

Abstract
PUMA (p53 unregulated modulator of apoptosis), a BH3-only Bcl-2 family member, can be induced by p53-dependent and p53-independent manners. It plays an important role as regulator of cellular apoptosis. Herein, we evaluate the effects of H1 (a derivative of tetrandrine) on induction of PUMA and underlie its potential mechanism in p53-independent cytotoxic response. Anti-proliferative activity and evidently cytotoxic activity of H1 were observed in wild-type and p53 null cells. Further studies demonstrated that H1 resulted in an increase of cleaved PARP, decease of survivin and elevation of p-H2AX. What is more, H1 significantly induced PUMA expression in a concentration- and time-dependent manner and caused an increase of Bax/Bcl-2 ratio in p53 null cells. Of note, knockdown of PUMA attenuated cytotoxic activity of H1. Further studies demonstrated that inhibition of AKT/FoxO3a signaling contributed to H1-mediated PUMA induction. Targeted suppression of AKT/FoxO3a signaling by siRNA could overcome H1-mediated PUMA induction. In addition, H1 significantly suppressed NF-κB activity and caused an increase of early apoptotic and late apoptotic cells, and elevated caspase-3 activity. Taken together, we found that inhibition of AKT/FoxO3a signaling may contribute to H1-mediated PUMA induction, suggesting that inhibition of AKT/FoxO3a signaling result in PUMA expression in response to p53-independent cytotoxic effects of H1.
Keywords: AKT/FoxO3a signaling, apoptosis, cytotoxicity, PUMA, p53, tetrandrine
Abbreviations
- FoxO3a
Forkhead Homeobox type O3a; NF-κB, Nuclear Factor Kappa-light-chain-enhancer of activated B cells; PARP, Poly ADP (Adenosine Diphosphate)-Ribose Polymerase; PUMA, p53 Unregulated Modulator of Apoptosis; Tet, Tetrandrine; TNF-α, Tumor Necrosis Factor alpha
Introduction
Apoptosis is a tightly regulated cellular process and abnormally regulation of apoptosis is a hallmark of human cancers. Targeting key apoptosis regulators aim to restore apoptosis in cancer cells has been known as cancer chemotherapeutic strategy.1 In addition, apoptosis is a main cytotoxic mechanism of chemotherapeutic agents, and defective apoptosis regulation in cancer cells lead to therapeutic resistance. p53 protein can induce the expression of numerous apoptotic genes that can contribute to the activation of both death-receptor and mitochondrial apoptosis pathways. p53 is a key tumor suppressor protein that induces apoptosis in response to oncogenic stress. Loss of p53 function or mutation in p53 gene (named TP53) leads to tumor malignant progression.2 TP53 is the most commonly mutated gene in human cancer, and inactivated p53 occurs in over 50% of all types of cancer.3 Loss function of p53 affects binding to DNA or fail to form correct folding or oligomerization of the tumor suppressor. In addition, loss of p53 function is due to overexpression of p53 regulatory proteins that suppress p53 activity, such as MDM2 and MDMX.4,5 p53 is activated in response to several malignancy associated stress signals, resulting in the inhibition of cancer cell growth. Several responses can be provoked by p53, including cell cycle arrest, senescence, differentiation and apoptosis.6,7 In addition, p53 can also affect the efficiency of cellular survival signaling.8-10
PUMA (p53 unregulated modulator of apoptosis) encodes a BH3-only protein that is induced by p53 tumor suppressor or other apoptotic stress.11 The pro-apoptotic activity of PUMA requires its interactions with other Bcl-2 family members and mitochondria localization.12 Previous studies demonstrated that PUMA induced apoptosis by activating the pro-apoptotic protein Bax through its interaction with anti-apoptotic Bcl-2 family members, thereby triggering mitochondrial dysfunction and caspase cascade.13 PUMA can also be induced in a p53-independent manner. Under given condition of nongenotoxic stimuli, for instance: inflammatory cytokines, growth factor deprivation, and kinase inhibitors, p53-independent PUMA induction can be mediated by different transcription factors, including p73, FoxO3a, NF-κB, and so on.14-16 After this induction, PUMA strongly induces apoptosis in cancer cells by acting on other Bcl-2 family members such as Bax and Bcl-2, leading to trigger caspase cascades.
Tetrandrine (Tet) is a bisbenzylisoquinoline, which is the main active component in the root Stephania tetrandra S. Moore.17 Based on the chemical structure of Tet, a novel derivative of Tet named H1 was synthesized. Our previous findings demonstrated that H1 exerted good anti-MDR activity both in vitro and in vivo. It could induce apoptosis in sensitive and resistant cancer cells. Further studies found that H1-treatment resulted in the increase of ROS generation, elevation of the Bax/Bcl-2 ratio, loss of mitochondrial transmembrane potential, release of cytochrome c and AIF from mitochondria into cytosol, and activation of caspase-9 and caspase-3, but had no effect on activation of caspase-8. In addition, H1 also inhibited survival pathways.18
In this study, we evaluate the effects of H1 on induction of PUMA. Furthermore, we explore the potential mechanism of PUMA induction in p53-independent cytotoxic response of H1.
