are the key host molecules in the regulation of the inflammatory response during CNS damage (Medzhitov, 2001). TLRs can recognize endogenous danger‐associated molecular patterns associated with tissue stress or injury that lead to the initiation of the inflammatory response (Medzhitov, 2001; Matzinger, 2002). So far, 11 human and 13 mouse TLRs have been identified, and TLR4 is recognized as an important component of the innate immunity of the CNS (Lehnardt et al., 2002). TLR4 also participates in the cerebral injury following ischaemic stroke (Caso et al., 2007; Kilic et al., 2008). TLR4 signalling pathway consists of a MyD88‐dependent and a MyD88‐independent pathways (Vallabhapurapu and Karin, 2009). In the more common MyD88‐dependent pathway, the IL‐1 receptor‐associated kinase (IRAK) is recruited to TLR4 through interaction with MyD88. IRAK is activated by phosphorylation and then associates with TRAF6, leading to the activation of downstream signalling pathways, including MAPKs and NF‐κB (Akira and Takeda, 2004). A critical role of NF‐κB is to regulate the genes that encode inflammation‐associated molecules and cytokines (Kim et al., 2009; Chan et al., 2010). Cinnamaldehyde suppressed lipopolysaccharide‐induced activation of TLR4 pro‐inflammatory signalling and inhibited the expression of high mobility group box 1, an endogenous ligand of TLR4 (Youn et al., 2008; Hwa et al., 2012). Cinnamaldehyde blocked the over‐expression of TLR4 and TRAF6 in macrophages (Zhao et al., 2008). Cinnamaldehyde also regulated NF‐κB activation by reacting with the free sulfhydryl groups of cysteine (Heiss et al., 2001). Therefore, we wanted to know whether the therapeutic benefit of cinnamaldehyde was associated with the TLR4/TRAF6/NF‐κB signalling pathway in our mouse model of pMCAO. We found that cinnamaldehyde inhibited the expression of TLR4 and TRAF6 and the nuclear translocation of NF‐κB after cerebral ischaemia, suggesting that the action of cinnamaldehyde could take place upstream of the activation of TLR4. We also observed the location of NF‐κB in three different types of cells including neurons, residential inflammatory cells, such as astrocytes, and infiltrated inflammatory cells, such as neutrophils. We found that NF‐κB was expressed mainly in neurons, with little expression in astrocytes and neutrophils. This is in accordance with earlier results showing that NF‐κB was activated after cerebral ischaemia, in neurons, astrocytes and infiltrating inflammatory cells (Ridder and Schwaninger, 2009). Thus, it is possible that cinnamaldehyde might cross the blood brain barrier (BBB) to exert anti‐inflammatory effects, in the CNS. The action of cinnamaldehyde could take place upstream of the activation of TLR4 to inhibit the expression of TLR4/TRAF6 and nuclear translocation of NF‐κB. However, cinnamaldehyde has exhibited a wide range of anti‐inflammatory effects in other pathologies. Does cinnamaldehyde exert its major effects by crossing BBB or by acting on circulating peripheral cells? Does cinnamaldehyde act through a completely different signalling pathway? Further research is still needed to understand the effects of cinnamaldehyde on the structure and function of the BBB, circulating peripheral cells and other signalling pathways.
In TLR4/TRAF6/NF‐κB pathway, as a critical signalling adapter molecule at the central convergence of different signal pathways, TRAF6 leads to the activation of downstream signalling pathways such as MAPKs and NF‐κB cascades. Miyahara et al. (2004) showed that an inflammatory signalling pathway containing TRAF6 contributed to intimal lesion formation in arterial injury model, indicating that TRAF6‐mediated signalling pathway might act as a therapeutic target for the treatment of vascular occlusive disease (Miyahara et al., 2004). Recently, TRAF6 was demonstrated to participate in the pathogenesis of cerebral ischaemia (Liu et al., 2012; Zhang et al., 2013). In our present study, the high level of TRAF6 following cerebral ischaemia was decreased by cinnamaldehyde, implying that TRAF6 was positively correlated with cerebral ischaemic injury and might be a novel therapeutic target for ischaemic stroke. The neuroprotective mechanism of cinnamaldehyde was at least in part linked to TRAF6 inhibition. However, the extent of the relationship between TRAF6 and ischaemic stroke, even TRAF6 and inflammation‐related disease, needs further research.
In conclusion, we have demonstrated that cinnamaldehyde may improve neurological deficit, alleviate brain oedema and reduce brain infarct volume by inhibiting the inflammatory response and this may occur by reducing the expression of TLR4and TRAF6, and the nuclear translocation of NF‐κB (Figure 7). Our findings suggest that cinnamaldehyde may serve as a novel candidate for stroke treatment.
Figure 7.
