

Abstract
Vestibular schwannoma (VS), the fourth most common intracranial tumor, arises from the Schwann cells of the vestibular nerve. Although several pathways have been independently implicated in VS pathobiology, interactions among these pathways have not been explored in depth. We have investigated the potential cross-talk between hepatocyte growth factor (HGF) and vascular endothelial growth factor-A (VEGF-A) in human VS, an interaction that has been described in other physiological and pathological cell types. We affirmed previous findings that VEGF-A signaling is aberrantly upregulated in VS, and established that expression of HGF and its receptor cMET is also significantly higher in sporadic VS than in healthy nerves. In primary human VS and Schwann cell cultures, we found that VEGF-A and HGF signaling pathways modulate each other. siRNAs targeting cMET decreased both cMET and VEGF-A protein levels, and siRNAs targeting VEGF-A reduced cMET expression. Additionally, siRNA-mediated knockdown of VEGF-A or cMET and pharmacologic inhibition of cMET decreased cellular proliferation in primary human VS cultures. Our data suggest cross-talk between these 2 prominent pathways in VS and highlight the HGF/cMET pathway as an additional important therapeutic target in VS.
Keywords: hepatocyte growth factor, Schwann cells, siRNA, cross-talk, vascular endothelial growth factor, vestibular schwannoma
Abbreviations
- BrdU
5-Bromo-2'-Deoxyuridine
- cMET
MNNG HOS transforming gene, hepatocyte growth factor receptor
- DMSO
Dimethyl sulfoxide
- GAN
Great auricular nerve
- HCl
Hydrochloric acid
- HGF
Hepatocyte growth factor
- HGF
Gene encoding HGF protein
- HRP
Horse-radish peroxidase
- KDR
Gene encoding vascular endothelial growth factor receptor 2
- MET
Gene encoding cMET protein
- mRNA
Messenger ribonucleic acid
- NF2
Neurofibromatosis type 2
- PBS
Phosphate buffered saline
- S100
Schwann cell/schwannoma cell marker
- SD
Standard deviation
- SEM
Standard error of mean
- siRNA
Small interfering ribonucleic acid
- VEGFA
Gene encoding VEGF-A protein
- VEGF-A
Vascular endothelial growth factor-A
- VEGFR2
Vascular endothelial growth factor receptor 2
- VS
Vestibular schwannoma
Introduction
Vestibular schwannomas (VSs), benign tumors arising from Schwann cells of vestibular nerves, are the fourth most common intracranial tumors.1 Although several pathways have been implicated in VS pathobiology, interactions among these pathways have been scarcely established. Levels of vascular endothelial growth factor-A (VEGF-A), a prominent mitogenic and angiogenic factor, and its receptor tyrosine kinase VEGFR1 correlate with VS growth rate.2 Administration of bevacizumab, a humanized VEGF-A antibody, to patients with Neurofibromatosis type 2 (NF2)-associated VS led to a volumetric decrease in 55% of the VSs.3,4 As investigators continue to elucidate all of the factors that interact with VEGF-A, it is important to explore the potential of VEGF-A to regulate and be regulated by other molecules that could be driving VS growth, providing us with new therapeutic targets and the ability to overcome potential drug resistance inevitable with monotherapies.
HGF, a potent angiogenic factor, and its receptor tyrosine kinase cMET have been implicated in several other cancers5 in addition to VS,6 though they have previously not been explored as therapeutic targets against VS. We investigated cross-talk between VEGF-A and HGF, an interaction that has been established in a few other cell types. The HGF/cMET signaling pathway has been shown to interact closely with the VEGF-A signaling pathway in other physiological signaling, such as in endothelial cells,7 and in pathological signaling, such as in adenocarcinoma8 and glioma cells.9 Specifically, previous research in endothelial cells shows that VEGF-A and HGF synergistically activate mitogen-activated protein kinases (MAPKs), stimulation with VEGF-A increases cMET levels, and stimulation with HGF elevates VEGFR2 levels.7 Exploring this cross-talk between VEGF-A and cMET in VS cells, compared to non-neoplastic Schwann cell controls, can provide insight into the mechanism for VEGF-A and HGF signaling-mediated VS growth. Further, with a focus on devising therapies aimed toward reducing functional HGF or VEGF-A signaling, we silenced cMET or VEGF-A and noted the resultant effect on the other factor. These experiments contrast previous work focused on establishing cross-talk through increased signaling of these pathways.