Results
Effect of H1 on growth of human colorectal cancer cells
Our previous results showed that H1 had good anti-MDR activity in vitro and in vivo, and its mechanism is associated with induction of apoptosis and inhibition of cell survival pathways.18 Herein, we evaluate the cytotoxic effects of H1 in human colorectal cancer cells, including p53 wild-type and p53 null cells. As seen in Figure 1B, the IC50 of H1 is a range from 2–5 μM in human colorectal cancer cells. Interestingly, cytotoxic effect of H1 in wild-type HCT116 cells and p53 null (HCT116 p53−/−) cells is similar, and IC50 is 2.02±0.43 and 2.45±0.22 μM, respectively. This result suggests that H1 may inhibit the proliferation of human colorectal cancer cells through p53-independent manner. We also tested the effect of H1 on growth of normal human colon cells, such as CCD 841. H1 exerted some toxic activity, and the IC50 is 3.17 μM. However, our previous studies showed that H1 did not affect the body weight of xgenogrft mice, and no obvious toxicity of H1 was observed.18
Figure 1.
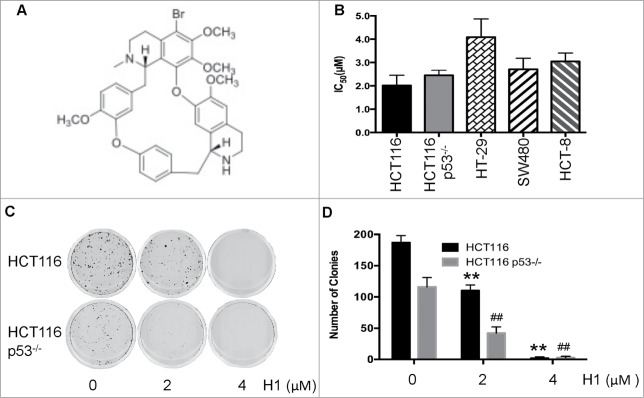
Effect of H1 on growth of human colorectal cancer cells. The chemical structure of H1 was shown in (A). HCT116, HCT116 p53−/−, HT-29, SW480 and HCT-8 cells were seeded into 96-well plate. After exposure to various concentrations of H1 for 72 h, cell viability was determined by MTT assay. Data from 3–5 independent experiments were used to calculate IC50 by Graphpad Prism 6.0 software (B). HCT116 and HCT116 p53−/− (C&D) were seeded into 6-well plates at concentration of 400 cells/mL. After an overnight incubation, cells were treated by H1 for an additional 24 h, then change to fresh medium. After 10–14 days incubation, colonies (>50 cells) were fixed and manually counted. A representative of 4 experiments is shown. **P < 0.01; ##P < 0.01 vs. each control group.
In addition, we determined the effect of H1 on clongentic. H1 significantly decreased the clone number of both HCT116 and HCT116 p53−/− cells in a concentration-dependent manner. Treatment of 2 μM H1 already decreased 50% of the colonies number, and 4 μM of H1 almost killed all the cells (Fig. 1C, D).
Effect of H1-induced apoptosis in HCT116 and HCT116 p53−/− cells
Based on anti-proliferative activity of H1 against HCT116 and HCT116 p53−/− cells, the effect of H1-induced early and late apoptosis was determined by flow cytometry. As seen in Figure 2A, H1 could significantly result in an increase of early and late apoptosis in both HCT116 and HCT116 p53−/− cells in a concentration-dependent manner. 4 μM of H1 significantly induced early and late apoptotic cells (Fig. 2B, C). Caspases family plays an important role in regulation of cell apoptosis. Apoptotic machinery is composed of upstream initiators and downstream effectors. For example, caspase-3 is a main caspase effector.19 Thus, we detected the effect of H1 on activity of caspase-3. As seen in Figure 2D, H1 significantly increased the caspase-3 activity in a concentration-dependent manner. These results suggest that H1 caused an increase of early apoptotic and late apoptotic cells, and elevated caspase-3 activity. Moreover, the potential of H1 on inducing apoptosis may through p53-independent manner.
Figure 2.

Effect of H1-induced apoptosis in wild-type and p53-null HCT116 cells. HCT116 and HCT116 p53−/− cells were treated by various concentrations of H1 for 24 h, cells were processed, stained, and the number of early apoptotic and late apoptotic cells was analyzed by FCM as descripted in the Methods. A representative of 3 experiments is shown (A, B and C). *P < 0.05, **P < 0.01; ##P < 0.01 vs. each control group. After treated with H1 for 24 h, cells were lysed. Protein concentration was determined by Bradford assay. According to instruction, caspase-3 activity was determined using Caspase-3 Assay Kit (D). Data from 4 independent experiments were used. *P < 0.05; **P < 0.01; #P < 0.05; ##P < 0.01 vs. each control group.
H1 caused p53-independent cytotoxic effect
Given that H1 could induce apoptosis in HCT116 and HCT116 p53−/− cells, we further detected the effects of H1 on several key mediators of cytotoxic response. After treatment of H1 for 24 h, the expression level of PARP, survivin, p53, p-H2AX, and total H2AX were determined by western blot analysis. As shown in Figure 3, H1 resulted in an increase of cleaved PARP, decrease of survivin, and elevation of p-H2AX. 4 µM of H1 significantly decreased the expression of survivin in both HCT116 and HCT116 p53−/− cells. Of note, we observed that H1 caused a decrease of p53 protein expression of HCT116 cells in a concentration-dependent manner.
Figure 3.
