

Abstract
Immunization of mice or rats with a "non-self" protein is a commonly used method to obtain monoclonal antibodies, and relies on the immune system's ability to recognize the immunogen as foreign. Immunization of an antigen with 100% identity to the endogenous protein, however, will not elicit a robust immune response. To develop antibodies to mouse proteins, we focused on the potential for breaking such immune tolerance by genetically fusing two independent T-cell epitope-containing sequences (from tetanus toxin (TT) and diphtheria toxin fragment A (DTA)) to a mouse protein, mouse ST2 (mST2). Wild-type CD1 mice were immunized with three mST2 tagged proteins (Fc, TT and DTA) and the specific serum response was determined. Only in mice immunized with the T-cell epitope-containing antigens were specific mST2 serum responses detected; hybridomas generated from these mice secreted highly sequence-diverse IgGs that were capable of binding mST2 and inhibiting the interaction of mST2 with its ligand, mouse interleukin (IL)-33 (mIL-33). Of the hundreds of antibodies profiled, we identified five potent antibodies that were able to inhibit IL-33 induced IL-6 release in a mast cell assay; notably one such antibody was sufficiently potent to suppress IL-5 release and eosinophilia infiltration in an Alternaria alternata challenge mouse model of asthma. This study demonstrated, for the first time, that T-cell epitope-containing tags have the ability to break tolerance in wild-type mice to 100% conserved proteins, and it provides a compelling argument for the broader use of this approach to generate antibodies against any mouse protein or conserved ortholog.
Keywords: Antibody generation, diphtheria toxin, hybridoma, immunological tolerance, mouse ST2, T-cell epitopes, tetanus toxin
Abbreviations
- TT
tetanus tosxin
- DTA
diphtheria toxin
- mST2
mouse ST2
- IL
interleukin
- ILC2
type 2 innate lymphoid cells
- SLE
systemic lupus erythematosus
- APC
antigen presenting cells
- TCR
T cell receptor
- MHC
major histocompatibility complex
- HLA
human leukocyte antigen
- PADRE
pan HLA-DR-binding T cell epitope
- CHO
Chinese hamster ovary
- HTRF
homogenous time-resolved fluorescence
- CDR
complementarity determining region
- DMEM
dulbecco's modified eagles' medium
- ELISA
enzyme-linked immunosorbent assay
- IgG
immunoglobulin G
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel
- VH
variable region of immunoglobulin heavy chain
- VL
variable region of immunoglobulin light chain
Introduction
The isolation of a monoclonal antibody to a protein of interest can be achieved through a number of different approaches; for example, by screening libraries of antibody fragments in vitro, or through the immunization of rodents and screening of subsequent hybridoma-derived antibodies. The immunization approach is attractive as it has the potential to provide high-affinity antibodies that have been matured in vivo, and can be both cost- and time-effective. This approach, however, is dependent on a divergence in sequence between the endogenous protein and the protein being immunized, to enable the immune system to recognize the immunogen as non-self. Thus, it is particularly challenging to generate antibodies in rodents to rodent proteins, which precludes the routine use of the immunization method. In this study, we sought to investigate whether introducing T-cell epitopes into a mouse immunogen would break tolerance in mice and enable generation and identification of large panels of potent functional antibodies.
We chose to use mouse ST2 (mST2) as our test protein because, despite the importance of this molecule to atopic responses, there were no functional (blocking) tool reagents available commercially. ST2 is the receptor for interleukin (IL)-33, and it is expressed at high levels on mast cells and type 2 innate lymphoid cells (ILC2s).1,2 IL-33, a member of the IL-1 family of cytokines expressed by some mucosal epithelial cells,3-6 is believed to act as an alarmin.3,4,7 Once released, IL-33 rapidly acts via ST2 to activate an inflammatory response by stimulating mast cells and ILC2s to release Th2 cytokines and chemokines.8-10 For example, the eosinophilia and IL-5 release caused by intranasal administration of the fungal allergen Alternaria alternate is abrogated in ST2-deficient mice.2 A function-blocking anti-mST2 antibody, sufficiently potent to work in vivo would provide an extremely valuable tool to investigate the importance of the ST2-IL-33 pathway in disease models of asthma, and provide significant advantages over other small molecule and genetic deletion approaches.
Overcoming the immune tolerance of mice to mouse proteins or those with high homology in order to generate such antibodies, however, is a challenge. To surmount this, a number of different approaches have been adopted. SLE-like mouse models (e.g., the NZB/W mouse strain) that display defective B cell tolerance have been successfully employed to generate antibodies to a number of self-antigens and closely-related proteins, but have not been universally successful.11,12 Alternatively, mice with a genetic knockout for a particular protein can be immunized with that protein exogenously to elicit an immune response. This approach has been used to develop antibodies to mouse and human butyrylcholinesterase13 and to mouse cellular prion protein.14 The lack of knock-out animals for many targets of interest prohibits the routine application of this strategy.
Another approach to improving the immunogenicity of an antigen is to incorporate T-cell epitopes either in the form of an immunogenic carrier protein15 or smaller T-cell epitope peptides.16 T cells are activated by specific foreign antigens via T-cell receptors (TCR) expressed on the cell surface. TCRs bind linear peptide fragments known as T-cell epitopes, which, when processed and displayed on MHC class II molecules by antigen-presenting cells (APCs), leads to the activation of T-helper cells. In secondary lymphoid organs, T and B cells interact and proliferate.17 T cells, via secreted cytokines, such as IL-4, induce B cell switch transcripts, resulting in switch recombination of the IgG heavy chain genes.18 B cells then differentiate to become fully mature antibody-secreting plasma cells or migrate to primary follicles and proliferate to form a germinal center. Here, the B cells undergo somatic hyper-mutation and a selection process that results in increased affinity of their antigen receptors and secreted antibodies for their antigen. The incorporation of T-cell epitopes into an immunogen is, therefore, predicted to enhance the immune response by boosting T cell help to B cells, to aid their subsequent maturation.
Chemical conjugation to an immunogenic carrier protein provides one means of introducing T-cell epitopes into an immunogen.19,20 Indeed, many marketed vaccines have employed chemical conjugation to various immunogenic carriers, including the subunits of bacteria, inactive bacterial toxoids or other proteins/peptides containing T-cell epitopes.21 While chemical conjugation is effective, there are a number of drawbacks. In the case of bacterial toxoids, the uncontrolled nature of the chemical (formaldehyde) treatment results in a heterogeneous preparation, and can alter the conformation of the protein, generating neo-epitopes and removing T-cell epitopes. In addition, the conjugation process itself requires optimization for each antigen molecule and is not a robust and reproducible process. Moreover, through conjugation to the carrier, key residues on the antigen may become occupied or obscured, which could affect the recognition of genuine B-cell epitopes on the antigen, compromising the production of diverse antibodies.