We found that HGF signaling, along with VEGF-A signaling, is significantly upregulated in VS, as measured through mRNA and secreted protein levels. In both primary human VS and SC cultures, we found that VEGF-A and cMET signaling pathways modulate each other. In VS cultures, siRNAs targeting cMET decreased VEGF-A and VEGFR2 protein levels, and targeting VEGF-A reduced cMET expression. Additionally, siRNA-mediated knockdown of VEGF-A or cMET, and pharmacologic inhibition of cMET led to decreased proliferation in primary VS cultures. In this study, by investigating the cross-talk between VEGF-A and cMET pathways in VS, we highlight cMET as an additional therapeutic target.
Materials and Methods
Specimen collection
Freshly-harvested human specimens of sporadic VSs and GANs were collected from indicated surgeries. The study protocols were approved by Human Studies Committee of Massachusetts General Hospital (Protocol No. 2004- P2297/2, PI: K.M.S) and the Massachusetts Eye and Ear Infirmary (Protocol No. 05–02–009X, PtdIns: K.M.S). Written informed consent was received from all subjects prior to inclusion in the study. Immediately after extraction, specimens were placed in saline solution and transported to the laboratory on ice. The specimen was rinsed with saline and divided for protein, RNA or culture work. Procedures were in accordance with the Helsinki Declaration of 1975.
Real time-quantitative polymerase chain reaction (RT-qPCR)
Expression of VEGF and HGF pathway was measured in VSs versus GANs (healthy nerve controls). Specifically, human VS or GAN tissue was placed in RNAlater (Qiagen, #76106) and stored at −20°C until RNA extraction. Total RNA was extracted using RNeasy Mini Kit (Qiagen, #74104) according to the manufacturer's protocol. Quantification and quality assessment of the RNA were performed using Agilent 2100 Bioanalyzer (Agilent Technologies) or NanoDrop (ThermoScientific). All samples yielded undegraded RNA as shown by electropherograms or through 260/280 nm absorbance ratios. Isolated RNA was stored at −80°C. The RNA was reverse-transcribed to cDNA with TaqMan Reverse Transcription Reagent Kit (Applied Biosystems, #4304134) following the manufacturer's protocol. The cDNA was stored at either 4°C for short term or −20°C for long term. qPCR was performed with TaqMan primers and 6-Carboxyfluorescein (6-FAM) linked fluorescent probes (Applied Biosystems) for VEGFA (#Hs00900055_m1), HGF (#Hs00300159_m1), KDR (#Hs00911700_m1) and MET (#Hs01565582_g1). The reference gene was rRNA 18s (#Hs9999901_s1). Statistical significance was determined using the 2-tailed t-test with a P < 0 .05 considered significant after a Benjamini-Hochberg correction for multiple hypotheses.
Protein extraction and immunoblotting
Translation and activation of the VEGF and HGF pathway components was investigated through western blot analysis. Total protein was extracted from freshly-harvested specimens of VS and GAN in radioimmunoprecipitation assay (RIPA) buffer supplemented with protease and phosphatase inhibitors on ice. The lysate was isolated by centrifugation at 10,000 RPM for 10 minutes at 4°C. The protein was stored at -80°C and was subjected to a maximum of 2 freeze/thaw cycles. Samples were loaded at a total protein concentration of 7.5–15 μg per lane, separated on a 4–20% Tris-Glycine Gel (Life Technologies, #EC6025BOX) and transferred onto Immobilon-P PVDF Membrane (Millipore, #IPVH00010). The membrane was blocked for an hour with 5% Bovine Serum Albumin/PBST (w/v) solution and probed with Santa Cruz antibodies against VEGF (#sc-152) and cMET (#sc-161) and Cell Signaling antibodies against phosphorylated (P-)-cMET (#3077) and VEGFR2 (#2479). Antibody against β-actin (Cell Signaling, #4970) served as an internal control. Membranes were visualized with an enhanced chemiluminescence detection system ChemiDoc Plus (BioRad Laboratories). Band densities were quantified using ImageJ and were normalized to β-actin for a given lane. Statistical significance of the data was determined using the 2-tailed t-test with a p < 0 .05 considered significant.