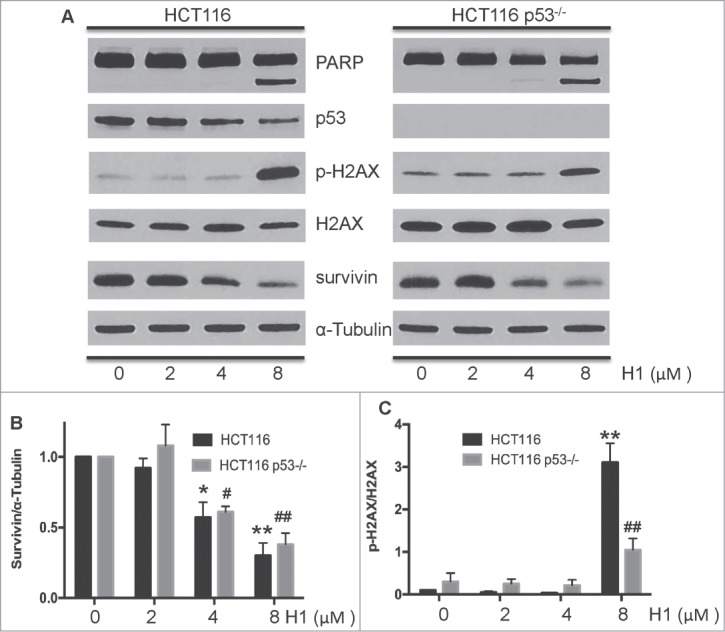
H1 caused p53-independent cytotoxic effect. Cells were treated with different concentrations of H1 for 24 h. Then, cells were rinsed and lysed containing freshly added Protease Inhibitor Cocktail. Expression level of cleaved PARP, survivin, p53, p-H2AX and total H2AX were determined by western blot analysis. A representative of 3–4 experiments is shown. *P < 0.05, **P < 0.01; #P < 0.05, ##P < 0.01 vs. each control group.
p53-independent induction of PUMA in response to cytotoxic effect of H1
PUMA encodes a BH3-only protein that is induced by p53-dependent and p53-independent manners. Then PUMA can bind to Bcl-2 and Bcl-xL, localize to the mitochondria, promote cyctochrome c release and induce apoptosis. PUMA plays a critical role in p53-dependent and p53-independent apoptosis induced by a variety of signals, and is regulated by transcription factors, not by post-translational modifications.20 After this activation, PUMA interacts with anti-apoptotic Bcl-2 family members, thus free Bax and/or Bak which are then able to initiated mitochondrial apoptosis pathway. Targeted disruption of the PUMA gene impairs p53-mediated apoptosis and tumor suppression.21 In addition, our previous studies demonstrated that H1 initiated intrinsic apoptosis pathway. Based on these findings, we determined the effect of H1 on inducing expression of PUMA. After treated HCT116 and HCT116 p53−/− cells with H1 for 24 h, the expression level of PUMA, Bax, and Bcl-2 were determined by western blot analysis. As seen in Figure 4A, H1 significantly induced PUMA expression in a concentration-dependent manner. 4 µM of H1 significantly elevated the expression level of PUMA. After this induction, PUMA triggers apoptosis in cancer cells by acting on other Bcl-2 family members such as Bax, Bcl-2, and Bcl-XL, leading to activate caspase cascades. We observed that H1 significantly elevated the ratio of Bax/Bcl-2 (Fig. 4C). HCT116 and HCT116 p53−/− cells were exposed to H1 (4 µM) at different time points (0, 4, 8, 12, 24 h). We observed that H1 induced the expression of PUMA in a time-dependent manner (Fig. 4D, E). Noticeably, H1 significantly caused PUMA induction after 8 h exposure. What is more, knockdown of PUMA by specific siRNA effectively attenuated the cytotoxic response of H1, while the control siRNA did not affect the PUMA expression and cell viability (Fig. 4F, G). Taken together, these results suggest that p53-independent induction of PUMA contribute to the cytotoxic effect of H1.
Figure 4.

(See previous page) . p53-independent induction of PUMA in response to cytotoxic effect of H1. HCT116 and HCT116 p53−/− cells were treated with various concentrations of H1 for 24 h, and cells were harvested. Induction of PUMA by H1 was determined by western blot analysis (A and B). Effect of H1 on the expression of Bax and Bcl-2 also detected (A and C). In addition, cells were exposed to H1 for different time points (0, 4, 8, 12, 24 h), after that PUMA expression was determined by western blot analysis (E and F). siRNA specific targeting to PUMA was mixed with Lipofectamine 2000 (LF2000, Invitrogen) in serum-free RPMI-1640 medium and added into plated cells. After 24 h incubation, cells were treated with H1 for additional 24 h. Then, cells were rinsed and lysed containing freshly added Protease Inhibitor Cocktail. Cell lysates were used for western blot analysis (F). HCT116 p53−/− cells were seeded into 96-well plate at density of 1000 cells per well, then transfected with 10nM siPUMA mixed with LF2000 added into the well. After 24 h transfection, H1 (4μM) was added and exposed for an additional 72 h, and then cell viability was determined by MTT assay (G). **P < 0.01; ##P < 0.01 vs. each control group.
