

Abstract
Cyclic adenosine monophosphate (cAMP) is one of the second messengers critically involved in the molecular mechanisms underlying memory formation. In the CNS, the availability of cAMP is tightly controlled by phosphodiesterase 4 (PDE4), a family of enzymes that degrades the cyclic nucleotide to inactive AMP. Among the different PDE4 isoforms, in the last few years PDE4D has been hogging the limelight due to accumulating evidence for its crucial role in cognitive processes, which makes this enzyme a promising target for therapeutic interventions in a variety of pathological conditions characterized by memory impairment, such as Alzheimer's disease. In this article, we review the role of the cAMP signal transduction pathway in memory formation with a particular focus on the recent progress in PDE4D research.
Abbreviations
- CNG
cyclic nucleotide gated
- CREB
cAMP responsive element binding protein
- Epac
exchange protein directly activated by cAMP
- LTP
long‐term potentiation
- MWM
Morris water maze
- NAMs
negative allosteric modulators
- ORT
object recognition task
- Rap
Ras‐related protein
- Ras
rat sarcoma
- Rho
Ras homologue gene family
- LTM
long‐term memory
Tables of Links
| TARGETS | ||
|---|---|---|
| Ion channels a | Enzymes b | |
| CNG channels | ACs | PDE4D |
| Other protein targets | Epac1 | PI3K |
| CREB | Epac2 | PKA |
| PDEs | PKB (Akt) |
| LIGANDS | |
|---|---|
| AMP | cAMP |
| Amyloid‐β | cGMP |
| Apremilast | Roflumilast |
| ATP | Rolipram |
| BDNF |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Introduction
Memory formation is one of the most fascinating processes of the brain, and understanding the molecular mechanisms involved in this phenomenon has been, and still is, a very challenging task for neuroscientists.
Although no single molecule can be regarded as the sole trigger, during the last 30 years, a large amount of evidence has demonstrated beyond any doubt that cAMP has a prominent role in memory. In the CNS, cAMP is synthesized by adenylyl cyclases (ACs), a family of membrane‐bound enzymes (AC1–AC9) structurally composed of six hydrophobic transmembrane helices and three cytoplasmic domains termed N, C1 and C2 (Chern, 2000), whose function is regulated by α subunits of either stimulatory or inhibitory G proteins (Gs and Gi/o, respectively), although each AC isoform can be controlled by way of distinct molecular mechanisms. Moreover, the Gβγ subunits of the trimeric G proteins are able to regulate the activity of some AC isoforms to integrate Gα‐mediated signals. Several studies have also shown that the function of the distinct AC isoenzymes can be positively or negatively modulated by calmodulin‐ and calcineurin‐dependent mechanisms, various protein kinases and phosphatases. In addition to transmembrane ACs, cAMP can also be synthesized by a soluble form of the cyclase enzyme (AC10) that is directly activated by calcium (Steegborn, 2014). As for their localization, while the majority of AC isoenzymes show a widespread distribution in the CNS, some of them are expressed in discrete brain regions where they regulate specific functions, as mentioned below.
Almost 10 years after the discovery of cAMP, PKA was identified as the downstream effector of the cyclic nucleotide (Corbin and Krebs, 1969), which binds to the two regulatory subunits of the enzyme, thus causing a conformational change that releases the two catalytic subunits and allows the phosphorylation of their substrates.
Later in the 1980s, it was found that cAMP modulates gene expression via PKA‐mediated phosphorylation of the cAMP responsive element binding protein (CREB), a nuclear factor that binds to the cAMP response element, a conserved sequence found in the promoter region of several genes (Montminy et al., 1986; Gonzalez et al., 1989).
