

Abstract
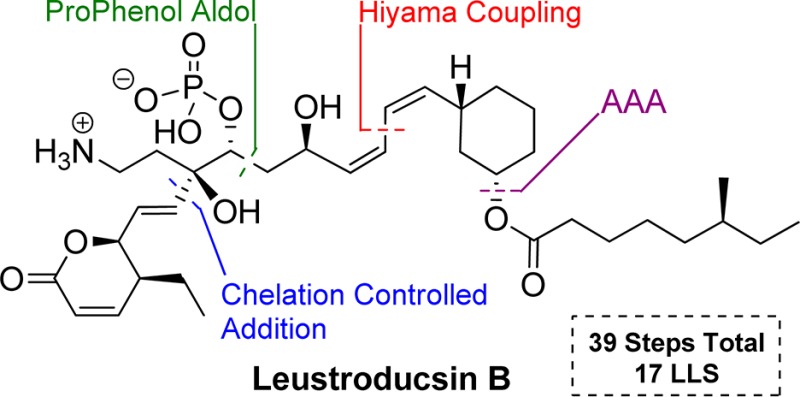
Leustroducsin B exhibits a large variety of biological activities and unique structural features. An efficient and highly convergent total synthesis of Leustroducsin B was achieved in 17 longest linear and 39 total steps by disconnecting the molecule into three fragments having similar levels of complexity. These pieces were connected via a highly efficient chelate-controlled addition of a vinyl zincate to an α-hydroxy ketone and a silicon-mediated cross-coupling. The stereochemistry of the central and western fragments was set catalytically in high yields and excellent de by a zinc-ProPhenol-catalyzed aldol reaction and a palladium-catalyzed asymmetric allylic alkylation.
Leustroducsins A–C were first identified and isolated from a culture broth of the soil bacterium Streptomyces platensis SANK 60191 in 1993 by Kohama et al. at Sankyo.1 These compounds belong to the phoslactomycin family2 and exhibit a range of potentially useful bioactivities, such as antibacterial, antifungal, and antitumor activity.3 They showed high potency and selectivity toward inhibition of protein serine/threonine phosphatase 2A, an enzyme that plays a central role in the regulation of cell growth and inhibition of metastasis.4 Leustroducsin B has specifically shown potent in vitro induction of cytokine production by KM-102 cells3d as well as an increase of host in vivo resistance to E. coli infections4a and thrombocytosis induction3b when administered to mice. These remarkable biological activities and its unique highly congested linear structure make Leustroducsin B a synthetic target of strong interest for the scientific community.1,5
Currently, two total syntheses,6 two formal syntheses,7 and a semisynthesis8 starting from leustroducsin H have been reported. These excellent efforts reveal the difficulty of the challenge, as the shortest prior synthesis required 37 linear and 64 total steps, therefore, a more efficient total synthesis of leustroducsin B remains of high interest. As part of our ongoing program aimed at synthesizing highly potent molecules bearing structural complexity in a step and atom economical fashion,9 we report herein a concise and highly convergent synthesis of leustroducsin B.
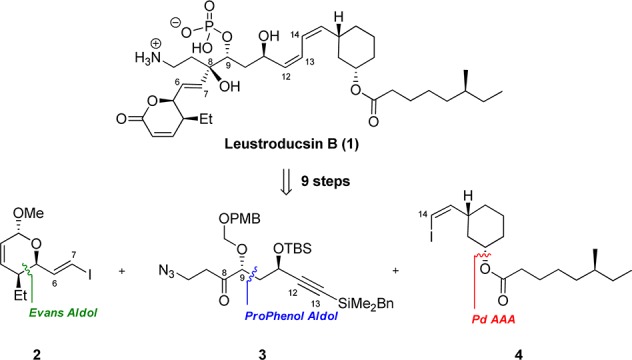
Our retrosynthetic approach takes advantage of a highly convergent strategy involving two key disconnections to provide three intermediates with similar size and structural complexity (Scheme 1). We envisioned that western fragment 2 and central fragment 3 could be coupled via α-alkoxy-directed diastereoselective vinyl zinc addition to generate the C7–C8 bond.10 Fragment 4 could be united to the rest of the molecule by the modified Hiyama11 silicon-based palladium-catalyzed cross coupling reaction (C13–C14 bond) developed in our group.12 To enhance the efficiency of the synthesis, the absolute and relative stereochemistry of fragments 3 and 4 is derived respectively from a zinc-ProPhenol-catalyzed aldol reaction13,14 and a palladium-catalyzed asymmetric allylic alkylation.15
Scheme 1. Retrosynthetic Analysis of Leustroducsin B.
