

Abstract
The advent of next generation sequencing has influenced every aspect of biological research. Many labs are now using whole genome sequencing in Arabidopsis thaliana as a means to quickly identify EMS-generated mutations present in isolated mutants. Following identification of these mutations, examination of T-DNA insertional alleles defective in candidate genes or complementation of the mutant phenotype with a wild type copy of candidate genes can be used to verify which mutation is causative for the phenotype of interest. Here, we discuss the benefits and pitfalls of using this method to identify mutations underlying phenotypes.
Keywords: abscisic acid, ethylmethane sulfonate, mutation mapping, next-generation sequencing
Abbreviations and Acronyms
- ABA
abscisic acid
- AR
ABA Root Resistance
- EMS
ethylmethane sulfonate
- NGS
next-generation sequencing
- PCR
polymerase chain reaction
- SNP
single nucleotide polymorphism
Over the past few decades, many techniques have been used to identify Arabidopsis thaliana causative mutations in mutant isolates. In the 1990's, many graduate students and postdocs spent their entire tenures carrying out the laborious process of ‘chromosome walking’, in which a physical map was built using yeast artificial chromosomes (YACs), and markers identified one-by-one as researchers narrowed the region that may contain the causative gene mutation.1 After this lengthy process, it was no small feat to sequence the region of interest in the mutant and wild type plant backgrounds, identify the causative mutation, and complement the phenotype by transformation to confirm causality.2,3
The release of the Arabidopsis genome sequence in 2000 was the advent of the ‘genomics’ era, and altered the speed with which mutations were identified.4 The release of the sequence for the first ecotype (Columbia-0, Col-0)4 was shortly followed by the release of the sequence of the ecotype Landsberg erecta (Ler-0).5 The sequence of these two ecotypes allowed researchers to compare the sequences and identify single nucleotide polymorphisms (SNPs), revolutionizing Arabidopsis gene mapping and allowing gene identification in a year or less, compared to 3-5 years with earlier chromosome walking methods.5
In the post-genomics era, and with the availability of next-generation sequencing, genomic data has become exponentially faster and cheaper. Further, the 1001 Genomes Project will sequence 1001 Arabidopsis ecotypes, providing a seemingly inexhaustible supply of natural variation data (www.1001genomes.org). Similarly, next-generation sequencing is currently used in a broad array of organisms to answer new biological questions and to quickly identify causative mutations in organisms from metazoans to microbes.6-8
We recently described a method using next-generation sequencing (NGS) to quickly identify causative EMS mutations. We used EMS mutagenesis and screened Arabidopsis seedlings to identify abscisic acid (ABA)-resistant root elongation (AR) mutants.9 For our experiments, we performed a genetic screen for our phenotype of interest (long roots in seedlings grown on ABA-containing growth medium) in EMS-mutagenized M2 seedlings. We then used two whole genome sequencing strategies to identify the causative mutations in these AR lines. For some of our AR lines, we retested M3 lines for the original phenotype, then crossed them to Col-0 to obtain plants heterozygous for every mutation. From this backcross, F2 progeny displaying the long root phenotype were identified and their F3 progeny retested and used for genomic DNA extraction, typically from 7-10 F3 pools of 500 seedlings each (Fig. 1). For other AR lines, we extracted genomic DNA from pooled M4 seedlings that had not been backcrossed to Col-0. For both of these strategies, we used Illumina-based next-generation sequencing to identify all SNPs. We narrowed our search for causative mutations by identifying exon-encoded, homozygous mutations that might be caused by EMS treatment (i.e. G-to-A or C-to-T mutations). We then used this list of EMS-related mutations as a starting point to determine which mutation might be causing the ABA resistance phenotype.
Figure 1.