Cytokine array
Detailed methods have been published previously.14 Briefly, VS and GAN secretions were collected by incubating freshly resected and washed tissue in PBS for 1 hour at 37°C with 5% CO2 levels. Human cytokine array membranes (RayBiotech, Inc.., custom order) were probed with 21 VS secretion samples, 7 GAN samples and 1 blank sterile PBS. Manufacturer's protocol was followed in conducting the experiment and data analysis. Samples were dialyzed twice with PBS. The membranes were exposed to the blocking buffer at room temperature for 1 hour, incubated with sample at 4ºC overnight, washed with wash buffer I and II at room temperature, incubated with biotin-conjugated antibodies at 4°C overnight, washed and incubated with HRP-conjugated streptavidin at room temperature for 1 h. The membranes were then exposed in ChemiDoc (BioRad Laboratories). The relative expression levels of HGF and VEGF were compared after densitometry analysis using Quantity One (BioRad Laboratories). Statistical significance was determined using ANOVA test with α set to 0.05.
Primary human Schwann cell and vestibular schwannoma cell culture
Detailed methods have been published previously.15 Briefly, using sterile technique under the hood, freshly harvested VS or GAN tissue was rinsed in sterile PBS trice and cut into 1 mm-sized pieces in Dulbecco's modified eagle's medium with Ham's F12 nutrient mixture (DMEM/F12), 10% fetal bovine serum, 1% Penicillin/Streptomycin (Pen/Strep) and 1% GlutaMAX (all purchased from Life Technologies). To obtain a more pure SC population, the epineurium was removed from the nerve tissue by tugging and removing the outer layers under a dissecting microscope. The specimen pieces in the media were centrifuged at 3000 g at 8ºC for 3 minutes. The media was aspirated and the tissue pellet was incubated in new media containing 5% Collagenase (Sigma-Aldrich, #C1639) and 0.5% Hyaluronidase (Sigma-Aldrich, #H3506) for 18–24 hours at 37ºC. The cells were plated in Poly-L-Ornithine and Laminin pre-coated 12-well culture dishes with 5 mm glass slides (BD Biosciences, #354087) in DMEM/F12 media with 10% FBS, 1% Pen/Strep and 1% glutamine. The cell cultures were maintained for 3 to 4 weeks and media was changed every 3 d
siRNA and pharmacologic treatment
To understand cross-talk between HGF and VEGF-A pathway, cultured VS cells were incubated with Ambion siRNAs targeting VEGF (#s461), MET (#s8700) or KDR (#s7824). To understand whether HGF signaling contributed to VS proliferation, cultured VS cells were treated with MET inhibitor SU11274 (Sigma-Aldrich, #S9820). Seventy-2 hours after siRNA treatment or 12 hours after treatment with 2 μM SU11274, cells were incubated with 10 μg/mL 5-Bromo-2´-Deoxyuridine (BrdU, Life Technologies, #B23151) for 20 hours. After treatment, cells were fixed with 4% paraformaldehyde. After cell membrane permeabilization by incubation in 1% Triton-X, cells were incubated in 1N HCl for 25 mins, blocked for 1 hour in normal horse serum (NHS, Sigma-Aldrich) and incubated with primary antibodies against BrdU (AbD Serotec, #OBT0030G) and S100 (Dako, #Z0311) diluted in NHS overnight at 4°C. After PBS washes, secondary antibodies (Alexa Fluor 555 anti-rat and Alexa Fluor 488 anti-rabbit, Life Technologies) were applied for 1 hour at room temperature and the cells’ nuclei were counterstained with Hoechst stain (Life Technologies, #H1399). The coverslips were mounted onto glass slides for fluorescence microscopy. Cells were counted by an investigator (D.S.R or SD) blinded to the treatment conditions. Nuclei were counted in ≥3 fields and the cell proliferation rate was calculated as BrdU positive nuclei over the total number of nuclei. The inhibitors’ effect on proliferation was normalized to the corresponding tumor's proliferation in culture receiving no treatment (DMSO only). A paired 2-tailed t-test was performed to compare the control and treatment groups with P < 0 .05 considered statistically significant.