Vinyl iodide fragment 2 was most efficiently synthesized from commercially available oxazolidinone 5 and propiolaldehyde 6 in five steps (Scheme 2). This synthesis is an alternative strategy to the ring-closing metathesis approach recurrently used in the preparation of the six-membered α,β-unsaturated lactone moiety found in the leustroducsin,7 phoslactomycin,16 fostriecyn,17 and cytostatin18 families. Alcohol 7 was obtained in high yield and excellent diastereomeric ratio following Evan’s aldol protocol,19 and the chiral auxiliary was removed using diisobutylaluminum hydride to afford aldehyde 8 as a solution in toluene upon workup.20 This aldehyde was treated under mild phase-transfer Wittig reaction conditions in the presence of tris[2-(2-methoxyethoxy)ethyl]amine and phosphonium bromide 9 to obtain a mixture of alcohols 10 and 11 in 56% yield over two steps.21 Ketalization using a catalytic amount of p-toluenesulfonic acid in methanol was followed by addition of potassium carbonate to deprotect the terminal alkyne in a one-pot procedure. Lastly, hydrozirconation of the terminal alkyne and addition of molecular iodine gave access to key intermediate 2 in 62% yield over two steps on a multigram scale.22
Scheme 2. Synthesis of Eastern Fragment 2.

With fragment 2 in hand, an efficient route to synthesize central key intermediate 3 was developed (Scheme 3). First, carbonyl addition of the enolate of ester 12 to methyl formate followed by acidic hydrolysis (pH 4) of the hemiacetal intermediate delivered aldehyde 13. The latter was immediately subjected to Wittig olefination conditions followed by reduction of the ester functionality to give aldehyde 14 in good yield. The zinc-ProPhenol-catalyzed aldol reaction between aldehyde 14 and ketone 15(17b) delivered adduct 16 in 78% yield and 99% ee.14 The 1,3-anti relationship of diol 17 was then set by a asymmetric transfer hydrogenation using Noyori’s catalyst (R,R)-TsDPEN-Ru(p-cymene)23 in 90% yield and >20:1 dr.17b Use of achiral reagent Me4NB(OAc)3H at −35 °C (Evans’ conditions)24 gave moderate 3:1 dr. Although the selectivity was lower, the two diastereomers were separable, and 1,3-anti diol 17 could be isolated in 61% yield. The undesired diastereomer could also be recycled back to the ynone 16 using manganese dioxide. Protection of the less sterically encumbered hydroxyl group with tert-butyldimethylsilyl chloride was followed by addition of alkyl chloride 18 to afford 19 in almost quantitative yield. Finally, one-pot deprotection of the diethoxyacetal and conversion of the resulting enone into azide 3 was accomplished in 91% yield.
Scheme 3. Synthesis of Central Fragment 3.