Inhibition of AKT/FoxO3a signaling mediated p53-independent expression of PUMA
Given that PUMA can be induced by H1 in a concentration- and time-dependent manner in p53 null cells, we further determined the effects of H1 on p53-indepndent transcription factors of PUMA expression, including NF-κB, E2F1, p73 and FoxO3a. NF-κB is a key transcription factor that can induce PUMA expression. Thus, we have investigated effect of H1 on activation of NF-κB signaling. As seen in Figure 5A, H1 significantly suppressed the activation of NF-κB in a dose-dependent manner. The result suggests that induction of PUMA may not through NF-κB activation. However, inhibition of NF-κB activity may contribute to cytotoxic effect of H1. In addition, we detected the effects of H1 on expression of other p53-independent transcription factors, such as p73, E2F1 and FoxO3a. As shown in Figure 5B, H1 caused a decrease of E2F1 and phosphorylated FoxO3a, while H1 did not affect p73 expression. Recently, You et al found that PUMA can be a FoxO3a downstream target to mediate a stress response when PI3K/AKT signaling was suppressed.22 Meanwhile, our previous findings demonstrated that H1 could inhibit the activation of AKT.18 It is possible that H1 induces PUMA expression through inhibition of AKT/FoxO3a pathway. Thus, we further determined the effect of H1 (4 µM) on the activation of AKT/FoxO3a pathway compared with 10 µM LY294002 (a pan-PI3K inhibitor). We observed that H1 or LY294002 significantly induced PUMA expression and effectively decreased expression of p-AKT and p-FoxO3a, but did not affect the total AKT and FoxO3a expression (Fig. 5C). Furthermore, Knockdown FoxO3a by specific siRNA could overcome effect of H1 on induction of PUMA (Fig. 5D). We also determined the mRNA level of PUMA by Real-Time PCR, and the similar result was observed in both HCT116 p53−/− and SW480 cells (Fig. 5E). Taken together, these results suggest that inhibition of AKT/FoxO3a signaling may be responsible to H1-mediated PUMA induction.
Figure 5.

p53-independent induction of PUMA via inhibition AKT/FoxO3a signaling. HCT116 p53−/− cells were transiently transfected with a luciferase plasmid under the control of a NF-κB response element. On the following day, cells were pretreated with H1 for 2 h, and then stimulated with TNF-α (50ng/mL) for an additional 5 h. NF-κB activity was determined by the dual-luciferase assay (A). *P <0.05, **P<0.01, vs. each positive group, respectively. HCT116 p53−/− cells were treated by H1 for 6 h. E2F1, p73, p-FoxO3a and total FoxO3a were determined by western blot analysis and a representative of 3 separate experiments is shown (B). H1 (4 µM) and LY294002 (10 µM) treated HCT16 p53−/− cells for 6 h, the expression level of PUMA, p-AKT, p-FoxO3a, total-AKT and total FoxO3a were determined by western blot analysis, a representative of 3 separate experiments is shown (C). HCT116 p53−/− and SW480 cells were transiently transfected with siRNA targeted to FoxO3a for 48 h, then the cells were treated with 4 µM H1 for another 6 h. The expression level of PUMA, p-FoxO3a and total FoxO3a were detected by western blot analysis (D). In addition, the mRNA expression level of PUMA was determined by Real-Time PCR analysis, data from 3 independent experiments were used. (E). **P < 0.05, **P < 0.01, ##P < 0.01 vs. each control group.
Discussion
Tetrandrine (Tet) and its derivatives displayed significant antitumor activities against various tumor types, including human hepatocarcinoma, lung cancer, neuroblastoma, breast cancer, and so on.23-26 Our previous findings demonstrated that H1 could directly kill cancer cells with MDR phenotype, and exhibit good anti-MDR activity both in vitro and in vivo.18 In this study, we evaluated the effects of H1 on induction of PUMA and underlie its potential mechanism in p53-independent cytotoxic response. Our results showed that H1 significantly induced PUMA expression in a dose- and time-dependent manner. What is more, PUMA induction may contribute to the cytotoxic effects of H1.
PUMA (p53 unregulated modulator of apoptosis), a BH3-only Bcl-2 family member, can be induced by p53 or other apoptotic signals.11 PUMA associates with the mitochondria and induces cell death when overexpressed in various cell lines and its apoptotic activity requires an intact BH3 domain. These in vitro studies, as well as findings derived from a somatic knockout tumor cell line 27, suggest that PUMA may play an important role as an in vivo regulator of apoptosis. PUMA has been implicated in key processes of tumorgenesis. PUMA may be a potential chemotherapeutic target, as activating PUMA inhibits tumor growth by restoring apoptosis in cancer cells. PUMA can also be induced in a p53-independent manner. Under given condition of nongenotoxic stimuli, for instance: inflammatory cytokines, growth factor deprivation, and kinase inhibitors, p53-independent PUMA induction can be mediated by different transcription factors, including E2F1, p73, FoxO3a, and NF-κB.14–16 After this induction, PUMA strongly induces apoptosis in cancer cells by acting on other Bcl-2 family members such as Bax and Bcl-2, and triggers caspase cascades. In this study, we found that H1 significantly induced PUMA expression in a dose- and time-dependent manner in wild-type and p53 null cells. After that H1 elevated the ratio of Bax/Bcl-2. Knockdown PUMA by specific siRNA effectively attenuated the cytotoxic effect of H1. These findings suggest that cytotoxic effect of H1 may be associated with p53-independent induction of PUMA.