Results
Increased expression and activation of cMET and VEGF-A signaling in VS
To investigate aberrant expression of HGF and VEGF-A signaling pathways in VSs, gene expression differences in HGF, VEGF-A, cMET, and VEGFR2 (gene KDR) were determined in human VSs in comparison to healthy nerves. Tumor specimens (n = 8 different specimens) had significantly elevated expression of the HGF and VEGF signaling pathways compared to healthy nerves (n = 7 different specimens), being 286.7–fold (p = 0.003) and 15.0–fold (p = 0.011) higher with a standard error range of 116.6 -705.4 and 7.1–31.6 for VEGFA and KDR, respectively (Fig. 1A). Healthy nerves had a range of 0.3–3.5 and 0.6–1.6 for VEGFA and KDR, respectively. HGF and its receptor MET were also significantly upregulated, being 15.4–fold (p = 0.043) and 632.0 –fold (p = 0.001) with a range of 5.4–43.7 and 286.0–1391.9, respectively. Healthy nerves had a standard error range of 0.6–1.7 and 0.3–3.4 for HGF and MET, respectively (Fig. 1A).
Figure 1.
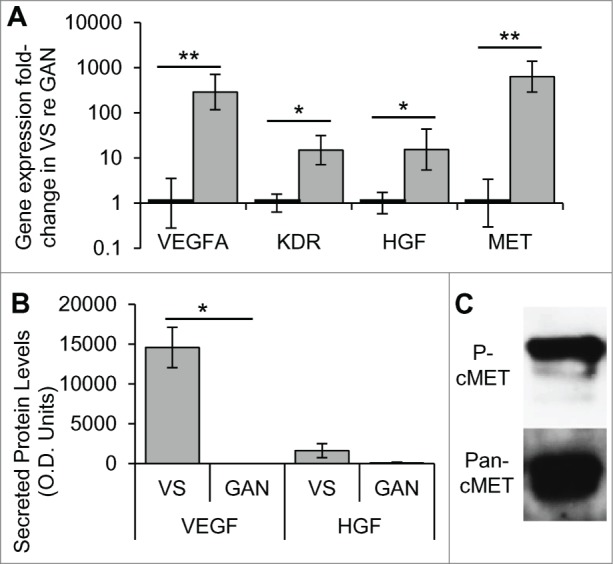
HGF and VEGF-A pathways are aberrantly expressed and activated in VS. (A) Gene expression of VEGFA and its receptor KDR, and HGF and its receptor MET, in human VS (n = 8 different tumors) normalized to great auricular nerves (GAN, n = 7 different nerves) as measured through qPCR. *P < 0 .05, **P < 0 .01. Error bars represent range. (B) VEGF and HGF protein levels in secretions from human VS (n = 21) and GAN (n = 8). *P < 0 .05. (C) Representative image of cMET expression and phosphorylation (Try 1234, P-cMet) in VS, as detected by protein gel blot (n = 5). re = in comparison to.
We sought to determine if VEGF-A or HGF secretion levels were higher in VSs than in healthy nerves. Measuring HGF and VEGF-A levels using a cytokine array, we found VEGF-A to be selectively secreted from VSs (n = 21 different tumors), with an average optical density (O.D.) value of 14,558 ± standard error of mean (SEM) of 2,527, and no detectable VEGF-A in great auricular nerve (GAN) secretions (n = 7 different nerves) (Fig. 1B). This differential level of secretion was highly significant (p = 0.003). HGF tended to be secreted at higher levels in VSs, with an O.D. value of 1,615 ± 885, than in GANs, which had an O.D. value of 87 ± 87, although the trend did not meet significance (p = 0.334, Fig. 1B).
To understand if cMET is activated in sporadic VSs, we investigated the phosphorylation status of c-MET at the Tyrosine 1234 site. Five independent sporadic VS tumors consistently demonstrated phosphorylation of cMET (Fig. 1C).
Cross-talk between cMET and VEGF-A signaling pathways in primary SCs
Previous studies in endothelial cells identified possible cell signaling cross-talk between the VEGF-A and HGF receptor signaling pathways.7,9 We have investigated this cross-talk in normal SCs with siRNAs targeting the VEGF-A and HGF signaling pathways. Comparing protein expression in SCs from the same culture treated with vehicle only, siRNAs were capable of knocking down VEGF-A and cMET substantially, to 32 ± 25% (n = 4 different cultures, p = 0.04) and 54 ± 22% (n = 5 different cultures, p = 0.003) of vehicle-only treated cells, respectively (Fig. 2A and B). Importantly, MET knockdown decreased VEGF-A levels in normal SCs significantly, reducing the protein levels to 43 ± 34% of vehicle only-treated cells (p = 0.02, n = 4 different cultures, Fig. 2A and B). MET knockdown also decreased VEGFR2 levels, reducing the protein levels to 50 ± 24% (n = 3 different cultures, Fig. 2A and B) of vehicle only-treated cells, although this trend did not meet significance (p = 0.07).