The synthesis of vinyl iodide 4 started from racemic lactone 20 (Scheme 4), which is easily accessible from commercially available 3-cyclohexene-1-carboxylic acid via an iodolactonization/elimination process.25 Lactone 20 was opened with benzyl alcohol, and the resulting hydroxyl group reacted with methylchloroformate to form racemic 21 in 70% yield over two steps. Carbonate 21 was treated with Pd(0), standard (S,S)-Trost ligand, and the sodium salt of carboxylic acid 22 (prepared in five synthetic steps from commercially available materials)26 to deliver ester 23 in 93% yield and 99% de.15 In this particular asymmetric alkylation, the intermediate π-allyl palladium complex is pseudomeso, thus both enantiomers of the starting material can be used in the alkylation. Previous studies showed a so-called memory effect indicating the nonsymmetrical nature of the π-allylpalladium intermediate.27 To ensure its full equilibration prior to nucleophilic attack, tetrahexylammonium bromide is added to slow the rate of carboxylate attack to maximize the effective symmetry and therefore ee. Alcohol 24 was then obtained by hydrogenolysis of ester 23 followed by selective reduction of the carboxylic acid using BH3·Me2S. Oxidation of alcohol 24 to the corresponding aldehyde was accomplished with Dess–Martin periodinane, and the resulting aldehyde was subject to a Stork–Zhao olefination to afford vinyl iodide 4 with high Z-selectivity.28
Scheme 4. Synthesis of Western Fragment 4.

The coupling of the western fragment 2 and central fragment 3 has been the most challenging part of this program (Scheme 5). In order to increase the convergent character of our approach for the preparation of leustroducsin B, we investigated the direct addition of vinyl iodide 2 to ketone 3 to generate tertiary allylic alcohol 26 with sensitive functional groups present in the molecule. The desired transformation required formation of metal vinyl species 25 generated from vinyl iodide 2 to add to β-azido ketone 3 in a diastereoselective fashion by chelation to the α-alkoxy group (27 to 28) to finally access alcohol 26.10 Extensive screening and reaction optimization29 revealed that the addition proceeded in high yield and remarkable diastereoselectivity when dimethylzinc30 was used to generate vinyl zincate complex 25 as the precursor for the chelation-controlled addition. The success of the zincate addition highly depends on the quality of the organometallic reagent employed, and the reaction temperature must be kept rigorously under −50 °C in order to avoid decomposition of ketone 3.
Scheme 5. Chelation Controlled Addition of 2 to 3.

Scheme 6. Completion of the Synthesis of Leustroducsin B.
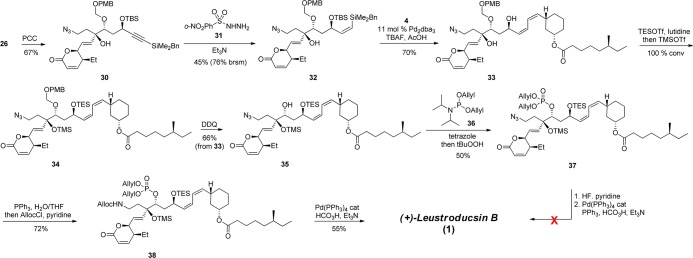
After we successfully coupled fragments 2 and 3, methyl ketal 26 was oxidized to lactone 30 using pyridinium chlorochromate (PCC). The alkyne moiety was reduced chemoselectively to cis-alkene 31 by use of diimide under mild basic conditions31,32 to avoid significant decomposition of starting material. Cross-coupling between lactone 32 and vinyl iodide 4 was then examined in the presence of a catalytic amount of Pd2dba3·CHCl3 and tetrabutylammonium fluoride (TBAF).12 To our delight, cis,cis-diene 33 was isolated in 70% yield. Addition of acetic acid was essential to the success of this transformation as it buffered the reaction mixture.3332 and 33 could easily decompose in the presence of residual hydroxide species present in commercial TBAF. The stability of the azide to this series of transformations is particularly noteworthy.