The recombinant expression and purification of a single antigen-carrier T-cell epitope fusion protein provides a simple and robust alternative means of generating potent immunogens. This approach has been adopted in a number of studies and has been shown to enhance the immune response to proteins with high homology to their mouse orthologues, or in the context of SLE-like mouse model strains.11 The use of such T-cell epitope tags for immunization of mouse proteins in wild-type mice, however, has not yet been explored. We sought to investigate the potential for this approach to provide antibodies to the mouse ST2 protein and chose two different T-cell epitope-containing sequences. In selecting carrier molecules or fusion partners, the ideal candidate would provide optimal T cell help, and contain minimal B-cell epitopes itself, so that the B cell response is not diverted in helping the generation of anti-carrier antibodies. The T-cell epitopes should be retained in the molecule in such a way that they are optimally processed within the APCs. Here, we employed two different T-cell epitope-containing carriers; either a non-toxic fragment of diphtheria toxin (DTA) or two tandem peptide epitopes of the tetanus toxin (TT). These carriers, based on bacterial toxin molecules, were chosen based on their widespread use in evoking immune stimulation in both humans and in mice,20,21 and because they differed in size and T-cell/B-cell epitope content.
Diphtheria toxin is secreted from the bacterium Corynebacterium diphtheria and consists of a single polypeptide chain that forms two subunits (A and B) linked by disulfide bridges, each subunit carrying a distinct function.22 Fragment B binds to the cell surface receptor, heparin-binding epidermal growth factor-like growth factor receptor, present on mammalian cells, enabling entry of the toxin into the cell, and fragment A provides the catalytic activity of the molecule through binding to elongation factor 2 and ablating protein synthesis.23 In this study, a non-toxic DTA fragment incorporating the K51E/E148K mutations24 was used as a fusion partner for mST2 to limit the toxicity to the cells secreting the antigen fusion protein.
Another vaccine carrier, tetanus toxin Fragment C is capable of eliciting the production of antibodies when used as a fusion partner for foreign antigens.25,26 It has been shown that the adjuvant potency of the tetanus toxin relies on the presence of promiscuous T-cell epitopes within the protein.27 The tetanus toxin molecule contains a number of T-cell epitopes that are represented by peptides including p2, p21, p23, p30 and p32. The p30 peptide (FNNFTVSFWLRVPKVSASHLEQY) is a strong immunogenic peptide that has been shown to consist of at least three distinct overlapping helper epitopes, where each epitope is recognized in association with multiple HLA class II alleles, including those found in mice.27 In addition to p30, other T-cell epitopes with the same ability to bind to the most common HLA molecules have been identified, including p2 (QYIKANSKFIGITE), which contains a single T-cell epitope. These two T-cell epitopes have been used previously in DNA vaccines to enhance the antigenicity of a number of antigens28 and were combined in tandem to provide an alternative, smaller, fusion partner for mST2.
In this study, we incorporated two distinct T-cell epitopes into a mouse immunogen, ST2, and used them to break immunological tolerance in wild-type mice to enable the generation of a panel of potent functional antibodies.
Results
Generation of active recombinant mST2 fusion proteins
The extracellular domain of mST2 containing either C-terminal Fc, DTA or TT tags was expressed in suspension-adapted CHO cells, and purified by virtue of a distal C-terminal hexahistidine tag. The recombinant proteins were analyzed by SDS-PAGE (Fig. S1) and migrate slightly slower than anticipated, likely due to glycosylation by the host cells. In addition, an unrelated recombinant protein was generated with the equivalent tag for each of the three fusions partners. The activity of the three mST2 fusion proteins was confirmed by assessing their ability to compete with biotinylated mST2-Fc fusion protein for binding to mIL-33 in a homogenous time-resolved fluorescence (HTRF) binding assay. Both the TT and DTA tagged mST2 proteins inhibit the binding of labeled mST2-Fc fusion to mIL-33, and this was comparable to that of the unlabelled mST2-Fc fusion protein (Fig. S2), suggesting that the three recombinant proteins had a similar conformation.
Mouse immunization and hybridoma generation
Mice were immunized with either Fc-, TT- or DTA-tagged mST2, dosed at 10 μg following a 28 day immunization schedule. The serum titers (pre-bleed and day 20) were measured using an ELISA that detected binding to the immunized mST2 fusion protein, and also to the unrelated protein with the equivalent tag. Although good serum titers for the mST2-Fc fusion protein were obtained at day 20, the levels were no higher than those that bound the equivalent unrelated Fc- fusion protein (Fig. 1, panel A). This suggests that the response was directed to the Fc region rather than the mST2 domain of the immunogen, so no further studies were conducted with these mice. In mice immunized with the mST2-DTA fusion protein, there appeared to be a modest increase in serum titers between the unrelated protein and mST2-DTA (Fig. 1, panel C), whereas for the mST2-TT protein, a significant increase in serum titer to the mST2 fusion protein was obtained, when compared to the unrelated protein (Fig. 1, panel B). This increase is conceivably due to the smaller size of the TT tag compared to that of the DTA and Fc fusion proteins. With a smaller tag, the number of B cell epitopes is likely to be negligible, enabling the immune system to more effectively generate antibodies to the antigen itself.
Figure 1.
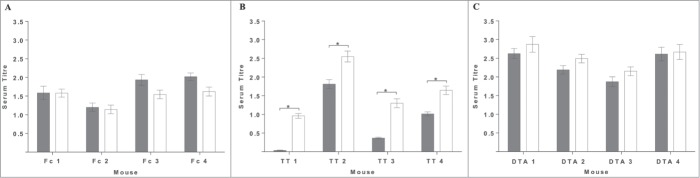
The mST2-specific immune response from mice immunized with T-cell epitope containing antigens. Sera from immunized mice taken at day 20, after 3 injections with antigen, were analyzed for anti-mST2 antibody response by ELISA. In panel A serum from mST2-Fc immunized mice was analyzed for binding to mST2-Fc (white bars) and a Fc-tagged unrelated protein (gray bars). In panel B serum from mST2-TT immunized mice was analyzed for binding to mST2-TT (white bars) and a TT tagged unrelated protein (gray bars). Likewise in panel C serum from mST2-DTA immunized mice was analyzed for binding to mST2-DTA (white bars) and a DTA tagged unrelated protein (gray bars). Serum titer is defined as the mean area under the curve for the 5 point titration normalized to the anti-his control antibody for each protein. Error bars correspond to ± 1 standard deviation. Significance was determined by an unpaired t test *<0.05.