In addition to PKA, cAMP can also transduce signals by directly activating cyclic nucleotide gated (CNG) channels or by stimulating the exchange protein directly activated by cAMP (Epac). CNG channels are a heterogeneous superfamily of ion channels with a binding domain for 3′,5′‐monoposphate cyclic nucleotides in their carboxy terminal region and are, therefore, activated by both cAMP and cGMP. Besides their localization in rod and cone photoreceptors and in olfactory sensory neurons, CNG channels are also present in other neurons (e.g. hippocampal neurons), both at the pre‐ and postsynaptic level, as well as in non‐neuronal tissues (Kaupp and Seifert, 2002; Podda and Grassi, 2014). The two Epac isoforms, Epac1 and Epac2, the latter predominantly expressed in the brain (De Rooij et al., 1998; Kawasaki et al, 1998), are characterized by a regulatory region that interacts with cAMP and a catalytic domain able to activate different effectors, such as Rap, Ras and Rho GTPases, MAPKs, PLC, PKB (Akt), PI3Ks (Roscioni et al, 2008).
cAMP signalling is then terminated by its degradation to AMP operated by phosphodiesterases, a superfamily of 11 different enzymes (PDE1–PDE11) encoded by 21 genes, most of which are expressed in multiple variants, thus leading to the production of up to 100 individual proteins. Of the 11 families, three are specific for cGMP (PDE5, PDE6 and PDE9), three are specific for cAMP (PDE4, PDE7 and PDE8) and five hydrolyze both cAMP and cGMP (PDE1, PDE2, PDE3, PDE10 and PDE11) although to a different extent (Conti and Beavo, 2007).
cAMP pathways, LTP and memory
The first demonstrations for the involvement of cAMP in learning and memory date back to the 1970s, when this feature began to be investigated in the simple learned behaviour model of Aplysia (Lee et al., 2008; Kandel 2012). Since then, an enormous body of evidence has accumulated demonstrating that the cAMP transduction pathway is also critically involved in the mechanisms underlying memory formation in mammals.
Undoubtedly, the most important milestone in memory formation mechanisms was the discovery of long‐term potentiation (LTP) in the hippocampus, a form of synaptic plasticity that was first hypothesised to serve for memory storage by Bliss and Lømo in 1973. Today, we know that all forms of LTP recorded in the three major glutamatergic synaptic pathways of the hippocampus (perforant pathway‐granule cells, granule cell mossy fibers‐CA3 pyramidal neurons, CA3 Schaffer Collateral‐CA1 pyramidal neurons) consist of two temporally distinct phases: early LTP (E‐LTP) that lasts 1–3 h and does not require gene expression and protein synthesis, and a transcription‐ and translation‐dependent late LTP (L‐LTP), which can be recorded for 6–8 h in vitro (actually, as long as the preparation is vital) and can last from days to weeks in vivo (Krug et al., 1984; Frey et al., 1988; Matthies et al., 1990; Bliss and Collingridge, 1993; Huang and Kandel, 1994; Huang, 1998). In addition, E‐LTP can switch into L‐LTP possibly via an intermediate, protein synthesis‐dependent procedure (Reymann and Frey, 2007).
In mammals, cAMP and its downstream effectors seem to be critical especially for the expression of hippocampal L‐LTP and hippocampal‐dependent long‐term memory (LTM; Poser and Storm, 2001). In fact, the late phase of CA1 LTP does not occur in hippocampal slices of AC1 and AC8 double knockout mice, an effect that is paralleled by significant deficits of LTM in passive avoidance and contextual learning, but not in cued learning and memory, which are amygdala‐dependent processes (Wong et al., 1999). In contrast, the overexpression of AC1 facilitates and potentiates hippocampal CA1 LTP, and improves recognition and spatial memory without affecting the ability to extinguish old memories (Wang et al., 2004; Zhang and Wang, 2013).
In addition, pharmacological and genetic manipulations of the cAMP‐activated PKA pathway do result in the alteration of L‐LTP (but not of E‐LTP) and behavioural deficits in LTM (Frey et al., 1993; Huang and Kandel, 1994; Abel et al., 1997; Koh et al., 2002; Young et al., 2006; Bollen et al., 2014).
Similarly, gain or loss of function of CREB, the protein generally accepted as the molecular switch between short‐term and long‐term forms of synaptic plasticity, facilitates or disrupts L‐LTP and LTM respectively (Barco et al., 2002; Pittenger et al., 2002; Suzuki et al., 2011; Kida, 2012).
