

Abstract
Purpose
Lenalidomide and rituximab (LR) are active agents in follicular lymphoma (FL). Combination regimens have not been previously assessed in randomized studies.
Patients and Methods
The Cancer and Leukemia Group B (Alliance) 50401 trial is a randomized phase II trial studying rituximab (375 mg/m2 weekly for 4 weeks), lenalidomide (15 mg per day on days 1 to 21, followed by 7 days of rest, in cycle 1 and then 20 mg per day on days 1 to 21, followed by 7 days of rest, in cycles 2 to 12), or LR. The rituximab-alone arm was discontinued as a result of poor accrual. Eligibility included recurrent FL and prior rituximab with time to progression of ≥ 6 months from last dose. Aspirin or heparin was recommended for patients at high thrombosis risk.
Results
Ninety-one patients (lenalidomide, n = 45; LR, n = 46) received treatment; median age was 63 years (range, 34 to 89 years), and 58% were intermediate or high risk according to the Follicular Lymphoma International Prognostic Index. In the lenalidomide and LR arms, grade 3 to 4 adverse events occurred in 58% and 53% of patients, with 9% and 11% of patients experiencing grade 4 toxicity, respectively; grade 3 to 4 adverse events included neutropenia (16% v 20%, respectively), fatigue (9% v 13%, respectively), and thrombosis (16% [n = 7] v 4% [n = 2], respectively; P = .157). Thirty-six percent of lenalidomide patients and 63% of LR patients completed 12 cycles. Lenalidomide alone was associated with more treatment failures, with 22% of patients discontinuing treatment as a result of adverse events. Dose-intensity exceeded 80% in both arms. Overall response rate was 53% (20% complete response) and 76% (39% complete response) for lenalidomide alone and LR, respectively (P = .029). At the median follow-up of 2.5 years, median time to progression was 1.1 year for lenalidomide alone and 2 years for LR (P = .0023).
Conclusion
LR is more active than lenalidomide alone in recurrent FL with similar toxicity, warranting further study in B-cell non-Hodgkin lymphoma as a platform for addition of novel agents.
INTRODUCTION
Despite high response rates to chemotherapy-based regimens, most patients with indolent non-Hodgkin lymphoma (NHL) develop recurrent or refractory disease, and many ultimately die from lymphoma-related complications. The anti-CD20 monoclonal antibody rituximab was originally approved by the US Food and Drug Administration for use in patients with relapsed and refractory follicular lymphoma (FL) and low-grade lymphoma, after a pivotal trial of 166 patients demonstrated an objective response rate of 48% (approximately 60% in FL), with a median time to progression (TTP) of 12 months in responders.1 For patients with indolent NHL who initially respond (complete or partial remission with a TTP of at least 6 months) and then experience relapse after single-agent rituximab therapy, re-treatment with rituximab alone or in combination with chemotherapy is commonly used.2 However, until recently,3 the effectiveness of rituximab single-agent treatment in patients with relapsed FL after rituximab-chemotherapy combination regimens was not well established although of clinical importance.
One approach to enhance the activity of rituximab is through the use of biologic agents to explore the potential for additive or synergistic activity. These include cytokines, other antibodies, and immunomodulatory or proapoptotic agents.4–6 Such combination regimens are particularly attractive to patients and clinicians who wish to avoid toxicities more typically associated with cytotoxic chemotherapy and offer alternative mechanisms of action against chemotherapy-resistant disease. One agent that may potentially augment the activity of rituximab in NHL is the immunomodulatory drug lenalidomide, a potent thalidomide derivative with immune, antiangiogenic, and direct antilymphoma effects.7 Lenalidomide has demonstrated antitumor activity in laboratory and clinical settings in lymphoid malignancies.8 With a dosing range of up to 25 mg per day administered orally on days 1 through 21 of a 28-day cycle, toxicities have included myelosuppression, rash, and thrombosis.9 Preclinical studies have suggested that the addition of lenalidomide to rituximab (LR) augments antitumor effects, providing rationale for further evaluation of this combination in patients with NHL.10
Given the importance of rituximab and the promise of rituximab-based combinations in lymphoma, the Cancer and Leukemia Group B (CALGB; Alliance) 50401 trial was designed as a randomized phase II study of rituximab alone, lenalidomide alone, or LR in patients with recurrent, rituximab-nonrefractory FL. The increasing use of rituximab maintenance in this population led to the removal of the rituximab-alone arm early in the study as a result of poor accrual. Here, we provide information on the clinical activity and safety of lenalidomide alone and the LR combination in recurrent FL, establishing a platform for further development of effective and tolerable combination biologic, chemotherapy-free treatment regimens for B-cell NHL.
PATIENTS AND METHODS
Eligibility Criteria
Patients had previously treated, histologically confirmed follicular center cell lymphoma, WHO classification grade 1, 2, or 3a, based on local review. Patients with transformed lymphoma were not eligible. Previous treatment was required with rituximab either alone or in combination with chemotherapy, with a TTP of ≥ 6 months from last rituximab dose. The most recent prior treatment regimen was not required to include rituximab. Other inclusion criteria included measurable disease, age ≥ 18 years, and Eastern Cooperative Oncology Group performance status of 0 to 2. Required initial laboratory values included absolute neutrophil count (ANC) of ≥ 1,000/μL, platelet count of ≥ 75,000/μL, serum creatinine of ≤ 1.5× the institutional upper limit of normal, estimated creatinine clearance ≥ 30 mL/min, and serum bilirubin ≤ 2× the institutional upper limit of normal.
Key exclusion criteria included corticosteroids within 2 weeks of study, except for maintenance therapy for a nonmalignant disease, radioimmunotherapy within 12 months before study entry, known CNS involvement by lymphoma, known HIV infection, and ongoing pregnancy (or nursing). Patients with a currently active second malignancy, other than nonmelanoma skin cancers, at the time of study entry were not eligible. Also excluded were patients with a history of deep venous thrombosis (DVT) or pulmonary embolus (PE) within 3 months before study entry, whereas patients greater than 3 months since DVT/PE were eligible but recommended to receive prophylaxis.