For antibody production, hybridomas were generated from lymphocytes extracted from four of the immunized mice (TT 2, TT 4, DTA 1 and DTA 3). Mice TT 2 and TT 4 were selected as they had the highest serum titer to mST2-TT (Fig. 1). Three of the mice from the DTA immunized group (DTA 1, DTA 2 and DTA 3) showed higher serum titers on the mST2-DTA compared with the irrelevant DTA tagged protein. Of these 3 mice DTA 1 and DTA 3 were selected to represent the mST2-DTA immunized group. Fusion material was plated into semi-solid media containing an anti-IgG fluorophore-labeled antibody to identify IgG-secreting colonies using the ClonePix FL. The percentage of hybridoma colonies secreting IgG was calculated and found to be greater in the mice immunized with mST2-DTA compared with mST2-TT; 8–11% of hybridoma colonies were IgG-secreting within the TT group and 38–39% for the DTA group (Table 1).
Identification of functional anti-mouse ST2 antibodies
A total of 1,422 IgG-secreting hybridoma colonies were grown in liquid culture for up to seven days and the IgG-containing conditioned media (supernatant) was collected. All supernatants were individually tested for binding to mST2-Fc in a HTRF direct binding assay, and screened for their ability to inhibit the binding of mST2-Fc to mIL-33. Two hundred and sixty of the 1422 IgG-containing supernatants bound mST2 and of these, 111 inhibited the interaction of mST2 with its ligand mIL-33 using single point analysis (Table 1). mST2 binding antibodies were identified from all four mice; however, it should be noted here that the percentage of mST2-binding IgGs from the TT 4 mouse was much lower compared with the other three mice, and subsequently no mST2:mIL-33 inhibitor antibodies were identified from mouse TT 4. Nevertheless, considering that mice were immunized with a mouse protein, the percentage of IgGs that bound mST2 (30, 2, 14 and 20%) was sufficient for further analysis.
Table 1.
Proportion of IgG secreting colonies, mST2 binding and mST2-mIL-33 inhibiting IgGs from mST2-TT and mST2-DTA immunized mice. Lymphocytes were fused from mice TT 2, TT 4, DTA 1 and DTA 3. Following 10-14 days of Azaserine-Hypoxanthine selection hybridoma colony number and IgG secreting colonies were evaluated using data generated from the ClonePix. All IgG-secreting hybridomas were picked and IgGs screened in the mST2 binding and the mST2: mIL-33 competition HTRF assays
| Mouse |
||||
|---|---|---|---|---|
| TT 2 | TT 4 | DTA 1 | DTA 3 | |
| Hybridoma colonies | 1261 | 1282 | 906 | 2147 |
| IgG secreting colonies | 143 | 107 | 357 | 815 |
| % of colonies IgG | 11 | 8 | 39 | 38 |
| mST2 binding IgGs | 43 | 2 | 49 | 166 |
| mST2:mIL-33 inhibitor IgGs | 15 | 0 | 16 | 80 |
| % of IgGs mST2 binding | 30 | 2 | 14 | 20 |
To assess the sequence diversity of the blocking antibodies obtained, the variable regions of all 111 IgGs were sequenced, and 81 unique sequences were identified. The 111 hybridoma supernatants that inhibited the mST2/mIL-33 interaction were then taken forward for IgG purification using a small-scale automated plate based method and confirmation of their inhibitory activity (mST2-mIL-33 interaction) was performed at a single sample dilution. From the 111 IgG preparations, 92 demonstrated ≥ 15% inhibition and 22 were identified that demonstrated ≥ 50% inhibition (Fig. 2A).
Figure 2.
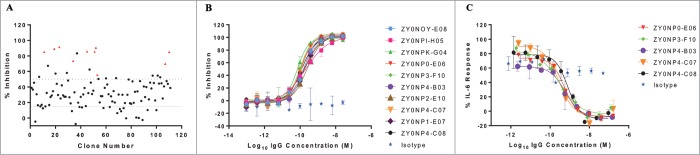
Selection of the most potent antibodies and characterization in biochemical and mouse mast cell assays. One hundred and eleven antibodies, identified from the primary high throughput assay to inhibit mST2 binding to mIL-33 were purified and tested single point at one dilution (0.006% sample) in the receptor ligand competition HTRF assay (A). The dotted lines represent 15% and 50% inhibition. Ten IgGs denoted as red triangles were taken forward for full characterization in the receptor ligand competition assay (B) and are shown to have IC50 values in this assay below 240 pM. These ten antibodies were then tested for their ability to block IL-6 release from MC/9 cells. Supernatants from MC/9 cells stimulated with mIL-33 overnight were assayed by ELISA for mIL-6 release. Data showing the percentage IL-6 response from cells treated with five inhibiting IgGs is presented, IC50 values in this assay are all sub-nanomolar (C). Isotype control data shown is mIgG1. Error bars correspond to ± 1 standard deviation (B, C).
From this panel of 22 IgGs, a subset of 10 that represented the output from 3 different immunizations (DTA 1, DTA 3 and TT 2) and were sequence-unique were chosen for further in vitro testing (Table S1). The purified IgGs were titrated into the mST2:mIL-33 HTRF competition assay (Fig. 2B) and demonstrated IC50 values between 70 and 240pM (Table S2). Subsequently, they were profiled in a cell-based functional assay measuring IL-6 release from a mouse mast cell line, (MC/9). Five of the 10 IgGs demonstrated dose-dependent inhibition (Fig. 2C) with IC50 values of 290–780 pM. Interestingly, all five inhibitory IgGs were isolated from mouse DTA 3, and exhibited high sequence identity compared to the remaining five IgGs which did not inhibit or demonstrated enhanced IL-6 release in the mouse mast cell assay (Table S3). This could suggest that the inhibitory antibodies bind a distinct epitope essential for inhibiting ST2 activity in a biological system. The bias observed for IgGs from the DTA 3 mouse in the final panel of 10 antibodies reflects the large number of IgG-secreting colonies obtained from this immunization arm (Fig. S3). Indeed, it is clear that, by starting with a larger proportion of IgG-secreting hybridomas, as was the case for the DTA 3 mouse, the ability to obtain functionally potent IgGs is increased. In selecting the final panel of 10 IgGs, however, we adopted highly stringent criteria to reduce the number of IgGs for further testing. This stringent choice led to the exclusion of other potentially potent and diverse IgGs from the 3 other responding mice that could also have provided a source of IgGs for prospective studies.