Secondary alcohol 33 was protected with triethylsilyltriflate followed by addition of trimethylsilyltriflate to selectively silylate the hindered tertiary alcohol. The crude material was directly subjected to 1,2-dichloro-4,5-dicyano-1,4-benzoquinone (DDQ) in wet methylene chloride to promote removal of the PMB-ketal functionality and afford secondary alcohol 35 in 66% yield over two steps. Introduction of the phosphate group was accomplished by treatment of alcohol 35 with phosphoramidite reagent 36 in the presence of tetrazole, followed by oxidation of the phosphite intermediate into phosphate 37 in 50% yield.6 We first attempted to reduce the azide during the final deallylation step using palladium (0) under acidic conditions and an excess of PPh3. Although NMR analysis revealed that the allyl groups had been removed, it did not result in the formation of leustroducsin B (1). To circumvent this difficulty, a stepwise protocol was pursued. Reduction of azide 37 using standard Staudinger reaction conditions was followed by in situ addition of allylchloroformate and pyridine to deliver carbamate 38 in 72% yield. Similarly to previously reported synthesis of leustroducsin B6 and to increase the conciseness of our synthesis, we intended to remove both silyl and allyl protecting groups at the last step using an excess of formic acid.34 Gratifyingly, final deallylation of the phosphonate and the carbamate as well as desilylation of the two hydroxyl groups was successfully achieved using a catalytic amount of Pd(PPh3)4, formic acid, and triethylamine to reveal deprotected leustroducsin B in 55% yield.6
In conclusion, we have reported a highly convergent total synthesis of (+)-leustroducsin B in only 39 total and 17 longest linear steps starting from commercially available material 12. In comparison to previously reported routes, our concise approach reduced the total number of steps by 40% and exhibits a strong convergent character. The number of longest linear steps has been cut to less than half compared to the previously reported total syntheses of leustroducsin B.6 Our strategy takes advantage of the preparation of three key intermediates of similar complexity in terms of size and stereochemistry. Particularly noteworthy is the high enantioselectivity of both our zinc-ProPhenol aldol reaction and palladium-catalyzed deracemization using a carboxylate nucleophile. This synthesis also features the exceptionally diastereoselective addition of vinyl zincate 25 to ketone 3 in the presence of sensitive functional groups and a high yielding silicon-based palladium-catalyzed cross-coupling between two highly functionalized molecules. Finally, the last step of the synthesis removed three allyl and two silyl groups all at once, revealing the mildness of these conditions. The brevity and convergent nature of the synthesis reported herein offers an opportunity to explore further the biological effects of chemical modifications in the structure of leustroducsin B.
Acknowledgments
We thank the National Science Foundation (CHE-1145236) and the National Institute of Health (GM033049) for their generous support of our programs and the American Cancer Society (PF-11-013-01-CDD) for a postdoctoral fellowship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b07438.