Study Plan
Patients were originally randomly allocated 2:1:1 to one of the following three treatment arms: arm A (rituximab alone), arm B (lenalidomide alone), or arm C (LR; Fig 1). After arm A closed (September 15, 2007, three patients enrolled), patients were randomly assigned 1:1 to lenalidomide alone or LR. No stratification factors were used.
Fig 1.
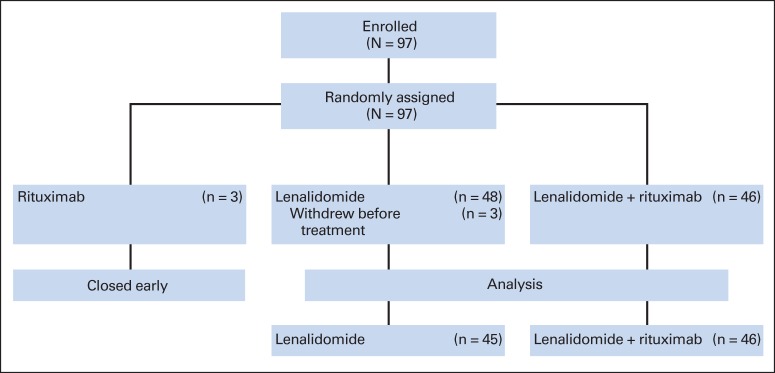
Cancer and Leukemia Group B (CALGB) 50401 consort diagram (flow of patients from enrollment to analysis).
Rituximab 375 mg/m2 per week was administered by intravenous infusion on days 1, 8, 15, and 22 (a total of four doses) to patients in arm A and on days 8, 15, 22, and 29 (beginning 1 week after initiation of lenalidomide) to patients in arm C. Premedication included diphenhydramine and acetaminophen. Standard infusion rates and adjustments were used.
For patients in arms B and C, lenalidomide was administered at the starting dose of 15 mg per day orally on days 1 through 21 followed by 7 days of rest, every 28 days. For cycle 2, the dose was escalated to 20 mg per day on days 1 through 21 in patients who did not require a delay and who had recovered from previous toxicity. For cycle 3 and beyond, the dose was escalated to 25 mg per day on days 1 through 21 of each 28-day cycle following similar guidelines. In the absence of intolerable toxicity or disease progression, lenalidomide was given for a total of 12 cycles. A new cycle of therapy could begin on the scheduled day 1 if ANC was ≥ 1,000/μL, platelet count was ≥ 50,000/μL, anemia was ≤ grade 3, any lenalidomide-related allergic reactions had resolved to ≤ grade 1, or other nonhematologic toxicity that may have occurred in a previous cycle had resolved to ≤ grade 1.
The protocol mandated that rituximab was to be discontinued for grade 3 or 4 hypersensitivity reactions. In the instance of ≥ grade 3 erythema multiforme, patients were to be removed from protocol therapy. Lenalidomide dose modifications occurred for several reasons, at 5-mg increments. No dose reductions of lenalidomide less than 5 mg were permitted. Lenalidomide therapy was delayed for ≥ grade 3 neutropenia on day 1 of a cycle, and CBCs were monitored weekly. If neutropenia resolved to ≤ grade 2 within 4 weeks, lenalidomide was resumed at the next lower dose level. If ≥ grade 3 neutropenia occurred during a cycle, lenalidomide was held for the remainder of the cycle and treatment could resume with one dose level reduction for subsequent cycles. If treatment was delayed for more than 4 weeks, protocol therapy was discontinued. Granulocyte colony-stimulating factor or granulocyte-macrophage colony-stimulating factor was not permitted to avoid dose reductions. In the instance of febrile neutropenia, lenalidomide was held and granulocyte colony-stimulating factor or granulocyte-macrophage colony-stimulating factor was administered until recovery (ANC ≥ 1,500/μL and absence of fever). When toxicity resolved to ≤ grade 2, lenalidomide was continued at a reduction of one dose level for subsequent cycles. If thrombocytopenia based on platelet count on day 1 or 15 worsened by one or more grade levels, lenalidomide was reduced by one dose level. If thrombocytopenia based on platelet count on day 1 or 15 worsened from grade 2 to 3, lenalidomide therapy was held until platelets were ≥ 50,000/μL and therapy could resume with a reduction of one dose level. In the case of drug-related anemia based on hemoglobin on day 1 or 15 that worsened by one or more grade levels, lenalidomide was reduced by one dose level; lenalidomide was discontinued for drug-related grade 4 hemoglobin. Once the dose of lenalidomide was decreased for any toxicity, it could not be increased.
Lenalidomide was held in the instance of a grade 2 allergic reaction and was resumed when toxicity resolved to ≤ grade 1. With ≥ grade 3 allergic reaction or any grade of desquamating rash, patients were removed from protocol therapy.
In the instance of grade 2 or 3 cardiac, thyroid, or other nonhematologic toxicity, lenalidomide was held until resolution to ≤ grade 1 and then resumed at the next lower dose level. Patients were removed from protocol therapy for grade 4 nonhematologic toxicity. Treatment was also held until resolution of grade 3 tumor lysis syndrome. For creatinine clearance less than 30 mL/min, lenalidomide was held. For creatinine clearance less than 60 mL/min but ≥ 30 mL/min, lenalidomide was reduced to the next lower dose level for all subsequent cycles.
Prophylactic aspirin or low molecular weight heparin was recommended for patients receiving lenalidomide with a high risk of developing DVT/PE or arterial thromboses unless contraindicated. High risk was defined as a history of DVT/PE, significant family history, Eastern Cooperative Oncology Group performance status greater than 2, smoking history, use of oral contraceptives, concurrent use of erythropoietin, or history of diabetes mellitus or coronary artery disease. Patients who developed signs or symptoms suggestive of thrombosis while on study treatment were evaluated and treated as clinically indicated. Lenalidomide was held for patients with venous thrombosis but could resume when adequately anticoagulated. Patients with recurrent thrombosis despite adequate anticoagulation were removed from protocol therapy.