In vivo efficacy
The five inhibitory IgGs demonstrated equivalent in vitro activity, and one of the five clones (ZY0NP0-E06) was empirically chosen to ascertain whether such an antibody was sufficiently potent to elicit an effect in a disease-relevant mouse model. Knock out studies have shown that the eosinophilia infiltration29 and induction of IL-530 following intranasal Alternaria alternate challenge is ST2-dependent. To confirm this finding and test the potency of the antibodies generated, BalB/c mice were prophylactically dosed with 30 mg/kg ZY0NP0-E06 or isotype control 1 day and 1 hour prior to challenge. The anti-mST2 antibody significantly reduced both the eosinophil infiltration and the induction of IL-5 in the bronchoalveolar lavage fluid (BALf) measured 24 hours post-challenge (Fig. 3). This clearly demonstrates that ZY0NP0-E06 is sufficiently potent to inhibit the ST2 axis in vivo even when dosed systemically, demonstrating that immunization with T-cell epitope-containing immunogens is sufficient to generate highly potent antibodies without the need for further in vitro optimization.
Figure 3.
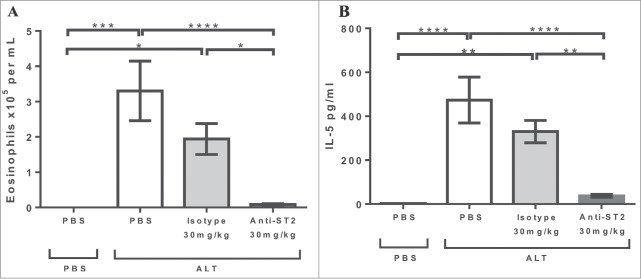
In vivo efficacy of antibody ZY0NP0-E06 in Alternaria alternata challenge model of asthma. ZY0NP0-E06 or isotype control (mIgG1) were dosed at 30 mg/kg intra-peritoneal 1 day and 1 hour prior to intranasal challenge with 25 μg Alternaria alternata extract. Eosinophil numbers (A) and IL-5 levels (B) were measured in the bronchoalveolar fluid 24 hr post-challenge. Error bars correspond to ± standard error of the mean, and represent the composite of 2 independent experiments with 7–12 mice per group. Significance was determined by 1 way ANOVA with a Tukey's multiple comparison test. * 0.05 > p > 0.01, ** 0.001 > p > 0.01, *** 0.0001> p >0.001 and ****0.0001 > p.
Discussion
In this study, we sought to investigate the feasibility of generating functional antibodies via immunization of wild-type mice to a self-protein, mST2, by genetically fusing it to a bacterial toxin-derived protein (DTA) or peptides (TT) known to potentiate immune responses. The serum response to these proteins was compared to that obtained through the immunization of a control mST2 protein fused to the Fc domain of human IgG1.
The fusion of a non-native protein/peptide sequence to the N- or C-terminus of a recombinant protein has the potential to affect the expression level of that protein. Evidence suggests that the fusion of proteins to maltose-binding protein or an IgG-derived Fc fragment for example, serves to improve the level of protein obtained in mammalian expression systems, whereas the addition of other proteins or peptides to different termini can have a detrimental impact on expression level.31 Although the two toxin-based fusion partners used in this study had the potential to negatively influence the expression level of the mST2 protein, the levels of antigen obtained were well within the acceptable range to enable sufficient material to be purified for immunization. In choosing a non-toxic domain of diphtheria toxin as a fusion partner for mST2, the likelihood of observing toxicity in the expression host cell was minimised. Indeed, no adverse effects on cell number or cell health were identified during expression of this fusion protein, suggesting that it could be used for other antigen fusion proteins. Another consideration for the addition of protein or peptide tags to antigens for immunization is the disruption of the antigen's tertiary structure, which could affect the presentation of appropriate B cell epitopes to the mouse immune system. This was evaluated prior to immunization by assessing the ability of the mST2 fusion proteins to compete for the binding to its ligand mIL-33, with no measurable impact of the two tags compared to the Fc-tagged protein (Fig. S2).
There is contradictory evidence in the literature regarding the relative merits of using a whole protein or domain vs defined T-cell epitope-containing peptides as immunogenic fusion partners, which is one consideration in choosing to investigate both DTA and TT options in this study. Despite eliciting a lower specific serum response than the TT-mST2 immunogen, the DTA-mST2 protein was able to induce a very high percentage of IgG-expressing hybridomas. This level of IgG secreting hybridomas obtained with the mouse ST2 DTA fusion protein suggests a role for the fusion protein in enhancing the immune response, and could be explained by a number of factors. The addition of a large protein domain to an antigen will increase the size of the immunogen, which could influence in vivo half-life via avoidance of hepatic clearance mechanisms.32 This, however, is probably not the most convincing argument considering that the Fc fusion protein (another large protein) demonstrated no specific serum titer levels. Another important consideration is that the context of the fusion partner may be important; in other words, the fusion of the 2 protein moieties may provide further T-cell epitopes that would depend both on the sequence of the antigen itself, the immunogenic carrier protein and any linker sequences. Moreover, the role of intracellular processing may be a critical one, in ensuring that the T-cell epitopes are processed appropriately to enable binding to the MHC class II molecule and recognition by the T-cell receptor.33 In presenting the intact DTA molecule to the antigen-presenting cell, the processing and presentation of the T-cell epitopes contained within the DTA molecule is likely optimal, increasing T cell help and thus IgG class-switching. An alternative explanation is that there is a larger number of, or more potent, T-cell epitopes within the intact DTA protein compared to the two tandem TT epitopes that are available to provide T cell help. It is likely the combination of all the factors described above explain why immunizations with the DTA fusion partner generated a higher number of IgG secreting hybridomas. The advantage of generating such highly enriched IgG-secreting hybridomas is self-evident; the larger the panel of IgGs screened, the higher the chance of successfully obtaining IgGs with the desired properties, as exemplified by the results obtained in this study (Fig. S3).