More recently, the cAMP‐Epac pathway has also been shown to participate in hippocampal synaptic plasticity and in memory formation and retrieval (Gelinas et al., 2008; Ma et al., 2009).
Type 4 phosphodiesterases, LTP and memory: spotlight on PDE4D
Some of the most compelling evidence for the involvement of cAMP in LTP and memory comes from studies on PDE4 enzymes, as, after the discovery of rolipram as a selective pan PDE4 inhibitor (PDE4‐I), a vast number of investigations has demonstrated that increasing cAMP, by blocking its PDE‐mediated breakdown, represents the molecular trigger to boost LTP and to improve memory formation and consolidation in rodents and non‐human primates (Figure 1).
Figure 1.
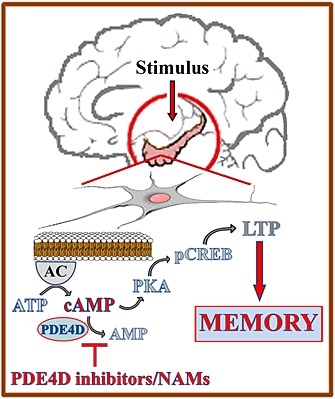
The cAMP pathway to memory. At the hippocampal level, salient stimuli to be stored in long‐term memory, trigger the cAMP/PKA/CREB‐dependent phase of late long‐term potentiation (LTP). Memory deficits can be prevented by enhancing cAMP intracellular levels using PDE4D inhibitors or negative allosteric modulators (NAMs).
Using a variety of behavioural tasks, these effects have been consistently proven under physiological conditions and in different models of pharmacologically‐induced cognitive deficits or in animal models of human pathologies, including Alzheimer's disease (Richter et al., 2013; Hansen III and Zhang, 2015; Heckman et al., 2015). Interestingly, it has been recently reported that the promnesic effects of PDE4‐I need several hours to manifest, again indicating the role of cAMP in switching a transient form of memory into a more stable one (Akkerman et al., 2014; Bollen et al., 2014).
Since the discovery that the PDE4 family consists of four isoforms (PDE4A to PDE4D) and 25 splice variants, neuroscientists have tried to unravel their functions in the brain, especially PDE4D in cognition, given its predominant expression in the hippocampus and its important role in hydrolyzing cAMP (Pérez‐Torres et al., 2000; Zhang et al., 2002).
To this purpose, given the lack of isoform selective inhibitors, the first studies took advantage of knock‐out (KO) strategies, thus demonstrating that PDE4D KO induces an enhancement of CA1 LTP in the hippocampus (Rutten et al., 2008). Surprisingly enough, it was found that PDE4D KO mice exhibited memory impairment, and not enhancement, when cued fear conditioned responses were assessed to examine associative learning and memory. However, the possibility that knocking out PDE4D might have caused developmental alterations in memory circuits should also be taken into account.
In fact, when in 2010 selective PDE4D negative allosteric modulators (NAMs; D158681, D159153, D159404, D159687) became available, their positive effects on recognition and spatial memory performance, assessed in the object recognition task (ORT) and in the Y‐maze task, respectively, clearly indicated a key role for this enzyme isoform in hippocampus‐dependent cognition (Burgin et al., 2010). Such important results were confirmed one year later, when it was shown that selective inhibition of PDE4D by the full inhibitor GEBR‐7b was indeed able to enhance both recognition and spatial memory in the ORT and in the object location test (OLT), respectively (Bruno et al., 2011). Notably, in the behavioural tasks, both allosteric modulators and GEBR‐7b were 3 to 10 times more potent than the pan PDE4 inhibitor rolipram. More recently, chronic administration of GEBR‐7b was also found to ameliorate spatial memory in a murine model of Alzheimer's disease (Sierksma et al., 2014) and NAMs proved to have significant pro‐cognitive effects in the object retrieval task in non‐human primates (Sutcliffe et al., 2014), a test that analyses multiple cognitive components (e.g. attention, response inhibition, planning) involving the prefrontal corticostriatal neuronal circuits rather than the hippocampus.