Female patients of childbearing potential were monitored with pregnancy tests before enrollment and periodically while on study, and either abstinence or contraceptive measures were mandated. It was recommended that patients at high risk of hepatitis B virus (HBV) infection be screened before starting treatment. Patients testing positive for HBV were to be closely monitored for evidence of active HBV infection and hepatitis during and for several months after rituximab treatment.
Patients were restaged by computed tomography scan (or magnetic resonance imaging) of neck/chest/abdomen/pelvis at months 2, 4, 6, 9, 12, 15, 18, and 24, and then yearly until disease progression or for a maximum of 10 years from study entry. Bone marrow biopsy was performed in patients in complete remission if marrow was involved with lymphoma at baseline. Responses were classified per the 1999 International Working Group criteria,11 with positron emission tomography scans not included.
Each participant signed an institutional review board–approved, protocol-specific informed consent in accordance with federal and institutional guidelines. Data collection and statistical analyses were conducted by the Alliance Statistics and Data Center. Data quality was ensured by review of data by the Alliance Statistics and Data Center and by the study chairperson following Alliance policies.
Statistical Considerations
The primary objective was to determine the response rates (overall response [OR] and complete response [CR]) within the lenalidomide-alone and LR treatment arms, independently. Secondary objectives included comparison of OR rates among treatment arms and assessments of TTP and overall survival (OS). TTP is calculated as the time from study entry until progression or death without progression. OS is calculated as the time from study entry until death. Additional goals were to determine the relative toxicity profile of the regimens.
The study was designed to randomly assign a total of 45 eligible patients each to both the lenalidomide and LR arms via two-stage design; for each arm, if five or more patients of the first 21 achieved response, then the arm would proceed to the second stage to accept the therapy if 11 or more patients responded out of the cumulative 45 patients. An observed OR rate of ≤ 15% for either arm would be considered as insufficient for further study, and an OR of ≥ 35% would be considered of strong interest. This two-stage design has a one-sided 4.8% type I error rate at 15% OR and 88.7% power at 35% OR for each arm. Assuming approximately 5% ineligibility, the target accrual for this study was 95 patients.
The true OR rates were estimated using the uniformly minimum unbiased estimator.12 The Jennison-Turnbull 95% CI will have a maximum length of 0.365. The overall response rates were compared among treatment arms using a two-sided Fisher's exact test for a difference in proportions. For each patient, dose-intensity for lenalidomide was computed as the ratio of the total dose given and total dose of lenalidomide expected for all cycles of treatment received. TTP (defined as the time from study entry until progression) and OS (defined as the time from study entry until death from any cause) were estimated using the Kaplan-Meier method. TTP and OS were compared among the lenalidomide-alone and LR arms using the log-rank test for univariable analyses and the Cox regression method. Toxicity data are summarized using frequency tables. All P values are two-sided except for the primary objective.
RESULTS