The data described herein also suggests that, despite the lower percentage of IgGs obtained from the mST2-TT hybridomas, the proportion of these that bound mST2 from mouse TT 2 was higher than both the mST2-DTA-treated mice. As the TT tag is significantly smaller than the DTA tag, there will be fewer “unproductive” B-cell epitopes to divert the immune system from generating mST2-specific IgGs, and this is reflected in the number of mST2-binding IgGs obtained from mouse TT 2. It could be argued that the ideal fusion partner would combine the benefits of both approaches used in this study, that is, the induction of class switching by powerful T-cell epitopes (as obtained via the DTA tag) and the reduction of non-specific IgGs via a smaller tag (as obtained with the TT tag; TT 2 mouse), enabling the generation of large panels of specific IgG-secreting hybridomas. To achieve this, the use of alternative peptide T-cell epitopes could be considered, for example, the PADRE epitope. This is a synthetic universal pan HLA-DR-binding T-cell epitope (PADRE), which binds with high affinity to most of the common HLA-DR alleles in humans and is more potent than the tetanus toxin p30 peptide.34 The sequence of PADRE is based on the core sequence of the ovalbumin master T-helper peptide (aa 323–339), and it has been adapted to enable binding to the most common human and mouse MHC class II molecules.35 In addition to the use of well-validated T-cell epitopes, there have been significant efforts both in vitro36 and in silico37 to identify the specific location of T-cell epitopes within proteins to enable the replacement of large carrier molecules with their minimally “active” peptide counterparts. These novel T-cell epitopes could prove to be even more potent than those already described and could provide the basis for alternative carrier molecules.
Nevertheless, by adopting the two alternative T-cell epitope-containing antigens in this study, we were successful in generating a large number of mST2 binding antibodies, and several highly potent antibodies that were active in biochemical and relevant cell-based assays, without the need for further in vitro affinity maturation. One of these antibodies also demonstrated sufficient potency to inhibit eosinophilia infiltration and the release of IL-5 in an in vivo disease model, enabling confirmation of the ST2/IL-33 pathway in atopic responses. In conducting this study, we have shown for the first time that, by incorporating a T-cell epitope-containing moiety into a mouse immunogen, the specific serum response that can be obtained via immunization in wild-type mice can be significantly increased, presumably through the engagement of optimal T cell help and subsequent class switching. We have not compared this approach to the use of other tags or alternative methods that are currently adopted for increasing the immune response during vaccinations or for the production of antibodies in mice. However, this is a straight-forward method, which circumvents the need for laborious and resource-intensive steps such as the generation of specific KO mouse models, and provides an alternative strategy that can be adopted widely for the generation of antibodies where breaking tolerance in the mouse is of critical importance. Moreover, it offers a potential way to increase the antibody response in mice to antigens such as integral membrane proteins that have limited extracellular epitopes. These proteins are often immunized in the context of a transfected cell and, due to the low relative expression level, the specific responses observed are very low. By using this approach to elicit a stronger immune response, the targeting of important classes of molecules such as these with functional and potent antibodies could be realized.
Materials and Methods
Generation of recombinant proteins
The cDNAs encoding the extracellular domain of mST2 (amino acid residues 1–332; Swiss-Prot Accession number P14719) or unrelated tagged proteins were optimized for mammalian expression and synthesized by Life Technologies. The cDNA fragment was cloned into a mammalian expression destination vector pDEST12.2 (Invitrogen) that had been modified to encode a tripeptide linker (Ala-Ser-Gly) followed by either the Fc domain of human IgG1, DTA (amino acid residues 26–218 of DT from corynebacteriophage; Swiss-Prot Accession number P00589) plus K51E/E148K mutations, or two tandem tetanus toxin epitopes (p2 and p30) separated by two amino acids as C-terminal fusions. In addition, the plasmids encoded a hexahistine tag at the very C-terminus, separated from the fusion partner by a Gly-Ser dipeptide linker, to enable facile purification. The fusion proteins were expressed in suspension-adapted CHO cells (mST2) or adherent HEK-EBNA cells (unrelated tagged proteins) using polyethylenamine (Polysciences) as a transfection reagent. Recombinant proteins were purified from cell culture supernatant using Ni-NTA (Histrap HP column (GE Healthcare)) affinity chromatography followed by size exclusion chromatography (Superdex 75 column (GE Healthcare)).
Synthetic DNA constructs encoding amino acids 97–266 of mIL-33 (Swiss Prot accession number Q8BVZ5) fused at the C-terminus to a FLAG 10His tag (DYKDDDDKAAHHHHHHHHHH) were made by DNA2.0 and cloned into the pJexpress404 expression vector. BL21(DE3) cells were transformed with the plasmid and protein expression induced with IPTG. Soluble protein was extracted using Bugbuster Reagent with Benzonase (Novagen) and purified using Ni-NTA (Histrap HP column (GE Healthcare)) affinity chromatography followed by size exclusion chromatography (Superdex 75 column (GE Healthcare)).
Immunization
Female CD1 mice (6–8 weeks) were injected subcutaneously with 10 μg recombinant mST2 in Freund's complete adjuvant (Sigma). Three subsequent boosts of 10 μg recombinant mST2 in Freund's incomplete adjuvant (Sigma) were administered through subcutaneous injection 7, 14 and 22 days after the initial prime. Two days after the fourth boost a final injection of 10 μg recombinant mST2 in PBS was administered intraperitoneally. Four days after the last injection mice were sacrificed and lymph nodes harvested. All work was carried out to UK Home Office ethical and husbandry standards under the authority of an appropriate project license.
Serum titer determination
Blood was collected via peripheral blood vessel on day 20 and serum was recovered by centrifugation using serum separator tubes (Microvette). Immune response to the antigen and immunogenic tags were assessed by enzyme-linked immuno-sorbent assay (ELISA). 96-well microtiter plates (Nunc) were coated with 5 μg/ml of antigen or the unrelated protein containing equivalent tags (Fc, DTA, TT) and incubated overnight at 4°C. The plates were blocked using 3% Marvel/PBS for 1 hr at room temperature (rt). Anti-His control mAb (Genscript) was prepared at 2 μg/ml and serum samples diluted 1/200 in 3% Marvel/PBS. After washing samples were serially diluted into the assay plates in duplicate over 5 points and incubated for 1 hr at rt. After washing, a 1:5000 dilution of horseradish peroxidase (HRP) conjugated goat anti-mouse IgG (γ chain specific) (Sigma) was added to each well and incubated at rt for 1 hr. Plates were washed before adding 50 μl of TMB substrate (Sigma) to each well and incubating for 10 min. The reaction was stopped by the addition of 50 μl 0.5 M sulphuric acid and absorbance read at 450 nm using the EnVision plate reader (PerkinElmer). Data was analyzed using Graph Pad Prism Software version 6.01 for Windows (GraphPad Software, La Jolla California USA, www.graphpad.com). Serum titer was determined, firstly by subtracting the average buffer only control for each protein from the sample. Area under the curve was then determined for each titration and the mean calculated. Finally values were normalized accordingly by dividing by the anti-his control for each plate.