Furthermore, mice with genetic deletion or miRNA‐mediated downregulation of PDE4D displayed an improvement in spatial and recognition memory in the radial arm maze, in the Morris water maze (MWM) and in the ORT (Li et al., 2011). In addition, administration of rolipram to these PDE4D‐deficient mice did not further improve memory, definitely demonstrating that, among the PDE4 isoforms, PDE4D is the most important in cognitive processes. Similar results have been obtained by knocking down PDE4D selectively in the prefrontal cortex, indicating that the lack of activity of this enzyme isoform is also beneficial for memory in that brain region (Wang et al., 2013).
Most importantly from a translational point of view, PDE4D silencing in the hippocampus was able to counteract amyloid β42‐induced cAMP decrease and memory deficit in the MWM and in the ORT; in addition, it also largely prevented the reduction of BDNF concentration and the increase of TNFα, IL‐1β and NF‐κB levels, suggesting that PDE4D loss of function might attenuate neuroinflammation and confer neuroprotection in Alzheimer's disease (Zhang et al., 2014).
However, all that glitters is not gold. In fact, PDE4D is also considered the isoform responsible for the emetic effects induced by pan PDE4 inhibitors such as rolipram, which have precluded their clinical use. Indeed, this enzyme isoform is localized in brain regions associated with emesis (e.g. area postrema and nucleus of the solitary tract; Cherry and Davis., 1999; Lamontagne et al., 2001; Mori et al., 2010) and its deletion in transgenic mice reduced the xylazine/ketamine‐induced anaesthesia, a test used to measure emetic potential in non‐vomiting species (Robichaud et al., 2002). Nevertheless, PDE4D NAMs and GEBR‐7b proved to possess a therapeutic index much higher than rolipram, improving memory at doses devoid of undesired emetic effects. In fact, D159404 and D159687 did not reduce the duration of the xylazine/ketamine‐induced anaesthesia in mice at doses 1000 times higher than those beneficial for cognition and were also 100 to 3000 less potent as an emetic than rolipram in vomiting species (Burgin et al., 2010). The absence of emetic adverse effects at pro‐cognitive doses was also confirmed in non‐human primates (Sutcliffe et al., 2014). Similarly, GEBR‐7b did not show emetic‐like effects in mice and rats at doses up to 100‐300 times higher than the pro‐cognitive ones, as evaluated using two different tests, the xylazine/ketamine test in mice and the taste reactivity test in rats (Bruno et al., 2011).
Thus, it emerges that a tailored inhibition of PDE4D activity can be achieved with selective modulators/inhibitors, which could, therefore, represent successful therapeutic agents without unwanted side effects. As a proof of concept, second generation PDE4 inhibitors, possessing selectivity for different isoforms, are much better tolerated than rolipram in humans, as indicated by clinical studies regarding asthma, inflammation and chronic obstructive pulmonary disease (COPD; Bruno et al., 2014; Gurney et al., 2015). Indeed, a more favourable therapeutic index has recently led to the approval of the first orally active PDE4 inhibitors roflumilast and apremilast for the treatment of COPD and psoriatic arthritis respectively.
Taken together, these results demonstrate the strategic role of PDE4D in the modulation of cognitive processes and indicate this enzyme isoform as a suitable molecular target to counteract memory deficits in a variety of pathological conditions, such as Alzheimer's disease. Will this be enough to remember? Of course, only controlled clinical trials on selective PDE4D inhibitors can answer this question, and it is hoped that such drugs will soon be available for human studies.
Conflict of interest
Authors declare patent application EP 14425015.6–1452 for selective PDE4D inhibitors.
Acknowledgement
We thank the University of Genoa for financial support.
Ricciarelli, R. , and Fedele, E. (2015) Phosphodiesterase 4D: an enzyme to remember. British Journal of Pharmacology, 172: 4785–4789. doi: 10.1111/bph.13257.