Between October 2006 and April 2011, 97 patients were accrued to the study (48 to lenalidomide alone, 46 to LR, and three to rituximab alone). Three patients randomly assigned to the lenalidomide-alone arm never began treatment, and the rituximab arm was discontinued early. The interim analysis revealed that after 21 patients were accrued to the lenalidomide-alone and LR arms, there were 12 (57%) and 16 (76%) responders, respectively. Therefore, the study proceeded to the second stage. The final analysis reported is from data collected through April 24, 2013. Patients randomly assigned to the lenalidomide-alone and LR arms were included in the final analysis (n = 91).
Key baseline characteristics of patients enrolled onto the lenalidomide-alone and LR arms are listed in Table 1. Median age was 63 years, and most patients had advanced-stage disease with low (42.2%) and intermediate (35.3%) risk by the Follicular Lymphoma International Prognostic Index criteria.13 Overall median TTP since last rituximab dose in this population with relapsed, nonrefractory FL was 1.4 years (range, 0.5 to 9.5 years); the median TTPs were 1.6 and 1.3 years in the lenalidomide-alone and LR arms, respectively.
Table 1.
Key Patient Characteristics
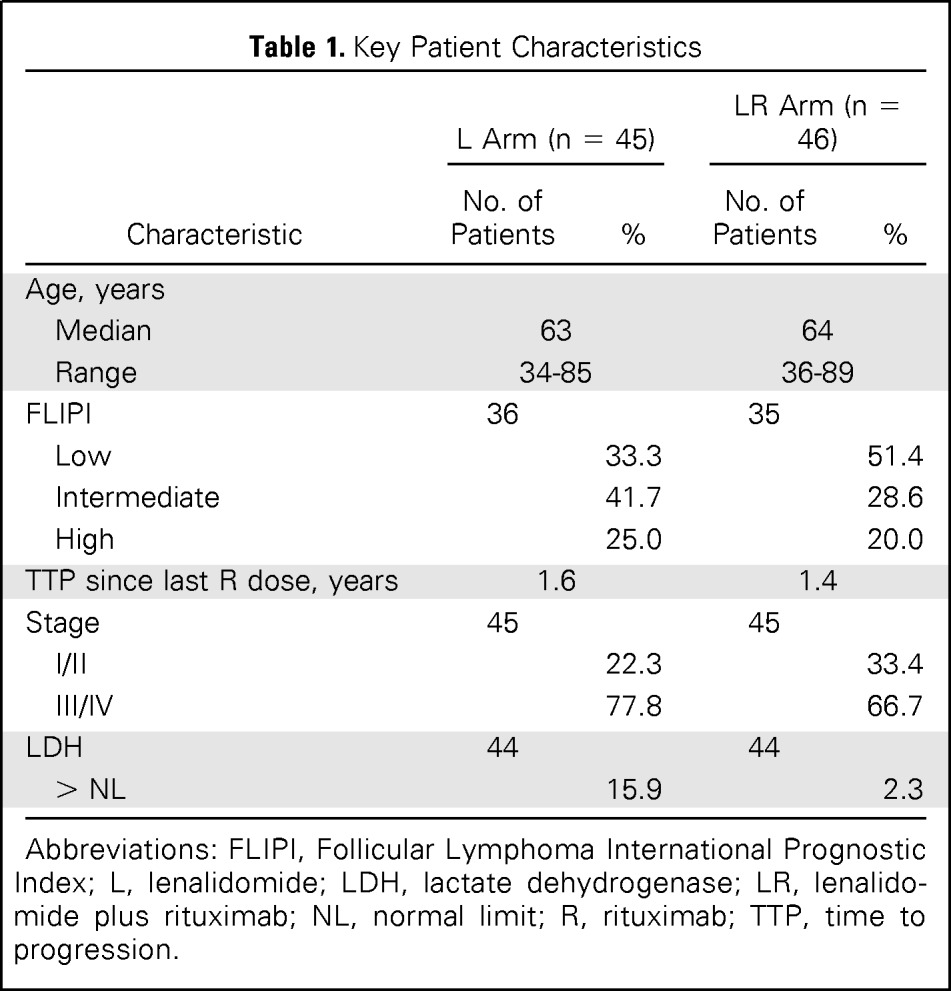
| Characteristic | L Arm (n = 45) |
LR Arm (n = 46) |
||
|---|---|---|---|---|
| No. of Patients | % | No. of Patients | % | |
| Age, years | ||||
| Median | 63 | 64 | ||
| Range | 34-85 | 36-89 | ||
| FLIPI | 36 | 35 | ||
| Low | 33.3 | 51.4 | ||
| Intermediate | 41.7 | 28.6 | ||
| High | 25.0 | 20.0 | ||
| TTP since last R dose, years | 1.6 | 1.4 | ||
| Stage | 45 | 45 | ||
| I/II | 22.3 | 33.4 | ||
| III/IV | 77.8 | 66.7 | ||
| LDH | 44 | 44 | ||
| > NL | 15.9 | 2.3 | ||
Abbreviations: FLIPI, Follicular Lymphoma International Prognostic Index; L, lenalidomide; LDH, lactate dehydrogenase; LR, lenalidomide plus rituximab; NL, normal limit; R, rituximab; TTP, time to progression.
The full treatment course was completed in 36% of patients in the lenalidomide-alone arm and 63% of patients in the LR arm, with the difference caused by more progressions or nonresponders in the lenalidomide-alone arm. In both arms, approximately 20% of patients discontinued therapy early as a result of adverse events, whereas 67% of patients in the lenalidomide-alone arm and 80% of patients in the LR arm had at least one dose modification. However, dose-intensity was greater than 80%, reflecting that reductions were modest and occurred in a minority of cycles. In the lenalidomide-alone and LR arms, grade 3 to 4 adverse events were reported in 58% and 52% of patients, respectively, including neutropenia (16% v 20%, respectively), fatigue (9% v 13%, respectively), and rash (4% v 4%, respectively). Grade 4 adverse events occurred in 9% of patients in the lenalidomide-alone arm and 11% of patients in the LR arm (Table 2). The incidence of thrombosis was compared among treatment arms and occurred in seven patients (16%) in the lenalidomide-alone arm and two patients (4%) in the LR arm (P = .157). Table 3 lists the thrombosis rates in specific risk groups. Nonsignificant trends toward higher rates of thrombosis were observed in groups receiving lenalidomide alone (v LR), those with diabetes, and those on anticoagulation (presumably associated with high-risk status). The sample size and nonrandomized (risk factor–driven) nature of prophylaxis and heterogeneity of the patient population limit correlations of risk factors and prophylaxis with risk of thrombosis.
Table 2.
Grade 3 to 4 Hematologic Adverse Events Occurring in > One Patient
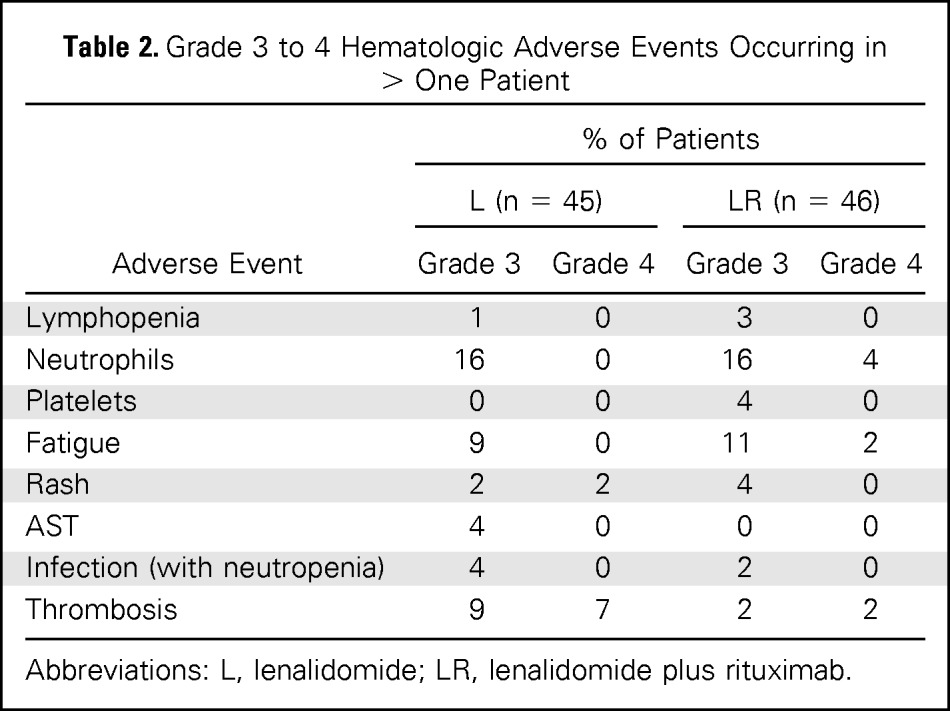
| Adverse Event | % of Patients |
|||
|---|---|---|---|---|
| L (n = 45) |
LR (n = 46) |
|||
| Grade 3 | Grade 4 | Grade 3 | Grade 4 | |
| Lymphopenia | 1 | 0 | 3 | 0 |
| Neutrophils | 16 | 0 | 16 | 4 |
| Platelets | 0 | 0 | 4 | 0 |
| Fatigue | 9 | 0 | 11 | 2 |
| Rash | 2 | 2 | 4 | 0 |
| AST | 4 | 0 | 0 | 0 |
| Infection (with neutropenia) | 4 | 0 | 2 | 0 |
| Thrombosis | 9 | 7 | 2 | 2 |
Abbreviations: L, lenalidomide; LR, lenalidomide plus rituximab.