Experiment details, compound characterization data, and spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Kohama T.; Enokita R.; Okazaki T.; Miyaoka H.; Torikata A.; Inukai M.; Kaneko I.; Kagasaki T.; Sakaida Y.; Satoh A.; Shiraishi A. J. Antibiot. 1993, 46, 1503–1511. 10.7164/antibiotics.46.1503. [DOI] [PubMed] [Google Scholar]; b Kohama T.; Nakamura T.; Kinoshita T.; Kaneko I.; Shiraishi A. J. Antibiot. 1993, 46, 1512–1519. 10.7164/antibiotics.46.1512. [DOI] [PubMed] [Google Scholar]
- Fushimi S.; Nishikawa S.; Shimazo A.; Seto H. J. Antibiot. 1989, 42, 1019–1025. 10.7164/antibiotics.42.1019. [DOI] [PubMed] [Google Scholar]; b Fushimi S.; Furihata K.; Seto H. J. Antibiot. 1989, 42, 1026–1036. 10.7164/antibiotics.42.1026. [DOI] [PubMed] [Google Scholar]
- a Kohama T.; Katayama T.; Inukai M.; Maeda H.; Shiraishi A. Microbiol. Immunol. 1994, 38, 741–745. 10.1111/j.1348-0421.1994.tb01850.x. [DOI] [PubMed] [Google Scholar]; b Kohama T.; Maeda H.; Imada Sakai J.; Shiraishi A.; Yamashita K. J. Antibiot. 1996, 49, 91–94. 10.7164/antibiotics.49.91. [DOI] [PubMed] [Google Scholar]; c Koishi R.; Serizawa N.; Kohama T. J. J. Interferon Cytokine Res. 1998, 18, 863–869. 10.1089/jir.1998.18.863. [DOI] [PubMed] [Google Scholar]; d Koishi R.; Yoshimura C.; Kohama T.; Serizawa N. J. J. Interferon Cytokine Res. 2002, 22, 343–350. 10.1089/107999002753675776. [DOI] [PubMed] [Google Scholar]; e Shimada K.; Koishi R.; Kohama T. Annu. Rep. Sankyo Res. Lab 2004, 56, 11–28. [Google Scholar]
- a Usui T.; Marriott G.; Inagaki G.; Swarup G.; Osada H. J. Biochem. 1999, 125, 960–965. 10.1093/oxfordjournals.jbchem.a022375. [DOI] [PubMed] [Google Scholar]; b Kawada M.; Kawatsu M.; Masuda T.; Ohba S.; Amemiya M.; Kohama T.; Ishizuka M.; Takeuchi T. Int. Immunopharmacol. 2003, 3, 179–188. 10.1016/S1567-5769(02)00231-X. [DOI] [PubMed] [Google Scholar]; c Faulkner E.; Lane B. R.; Bock P. J.; Markovitz D. M. J. Virol. 2003, 77, 2276–2281. 10.1128/JVI.77.3.2276-2281.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T.; Kurihara S.; Yoda K.; Haruyama H. Tetrahedron 1995, 51, 11999–12012. 10.1016/0040-4020(95)00759-2. [DOI] [Google Scholar]
- a Shimada K.; Kaburagi Y.; Fukuyama T. J. Am. Chem. Soc. 2003, 125, 4048–4049. 10.1021/ja0340679. [DOI] [PubMed] [Google Scholar]; b Miyashita K.; Tsunemi T.; Hosokawa T.; Ikejiri M.; Imanishi T. Tetrahedron Lett. 2007, 48, 3829–3833. 10.1016/j.tetlet.2007.03.152. [DOI] [Google Scholar]; c Miyashita K.; Tsunemi T.; Hosokawa T.; Ikejiri M.; Imanishi T. J. Org. Chem. 2008, 73, 5360–5370. 10.1021/jo8005599. [DOI] [PubMed] [Google Scholar]
- a Mise J.; Sonawane R. P.; Corsi C.; Wendeborn S. V.; Arseniyadis S.; Cossy J. Synlett 2008, 2008, 2617–2620. 10.1055/s-0028-1083511. [DOI] [Google Scholar]; b Druais V.; Hall M. J.; Corsi C.; Wenderborn S. V.; Meyer C.; Cossy J. Tetrahedron 2010, 66, 6358–6375. 10.1016/j.tet.2010.05.050. [DOI] [Google Scholar]; c Grezler S. N.; Malinowski J. T.; Johnson J. S. Org. Lett. 2011, 13, 3206–3209. 10.1021/ol2011192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuhashi H.; Shimada K. Tetrahedron 2002, 58, 5619–5626. 10.1016/S0040-4020(02)00528-8. [DOI] [Google Scholar]
- a Trost B. M. Science 1991, 254, 1471–1477. 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]; b Trost B. M. Angew. Chem., Int. Ed. Engl. 1995, 34, 259–281. 10.1002/anie.199502591. [DOI] [Google Scholar]; c Wender P. A.; Verma V. A.; Paxton T. J.; Pillow T. H. Acc. Chem. Res. 2008, 41, 40–49. 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- For a review on substrate-directable reactions, see:Hoveyda A. H.; Evans D. A.; Fu G. C. Chem. Rev. 1993, 93, 1307–1370. 10.1021/cr00020a002. [DOI] [Google Scholar]