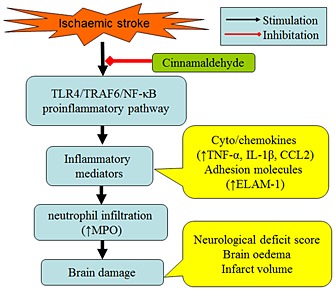
Diagram summarizing the neuroprotective mechanisms of cinnamaldehyde in ischemic stroke. Cinnamaldehyde suppresses the brain damage and inflammatory response in ischaemic stroke by reducing the expression of TLR4 and TRAF6 and the nuclear translocation of NF‐κB.
Author contributions
J. R. Zhao, L. P. Dong, Y. Wen and X. F. Zheng performed the research. J. R. Zhao designed the research study. X. J. Zhang, C. Zhang, Y. Zhang, Y. R. Li, T. T. He and X. Y. Zhu contributed essential reagents or tools. J. R. Zhao, R. Chen and L. T. Li analysed the data. J. R. Zhao wrote the paper.
Conflict of interest
None.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (grant no. 81371287). The authors thank technicians Ruichun Liu and Hongran Wu for their technical assistance and Drs Yansu Guo and Weisong Duan for providing valuable suggestions.
Zhao, J. , Zhang, X. , Dong, L. , Wen, Y. , Zheng, X. , Zhang, C. , Chen, R. , Zhang, Y. , Li, Y. , He, T. , Zhu, X. , and Li, L. (2015) Cinnamaldehyde inhibits inflammation and brain damage in a mouse model of permanent cerebral ischaemia. British Journal of Pharmacology, 172: 5009–5023. doi: 10.1111/bph.13270.
References
- Akira S, Takeda K (2004). Toll‐like receptor signalling. Nat Rev Immunol 4: 499–511. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013a). The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol 170: 1676–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN et al. (1997). Tumor necrosis factor‐alpha. A mediator of focal ischemic brain injury. Stroke 28: 1233–1244. [DOI] [PubMed] [Google Scholar]
- Barone FC, Feuerstein GZ (1999). Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab 19: 819–834. [DOI] [PubMed] [Google Scholar]
- Barone FC, Schmidt DB, Hillegass LM, Price WJ, White RF, Feuerstein GZ et al. (1992). Reperfusion increases neutrophils and leukotriene B4 receptor binding in rat focal ischemia. Stroke 23: 1337–1347. [DOI] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H (1986a). Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 17: 472–476. [DOI] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Germano SM, Nishimura MC, Davis RL, Bartkowski HM (1986b). Evaluation of 2,3,5‐triphenyltetrazolium chloride as stain for detection and quantification of experimental cerebral infarction in rats. Stroke 17: 1304–1308. [DOI] [PubMed] [Google Scholar]
- Breckwoldt MO, Chen JW, Stangenberg L, Aikawa E, Rodriguez E, Qiu S et al. (2008). Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc Natl Acad Sci U S A 105: 18584–18589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I (2007). Toll‐like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation 115: 1599–1608. [DOI] [PubMed] [Google Scholar]
- Chan SJ, Wong WS, Wong PT, Bian JS (2010). Neuroprotective effects of andrographolide in a rat model of permanent cerebral ischaemia. Br J Pharmacol 161: 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danton GH, Dietrich WD (2003). Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol 62: 127–136. [DOI] [PubMed] [Google Scholar]
- Dong L, Qiao H, Zhang X, Zhang X, Wang C, Wang L et al. (2013). Parthenolide is neuroprotective in rat experimental stroke model: downregulating NF‐κB, phospho‐p38MAPK, and caspase‐1 and ameliorating BBB permeability. Mediators Inflamm 2013: 370804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnan GA, Fisher M, Macleod M, Davis SM (2008). Stroke. Lancet 371: 1612–1623. [DOI] [PubMed] [Google Scholar]
- Downes CE, Crack PJ (2010). Neural injury following stroke: are toll‐like receptors the link between the immune system and the CNS? Br J Pharmacol 160: 1872–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Li L, Zhang X, Liu Y, Yang C, Yang Y et al. (2009). Oxymatrine downregulates TLR4, TLR2, MyD88, and NF‐kappaB and protects rat brains against focal ischemia. Mediators Inflamm 2009: 704706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatashita S, Hoff JT, Salamat SM (1988). Ischemic brain edema and the osomotic gradient between blood and brain. J Cereb Blood Flow Metab 8: 552–559. [DOI] [PubMed] [Google Scholar]