Hybridoma generation
Cells were isolated from lymph nodes by mechanical disruption using the gentleMACS dissociator (Miltenyi) and fused with SP2/0 myeloma cells (ATCC) using a BTX electrofuser (ECM2001). Following electrofusion cells were resuspended in semi-solid selection media containing FITC conjugated anti-mouse IgG monoclonal antibody (CloneMatrix concentrate (Genetix), DMEM powder (Gibco), 20% FCS (SAFC), 2% glutaMAX (Gibco), 1% sodium pyruvate (Sigma), 1% penicillin/streptomycin (Gibco), 10% hybridoma cloning factor (Roche), 2% oxaloacetate/pyruvate/insulin (Sigma), 2% hypoxanthine/azaserine (Sigma), 11 μg/ml goat anti-mouse IgG-FITC (Jackson ImmunoResearch)). Media containing cells was divided into omni trays (Nunc) and cells were allowed to grow at 37°C under a 7.5% CO2-enriched atmosphere. 13–16 days following fusion IgG secreting hybridomas were detected and picked into 96 well plates (Costar) containing media (DMEM, 20% FCS, 2% glutaMAX, 1% penicillin/streptomycin, 10% hybridoma cloning factor, 2% oxaloacetate/pyruvate/insulin, 2% hypoxanthine and thymidine (Sigma)) using the ClonePix robot (Molecular Devices). Hybridomas were allowed to grow for 3–7 d before supernatant was harvest from each well using the MiniTrak robot (Perkin Elmer).
cDNA preparation and variable chain sequencing
Hybridoma mRNA for heavy and light chain variable regions were isolated using Oligo (dT)25 magnetic beads (Novagen) in combination with the kingfisher 96 automated magnetic separator (Thermo Scientific). Contaminating SP2/0 MOPC abVκ mRNA was removed by targeted digestion with RNaseH (NEB) at 37˚C for 1 hr. Purified mRNA was transcribed to cDNA using superscript III reverse transcriptase (Invitrogen) at 44°C for 1 hr and then tailed with poly(G) by incubation with excess dGTP and terminal transferase (NEB) at 37°C for 1 hr. Variable light and heavy chain genes were amplified using Taq polymerase (Thermo) in separate reactions using oligo(dC)25 and specific primers to either the CH1 or kappa constant domain. Chain termination method was used to sequence the PCR product.
IgG purification
Selected hybridomas were cultured in serum free media (HL-1(Lonza), 2% HybER-Zero (Statins Serum Institute), 2% glutaMAX) for 10 d at 37°C under a 7.5% CO2-enriched atmosphere. IgG was purified from the culture supernatants using ProPlus resin bed Phytips (Phynexus) on the MiniTrak robot. Absorbance of purified material was read using the EnVision plate reader at 450 nm and IgG concentration determined using mouse IgG isotype calibration curves. For column based purifications hybridomas were grown as above in 50 ml volumes, supernatant was harvested and filtered prior to purification using Protein G chromatography. Supernatants were loaded onto a 1 ml HiTrap Protein G column (GE Healthcare) and IgG eluted from the column using 0.1 M glycine-HCl pH 2.7. Eluted IgGs were then buffer exchanged into 1x DPBS using a Nap10 columns (GE Healthcare) and the concentration of IgG was determined spectrophotometrically.
mST2:mIL-33 receptor ligand competition HTRF assay
A biochemical assay using homogeneous time resolved fluorescence resonance energy transfer (HTRF) technology was established to measure the binding of mIL-33 with a FLAG tag to mST2 with a human Fc tag and labeled with biotin. Europium cryptate conjugated anti FLAG IgG (Cisbio) is used as the donor and streptavidin-XLent! (Cisbio) is used as the acceptor. Test samples were incubated with 1 nM biotinylated mST2-Fc pre-mixed with 5 nM streptavidin-XLent! and 0.3 nM FLAG tagged mIL-33 pre-mixed with 0.5 nM europium cryptate conjugated anti FLAG IgG for 4 hr at rt followed by 15 hr at 4°C. All reagents were prepared in PBS, 0.8 M potassium fluoride and 0.1% bovine serum albumin (BSA). The fluorescence was measured on an EnVision plate reader using an excitation 320 nm filter with the emission filters 665 nm and 590 nm. The raw data was initially analyzed using the equation 665 nm / 590 nm * 10,000 and was then expressed as %DELTA F using the equation (sample ratio - negative control ratio / negative control ratio * 100). The % inhibition is calculated from the %DELTA F values using the following equation % inhibition = 100−((Sample − negative control / Total − negative control) * 100). In the hybridoma supernatant assay, any IgG positive in the mST2 binding HTRF showing greater than 30% inhibition was taken forward for purification. For IC50 determination purified IgG samples were serially diluted into the assay using 3-fold dilutions (in duplicate) over 11 points. The data was then analyzed using GraphPad Prism with the following 4 parameter equation; Y = Bottom + (Top − Bottom) / (1 + 10^ ((LogIC50 − X) * HillSlope)). Where X is the logarithm of the sample concentration and Y is the % inhibition.
mST2 binding HTRF assay
A biochemical assay using HTRF technology was established to measure the binding of IgG test samples to mST2 expressed with a human Fc tag using europium cryptate conjugated anti human Fc IgG (Cisbio) as the donor and DyLight650 conjugated anti mouse Fc IgG (Jackson ImmunoResearch) as the acceptor. Test samples were incubated with 5 nM mST2-Fc pre-mixed with 5 nM DyLight650 conjugated anti mouse Fc IgG and 0.9 nM europium cryptate conjugated anti human Fc IgG for 4 hr at rt followed by 15 hr at 4°C. All reagents were prepared in PBS containing, 0.53 M potassium fluoride and 0.13% BSA. The fluorescence was measured on an EnVision plate reader using an excitation 320 nm filter with the emission filters 665 nm and 590 nm. The raw data was initially analyzed using the equation 665 nm / 590 nm * 10,000 and was then expressed as % DELTA F using the equation (sample ratio - negative control ratio / negative control ratio * 100). Samples with a binding signal greater than 100% DELTA F were taken forward for further analysis.