- a Hatanaka Y.; Hiyama T. J. Org. Chem. 1988, 53, 918–920. 10.1021/jo00239a056. [DOI] [Google Scholar]; b For a recent review on Pd-catalyzed cross coupling using organosilicon reagent, see: Sore F.; Galloway W. R. J.D.; Spring D. R. Chem. Soc. Rev. 2012, 41, 1845–1866. 10.1039/C1CS15181A. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Machacek M. R.; Ball Z. T. Org. Lett. 2003, 5, 1895–1898. 10.1021/ol034463w. [DOI] [PubMed] [Google Scholar]
- For a recent review on ProPhenol-catalyzed asymmetric additions, see:Trost B. M.; Barlett M. J. Acc. Chem. Res. 2015, 48, 688–701. 10.1021/ar500374r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Fettes A.; Shireman B. T. J. Am. Chem. Soc. 2004, 126, 2660–2661. 10.1021/ja038666r. [DOI] [PubMed] [Google Scholar]
- For reviews on palladium-catalyzed allylic asymmetric alkylation, see:; a Trost B. M.; Van Vranken D. L. Chem. Rev. 1996, 96, 395–422. 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; b Trost B. M.; Crawley M. L. Chem. Rev. 2003, 103, 2921–2944. 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; c Trost B. M. J. Org. Chem. 2004, 69, 5813–5837. 10.1021/jo0491004. [DOI] [PubMed] [Google Scholar]; d Lu Z.; Ma S. Angew. Chem., Int. Ed. 2008, 47, 258–297. 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]; e Trost B. M.; Zhang T.; Sieber J. D. Chem. Sci. 2010, 1, 427–440. 10.1039/c0sc00234h. [DOI] [Google Scholar]; f Trost B. M. Org. Process Res. Dev. 2012, 16, 185–194. 10.1021/op200294r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shibahara S.; Fujino M.; Tashiro Y.; Takahashi K.; Ishihara J.; Hatakeyama S. Org. Lett. 2008, 10, 2139–2142. 10.1021/ol8004672. [DOI] [PubMed] [Google Scholar]; b Sarkar S. M.; Wanzala E. N.; Shibahara S.; Takahashi K.; Ishihara J.; Hatakeyama S. Chem. Commun. 2009, 5907–5909. 10.1039/b912267b. [DOI] [PubMed] [Google Scholar]; c Wang Y.-G.; Takeyama R.; Kobayashi Y. Angew. Chem., Int. Ed. 2006, 45, 3320–3323. 10.1002/anie.200600458. [DOI] [PubMed] [Google Scholar]; d König C. M.; Gebhardt B.; Schleth C.; Dauber M.; Koert U. Org. Lett. 2009, 11, 2728–2731. 10.1021/ol900757k. [DOI] [PubMed] [Google Scholar]; e Gebhardt B.; König C. M.; Schleth C.; Dauber M.; Koert U. Chem. - Eur. J. 2010, 16, 5934–5941. 10.1002/chem.201000104. [DOI] [PubMed] [Google Scholar]
- a Wang Y.-G.; Kobayashi Y. Org. Lett. 2002, 4, 4615–4618. 10.1021/ol020193q. [DOI] [PubMed] [Google Scholar]; b Trost B. M.; Frederiksen M. U.; Papillon J. P. N.; Harrington P. E.; Shin S.; Shireman B. T. J. Am. Chem. Soc. 2005, 127, 3666–3667. 10.1021/ja042435i. [DOI] [PubMed] [Google Scholar]; c Robles O.; McDonald F. E. Org. Lett. 2009, 11, 5498–5501. 10.1021/ol902365n. [DOI] [PubMed] [Google Scholar]; d Gao D.; O’Doherty A. Org. Lett. 2010, 12, 3752–3755. 10.1021/ol101340n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawhorn B. G.; Boga S. B.; Wolkenberg S. E.; Colby D. A.; Gauss C.-M.; Swingle M. R.; Amable L.; Honkanen R. E.; Bober D. L. J. Am. Chem. Soc. 2006, 128, 16720–16732. 10.1021/ja066477d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A.; Dehn R.; Schlager N.; Dieter B.; Firschning A. Org. Lett. 2014, 16, 568–571. 10.1021/ol403441c. [DOI] [PubMed] [Google Scholar]
- Compound 8 was stable in solution at room temperature. Extensive decomposition occurred when the solution was concentrated.