References
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R (1997). Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus‐based long‐term memory. Cell 88: 615–626. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA et al. (2013a). The concise guide to pharmacology 2013/14: ion channels. Br J Pharmacol 170: 1607–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013b). The concise guide to pharmacology 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkerman S, Blokland A, Prickaerts J (2014). Mind the gap: delayed manifestation of long‐term object memory improvement by phosphodiesterase inhibitors. Neurobiol Learn Mem 109: 139–143. [DOI] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER (2002). Expression of constitutively active CREB protein facilitates the late phase of long‐term potentiation by enhancing synaptic capture. Cell 108: 689–703. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993). A synaptic model of memory: long‐term potentiation in the hippocampus. Nature 361: 31–39. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lømo T (1973). Long‐lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant pathway. J Physiol 232: 331–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollen E, Puzzo D, Rutten K, Privitera L, De Vry J, Vanmierlo T et al. (2014). Improved long‐term memory via enhancing cGMP‐PKG signaling requires cAMP‐PKA signaling. Neuropsychopharmacology 39: 2497–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno O, Fedele E, Prickaerts J, Parker LA, Canepa E, Brullo C et al. (2011). GEBR‐7b, a novel PDE4D selective inhibitor that improves memory in rodents at non‐emetic doses. Br J Pharmacol 164: 2054–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno O, Ricciarelli R, Prickaerts J, Parker LA, Fedele E (2014). PDE4D inhibitors: a potential strategy for the treatment of memory impairment? Neuropharmacology 85: 290–292. [DOI] [PubMed] [Google Scholar]
- Burgin AB, Magusson OT, Singh J, Witte P, Staker BL, Bjornsson JM et al. (2010). Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat Biotechnol 28: 63–70. [DOI] [PubMed] [Google Scholar]
- Chern Y (2000). Regulation of adenylyl cyclase in the central nervous system. Cell Signal 12: 195–204. [DOI] [PubMed] [Google Scholar]
- Cherry JA, Davis RL (1999). Cyclic AMP phosphodiesterases are localized in regions of the mouse brain associated with reinforcement, movement, and affect. J Comp Neurol 407: 287–301. [PubMed] [Google Scholar]
- Conti M, Beavo J (2007). Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 76: 481–511. [DOI] [PubMed] [Google Scholar]
- Corbin JD, Krebs EG (1969). A cyclic AMP‐stimulated protein kinase in adipose tissue. Biochem Biophys Res Commun 36: 328–336. [DOI] [PubMed] [Google Scholar]
- De Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A et al. (1998). Epac is a Rap1 guanine‐nucleotide‐exchange factor directly activated by cyclic AMP. Nature 396: 474–477. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H (1988). Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res 452: 57–65. [DOI] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER (1993). Effects of cAMP simulate a late stage of LTP in hippocampal neurons. Science 260: 1661–1664. [DOI] [PubMed] [Google Scholar]
- Gelinas JN, Banko JL, Peters MM, Klann E, Weeber WJ, Nguyen PV (2008). Activation of exchange protein activated by cyclic‐AMP enhances long‐lasting synaptic potentiation in the hippocampus. Learn Mem 15: 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez GA, Yamamoto KK, Fischer WH, Karr D, Menzel P, Biggs W (1989). A cluster of phosphorylation sites on the cyclic AMP‐regulated nuclear factor CREB predicted by its sequence. Nature 337: 749–752. [DOI] [PubMed] [Google Scholar]
- Gurney ME, D'Amato EC, Burgin AB (2015). Phosphodiesterase‐4 (PDE4) molecular pharmacology and Alzheimer's disease. Neurotherapeutics 12: 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RT III, Zhang HT (2015). Phosphodiesterase‐4 modulation as potential therapeutic for cognitive loss in pathological and non‐pathological aging: possibilities and pitfalls. Curr Pharm Des 21: 291–302. [DOI] [PubMed] [Google Scholar]