Table 3.
Thrombosis Risk Factors and Development of Grade 3 or 4 Thrombosis on Treatment
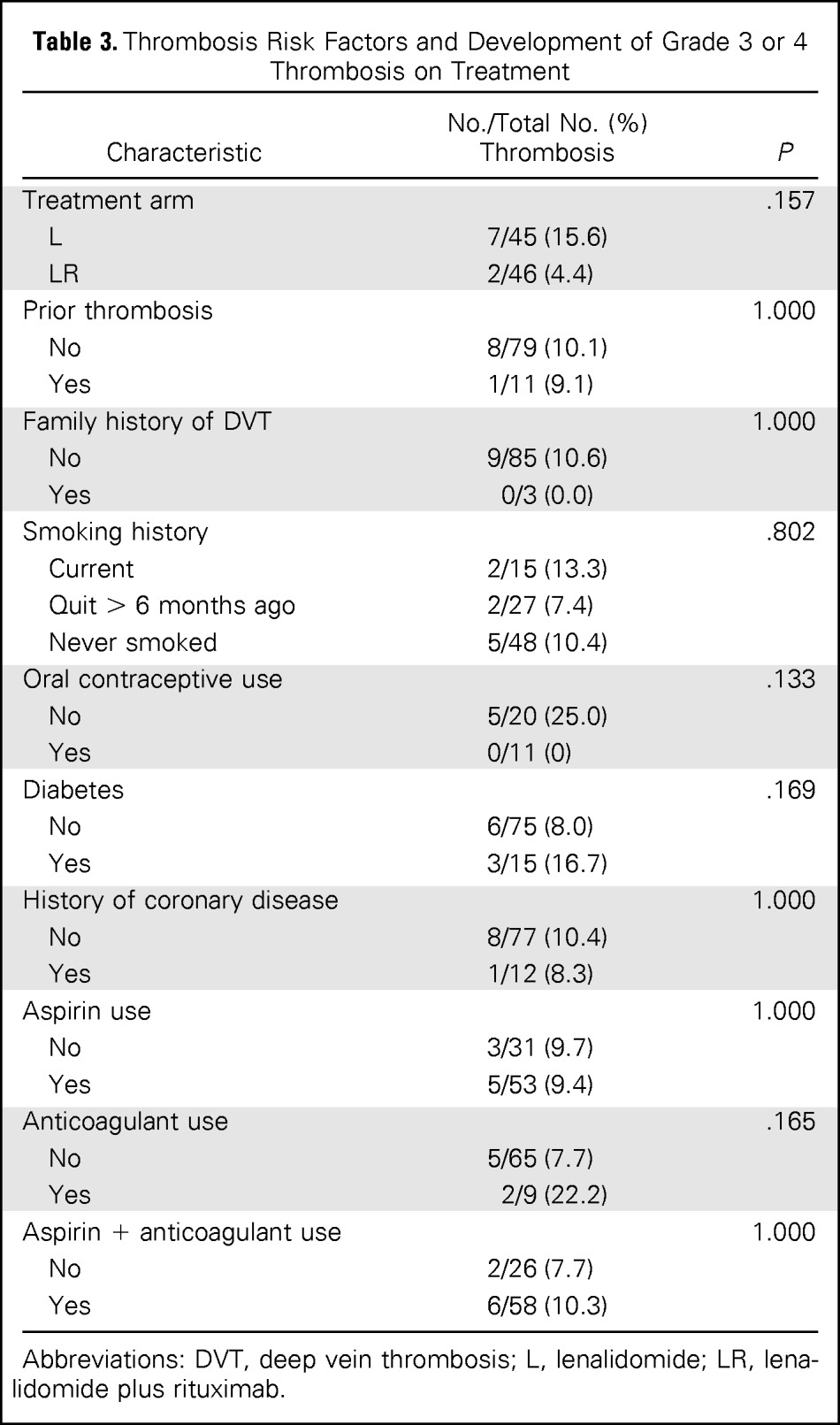
| Characteristic | No./Total No. (%)Thrombosis | P |
|---|---|---|
| Treatment arm | .157 | |
| L | 7/45 (15.6) | |
| LR | 2/46 (4.4) | |
| Prior thrombosis | 1.000 | |
| No | 8/79 (10.1) | |
| Yes | 1/11 (9.1) | |
| Family history of DVT | 1.000 | |
| No | 9/85 (10.6) | |
| Yes | 0/3 (0.0) | |
| Smoking history | .802 | |
| Current | 2/15 (13.3) | |
| Quit > 6 months ago | 2/27 (7.4) | |
| Never smoked | 5/48 (10.4) | |
| Oral contraceptive use | .133 | |
| No | 5/20 (25.0) | |
| Yes | 0/11 (0) | |
| Diabetes | .169 | |
| No | 6/75 (8.0) | |
| Yes | 3/15 (16.7) | |
| History of coronary disease | 1.000 | |
| No | 8/77 (10.4) | |
| Yes | 1/12 (8.3) | |
| Aspirin use | 1.000 | |
| No | 3/31 (9.7) | |
| Yes | 5/53 (9.4) | |
| Anticoagulant use | .165 | |
| No | 5/65 (7.7) | |
| Yes | 2/9 (22.2) | |
| Aspirin + anticoagulant use | 1.000 | |
| No | 2/26 (7.7) | |
| Yes | 6/58 (10.3) |
Abbreviations: DVT, deep vein thrombosis; L, lenalidomide; LR, lenalidomide plus rituximab.