Inhibition of mST2 in a mouse mast cell line
IL-6 release from the mouse mast cell line MC/9 can be achieved via IL-33 binding to mST2.38-40 The inhibition of IL-6 release by anti-ST2 antibodies was considered an appropriate assay to assess functional activity in a cell line expressing native mST2. MC/9 cells were cultured as per the manufacturer's instruction (ATCC). Media was changed on the MC/9 the day prior to use. MC/9 cells were pre-incubated for 30 min with anti-mST2 IgGs then stimulated overnight with 0.1 nM mIL-33 (Peprotech). Conditions were applied to duplicate wells. Supernatants were collected and assayed by ELISA for mIL-6 (R&D Systems). This was performed to manufacturer's instructions but used a Perkin Elmer Europium based detection. Briefly, Eu-N1 streptavidin (Perkin Elmer) was diluted in DELFIA Assay Buffer (Perkin Elmer) and incubated for 40 min at rt protected from the light. Following washing plates were incubated for 10 min in DELFIA Enhancement Solution (Perkin Elmer). The fluorescence was then measured on an EnVision plate reader using excitation and emission filters 340 and 615 nm, respectively. Data was transformed to percentage mIL-6 response using Microsoft Excel and IC50 values were generated from 4-PL curves plotted using GraphPad Prism, where X is log IgG concentration and Y is percentage mIL-6 release.
Alternaria alternata challenge mouse model
Female BALB/c mice (6–8 weeks) received either 25 μg of Alternaria alternata (Greer, Lenoir, NC) or 50 μl vehicle, intranasally. Mice were treated intraperitoneally, with anti mST2 antibody (ZY0NP0-E06, 30 mg/kg), isotype control (NIP228, 30 mg/kg) or vehicle (PBS, 10 ml/kg), at 24 hr and 1 hr prior to intranasal challenge. Mice were culled 24 hr after challenge. Bronchoalveolar lavage fluid (BALf) was collected by lavage (3 × 0.3 ml) via tracheal cannula. BALf was centrifuged, cells counted and supernatant was analyzed for cytokines by ELISA (Meso Scale Discovery, Rockville, MD). Differential cell counts (200 cells/slide) were performed on cytospin preparations stained with Diff-Quik (Fisher Scientific, UK). All work was carried out to UK Home Office ethical and husbandry standards under the authority of an appropriate project license.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Lisa Bamber and David Bannister for protein expression and purification, Ling Huang and Jayne Hammersley for help with hybridoma generation and Kate Goode for performing the sequencing reactions. We would like to acknowledge the technical expertise of our staff in both our animal facility and tissue culture unit.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Moritz DR, Rodewald HR, Gheyselinck J, Klemenz R. The IL-1 receptor-related T1 antigen is expressed on immature and mature mast cells and on fetal blood mast cell progenitors. J Immunol 1998; 161:4866-74; PMID:9794420 [PubMed] [Google Scholar]
- 2. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TKA, Bucks C, Kane CM, Fallon PG, Pannell R, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010; 464:1367-70; PMID:20200518; http://dx.doi.org/ 10.1038/nature08900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: A novel 'alarmin'? PLoS One 2008; 3:e3331; PMID:18836528; http://dx.doi.org/ 10.1371/journal.pone.0003331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Enoksson M, Lyberg K, Moller-Westerberg C, Fallon PG, Nilsson G, Lunderius-Andersson C. Mast cells as sensors of cell injury through IL-33 recognition. J Immunol 2011; 186:2523-8; PMID:21239713; http://dx.doi.org/ 10.4049/jimmunol.1003383 [DOI] [PubMed] [Google Scholar]
- 5. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005; 23:479-90; PMID:16286016; http://dx.doi.org/ 10.1016/j.immuni.2005.09.015 [DOI] [PubMed] [Google Scholar]
- 6. Prefontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, Lemière C, Martin JG, Hamid Q. Increased expression of IL-33 in severe asthma: Evidence of expression by airway smooth muscle cells. J Immunol 2009; 183:5094-103; PMID:19801525; http://dx.doi.org/ 10.4049/jimmunol.0802387 [DOI] [PubMed] [Google Scholar]
- 7. Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci U S A 2009; 106:9021-6; PMID:19439663; http://dx.doi.org/ 10.1073/pnas.0812690106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol 2007; 179:2051-4; PMID:17675461; http://dx.doi.org/ 10.4049/jimmunol.179.4.2051 [DOI] [PubMed] [Google Scholar]
- 9. Iikura M, Suto H, Kajiwara N, Oboki K, Ohno T, Okayama Y, Saito H, Galli SJ, Nakae S. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab Invest 2007; 87:971-8; PMID:17700564; http://dx.doi.org/ 10.1038/labinvest.3700663 [DOI] [PubMed] [Google Scholar]
- 10. Silver MR, Margulis A, Wood N, Goldman SJ, Kasaian M, Chaudhary D. IL-33 synergizes with IgE-dependent and IgE-independent agents to promote mast cell and basophil activation. Inflamm Res 2010; 59:207-18; PMID:19763788; http://dx.doi.org/ 10.1007/s00011-009-0088-5 [DOI] [PubMed] [Google Scholar]
- 11. Zhou H, Wang Y, Wang W, Jia J, Li Y, Wang Q, Wu Y, Tang J. Generation of monoclonal antibodies against highly conserved antigens. PLoS One 2009; 4:e6087; PMID:19564921; http://dx.doi.org/ 10.1371/journal.pone.0006087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Andrievskaia O, McRae H, Elmgren C, Huang H, Balachandran A, Nielsen K. Generation of antibodies against bovine recombinant prion protein in various strains of mice. Clin Vaccine Immunol 2006; 13:98-105; PMID:16426006; http://dx.doi.org/ 10.1128/CVI.13.1.98-105.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hrabovska A, Bernard V, Krejci E. A novel system for the efficient generation of antibodies following immunization of unique knockout mouse strains. PLoS One 2010; 5:e12892; PMID:20886120; http://dx.doi.org/ 10.1371/journal.pone.0012892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nitschke C, Flechsig E, van den Brandt J, Lindner N, Lührs T, Dittmer U, Klein MA. Immunisation strategies against prion diseases: Prime-boost immunisation with a PrP DNA vaccine containing foreign helper T-cell epitopes does not prevent mouse scrapie. Vet Microbiol 2007; 123:367-76; PMID:17499458; http://dx.doi.org/ 10.1016/j.vetmic.2007.03.032 [DOI] [PubMed] [Google Scholar]