The transcription factor NF-κB is strongly implicated in a variety of hematologic and solid tumor malignancies. In cancer cells, NF-κB takes part in regulation of cell proliferation, control of apoptosis, promotion of angiogenesis, and stimulation of invasion/metastasis.28 Abnormally activation of NF-κB pathway contributes to tumor progression, chemoresistance, and radioresistance.29 Recent studies demonstrated that activation of NF-κB caused induction of PUMA. Thus, we detected effect of H1 on the activation of NF-κB. H1 significantly suppressed the activation of NF-κB in a concentration-dependent manner (Fig. 5A). It suggests that induction of PUMA may not through NF-κB pathway. However, inhibition of NF-κB activity may contribute to cytotoxic effect of H1.
Recently, some groups demonstrated that FoxO3a-mediated PUMA induction through suppression of PI3K/AKT signaling pathway. For example, multi-kinases inhibitor sunitinib can induce PUMA transcription via the AKT/FoxO3a axis in human colorectal cancer.30 Under cytokine or growth factor deprivation condition, FoxO3a directly up-regulates PUMA expression when PI3K/AKT signaling is blocked.22 Zhao et al found that PUMA expression through AKT/FoxO3a signaling in response to apoptosis of cisplatin-resistant ovarian cancer cells.31 Given that H1 could suppress the activation of AKT, it is possible that inhibition of AKT/FoxO3a signaling by H1 lead to PUMA induction. Thus, we treated HCT116 p53−/− cells with H1 and a pan-PI3K inhibitor (LY294002) and observed that H1 and LY294002 significantly blocked AKT/FoxO3a pathway and induced PUMA expression. Meanwhile, knockdown FoxO3a attenuated the effect of H1 on PUMA induction (Fig. 5D, E). These findings suggest that inhibition of AKT/FoxO3a signaling may contribute to p53-independent induction of PUMA by H1.
In summary, H1 exerts potent inhibitory effect against human colorectal cancer cells and similarly cytotoxic activity in wild-type and p53 null cells. Further studies demonstrated that H1 significantly elevated cleavage of PARP, deceased of survivin expression, activated H2AX and suppressed NF-κB activity in p53 wild-type and p53 null cells. What is more, H1 significantly induced PUMA expression in a concentration- and time-dependent manner, and then resulted in an increase of Bax/Bcl-2 ratio. Our results demonstrated that inhibition of AKT/FoxO3a signaling could be responsible to H1-mediated PUMA expression. Consequently, H1 caused an increase of early apoptosis, late apoptosis and caspase-3 activity in both wild-type and p53 null cells. Taken together, our results suggest that inhibition of AKT/FoxO3a signaling cause PUMA induction in response to p53-independent cytotoxic effect of H1 (Fig 6).
Figure 6.
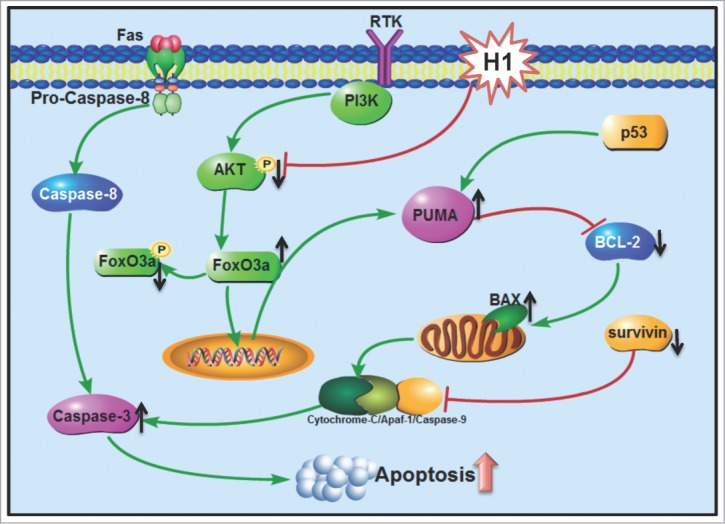
Schematic of p53-independent induction of PUMA via inhibition of AKT/FoxO3a signaling in response to cytotoxic effect of H1. H1 significantly induced PUMA expression in a concentration- and time-dependent manner, and then resulted in an increase of Bax/Bcl-2 ratio. Further studies demonstrated that inhibition of AKT/FoxO3a signaling could be responsible to H1-induced PUMA expression. Consequently, H1 caused an increase of early apoptosis, late apoptosis and caspase-3 activity in both wild-type and p53 null cells. Taken together, our results suggest that inhibition of AKT/FoxO3a signaling cause PUMA induction in response to p53-independent cytotoxic effects of H1.
Materials and Methods
Drugs and chemicals
H1, a new derivative of tetrandrine, was kindly provided by Professor Feng-Peng Wang (Department of Chemistry of Medicinal Natural Products, West College of Pharmacy, Sichuan University, Chengdu, China). It is a slight yellow powder with 99.0% purity, and freshly solved in dimethyl suifoxide (DMSO) before use. The final concentration of DMSO is less than 0.1% in all the experiments. The chemistry structure is shown in Figure 1A. Molecular formula: C27H40N2O6Br, and molecular weight is 690 Da. LY294002 (a pan-PI3K inhibitor), 1-(4, 5-dimethylthiazol-2-yl)-3, 5-diphenyformazan (MTT) and other chemicals were purchased from Sigma chemical Co. (St. Louis, MO).