Efficacy data for both arms are listed in Table 4. Among patients receiving lenalidomide alone, 24 (53%) achieved an objective response (nine CRs [20%]), where 35 patients (76%) receiving LR were responders (18 CRs [39%]). The OR rate of patients receiving LR was significantly higher than that of patients receiving lenalidomide alone (P = .029). With a median follow-up time of 2.5 years (range, 0.1 to 4.8 years), median TTP was 1.1 year for lenalidomide alone and 2 years for LR (P = .002, log-rank test; Fig 2). OS was similar in the two arms and was 4.5 years for lenalidomide alone and not reached in LR (P = .149; Fig 3).
Table 4.
Response Rate and Progression-Free Survival
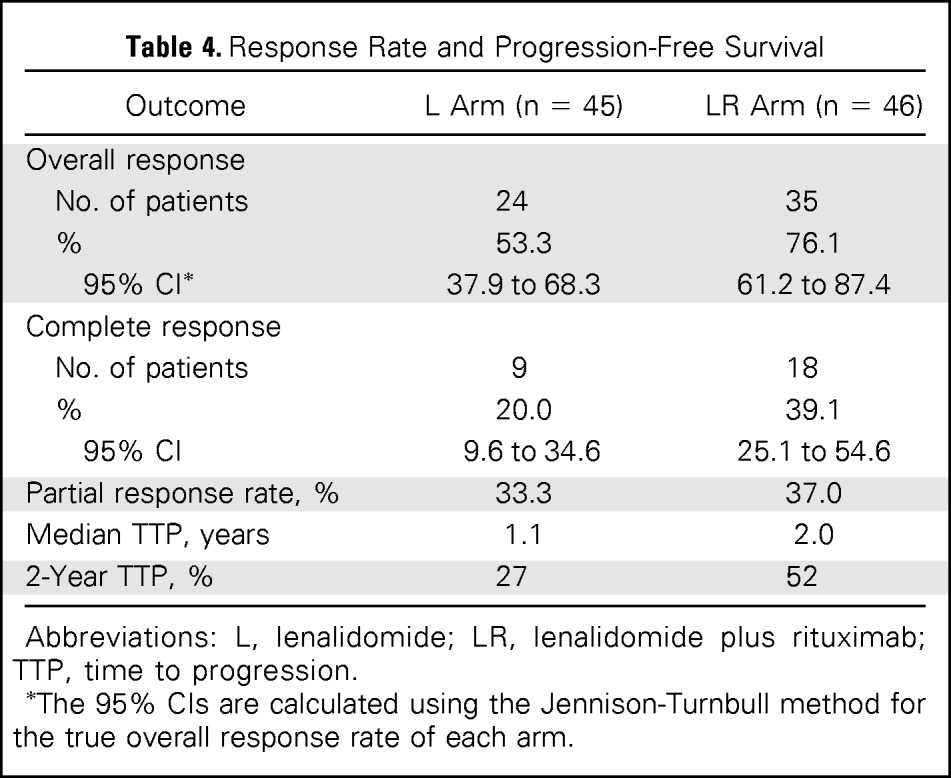
| Outcome | L Arm (n = 45) | LR Arm (n = 46) |
|---|---|---|
| Overall response | ||
| No. of patients | 24 | 35 |
| % | 53.3 | 76.1 |
| 95% CI* | 37.9 to 68.3 | 61.2 to 87.4 |
| Complete response | ||
| No. of patients | 9 | 18 |
| % | 20.0 | 39.1 |
| 95% CI | 9.6 to 34.6 | 25.1 to 54.6 |
| Partial response rate, % | 33.3 | 37.0 |
| Median TTP, years | 1.1 | 2.0 |
| 2-Year TTP, % | 27 | 52 |
Abbreviations: L, lenalidomide; LR, lenalidomide plus rituximab; TTP, time to progression.
The 95% CIs are calculated using the Jennison-Turnbull method for the true overall response rate of each arm.
Fig 2.
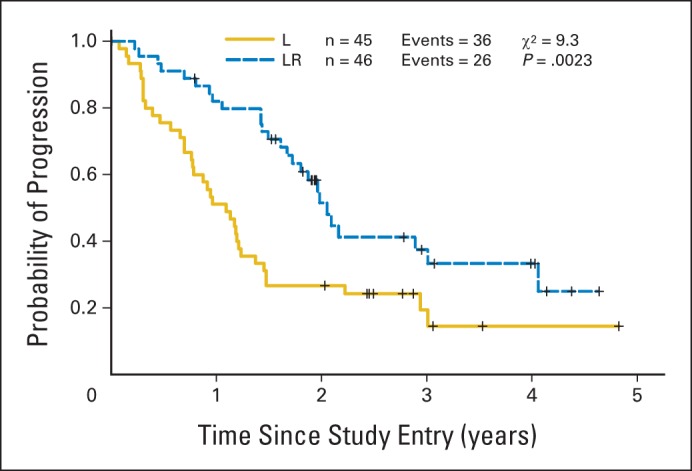
Kaplan-Meier curve for time to progression by treatment arm (arm B = lenalidomide [L], arm C = lenalidomide and rituximab [LR]).
Fig 3.
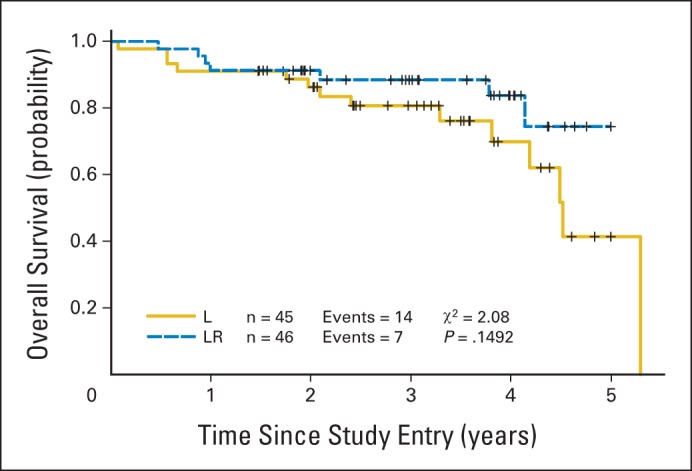
Kaplan-Meier curves for overall survival by treatment arm (arm B = lenalidomide [L], arm C = lenalidomide and rituximab [LR]).