- 15. Mitchison NA. The carrier effect in the secondary response to hapten-protein conjugates. II. cellular cooperation. Eur J Immunol 1971; 1:18-27; PMID:14978857; http://dx.doi.org/ 10.1002/eji.1830010104 [DOI] [PubMed] [Google Scholar]
- 16. Bixler GS, Jr, Pillai S. The cellular basis of the immune response to conjugate vaccines. Contrib Microbiol Immunol 1989; 10:18-47; PMID:2684506 [PubMed] [Google Scholar]
- 17. Liu YJ, Zhang J, Lane PJ, Chan EY, MacLennan IC. Sites of specific B cell activation in primary and secondary responses to T cell-dependent and T cell-independent antigens. Eur J Immunol 1991; 21:2951-62; PMID:1748148 [DOI] [PubMed] [Google Scholar]
- 18. Toellner KM, Gulbranson-Judge A, Taylor DR, Sze DM, MacLennan IC. Immunoglobulin switch transcript production in vivo related to the site and time of antigen-specific B cell activation. J Exp Med 1996; 183:2303-12; PMID:8642339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Siegrist CA. Section 1: General aspects of vaccination; vaccine immunology. Vaccines 2008:17-36. [Google Scholar]
- 20. Tontini M, Berti F, Romano MR, Proietti D, Zambonelli C, Bottomley MJ, De Gregorio E, Del Giudice G, Rappuoli R, Costantino P, et al. Comparison of CRM197, diphtheria toxoid and tetanus toxoid as protein carriers for meningococcal glycoconjugate vaccines. Vaccine 2013; 31:4827-33; PMID:23965218 [DOI] [PubMed] [Google Scholar]
- 21. National Institutes of Health The jordan report: Accelerated development of vaccines 2007. 2007. [Google Scholar]
- 22. Gill DM, Dinius LL. Observations on the structure of diphtheria toxin. J Biol Chem 1971; 246:1485-91; PMID:5545090 [PubMed] [Google Scholar]
- 23. Pappenheimer AM, Jr. Diphtheria toxin. Annu Rev Biochem 1977; 46:69-94; PMID:20040 [DOI] [PubMed] [Google Scholar]
- 24. Kimura Y, Saito M, Kimata Y, Kohno K. Transgenic mice expressing a fully nontoxic diphtheria toxin mutant, not CRM197 mutant, acquire immune tolerance against diphtheria toxin. J Biochem 2007; 142:105-12; PMID:17522091 [DOI] [PubMed] [Google Scholar]
- 25. Spellerberg MB, Zhu D, Thompsett A, King CA, Hamblin TJ, Stevenson FK. DNA vaccines against lymphoma: Promotion of anti-idiotypic antibody responses induced by single chain fv genes by fusion to tetanus toxin fragment C. J Immunol 1997; 159:1885-92; PMID:9257853 [PubMed] [Google Scholar]
- 26. Khan CM, Villarreal-Ramos B, Pierce RJ, Riveau G, Demarco de Hormaeche R, McNeill H, Ali T, Fairweather N, Chatfield S, Capron A. Construction, expression, and immunogenicity of the schistosoma mansoni P28 glutathione S-transferase as a genetic fusion to tetanus toxin fragment C in a live aro attenuated vaccine strain of salmonella. Proc Natl Acad Sci U S A 1994; 91:11261-5; PMID:7972044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Panina-Bordignon P, Tan A, Termijtelen A, Demotz S, Corradin G, Lanzavecchia A. Universally immunogenic T cell epitopes: Promiscuous binding to human MHC class II and promiscuous recognition by T cells. Eur J Immunol 1989; 19:2237-42; PMID:2481588 [DOI] [PubMed] [Google Scholar]
- 28. Chambers RS, Johnston SA. High-level generation of polyclonal antibodies by genetic immunization. Nat Biotechnol 2003; 21:1088-92; PMID:12910245 [DOI] [PubMed] [Google Scholar]
- 29. Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage -CD25 +CD44 hi lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol 2012; 188:1503-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kouzaki H, Iijima K, Kobayashi T, O'Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol 2011; 186:4375-87; PMID:21357533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Terpe K. Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol 2003; 60:523-33; PMID:12536251 [DOI] [PubMed] [Google Scholar]
- 32. Xuemin C, Danya L, Baoyan W. Induction of anti-HMW-MAA immunity by anti-idiotypic MAB MK2-23-IL-2 fusion protein. Journal of Xi'an Medical University, English Edition 1998; 10:125-32 [Google Scholar]
- 33. Kjerrulf M, Lowenadler B, Svanholm C, Lycke N. Tandem repeats of T helper epitopes enhance immunogenicity of fusion proteins by promoting processing and presentation. Mol Immunol 1997; 34:599-608; PMID:9393963; http://dx.doi.org/ 10.1016/S0161-5890(97)00078-3 [DOI] [PubMed] [Google Scholar]
- 34. Alexander J, del Guercio MF, Maewal A, Qiao L, Fikes J, Chesnut RW, Paulson J, Bundle DR, DeFrees S, Sette A. Linear PADRE T helper epitope and carbohydrate B cell epitope conjugates induce specific high titer IgG antibody responses. J Immunol 2000; 164:1625-33; PMID:10640784; http://dx.doi.org/ 10.4049/jimmunol.164.3.1625 [DOI] [PubMed] [Google Scholar]
- 35. del Guercio MF, Alexander J, Kubo RT, Arrhenius T, Maewal A, Appella E, Hoffman SL, Jones T, Valmori D, Sakaguchi K, et al. Potent immunogenic short linear peptide constructs composed of B cell epitopes and pan DR T helper epitopes (PADRE) for antibody responses in vivo. Vaccine 1997; 15:441-8; PMID:9141216; http://dx.doi.org/ 10.1016/S0264-410X(97)00186-2 [DOI] [PubMed] [Google Scholar]
- 36. Li Pira G, Ivaldi F, Moretti P, Manca F. High throughput T epitope mapping and vaccine development. J Biomed Biotechnol 2010; 2010; PMID:20617148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Patronov A, Doytchinova I. T-cell epitope vaccine design by immunoinformatics. Open Biol 2013; 3:120139; PMID:23303307; http://dx.doi.org/ 10.1098/rsob.120139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hsu CL, Neilsen CV, Bryce PJ. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS One 2010; 5:e11944; PMID:20689814; http://dx.doi.org/ 10.1371/journal.pone.0011944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moulin D, Donze O, Talabot-Ayer D, Mezin F, Palmer G, Gabay C. Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine 2007; 40:216-25; PMID:18023358; http://dx.doi.org/ 10.1016/j.cyto.2007.09.013 [DOI] [PubMed] [Google Scholar]
- 40. Ho LH, Ohno T, Oboki K, Kajiwara N, Suto H, Iikura M, Okayama Y, Akira S, Saito H, Galli SJ, et al. IL-33 induces IL-13 production by mouse mast cells independently of IgE-FcepsilonRI signals. J Leukoc Biol 2007; 82:1481-90; PMID:17881510; http://dx.doi.org/ 10.1189/jlb.0407200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.