Cell lines and cell culture
HCT116 p53+/+ and HCT116 p53−/− cell lines were kindly provided by Dr. Bert Vogelstein.32 HT-29, SW480 and HCT-8 cells were purchased from KeyGEN BioTECH Company. CCD 841 cells was purchased from ATCC.33 All the cells were all grown in RPMI1640 (GIBCO) medium supplemented with 10% heat-inactivated newborn calf serum, 100U/mL penicillin, and 100 µg/mL streptomycin.
Cell proliferation assay
Cell viability was determined by MTT assay. As our previous described,17 cells were seeded in 96-well plates. After an overnight incubation (37°C with 5% CO2), various concentrations of H1 was added into wells and incubated for additional 72 h. Thereafter, 100 µL of 0.5 µg/mL MTT was added to each well after withdraw the culture medium and incubated for an additional 4 h. The resulting formazan was dissolved in 150 µL DMSO after aspiration of the culture medium. Plates were placed on a plate shaker for 30 min and read immediately at 570 nm using a micro-plate reader (Bio-Rad Model 450). The IC50 was determined in duplicates and each experiment was repeated 3–5 times under identical conditions. IC50 value was defined as the drug concentration that inhibits 50% cell growth compared with the untreated controls and calculated by Graphpad Prism 6.0 software.
Clonogenic assay
HCT116 or HCT116 p53−/− cells were seeded in 6-well plates at density of 400 cells per well. On the following day, cells were exposed to various concentrations of H1 for 24 h. After that, the growth medium was then replaced with fresh medium. After 10–14 days, cell colonies were fixed with trypan blue solution (75% methanol/25% acetic acid/0.25% trypan blue) for 15 min, washed with PBS twice, and air-dried before counting colonies >50 cells.
AnnexinV-propidium iodide binding assay
Briefly, following 24 h treatment of H1, HCT116 and HCT116 p53−/− cells were resuspended with the cold binding buffer. According to the manufacture's instruction (KeyGEN Biotech Inc., China), 5 µL of Annexin-V-FITC and 5 µL of propidium iodide (PI) were added and the cells were incubated for 10 min in dark at room temperature. Flow cytometry analysis was performed using a FACS (Beckman coulter, USA). Annexin-V- and PtdIns- double negative cells were defined as live cells. Annexin-V-positive, PI-negative cells were defined as early apoptotic cells and Annexin-V- and PtdIns-double-positive cells were defined as late arising apoptotic cells.
Caspase -3 colorimetric assay
Effect of H1 on caspase-3 activity was determined by caspase-3 colorimetric assay Kit (KeyGEN Biotech Inc., China). Briefly, after treatment of H1 for 24 h, cells were lysed. Protein concentration was determined by Bradford assay. The lysate containing 100 μg of protein in 90 μL assay buffer were mixed with 10 μL caspase-3 specific synthetic fluorescent substrates. After incubation at 37 °C for 4 h, yellowish color from the pNA released from the substrates by the action of active caspase-3 was determined at 405 nm on Fluorescence Reader (Bio-TEK INSTRUMENTS Inc.., USA).
NF-κB activity assay
HCT116 or HCT116 p53−/− cells were seeded in 24-well plates at a density of 1.2×105 cells/well. On the following day, pGL3-Luc-NF-κB or pGL3-Luc DNA (0.5 µg) was co-transfected with 0.1 μg of renilla luciferase plasmid DNA into cells using Lipofectamine 2000 according to the manufacturer's instruction (Invitrogen). The renilla luciferase plasmid DNA was used as an internal control for transfection efficiency. After 6 h, transfection medium was changed, and on the following day, cells were incubated for 2 h with various concentrations of H1 followed by addition of 50 ng/mL TNF-α for an additional 5 h. Firefly luciferase values were normalized with renilla activity and the reporter assays were performed in triplicate.34
RNA interference
HCT116 or HCT116 p53−/− cells were plated at a density of 2.0×105 cells/well. On the following day, specific siRNA targeting to PUMA (UCUCAUCAUGGGACUCCUG, Dharmacon; Chicago, IL) or FoxO3a (ACUCCGGGUCCAGCUCCAC, Dharmacon; Chicago, IL) was mixed with Lipofectamine 2000 in serum-free RPMI-1640 medium and added to the plated cells. On the following day, cells were treated with H1 (4 µM) for another 24 h. After that, cells were rinsed with PBS and scraped in cell lysis buffer containing freshly added Protease Inhibitor Cocktail (Sigma; St. Louis, MO). Lysates were centrifuged at 12,000 rpm for 20 min at 4°C to remove debris. Cell lysates were stored at -80°C for western blots analysis. For the proliferation assay, cells were transfected with 10nM siPUMA for 24 h, and then added H1 (4 µM) for another 72 h. Cell viability was determined by MTT assay.
RNA extracts and Real-Time PCR analysis
According to the manufacturer's instructions, total RNA was extracted using the TRIzol RNA kit (Invitrogen, CA, USA). The first strand cDNA (cDNA) was prepared from total RNA using PrimeScript™ RT reagent kit (Takara Bio, Inc., Japan). PCR was performed in triplicate using the SsoFastTM Probes Supermix (Bio-Rad) in a final reaction volume of 20 µL with gene-specific primer/probe sets, and a standard thermal cycling procedure (40 cycles) on a Bio-Rad CFX96TM Real-Time PCR System. PUMA and β-actin (probe ID: Hs00248075_m1 and Mm00607939_s1) mRNA levels were assessed using TaqMan Gene Expression Real-Time PCR assays. Result was expressed as the threshold cycle (Ct). The relative quantification of the target transcripts was determined by the comparative Ct method (ΔΔCt) according to the manufacturer's protocol. The 2−ΔΔCt method was used to analyze the relative changes in gene expression. Control experiments were conducted without reverse transcription to confirm that the total RNA was not contaminated with genomic DNA. β-actin was used as an internal control gene in order to normalize.