DISCUSSION
CALGB (Alliance) 50401 represents the largest multicenter experience of single-agent lenalidomide in patients with relapsed (non–rituximab-refractory) FL. In 23 patients, largely with rituximab-refractory disease, Witzig et al9 reported a 27% OR rate. We observed a 53% OR rate and median TTP of greater than 1 year in a less refractory population. Furthermore, on the basis of a strong preclinical rationale, we developed the combination of LR to explore the potential of biologic doublets in FL and other lymphomas. Although there are modest differences in the patient characteristics in the two arms, our data demonstrate that the addition of rituximab to lenalidomide in this population significantly increases the OR rate (76%; P = .029) and TTP (2.0 years; P = .002) compared with lenalidomide alone.
This trial helps to establish the safety profile of single-agent lenalidomide in FL, and the randomized nature also allows a direct assessment of potential toxicity resulting from the addition of rituximab to lenalidomide. Both lenalidomide alone and LR were well tolerated, and the principal grade 3 and 4 toxicities included cytopenias, fatigue, and thrombosis. Dose adjustments were not uncommon, but dose-intensity was greater than 80% in both arms. There was no evidence of increased toxicity from the LR combination compared with lenalidomide alone. In fact, the addition of rituximab allowed for a longer duration of therapy (as a result of fewer treatment failures). Interestingly, there was a trend toward less thrombosis in the LR arm, which we speculate may be a result of better lymphoma control, reducing venous obstruction and other risks for clot. It is important to stress that the heterogeneous nature of the patient population with respect to thrombosis risk (and associated use of aspirin and anticoagulants as prophylaxis) makes it difficult to assess the effectiveness of ancillary care measures in preventing this complication.
Since the initiation of this trial, other groups have begun to evaluate the LR combination for indolent lymphoma. To our knowledge, our trial is the first randomized, multicenter cooperative group experience and provides a median follow-up time of 2.5 years (with > 4 years in some patients). The high frequency and durability of responses justify further study of the LR regimen, including as initial treatment. CALGB (Alliance) 50803 is a phase II multicenter trial of LR as first-line therapy for FL.14 Among 54 evaluable patients, the preliminary OR rate was 93% and CR rate was 72%. As in the study reported here, principal toxicities included cytopenias, rash, and fatigue. Similar data have been preliminarily reported in abstract form from a single-institution experience.15 This work has led to the development of the RELEVANCE study (ClinicalTrials.gov identifier: NCT01476787), an international randomized phase III trial of LR versus chemotherapy plus rituximab as initial treatment of FL. In addition, on the basis of our study, the AUGMENT (ClinicalTrials.gov identifier: NCT01938001) international randomized phase III study of LR versus rituximab alone is under way in patients with relapsed/refractory indolent NHL including FL.
Our group believes that the results from CALGB (Alliance) 50401 provide sufficient safety and efficacy data to support the use of LR as a backbone to move toward combination biologic triplet therapy as we explore our next generation of chemotherapy-free regimens in indolent and other lymphomas. One example is Alliance 051103 (ClinicalTrials.gov identifier: NCT01829568), a trial of rituximab, lenalidomide, and the Bruton tyrosine kinase inhibitor ibrutinib in patients with previously untreated FL. Such combinations of targeted agents offer the potential to further improve efficacy while moving us further in the direction of more rationally designed chemotherapy-free therapeutic regimens.
Appendix
The following institutions participated in this study: Georgetown University Medical Center, Washington, DC, Bruce Cheson, MD, supported by Grant No. CA77597; Greenville Community Clinical Oncology Program (CCOP), Greenville, SC, Jeffrey K. Giguere, MD, supported by Grant No. CA29165; Illinois Oncology Research Association, Peoria, IL, John W. Kugler, MD, supported by Grant No. CA35113; Kansas City Community Clinical Oncology Program CCOP, Kansas City, MO, Rakesh Gaur, MD, Mount Sinai Medical Center, Miami, FL, Michael A. Schwartz, MD, supported by Grant No. CA45564; New Hampshire Oncology-Hematology PA, Concord, NH, Douglas J. Weckstein, MD, The Ohio State University Medical Center, Columbus, OH, Clara D. Bloomfield, MD, supported by Grant No. CA77658; Roswell Park Cancer Institute, Buffalo, NY, Ellis Levine, MD, supported by Grant No. CA59518; Southeast Cancer Control Consortium CCOP, Goldsboro, NC, James N. Atkins, MD, supported by Grant No. CA45808; University of California at San Diego, San Diego, CA, Barbara A. Parker, MD, supported by Grant No. CA11789; University of Chicago, Chicago, IL, Hedy L. Kindler, MD, supported by Grant No. CA41287; University of Minnesota, Minneapolis, MN, Bruce A. Peterson, MD, supported by Grant No. CA16450; University of Vermont, Burlington, VT, Steven M. Grunberg, MD, supported by Grant No. CA77406; Wake Forest University School of Medicine, Winston-Salem, NC, David D. Hurd, MD, supported by Grant No. CA03927; Walter Reed Army Medical Center, Washington, DC, David C. Van Echo, MD, supported by Grant No. CA26806; Washington University School of Medicine, St Louis, MO, Nancy Bartlett, MD, supported by Grant No. CA77440; Weill Medical College of Cornell University, New York, NY, John Leonard, MD, supported by Grant No. CA07968.
Footnotes
Supported, in part, by grants from the National Cancer Institute to the Alliance for Clinical Trials in Oncology (Grant No. CA31946; Monica M. Bertagnolli, MD, chair) and to the Alliance Statistics and Data Center (Grant No. CA33601; Daniel J. Sargent, PhD).
Presented in part at the 48th Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, June 1-5, 2012.