Figure 2.
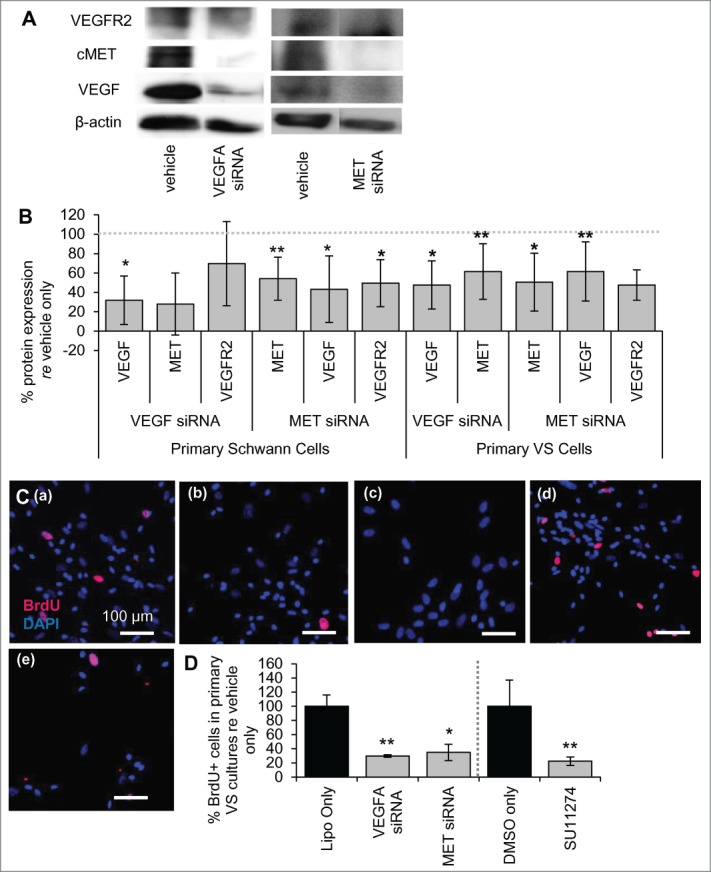
VEGF-A and cMET pathways interact at the molecular level. (A) Representative image of western blot showing expression of VEGFR2, cMET and VEGF-A for vehicle only and for siRNAs targeting VEGFA and MET genes in primary human SCs. (B) Protein expression of VEGF-A, cMET and VEGFR2 after VEGFA and MET siRNA treatment of cultured human SCs (n = 2–4 different cultures) and VS cells (n = 5 different cultures) quantified through protein gel blot analysis. All levels are normalized to vehicle only protein expression, being 100% (dashed line). *P < 0 .05 **P < 0 .01. Error bars represent SEM. (C) Representative pictures of primary human VS cells treated with (a) vehicle only, (b) VEGFA siRNA, (c) MET siRNA, (d) DMSO only or (e) SU11274. BrdU in nuclei (red) marks proliferating cells, nuclei are labeled with DAPI. Scale bar = 100 μm for all images. (D) Quantification of proliferation changes after siRNA (n = 5 different cultures) and after SU11274 (n = 4 different cultures) treatment of primary VS cells normalized to proliferation in control non-treated cells. *P < 0 .05, **P < 0 .01. re = in comparison to. Error bars represent SEM.
VEGFA knockdown did not lead to a significant decrease in cMET levels, at 28 ± 32% of vehicle only controls (n = 2 different cultures, p = 0.19), or of VEGFR2 levels, at 70 ± 43% with VEGFA siRNA treatment of vehicle only controls (n = 3 different cultures, p = 0.35) (Fig. 2A and B). Although VEGFR2 level changes were not consistent between experiments with VEGFA siRNA leading to a large variability, c-MET levels were consistently decreased in the 2 experiments. More experiments with VEGFA siRNA in SCs will be helpful in establishing the protein expression changes with confidence.