- Heckman PR, Blokland A, Ramaekers J, Prickaerts J (2015). Phosphodiesterase inhibitors as a target for cognition enhancement in aging and Alzheimer's disease: a translational overview. Curr Pharm Des 21: 317–331. [DOI] [PubMed] [Google Scholar]
- Huang EP (1998). Synaptic plasticity: going through phases with LTP. Neuron 8: R350–352. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER (1994). Recruitment of long‐lasting and protein kinase A‐dependent long‐term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn Mem 1: 74–82. [PubMed] [Google Scholar]
- Kandel ER (2012). The molecular biology of memory: cAMP, PKA, CRE, CREB‐1, CREB‐2, and CPEB. Mol Brain 5: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupp UB, Seifert R (2002). Cyclic nucleotide‐gated ion channels. Physiol Rev 82: 769–824. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizucki N, Toki S, Nakaya M, Matsuda M et al. (1998). A family of cAMP‐binding proteins that directly activate Rap1. Science 282: 2275–2279. [DOI] [PubMed] [Google Scholar]
- Kida S (2012). A functional role for CREB as a positive regulator of memory formation and LTP. Exp Neurobiol 21: 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh MT, Thiele TE, Bernstein IL (2002). Inhibition of protein kinase A activity interferes with lon‐term, but not short‐term, memory of conditioned taste aversions. Behav Neurosci 116: 1070–1074. [PubMed] [Google Scholar]
- Krug M, Lössner B, Ott T (1984). Anisomycin blocks the late phase of long‐term potentiation in the dentate gyrus of freely moving rats. Brain Res Bull 13: 39–42. [DOI] [PubMed] [Google Scholar]
- Lamontagne S, Meadows E, Luke P, Normandin D, Muise E, Boulet L et al. (2001). Localization of phosphodiesterases‐4 isoforms in the medulla and nodose ganglion of the squirrel monkey. Brain Res 920: 84–96. [DOI] [PubMed] [Google Scholar]
- Lee YS, Bailey CH, Kandel ER, Kaang BK (2008). Transcrptional regulation of long‐term memory in the marine snail Aplysia. Mol Brain 1: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YF, Cheng YF, Huang Y, Conti M, Wilson SP, O'Donnel JM et al. (2011). Phosphodiesterase‐4D knock‐out and RNA interference‐mediated knock‐down enhance memory and increase hippocampal neurogenesis via increased cAMP signaling. J Neurosci 31: 172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma N, Abel T, Hernadez PJ (2009). Exchange protein activated by cAMP enhances long‐term memory formation independent of protein kinase A. Learn Mem 16: 367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthies H, Frey U, Reymann KG, Krug M, Jork R, Schroeder H (1990). Different mechanisms and multiple stages of LTP. Adv Exp Med Biol 268: 359–368. [DOI] [PubMed] [Google Scholar]
- Montminy MR, Sevarino KA, Wagner JA, Mandel G, Goodman RH (1986). Identification of a cyclic AMP‐responsive element within the rat somatostatin gene. Proc Natl Acad Sci U S A 83: 6682–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori F, Perez‐Torres S, De Caro R, Porzionato A, Macchi V, Beleta J et al. (2010). The human area postrema and other nuclei related to the emetic reflex express cAMP phosphodiesterases 4B and 4D. J Chem Neuroanat 40: 36–42. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ; NC‐IUPHAR. (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Torres S, Mirò X, Palacios JM, Cortes R, Puigdoménech P, Mengod G (2000). Phosphodiesterase type 4 isoenzymes expression in human brain examined by in situ hybridization hystochemistry and [3H]rolipram binding autoradiography: comparison with monkey and rat brain. J Chem Neuroanat 20: 349–374. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S et al. (2002). Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus‐dependent spatial memory. Neuron 34: 447–462. [DOI] [PubMed] [Google Scholar]