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
Clinical trial information: NCT00238238.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: John P. Leonard, Sin-Ho Jung, Bruce D. Cheson
Provision of study materials or patients: Bruce D. Cheson
Collection and assembly of data: John P. Leonard, Sin-Ho Jung, Jeffrey Johnson, Nancy L. Bartlett, Kristie A. Blum, Myron Czuczman, Jeffrey K. Giguere, Bruce D. Cheson
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Randomized Trial of Lenalidomide Alone Versus Lenalidomide Plus Rituximab in Patients With Recurrent Follicular Lymphoma: CALGB 50401 (Alliance)
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
John P. Leonard
Consulting or Advisory Role: Genentech, Celgene
Research Funding: Celgene
Travel, Accommodations, Expenses: Celgene, Genentech
Sin-Ho Jung
No relationship to disclose
Jeffrey Johnson
Research Funding: Celgene
Brandelyn N. Pitcher
No relationship to disclose
Nancy L. Bartlett
Consulting or Advisory Role: Seattle Genetics
Research Funding: Seattle Genetics (Inst), Millennium (Inst), Pfizer (Inst), Pharmacyclics (Inst), MedImmune (Inst), Celgene (Inst), Genentech (Inst), Janssen Research Foundation (Inst)
Kristie A. Blum
No relationship to disclose
Myron Czuczman
Honoraria: Mundipharma
Consulting or Advisory Role: Algeta, Celgene, Teva, Boehringer Ingelheim, Mundipharma, Gilead Sciences, TG Therapeutics, Millennium Takeda, MorphoSys
Jeffrey K. Giguere
No relationship to disclose
Bruce D. Cheson
Consulting or Advisory Role: Celgene, Pharmacyclics, Gilead Sciences, Roche-Genentech
Research Funding: Celgene (Inst), Gilead Sciences (Inst), Pharmacyclics (Inst), Roche-Genentech (Inst), Acerta Pharma (Inst)
REFERENCES
- 1.McLaughlin P, Grillo-López AJ, Link BK, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: Half of patients respond to a four-dose treatment program. J Clin Oncol. 1998;16:2825–2833. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 2.Davis TA, Grillo-López AJ, White CA, et al. Rituximab anti-CD20 monoclonal antibody therapy in non-Hodgkin's lymphoma: Safety and efficacy of re-treatment. J Clin Oncol. 2000;18:3135–3143. doi: 10.1200/JCO.2000.18.17.3135. [DOI] [PubMed] [Google Scholar]
- 3.Coiffier B, Osmanov EA, Hong X, et al. Bortezomib plus rituximab versus rituximab alone in patients with relapsed, rituximab-naive or rituximab-sensitive, follicular lymphoma: A randomised phase 3 trial. Lancet Oncol. 2011;12:773–784. doi: 10.1016/S1470-2045(11)70150-4. [DOI] [PubMed] [Google Scholar]
- 4.Cartron G, Zhao-Yang L, Baudard M, et al. Granulocyte-macrophage colony-stimulating factor potentiates rituximab in patients with relapsed follicular lymphoma: Results of a phase II study. J Clin Oncol. 2008;26:2725–2731. doi: 10.1200/JCO.2007.13.7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leonard JP, Schuster SJ, Emmanouilides C, et al. Durable complete responses from therapy with combined epratuzumab and rituximab: Final results from an international multicenter, phase 2 study in recurrent, indolent, non-Hodgkin lymphoma. Cancer. 2008;113:2714–2723. doi: 10.1002/cncr.23890. [DOI] [PubMed] [Google Scholar]
- 6.Pro B, Leber B, Smith M, et al. Phase II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in combination with rituximab in patients with recurrent B-cell non-Hodgkin lymphoma. Br J Haematol. 2008;143:355–360. doi: 10.1111/j.1365-2141.2008.07353.x. [DOI] [PubMed] [Google Scholar]
- 7.Chanan-Khan AA, Cheson BD. Lenalidomide for the treatment of B-cell malignancies. J Clin Oncol. 2008;26:1544–1552. doi: 10.1200/JCO.2007.14.5367. [DOI] [PubMed] [Google Scholar]
- 8.Ramsay AG, Clear AJ, Kelly G, et al. Follicular lymphoma cells induce T-cell immunologic synapse dysfunction that can be repaired with lenalidomide: Implications for the tumor microenvironment and immunotherapy. Blood. 2009;114:4713–4720. doi: 10.1182/blood-2009-04-217687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Witzig TE, Wiernik PH, Moore T, et al. Lenalidomide oral monotherapy produces durable responses in relapsed or refractory indolent non-Hodgkin's Lymphoma. J Clin Oncol. 2009;27:5404–5409. doi: 10.1200/JCO.2008.21.1169. [DOI] [PubMed] [Google Scholar]
- 10.Reddy N, Hernandez-Ilizaliturri FJ, Deeb G, et al. Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br J Haematol. 2008;140:36–45. doi: 10.1111/j.1365-2141.2007.06841.x. [DOI] [PubMed] [Google Scholar]
- 11.Cheson BD, Horning SJ, Coiffier B, et al. Report of an international workshop to standardize response criteria for non-Hodgkin's lymphomas: NCI Sponsored International Working Group. J Clin Oncol. 1999;17:1244. doi: 10.1200/JCO.1999.17.4.1244. [DOI] [PubMed] [Google Scholar]
- 12.Jung SH, Kim KM. On the estimation of the binomial probability in multistage clinical trials. Stat Med. 2004;23:881–896. doi: 10.1002/sim.1653. [DOI] [PubMed] [Google Scholar]
- 13.Solal-Céligny P, Roy P, Colombat P, et al. Follicular lymphoma international prognostic index. Blood. 2004;104:1258–1265. doi: 10.1182/blood-2003-12-4434. [DOI] [PubMed] [Google Scholar]
- 14.Martin P, Jung S, Johnson J, et al. CALGB 50803 (Alliance): A phase 2 trial of lenalidomide plus rituximab in patients with previously untreated follicular lymphoma. Hematol Oncol. 2013;(suppl 1):31. abstr 63. [Google Scholar]
- 15.Fowler NH, McLaughlin P, Hagemeister FB, et al. Complete response rates with lenalidomide plus rituximab for untreated indolent B-cell non-Hodgkin's lymphoma. J Clin Oncol. 2010;(suppl 15s):28. abstr 8036. [Google Scholar]