- He ZD, Qiao CF, Han QB, Cheng CL, Xu HX, Jiang RW et al. (2005). Authentication and quantitative analysis on the chemical profile of cassia bark (cortex cinnamomi) by high‐pressure liquid chromatography. J Agric Food Chem 53: 2424–2428. [DOI] [PubMed] [Google Scholar]
- Heiss E, Herhaus C, Klimo K, Bartsch H, Gerhauser C (2001). Nuclear factor‐κappa B is a molecular target for sulforaphane‐mediated anti‐inflammatory mechanisms. J Biol Chem 276: 32008–32015. [DOI] [PubMed] [Google Scholar]
- Ho SC, Chang KS, Chang PW (2013). Inhibition of neuroinflammation by cinnamon and its main components. Food Chem 138: 2275–2282. [DOI] [PubMed] [Google Scholar]
- Hughes PM, Allegrini PR, Rudin M, Perry VH, Mir AK, Wiessner C (2002). Monocyte chemoattractant protein‐1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab 22: 308–317. [DOI] [PubMed] [Google Scholar]
- Hwa JS, Jin YC, Lee YS, Ko YS, Kim YM, Shi LY et al. (2012). 2‐methoxycinnamaldehyde from Cinnamomum cassia reduces rat myocardial ischemia and reperfusion injury in vivo due to HO‐1 induction. J Ethnopharmacol 139: 605–615. [DOI] [PubMed] [Google Scholar]
- Jiang T, Gao L, Guo J, Lu J, Wang Y, Zhang Y (2012). Suppressing inflammation by inhibiting the NF‐kappaB pathway contributes to the neuroprotective effect of angiotensin‐(1–7) in rats with permanent cerebral ischaemia. Br J Pharmacol 167: 1520–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Yu JT, Zhu XC, Wang HF, Tan MS, Cao L et al. (2014). Acute metformin preconditioning confers neuroprotection against focal cerebral ischemia by pre‐activation of AMPK‐dependent autophagy. Br J Pharmacol 171: 3146–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N, Moyle M, Soule HR, Rote WE, Chopp M (1995). Neutrophil inhibitory factor is neuroprotective after focal ischemia in rats. Ann Neurol 38: 935–942. [DOI] [PubMed] [Google Scholar]
- Kilic U, Kilic E, Matter CM, Bassetti CL, Hermann DM (2008). TLR4 deficiency protects against focal cerebral ischemia and axotomy‐induced neurodegeneration. Neurobiol Dis 31: 33–40. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Jin YC, Kim YM, Rhie S, Kim HJ, Seo HG et al. (2009). Daidzein administration in vivo reduces myocardial injury in a rat ischemia/reperfusion model by inhibiting NF‐κappaB activation. Life Sci 84: 227–234. [DOI] [PubMed] [Google Scholar]
- Kleinig TJ, Vink R (2009). Suppression of inflammation in ischemic and hemorrhagic stroke: therapeutic options. Curr Opin Neurol 22: 294–301. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Walsh MC, Choi Y (2004). The role of TRAF6 in signal transduction and the immune response. Microbes Infect 6: 1333–1338. [DOI] [PubMed] [Google Scholar]
- Lee JY, Kang HS, Park BE, Moon HJ, Sim SS, Kim CJ (2009). Inhibitory effects of Geijigajakyak‐Tang on trinitrobenzene sulfonic acid‐induced colitis. J Ethnopharmacol 126: 244–251. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE et al. (2002). The toll‐like receptor TLR4 is necessary for lipopolysaccharide‐induced oligodendrocyte injury in the CNS. J Neurosci 22: 2478–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, He D, Zhang X, Li Y, Zhu C, Dong L et al. (2012). Neuroprotective effect of early and short‐time applying sophoridine in pMCAO rat brain: down‐regulated TRAF6 and up‐regulated p‐ERK1/2 expression, ameliorated brain infarction and edema. Brain Res Bull 88: 379–384. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R (1989). Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20: 84–91. [DOI] [PubMed] [Google Scholar]
- Matzinger P (2002). The danger model: a renewed sense of self. Science 296: 301–305. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R (2001). Toll‐like receptors and innate immunity. Nat Rev 1: 135–145. [DOI] [PubMed] [Google Scholar]
- Miyahara T, Koyama H, Miyata T, Shigematsu H, Inoue J, Takato T et al. (2004). Inflammatory signaling pathway containing TRAF6 contributes to neointimal formation via diverse mechanisms. Cardiovasc Res 64: 154–164. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C (2010). The science of stroke: mechanisms in search of treatments. Neuron 67: 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy NJ, Ross J, Rothwell NJ, Loddick SA (2003). Delayed administration of interleukin‐1 receptor antagonist protects against transient cerebral ischaemia in the rat. Br J Pharmacol 140: 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA (2003). The role of MyD88‐like adapters in toll‐like receptor signal transduction. Biochem Soc Trans 31: 643–647. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyo JH, Jeong YK, Yeo S, Lee JH, Jeong MY, Kim SH et al. (2013). Neuroprotective effect of trans‐cinnamaldehyde on the 6‐hydroxydopamine‐induced dopaminergic injury. Biol Pharm Bull 36: 1928–1935. [DOI] [PubMed] [Google Scholar]