- Daubresse N.; Francesch C.; Rolando C. Tetrahedron 1998, 54, 10761–10770. 10.1016/S0040-4020(98)00631-0. [DOI] [Google Scholar]
- Liu X.; Ready J. M. Tetrahedron 2008, 64, 6955–6960. 10.1016/j.tet.2008.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura K.; Hashiguchi S.; Ikariya T.; Noyori R. J. Am. Chem. Soc. 1997, 119, 8738–8739. 10.1021/ja971570a. [DOI] [Google Scholar]
- Evans D. A.; Chapman K. T.; Carreira E. M. J. Am. Chem. Soc. 1988, 110, 3560–3578. 10.1021/ja00219a035. [DOI] [Google Scholar]
- Trost B. M.; Richardson J.; Yong K. J. Am. Chem. Soc. 2006, 128, 2540–2541. 10.1021/ja057163d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The synthesis of (6S)-methyloctanoic acid 22 is described in the Supporting Information.
- For leading references on the equilibration of π-allyl palladium complexes, see:; a Trost B. M.; Bunt R. C. J. Am. Chem. Soc. 1994, 116, 4089–4090. 10.1021/ja00088a059. [DOI] [Google Scholar]; b Trost B. M.; Bunt R. C. J. Am. Chem. Soc. 1996, 118, 235–236. 10.1021/ja9530782. [DOI] [Google Scholar]; c Trost B. M.; Bunt R. C. J. Am. Chem. Soc. 1998, 120, 70–79. 10.1021/ja9726522. [DOI] [Google Scholar]; d Lloyd-Jones G. C.; Stephen S. C.; Fairlamb I. J. S.; Martorell A.; Dominguez B.; Tomlin P. M.; Murray M.; Fernandez J. M.; Jeffery J. C.; Riis-Johannessen T.; Guerziz T. Pure Appl. Chem. 2004, 76, 589–601. 10.1351/pac200476030589. [DOI] [Google Scholar]
- Stork G.; Zhao K. Tetrahedron Lett. 1989, 30, 2173–2174. 10.1016/S0040-4039(00)99640-0. [DOI] [Google Scholar]
- For an optimization table of selected reaction conditions screened, see Table S-1 in Supporting Information.
- For examples of diastereoselective chelation controlled additions using Zn(II) species, see:; a Stanton G. R.; Koz G.; Walsh P. J. J. Am. Chem. Soc. 2011, 133, 7969–7976. 10.1021/ja201629d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Stanton G. R.; Norrby P.-O.; Carroll P. J.; Walsh P. J. J. Am. Chem. Soc. 2012, 134, 17599–17604. 10.1021/ja306781z. [DOI] [PubMed] [Google Scholar]; c Stanton G. R.; Kauffman M. C.; Walsh P. J. Org. Lett. 2012, 14, 3368–3371. 10.1021/ol301354w. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Raffier L.; Stanton G. R.; Walsh P. J. Org. Lett. 2013, 15, 6174–6177. 10.1021/ol4030259. [DOI] [PubMed] [Google Scholar]
- Myers A. G.; Zheng B.; Movassaghi M. J. Org. Chem. 1997, 62, 7507–7507. 10.1021/jo9710137. [DOI] [PubMed] [Google Scholar]
- Onyango E. O.; Tsurumoto J.; Imai N.; Takahashi K.; Ishihira J.; Hatakeyama S. Angew. Chem., Int. Ed. 2007, 46, 6703–6705. 10.1002/anie.200702229. [DOI] [PubMed] [Google Scholar]
- When the reaction was run without acetic acid, only traces of product were observed.
- Chandra T.; Broderick W. E.; Broderick J. B. Nucleosides, Nucleotides Nucleic Acids 2009, 28, 1016–1029. 10.1080/15257770903362172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.