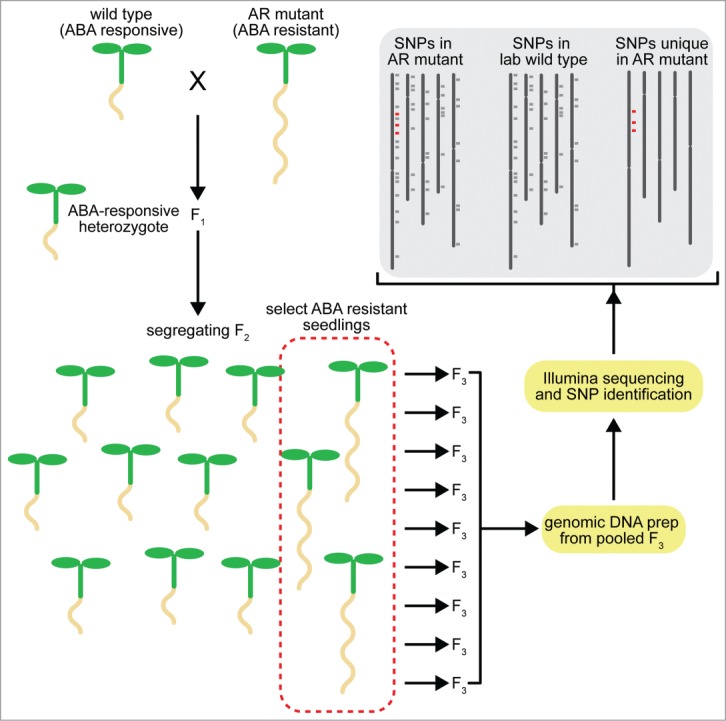
Scheme for identifying potential causative mutations in Arabidopsis with NGS. In this example, we cross an ABA-resistant (AR) mutant created by ethyl methanesulfonate (EMS) treatment to wild type (Columbia-0). The resultant F1 is self-pollinated to create a segregating F2 population. Individuals displaying the phenotype of interest (ABA resistance in root elongation) are selected and allowed to self-pollinate to create F3 progeny. These progeny are retested for the phenotype of interest, tissue from multiple retested F3 lines are pooled, and bulk genomic DNA is sequenced using Illumina technology. The resulting reads are aligned to the reference sequence (TAIR v10) using Novoalign (Novocraft; http://novocraft.com) and SNPs identified by SAMtools20 and annotated using snpEFF.21 We then compare identified canonical EMS-induced changes (G-to-A or C-to-T) from our mutant to our lab wild type Col-0 strain to identify mutations unique to the mutant.
From the data generated by sequencing pooled bulk backcrossed segregants, the list of potential mutations was reasonably short (typically 3 to 10 genes), allowing for quick identification of likely causative mutations. In one example, we found AR116 had mutations in the coding sequences of 8 genes, one of which was in a gene encoding the well-characterized auxin transporter AUXIN RESISTANT1 (AUX1). Because we had additional aux1 alleles in the laboratory, we were able to quickly perform an F1 non-complementation test to determine that the mutation in AUX1 was causative in this particular isolate. In other lines, we identified mutations in genes not previously associated with hormone responses; we have ordered T-DNA insertional alleles10 to test for the ABA-resistant root phenotype and for F1 non-complementation tests.
For the non-backcrossed M4 AR lines sequenced, we uncovered between one hundred and two hundred exonic, homozygous, EMS-associated mutations. We scanned these lists of genes for known genes involved in hormone signaling. For example, from the list of mutations identified in isolate AR211, a mutation in ETHYLENE INSENSITIVE ROOT1/PIN-FORMED2 seemed a likely candidate for a causative mutation. We therefore crossed AR211 to eir1-1 for an F1 non-complementation test and simultaneously used the identified AR211 SNPs to determine linkage between these SNPs and the phenotype using PCR-based genotyping in a segregating AR211 backcrossed population. We found that the phenotype was linked to EIR1 and that eir1-1 failed to complement the AR211 phenotype, confirming that the EIR1 mutation was causative for the ABA resistance phenotype in AR211.
The method we described in Thole et al. (2014) worked well for quickly identifying a handful of genes with known roles in hormone signaling, which we could then use to demonstrate that both auxin and ethylene responsiveness are required for a ABA-responsive inhibition of root elongation. From the time that a mutant phenotype of interest is identified to the time that F1 complementation tests were carried out to verify the causative mutation took between 2 to 4 months. This strategy is much faster than the traditional map-based cloning strategy, which takes approximately one year.5 An added benefit is the effort required using next generation sequencing is considerably less than positional cloning, in which large mapping populations of hundreds of plants and hundreds of PCR genotyping experiments are required to narrow the chromosomal region containing the causative mutation.
This strategy does not come without its pitfalls. Notably, our strategy involved identifying EMS-related changes (C-to-T and G-to-A11) in exons. However, mutations in introns and other non-coding regions can be causative and splice site mutations can lead to intron retention, resulting in a non-functional protein (for example, Thole et al.12). In addition, spontaneous, non-EMS mutations may be causative, even in EMS-mutagenized individuals. In these cases, re-examination of previously ignored data (from non-coding regions and including all SNPs) would be required to identify the causative mutation. An additional limitation of using NGS to identify mutations is that these technologies are particularly ineffective at identifying insertions and deletions,13 making it an unsuitable choice for identification of mutations caused by fast neutron or T-DNA insertion. Perhaps, as this technology improves and read lengths increase, NGS will become more amenable to identifying indel mutants.