- Qiao H, Zhang X, Zhu C, Dong L, Wang L, Zhang X et al. (2012). Luteolin downregulates TLR4, TLR5, NF‐κB and p‐p38MAPK expression, upregulates the p‐ERK expression, and protects rat brains against focal ischemia. Brain Res 1448: 71–81. [DOI] [PubMed] [Google Scholar]
- Rausch PG, Pryzwansky KB, Spitznagel JK (1978). Immunocytochemical identification of azurophilic and specific granule markers in the giant granules of Chediak‐Higashi neutrophils. N Engl J Med 298: 693–698. [DOI] [PubMed] [Google Scholar]
- Reddy AM, Seo JH, Ryu SY, Kim YS, Kim YS, Min KR et al. (2004). Cinnamaldehyde and 2‐methoxycinnamaldehyde as NF‐κappaB inhibitors from Cinnamomum cassia . Planta Med 70: 823–827. [DOI] [PubMed] [Google Scholar]
- Ridder DA, Schwaninger M (2009). NF‐kappaB signaling in cerebral ischemia. Neurosci 158: 995–1006. [DOI] [PubMed] [Google Scholar]
- Tatlisumak T, Carano RA, Takano K, Opgenorth TJ, Sotak CH, Fisher M (1998). A novel endothelin antagonist, A‐127722, attenuates ischemic lesion size in rats with temporary middle cerebral artery occlusion: a diffusion and perfusion MRI study. Stroke 29: 850–857. [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S, Karin M (2009). Regulation and function of NF‐κappaB transcription factors in the immune system. Annu Rev Immunol 27: 693–733. [DOI] [PubMed] [Google Scholar]
- Wang X, Yue TL, Barone FC, Feuerstein GZ (1995). Demonstration of increased endothelial leukocyte adhesion molecule I mRNA expression in rat ischemic cortex. Stroke 26: 1665–1669. [DOI] [PubMed] [Google Scholar]
- Wang YC, Lin S, Yang QW (2011). Toll‐like receptors in cerebral ischemic inflammatory injury. J Neuroinflammation 8: 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston RM, Jarrott B, Ishizuka Y, Callaway JK (2006). AM‐36 modulates the neutrophil inflammatory response and reduces breakdown of the blood brain barrier after endothelin‐1 induced focal brain ischaemia. Br J Pharmacol 149: 712–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston RM, Jones NM, Jarrott B, Callaway JK (2007). Inflammatory cell infiltration after endothelin‐1‐induced cerebral ischemia: histochemical and myeloperoxidase correlation with temporal changes in brain injury. J Cereb Blood Flow Metab 27: 100–114. [DOI] [PubMed] [Google Scholar]
- Williams AJ, Berti R, Dave JR, Elliot PJ, Adams J, Tortella FC (2004). Delayed treatment of ischemia/reperfusion brain injury: extended therapeutic window with the proteasome inhibitor MLN519. Stroke 35: 1186–1191. [DOI] [PubMed] [Google Scholar]
- Xu Z, Croslan DR, Harris AE, Ford GD, Ford BD (2006). Extended therapeutic window and functional recovery after intraarterial administration of neuregulin‐1 after focal ischemic stroke. J Cereb Blood Flow Metab 26: 527–535. [DOI] [PubMed] [Google Scholar]
- Youn HS, Lee JK, Choi YJ, Saitoh SI, Miyake K, Hwang DH et al. (2008). Cinnamaldehyde suppresses toll‐like receptor 4 activation mediated through the inhibition of receptor oligomerization. Biochem Pharmacol 75: 494–502. [DOI] [PubMed] [Google Scholar]
- Zhang J, Fu B, Zhang X, Chen L, Zhang L, Zhao X et al. (2013). Neuroprotective effect of bicyclol in rat ischemic stroke: down‐regulates TLR4, TLR9, TRAF6, NF‐ κ B, MMP‐9 and up‐regulates claudin‐5 expression. Brain Res 1528: 80–88. [DOI] [PubMed] [Google Scholar]
- Zhao BS, Huo HR, Ma YY, Liu HB, Li LF, Sui F et al. (2008). Effects of 3‐phenyl‐propenal on the expression of toll‐like receptors and downstream signaling components on raw264.7 murine macrophages. Am J Chin Med 36: 159–169. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Yenari MA (2004). Post‐ischemic inflammation: molecular mechanisms and therapeutic implications. Neurol Res 26: 884–892. [DOI] [PubMed] [Google Scholar]