Cross-talk between cMET and VEGF-A signaling pathways in primary VS cells
Knockdown of MET also led to decreased VEGF-A and VEGFR2 in primary VS cultures as noted in SCs. Comparing protein expression in VS cells from the same culture treated with vehicle only, siRNAs were capable of knocking down VEGFA and MET to a significant extent, with 48 ± 25% (n = 5 different cultures, p = 0.009) and 51 ± 30% (n = 6, p = 0.005) knockdown achieved, respectively (Fig. 2B). MET knockdown led to a decrease in VEGF-A and VEGFR2 levels, with reduction to 62 ± 31% (p = 0.03) and 48 ± 16% respectively (n = 5 different cultures, p = 0.007, Fig. 2B). VEGFA knockdown did cause a significant decrease in cMET expression in VS cells, reducing cMET levels to 62 ± 29% of the controls (p = 0.04, Fig. 2B).
Decreased VS cell proliferation with molecular VEGFA and MET or pharmacologic cMET inhibition
To understand the implications of silencing VEGFA and MET in VS cells, cell proliferation studies were performed in primary VS cells. Basal cell proliferation for VS cells treated with vehicle only was 14% on average, with a range of 2.2 to 7.0% (Fig. 2C (a)). Silencing VEGFA or MET reduced VS cell proliferation to 29.7 ± 1.8% (SEM) (n = 5 different cultures, p < 0 .01, Fig. 2C (b), 1D) and 34.8 ± 11.4% (n = 5 different cultures, p = 0.02, Fig. 2C (c), 2D) of control. Similarly, specific inhibition of cMET signaling with the 2 μM cMET inhibitor SU11274 reduced proliferation to 22.4 ± 11.7% (Fig. 2C (e), 2D) of VS cells treated with DMSO only (9.1 ± 3.4%) (n = 4 different cultures, p < 0 .01, Fig. 2C (d)).
Discussion
To gain a deeper understanding of the VS pathobiological interactome, we have focused on investigating the relationship between the HGF and VEGF-A signaling pathways. Our work establishes abnormal upregulation and activation of the HGF pathway in VS pathobiology. We found significantly higher levels of HGF and cMET being transcribed in comparison to healthy nerves, activated phosphorylated cMET in all tumors tested, and relatively higher levels of secreted HGF. Our findings further expand on previous work showing that HGF and cMET are present in VSs based on immunohistochemical staining of human VS surgical specimens.6 Importantly, we found that siRNA-mediated MET silencing or pharmacological inhibition of cMET led to significantly decreased primary VS cell proliferation, demonstrating cMET as a novel therapeutic target. Targeting of cMET and HGF may provide an alternate or adjuvant therapy to other pharmacological approaches being investigated to modulate VS growth.
In this work, we also affirmed aberrant VEGF-A signaling as VEGF-A and VEGFR2 were expressed at significantly higher levels in VS and VEGF-A was secreted at significant higher, albeit variable, levels in VS in comparison to healthy nerves. We also confirmed VEGF-A's role in VS growth as siRNA-mediated VEGFA silencing inhibited primary VS cell proliferation. This observation is in agreement with previous work showing the therapeutic efficacy of bevacizumab in VS patients with neurofibromatosis type 2 (NF2) and in mice cranially xenografted with the NF2 cell line HEI-193.10 We also show efficacy of anti-VEGF-A therapy against sporadic VS cells.
Along with exploring VEGF-A and cMET's role in VS separately, we explored cross-talk between these 2 pathways. Previous exploration of the cross-talk in other cell types7,9 focused on changes due to the elevation of either HGF or VEGF-A in cultures. With a focus on devising therapies and therefore reducing functional HGF or VEGF-A signaling, we designed our study to silence cMET or VEGF-A and note the effect on the other factor. VEGF-A and cMET have a direct regulatory relationship, rather than inverse, in VS and SCs. The trends discovered in VS and SC cultures are in line with previous studies in other cell types. Further, since decreases in VEGFR2 after VEGF siRNA treatment were not observed in SCs, it suggests that the siRNA targets its intended target specifically.
The stage of VEGF and cMET cross-regulation seems to be at the transcriptional level, in which VEGF-A and HGF signaling regulates downstream gene expression ultimately leading to modulation of the other pathway. Sulpice et al. demonstrated that, both VEGF-A and HGF slightly increased the MET and KDR mRNA levels, respectively, in endothelial cells.7 Moriyama et al. also found a similar pattern in which treatment of the glioma cells with HGF lead to increased secretion of VEGF proteins accompanying increased transcription of VEGF mRNA in a dose-dependent fashion.9 This relationship could also be potentially investigated in SC and VS cultures by measuring VEGFR2 and MET mRNA levels after HGF and VEGFA siRNA treatment.