Another difficulty in sequencing non-backcrossed lines arises if identified mutations do not reveal obvious candidates, in which case the researcher is left with a list of possibly hundreds of genes to examine. In this situation, one would likely backcross the line and use linkage between identified mutations and phenotype to narrow the list of candidate genes. We have encountered this issue and have used this mapping strategy to identify genes previously not known to be involved in ABA response. In one specific example, not included in our publication, we sequenced AR165 and identified a mutation in PLEIOTROPIC DRUG RESISTANCE3 (PDR3/ABCG31, At2g29940). Because a previous study reported that a close family member, PDR12/ABCG40 is a plasma membrane ABA uptake transporter,14 we were naturally excited about the possibility that PDR3 could play a similar role in ABA transport. Indeed, we found that pdr3 insertional alleles exhibited resistance to ABA in root elongation assays. We then performed F1 non-complementation tests with AR165 and the pdr3 insertional allele and found that the heterozygote displayed milder ABA resistance than the AR165 isolate. Additionally, PDR3 overexpression in the AR165 background only partially restored ABA responsiveness (data not shown). It did not escape our attention that AR165 also carried a mutation in AUXIN RESPONSE1 (AUX1). Unfortunately, both AUX1 and PDR3 are tightly linked on the same arm of Chromosome 2, and we were unable to separate these mutations in segregating populations. In other lines in which it appears that more than one gene contributes to the ABA-resistant root elongation phenotype, we will identify individuals segregating for different possible causative mutations, and further investigate their roles individually. These approaches may allow us to identify the causative mutation in our sequenced mutants that did not yield obvious candidate mutations.
We are not the first to use next-generation sequencing to quickly identify mutations in Arabidopsis, and many laboratories are using various next-generation approaches to identify causative mutations. Ashelford et al.15 took a similar approach to identify EMS-caused SNPs in the genome of a mutant of interest, however, they narrowed down their candidate genes using a functional genomic approach. These researchers rough-mapped their causative mutation, then used gene expression data to look for genes that normally had a rhythmic expression pattern, as the mutant of interest had a circadian clock phenotype. Ultimately, these researchers verified their phenotype by identifying a T-DNA insertional mutant of the gene of interest with the same phenotype.15 This method to identify candidate genes, facilitated by publicly available transcriptomic data, will likely be valuable to many researchers. Additional studies using next generation sequencing of backcrossed bulk segregants include Hartwig et al., 2012,16 Abe et al., 2012,17 and Lindner et al., 2012.18
Austin et al.19 took a different approach that they named Next Generation Mapping (NGM), in which they pooled genomic DNA from 80 outcrossed F2 lines, with an equal mix of mutant and parental ecotypes (Col-0 and Ler), allowing for identification of large ‘SNP deserts’ created by linkage to the mutation of interest. Discordant chastity statistics and probability estimates then narrowed the list of possible mutant sites, reducing the number of candidate mutations to between one and five SNPs. Within these candidates, these researchers identified new alleles for two mutants previously associated with their phenotype of interest; for the third mutant isolate, the researchers examined a T-DNA insertional allele in a candidate gene and observed the same phenotype as their original isolate, confirming the causative mutation.19 The approach of sequencing mapping populations is more intensive than our approach because of the large number of F2 pools required and the specialized bioinformatics and statistical analysis. Interestingly, this group has developed web-based application to perform the described analyses (http://bar.utoronto.ca/NGM).
In conclusion, our method of sequencing backcrossed bulk segregants is particularly time-effective and useful in creating a short list of candidate mutations. Given the ease and affordability of next-generation sequencing, this method is worth trying, and is particularly cost-effective when sequencing multiple mutants with indexing. For an in-depth review of using next generation sequencing technology to identify mutations, please see Schneeberger, 2014.8
Acknowledgments
We are grateful to Tara Enders, Elizabeth Frick, David Korasick, Marta Michniewicz, and Samantha Powers for critical comments on the manuscript. This research was supported by the National Institutes of Health (R00 GM089987 to L.C.S.).