- Podda MV, Grassi C (2014). New perspective in cyclic nucleotide‐mediated functions in the CNS: the emerging role of cyclic nucleotide‐gated (CNG) channles. Pflugers Arch‐Eur J Physiol 466: 1241–1257. [DOI] [PubMed] [Google Scholar]
- Poser S, Storm DR (2001). Role of Ca2+‐stimulated adenylyl cyclase in LTP and memory formation. Int J Dev Neurosci 19: 387–94. [DOI] [PubMed] [Google Scholar]
- Reymann KG, Frey JU (2007). The late maintenance of hippocampal LTP: requirements, phases, ‘synaptic tagging’, ‘late‐associativity’ and implications. Neuropharmacology 52: 24–40. [DOI] [PubMed] [Google Scholar]
- Richter W, Menniti FS, Zhang HT, Conti M (2013). PDE4 as target for cognition enhancement. Expert Opin Ther Targets 17: 1011–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaud A, Stamatiou PB, Lin SL, Lachance N, MacDonald D, Laliberté F et al. (2002). Deletion of phosphodiesterase 4D in mice shortens alpha(2)‐adrenoceptor‐mediated anesthesia, a behavioral correlate of emesis. J Clin Invest 110: 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscioni SS, Elzinga CR, Schmidt M (2008). Epac: effectors and biological functions. Naunyn Schmiedebergs Arch Pharmacol 377: 345–357. [DOI] [PubMed] [Google Scholar]
- Rutten K, Misner DL, Works M, Blokland A, Novak TJ, Santarelli L et al. (2008). Enhanced long‐term potentiation and impaired learning in phosphodiesterase 4D‐knockout (PDE4D‐/‐) mice. Eur J Neurosci 28: 625–632. [DOI] [PubMed] [Google Scholar]
- Sierksma AS, van den Hove DL, Pfau F, Philippens M, Bruno O, Fedele E et al. (2014). Improvement of spatial memory function in APPswe/PS1dE9 mice after chronic inhibition of phosphodiesterase type 4D. Neuropharmacology 77: 120–130. [DOI] [PubMed] [Google Scholar]
- Steegborn C (2014). Structure, mechanism, and regulation of soluble adenylyl cyclase‐ similarities and differences to transmembrane adenylyl cyclase. Biochim Biophys Acta 1842: 2535–2547. [DOI] [PubMed] [Google Scholar]
- Sutcliffe JS, Beaumont V, Watson JM, Chew CS, Beconi M, Hutcheson DM et al. (2014). Efficacy of selective PDE4D negative allosteric modulators in the object retrieval task in female cynomolgus monkeys (Macaca fascicularis). PLoS One 9: e102449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Fukushima H, Mukawa T, Toyoda H, Wu LJ, Zhao MG et al. (2011). Upregulation of CREB‐mediated transcription enhances both short‐ and long‐term memory. J Neurosci 31: 8786–8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ferguson GD, Pineda VV, Cundiff PE, Storm DR (2004). Overexpression of type‐1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat Neurosci 7: 635–642. [DOI] [PubMed] [Google Scholar]
- Wang ZZ, Zhang Y, Liu YQ, Zhao N, Zhang YZ, Yuan L et al. (2013). RNA interference‐mediated phosphodiesterase‐4D splice variants knock‐down in the prefrontal cortex produces antidepressant‐like and cognition‐enhancing effects. Br J Pharmacol 168: 1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC et al. (1999). Calcium‐stimulated adenylyl cyclase activity is critical for hippocampus‐dependent long‐term memory and late phase LTP. Neuron 23: 787–798. [DOI] [PubMed] [Google Scholar]
- Young JZ, Isiegas C, Abel T, Nguyen PV (2006). Metaplasticity of the late phase of long‐term potentiation: a critical role for protein kinase A in synaptic tagging. Eur J Neurosci 23: 1784–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Cheng Y, Wang H, Wang C, Wilson SP, Xu J et al. (2014). RNA interference‐mediated knockdown of long‐form phosphodiesterase‐4D (PDE4D) enzyme reverses amyloid‐β42‐induced memory deficits in mice. J Alzheimers Dis 38: 269–280. [DOI] [PubMed] [Google Scholar]
- Zhang HT, Huang Y, Jin SLC, Frith S, Suvarna N, Conti M et al. (2002). Antidepressant‐like profile and reduced sensitivity to rolipram in mice deficient in the PDE4D phosphodiesterase enzyme. Neuropsychopharmcology 27: 587–595. [DOI] [PubMed] [Google Scholar]
- Zhang M, Wang H (2013). Mice overexpressing type 1 adenylyl cyclase show enhanced spatial memory flexibility in the absence of intact synaptic long‐term depression. Learn Mem 20: 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]