We also found that MET siRNA led to a decrease in VEGFR2 levels in both primary SCs and VS cells, a correlation that has not been published for any cell type thus far. This result is intriguing because through its regulation of protein kinase B, p38 and other kinases downstream, the HGF/cMET signaling pathway may modulate several transcription factors that could in turn regulate many genes including VEGFA and KDR.7,9 Our findings could explain, at least partially, how neoplastic cells sustain growth and survival in an autocrine manner.
The cross-talk between VEGF and HGF pathways in VS is fascinating as it suggests that decreasing VEGF-A levels through pharmacological means, such as bevacizumab, could modulate cMET levels. VEGF-A has been known to cross-talk with several other biological molecules, such as fibroblast growth factor 2.11 VEGF-A inhibition may also affect several of these pathways in VS cells, with the cumulative effect of relatively high therapeutic efficacy of anti-VEGF therapy against VS, as seen with the clinical use of bevacizumab. To understand bevacizumab's effect on the entire VS pathobiological interactome, future studies are needed to assess proteomic and transcriptomic changes in VS after bevacizumab treatment,
Finally, our work provides insight into potential drug resistance in VSs. Resistance against bevacizumab has been noted in other tumors and cancers, but has yet to be explored after long-term use in VS.12 A potential mechanism of resistance could be the loss of HGF/cMET regulation by VEGF-A, which would lead to uncontrolled HGF-regulated growth in spite of VEGF-A inhibition.13 In this scenario, utilizing a cMET inhibitor would be effective in overcoming resistance to bevacizumab.
Overall, we discovered cross-talk between upregulated and activated angiogenic pathways in VS, namely the VEGF-A and HGF pathways. Specifically, we found that siRNA-mediated knockdown of VEGFA led to a decrease in cMET expression, and knockdown of MET led to a decrease in VEGF-A and VEGFR2 levels in SCs and VS cells. Our findings are in agreement with previous work that outlines these interactions in other cell types, such as endothelial cells. Through establishing cross-talk between VEGF-A and cMET, 2 molcules typically studied independently in VS, our work can provide new ways to understand and manipulate VS pathobiology. With this novel understanding, we can design more effective pharmacotherapies, including combination therapies targets VEGF-A and cMET.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
We would like to thank Drs. Kevin Emerick, Michael McKenna and Daniel Lee at Massachusetts Eye and Ear Infirmary and Drs. Frederick Barker and Robert Martuza at Massachusetts General Hospital for assistance in human sample collection.
Funding
This study was supported by the National Institute on Deafness and Other Communication Disorders Grants T32 DC00038 (SD, KMS) and K08DC010419 (KMS), the Bertarelli Foundation (KMS) and the US. Department of Defense Grant W81XWH-14–1–0091 (KMS).
References
- 1. Mahaley MSJ, Mettlin C, Natarajan N, Laws ERJ, Peace BB, Analysis of patterns of care of brain tumor patients in the United States: A study of the brain tumor section of the AANS and the CNS and the commission on cancer of the ACS. Clin Neurosurg 1990; 36:347-5; PMID:2295209 [PubMed] [Google Scholar]
- 2. Caye-Thomasen P, Werther K, Nalla A, Bøg-Hansen TC, Nielsen HJ, Stangerup SE, Thomsen J, VEGF and VEGF receptor-1 concentration in vestibular schwannoma homogenates correlates to tumor growth rate. Otol Neurotol 2005; 26(1):98-101; PMID:15699727 [DOI] [PubMed] [Google Scholar]