Western blot analysis
Cells were harvested and rinsed with PBS, and lyzed in denaturing lysis buffer (Applygen Technologies Inc., China) for 30 min on ice, centrifuged 12000 g for 20 min at 4°C. Protein concentrations were determined by Bradford assay. Equal quantities (30 µg of protein) of cell extract were resolved by 10% SDS-PAGE, the resolved protein were electrophoretically transferred to PVDF membrane, and blocked with 5% fat-free dry milk in TBST for 1 h at room temperature. The membrane was immunoblotted with anti-PARP, anti-p-H2AX, anti-H2AX, anti-survivin, anti-p73, anti-PUMA, anti-p-FoxO3a, anti-FoxO3a, anti-E2F1 (Cell Signaling Technology, USA), anti-p53, anti-Bax, anti-Bcl-2, anti-α-tubulin (Santa Cruz, USA) antibodies in 5% milk TBST, at 4°C overnight. The membranes were washed 3 times, incubated with HRP-conjugated secondary antibodies for 1 h at room temperature, and washed extensively before detection. The membranes were subsequently developed using ECL (FujiFilm, Japan) reagent (Applygen Technologies Inc., China) and exposed to film according to the manufacturer's protocol.
Statistical analysis
Data were shown as mean ± SD. Statistical analysis of the data was performed using the one-way ANOVA by SPSS software. P < 0.05 was considered statistically significant.
Funding Statement
This work was supported by Grants (No. 81202556) from China National Natural Sciences Foundation and Liaoning Provincial Natural Science Foundation of China (No. 2013022015).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57–70; PMID:10647931; http://dx.doi.org/ 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- 2.Balint EE, Vousden KH. Activation and activities of the p53 tumour suppressor protein. Br J Cancer 2001; 85:1813–23; PMID:11747320; http://dx.doi.org/ 10.1054/bjoc.2001.2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307–10; PMID:11099028; http://dx.doi.org/ 10.1038/35042675 [DOI] [PubMed] [Google Scholar]
- 4.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995; 378:206–8; PMID:7477327; http://dx.doi.org/ 10.1038/378206a0 [DOI] [PubMed] [Google Scholar]
- 5.Matijasevic Z, Steinman HA, Hoover K, Jones SN. MdmX promotes bipolar mitosis to suppress transformation and tumorigenesis in p53-deficient cells and mice. Mol Cell Biol 2008; 28:1265–73; PMID:18039860; http://dx.doi.org/ 10.1128/MCB.01108-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuler M, Green DR. Mechanisms of p53-dependent apoptosis. Biochem Soc Trans 2001; 29:684–8; PMID:11709054; http://dx.doi.org/ 10.1042/BST0290684 [DOI] [PubMed] [Google Scholar]
- 7.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer 2002; 2:594–604; PMID:12154352; http://dx.doi.org/ 10.1038/nrc864 [DOI] [PubMed] [Google Scholar]
- 8.Gottlieb TM, Leal JFM, Seger R, Taya Y, Oren M. Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene 2002; 21:1299–303; PMID:11850850; http://dx.doi.org/ 10.1038/sj.onc.1205181 [DOI] [PubMed] [Google Scholar]
- 9.Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov 2014; 13:217–36; PMID:24577402; http://dx.doi.org/ 10.1038/nrd4288 [DOI] [PubMed] [Google Scholar]
- 10.Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013; 493:542–6; PMID:23242140; http://dx.doi.org/ 10.1038/nature11743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene 2008; 27 Suppl 1:S71–83; http://dx.doi.org/ 10.1038/onc.2009.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7:683–94; PMID:11463392; http://dx.doi.org/ 10.1016/S1097-2765(01)00214-3 [DOI] [PubMed] [Google Scholar]
- 13.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001; 7:673–82; PMID:11463391; http://dx.doi.org/ 10.1016/S1097-2765(01)00213-1 [DOI] [PubMed] [Google Scholar]
- 14.Ming L, Sakaida T, Yue W, Jha A, Zhang L, Yu J. Sp1 and p73 activate PUMA following serum starvation. Carcinogenesis 2008; 29:1878–84; PMID:18579560; http://dx.doi.org/ 10.1093/carcin/bgn150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudgeon C, Wang P, Sun X, Peng R, Sun Q, Yu J, Zhang L. PUMA induction by FoxO3a mediates the anticancer activities of the broad-range kinase inhibitor UCN-01. Mol Cancer Ther 2010; 9:2893–902; PMID:20978166; http://dx.doi.org/ 10.1158/1535-7163.MCT-10-0635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang P, Qiu W, Dudgeon C, Liu H, Huang C, Zambetti GP, Yu J, Zhang L. PUMA is directly activated by NF-kappaB and contributes to TNF-alpha-induced apoptosis. Cell Death Differ 2009; 16:1192–202; PMID:19444283; http://dx.doi.org/ 10.1038/cdd.2009.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei N, Sun H, Wang FP, Liu GT. H1, a novel derivative of tetrandrine reverse P-glycoprotein-mediated multidrug resistance by inhibiting transport function and expression of P-glycoprotein. Cancer Chemoth Pharm 2011; 67:1017–25; http://dx.doi.org/ 10.1007/s00280-010-1397-7 [DOI] [PubMed] [Google Scholar]