References
- 1. Hwang I, Kohchi T, Hauge B, Goodman H. Identification and map position of YAC clones comprising one-third of the Arabidopsis genome. Plant J 1991; 1:367-74; PMID:1844889; http://dx.doi.org/ 10.1046/j.1365-313X.1991.t01-5-00999.x [DOI] [PubMed] [Google Scholar]
- 2. Arondel V, Lemieux B, Hwang I, Gibson S, Goodman HM, Somerville CR. Map-based cloning of a gene controlling omega-3 fatty acid desaturation in Arabidopsis. Science 1992; 258:1353-5; PMID:1455229; http://dx.doi.org/ 10.1126/science.1455229 [DOI] [PubMed] [Google Scholar]
- 3. Giraudat J, Hauge BM, Valon C, Smalle J, Parcy F, Goodman HM. Isolation of the Arabidopsis ABI3 gene by positional cloning. Plant Cell 1992; 4:1251-61; PMID:1359917; http://dx.doi.org/ 10.1105/tpc.4.10.1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Initiative AG. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000; 408:796-815; PMID:11130711; http://dx.doi.org/ 10.1038/35048692 [DOI] [PubMed] [Google Scholar]
- 5. Jander G, Norris SR, Rounsley SD, Bush DF, Levin IM, Last RL. Arabidopsis map-based cloning in the post-genome era. Plant Physiol 2002; 129:440-50; PMID:12068090; http://dx.doi.org/ 10.1104/pp.003533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blumenstiel JP, Noll AC, Griffiths JA, Perera AG, Walton KN, Gilliland WD, Hawley RS, Staehling-Hampton K. Identification of EMS-induced mutations in drosophila melanogaster by whole-genome sequencing. Genetics 2009; 182:25-32; PMID:19307605; http://dx.doi.org/ 10.1534/genetics.109.101998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schneeberger K, Weigel D. Fast-forward genetics enabled by new sequencing technologies. Trends Plant Sci 2011; 16:282-8; PMID:21439889; http://dx.doi.org/ 10.1016/j.tplants.2011.02.006 [DOI] [PubMed] [Google Scholar]
- 8. Schneeberger K. Using next-generation sequencing to isolate mutant genes from forward genetic screens. Nat Rev Genet 2014; 15:662-76; PMID:25139187; http://dx.doi.org/ 10.1038/nrg3745 [DOI] [PubMed] [Google Scholar]
- 9. Thole JM, Beisner ER, Liu J, Venkova SV, Strader LC. Abscisic acid regulates root elongation through the activities of auxin and ethylene in Arabidopsis thaliana. G3 2014; 4:1259-74; PMID:24836325; http://dx.doi.org/full_text [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alonso JM, Stepanova AN, Leisse TJ, Kim CJ, Chen H, Shinn P, Stevenson DK, Zimmerman J, Barajas P, Cheuk R, et al. . Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 2003; 301:653-7; PMID:12893945; http://dx.doi.org/ 10.1126/science.1086391 [DOI] [PubMed] [Google Scholar]
- 11. Burns PA, Allen FL, Glickman BW. DNA sequence analysis of mutagenicity and site specificity of ethyl methanesulfonate in Uvr+ and UvrB- strains of Escherichia coli. Genetics 1986; 113:811-9; PMID:3527868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thole JM, Vermeer JE, Zhang Y, Gadella TW, Nielsen E. Root hair defective4 encodes a phosphatidylinositol-4-phosphate phosphatase required for proper root hair development in Arabidopsis thaliana. Plant Cell 2008; 20:381-95; PMID:18281508; http://dx.doi.org/ 10.1105/tpc.107.054304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet 2010; 11:31-46; PMID:19997069; http://dx.doi.org/ 10.1038/nrg2626 [DOI] [PubMed] [Google Scholar]
- 14. Kang J, Hwang JU, Lee M, Kim YY, Assmann SM, Martinoia E, Lee Y. PDR-type ABC transporter mediates cellular uptake of the phytohormone abscisic acid. Proc Natl Acad Sci U S A 2010; 107:2355-60; PMID:20133880; http://dx.doi.org/ 10.1073/pnas.0909222107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ashelford K, Eriksson ME, Allen CM, D'Amore R, Johansson M, Gould P, Kay S, Millar AJ, Hall N, Hall A. Full genome re-sequencing reveals a novel circadian clock mutation in Arabidopsis. Genome Biol 2011; 12:R28; PMID:21429190; http://dx.doi.org/ 10.1186/gb-2011-12-3-r28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hartwig B, James GV, Konrad K, Schneeberger K, Turck F. Fast isogenic mapping-by-sequencing of ethyl methanesulfonate-induced mutant bulks. Plant Physiol 2012; 160:591-600; PMID:22837357; http://dx.doi.org/ 10.1104/pp.112.200311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, et al. . Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 2012; 30:174-8; PMID:22267009; http://dx.doi.org/ 10.1038/nbt.2095 [DOI] [PubMed] [Google Scholar]
- 18. Lindner H, Raissig MT, Sailer C, Shimosato-Asano H, Bruggmann R, Grossniklaus U. SNP-Ratio Mapping (SRM): identifying lethal alleles and mutations in complex genetic backgrounds by next-generation sequencing. Genetics 2012; 191:1381-6; PMID:22649081; http://dx.doi.org/ 10.1534/genetics.112.141341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Austin RS, Vidaurre D, Stamatiou G, Breit R, Provart NJ, Bonetta D, Zhang J, Fung P, Gong Y, Wang PW, et al. . Next-generation mapping of Arabidopsis genes. Plant J 2011; 67:715-25; PMID:21518053; http://dx.doi.org/ 10.1111/j.1365-313X.2011.04619.x [DOI] [PubMed] [Google Scholar]
- 20. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25:2978-2079; PMID:19505943; http://dx.doi.org/ 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEFF: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012; 6(2):1-13; PMID:22728672; http://dx.doi.org/ 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]