- 3. Plotkin SR, Stemmer-Rachamimov A, Barker FG, Halpin C, Padera TP, Tyrrell A, Sorensen AG, Jain RK, di Tomaso E, Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med 2009; 361(4):358-367; PMID:19587327; http://dx.doi.org/ 10.1056/NEJMoa0902579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Plotkin SR, Merker VL, Halpin C, Jennings D, McKenna MJ, Harris GJ, Barker FG 2nd. Bevacizumab for progressive vestibular schwannoma in neurofibromatosis type 2: A retrospective review of 31 patients. Otol Neurotol 2012; 33(6):1046-52; PMID:22805104; http://dx.doi.org/ 10.1097/MAO.0b013e31825e73f5 [DOI] [PubMed] [Google Scholar]
- 5. Gherardi E, Birchmeier W, Birchmeier C, Woude GV. Targeting MET in cancer: Rationale and progress. Nat Rev Cancer 2012; 12(2):89-103; PMID:22270953; http://dx.doi.org/ 10.1038/nrc3205 [DOI] [PubMed] [Google Scholar]
- 6. Moriyama T, Kataoka H, Kawano H, Yokogami K, Nakano S, Goya T, Uchino H, Koono M, Wakisaka S. Comparative analysis of expression of hepatocyte growth factor and its receptor, c-met, in gliomas, meningiomas and schwannomas in humans. Cancer Lett 1998; 124(2):149-155; PMID:9500204; http://dx.doi.org/ 10.1016/S0304-3835(97)00469-2 [DOI] [PubMed] [Google Scholar]
- 7. Sulpice E, Ding S, Muscatelli-Groux B, Bergé M, Han ZC, Plouet J, Tobelem G, Merkulova-Rainon T. Cross-talk between the VEGF-A and HGF signalling pathways in endothelial cells. Biol Cell 2009; 101(9):525-539; PMID:19281453; http://dx.doi.org/ 10.1042/BC20080221 [DOI] [PubMed] [Google Scholar]
- 8. Matsumura A, Kubota T, Taiyoh H, Fujiwara H, Okamoto K, Ichikawa D, Shiozaki A, Komatsu S, Nakanishi M, Kuriu Y, et al. . HGF regulates VEGF expression via the c-met receptor downstream pathways, PI3K/Akt, MAPK and STAT3, in CT26 murine cells. Int J Oncol 2013; 42(2):535-542; PMID:23233163; http://dx.doi.org/ 10.3892/ijo.2012.1728 [DOI] [PubMed] [Google Scholar]
- 9. Moriyama T, Kataoka H, Hamasuna R, Yokogami K, Uehara H, Kawano H, Goya T, Tsubouchi H, Koono M, Wakisaka S. Up-regulation of vascular endothelial growth factor induced by hepatocyte growth Factor/Scatter factor stimulation in human glioma cells. Biochem Biophys Res Commun 1998; 249(1):73-77; PMID:9705834; http://dx.doi.org/ 10.1006/bbrc.1998.9078 [DOI] [PubMed] [Google Scholar]
- 10. Wong HK, Lahdenranta J, Kamoun WS, Chan AW, McClatchey AI, Plotkin SR, Jain RK, di Tomaso E, Anti-Vascular endothelial growth factor therapies as a novel therapeutic approach to treating neurofibromatosis-related tumors. Cancer Res 2010; 70(9):3483-3493; PMID:20406973; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tokuda H, Takai S, Hanai Y, Harada A, Matsushima-Nishiwaki R, Kato H, Ogura S, Kozawa O. Potentiation by platelet-derived growth factor-BB of FGF-2-stimulated VEGF release in osteoblasts. J Bone Miner Metab 2008; 26(4):335-341; PMID:18600399; http://dx.doi.org/ 10.1007/s00774-007-0829-x [DOI] [PubMed] [Google Scholar]
- 12. Lieu CH, Tran H, Jiang Z, Mao M, Overman MJ, Lin E, Eng C, Morris J, Ellis L, Heymach JV, et al. . The association of alternate VEGF ligands with resistance to anti-VEGF therapy in metastatic colorectal cancer. PLoS ONE 2013; 8(10):e77117; PMID:24143206; http://dx.doi.org/ 10.1371/journal.pone.0077117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McCarty JH. Glioblastoma resistance to anti-VEGF therapy: has the challenge been MET? Clin Cancer Res 2013; 19(7):1631-1633; PMID:23403631; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-0051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dilwali S, Lysaght A, Roberts D, Barker FG,II, McKenna MJ, Stankovic KM. Sporadic vestibular schwannomas associated with good hearing secrete higher levels of fibroblast growth factor 2 than those associated with poor hearing irrespective of tumor size. Otol Neurotol 2013; 34(4):748-754; PMID:23512073; http://dx.doi.org/ 10.1097/MAO.0b013e31828048ec [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dilwali S, Patel PB, Roberts DS, Basinsky GM, Harris GJ, Emerick KS, Stankovic KM. Primary culture of human schwann and schwannoma cells: Improved and simplified protocol. Hear Res 2014; 315:25-33; PMID:24910344; http://dx.doi.org/ 10.1016/j.heares.2014.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]