- 18.Wei N, Liu GT, Chen XG, Liu Q, Wang FP, Sun H. H1, a derivative of Tetrandrine, exerts anti-MDR activity by initiating intrinsic apoptosis pathway and inhibiting the activation of Erk1/2 and Akt1/2. Biochem Pharmacol 2011; 82:1593–603; PMID:21864508; http://dx.doi.org/23732469 10.1016/j.bcp.2011.08.012 [DOI] [PubMed] [Google Scholar]
- 19.Parrish AB, Freel CD, Kornbluth S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol 2013; 5; pii: a008672; PMID:23732469; http://dx.doi.org/ 10.1101/cshperspect.a008672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeffers JR, Parganas E, Lee Y, Yang CY, Wang JL, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al.. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003; 4:321–8; PMID:14585359; http://dx.doi.org/ 10.1016/S1535-6108(03)00244-7 [DOI] [PubMed] [Google Scholar]
- 21.Vela L, Gonzalo O, Naval J, Marzo I. Direct Interaction of Bax and Bak Proteins with Bcl-2 Homology Domain 3 (BH3)-only Proteins in Living Cells Revealed by Fluorescence Complementation. J Biol Chem 2013; 288:4935–46; PMID:23283967; http://dx.doi.org/ 10.1074/jbc.M112.422204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, Villunger A, Mak TW. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med 2006; 203:1657–63; PMID:16801400; http://dx.doi.org/ 10.1084/jem.20060353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Chen JC, Tseng SH. Effects of tetrandrine plus radiation on neuroblastoma cells. Anticancer Res 2009; 29:3163–71; PMID:19661330 [PubMed] [Google Scholar]
- 24.Fu LW, Zhang YM, Liang YJ, Yang XP, Pan QC. The multidrug resistance of tumour cells was reversed by tetrandrine in vitro and in xenografts derived from human breast adenocarcinoma MCF-7/adr cells. Eur J Cancer 2002; 38:418–26; PMID:11818209; http://dx.doi.org/ 10.1016/S0959-8049(01)00356-2 [DOI] [PubMed] [Google Scholar]
- 25.Liu W, Zhang J, Ying C, Wang Q, Yan C, Jingyue Y, Zhaocai Y, Yan X, Heng-Jun S, Lin J. Tetrandrine combined with gemcitabine and Cisplatin for patients with advanced non-small cell lung cancer improve efficacy. Int J Biomed Sci 2012; 8:28–35; PMID:23675254 [PMC free article] [PubMed] [Google Scholar]
- 26.Oh SH, Lee BH. Induction of apoptosis in human hepatoblastoma cells by tetrandrine via caspase-dependent Bid cleavage and cytochrome c release. Biochem Pharmacol 2003; 66:725–31; PMID:12948852; http://dx.doi.org/ 10.1016/S0006-2952(03)00397-6 [DOI] [PubMed] [Google Scholar]
- 27.Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci U S A 2003; 100:1931–6; PMID:12574499; http://dx.doi.org/ 10.1073/pnas.2627984100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erstad DJ, Cusack JC Jr. Targeting the NF-kappaB pathway in cancer therapy. Surg Oncol Clin N Am 2013; 22:705–46; PMID:24012396; http://dx.doi.org/ 10.1016/j.soc.2013.06.011 [DOI] [PubMed] [Google Scholar]
- 29.Li F, Sethi G. Targeting transcription factor NF-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim Biophys Acta 2010; 1805:167–80; PMID:20079806 [DOI] [PubMed] [Google Scholar]
- 30.Sun J, Sun Q, Brown MF, Dudgeon C, Chandler J, Xu X, Shu Y, Zhang L, Yu J. The multi-targeted kinase inhibitor sunitinib induces apoptosis in colon cancer cells via PUMA. PloS One 2012; 7:e43158; PMID:22912816; http://dx.doi.org/ 10.1371/journal.pone.0043158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Z, Wang J, Tang J, Liu X, Zhong Q, Wang F, Hu W, Yuan Z, Nie C, Wei Y. JNK- and Akt-mediated Puma expression in the apoptosis of cisplatin-resistant ovarian cancer cells. Biochem J 2012; 444:291–301; PMID:22394200; http://dx.doi.org/ 10.1042/BJ20111855 [DOI] [PubMed] [Google Scholar]
- 32.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497–501; PMID:9822382; http://dx.doi.org/ 10.1126/science.282.5393.1497 [DOI] [PubMed] [Google Scholar]
- 33.Wei N, Chu E, Wu SY, Wipf P, Schmitz JC. The cytotoxic effects of regorafenib in combination with protein kinase D inhibition in human colorectal cancer cells. Oncotarget 2015; 6:4745-56.PMID: 2554476524634417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei N, Chu E, Wipf P, Schmitz JC. Protein kinase d as a potential chemotherapeutic target for colorectal cancer. Mol Cancer Ther 2014; 13:1130–41; PMID:24634417; http://dx.doi.org/ 10.1158/1535-7163.MCT-13-0880 [DOI] [PMC free article] [PubMed] [Google Scholar]