

Abstract
It is widely accepted that the increased use of antibiotics has resulted in bacteria with developed resistance to such treatments. These organisms are capable of forming multi-protein structures that bridge both the inner and outer membrane to expel diverse toxic compounds directly from the cell. Proteins of the resistance nodulation cell division (RND) superfamily typically assemble as tripartite efflux pumps, composed of an inner membrane transporter, a periplasmic membrane fusion protein, and an outer membrane factor channel protein. These machines are the most powerful antimicrobial efflux machinery available to bacteria. In Escherichia coli, the CusCFBA complex is the only known RND transporter with a specificity for heavy metals, detoxifying both Cu+ and Ag+ ions. In this review, we discuss the known structural information for the CusCFBA proteins, with an emphasis on their assembly, interaction, and the relationship between structure and function.
Keywords: multidrug resistance, heavy metal resistance, resistance-nodulation-cell division, CusCFBA efflux system
Introduction
Antibiotics have been used to treat infections for over a century. However, it is widely accepted that the increased use of these drugs has resulted in bacteria with developed resistance to such treatments, representing a major public health crisis. Recently, the World Health Organization reported that more than 50% of infections by the common pathogens Escherichia coli, Klebsiella pneumoniae, and Staphylococcus aureus were resistant to routine antibacterial drugs, in many settings.1 The complete failure of both gonorrhea and tuberculosis treatments, due to resistance to all available drugs, is already a reality.1
One of the major means by which bacteria exhibit antibiotic resistance is the expression of multidrug efflux pumps, which detoxify diverse antimicrobials by directly exporting them from the cell.2,3 Antibiotic resistance systems of this type have been found in every bacterial species tested.4 Based on sequence similarity, function, and energy source these pumps can be divided into five families:5.6 (i) the adenosine triphosphate (ATP)-binding cassette (ABC) superfamily7 (TC 3.A.1); (ii) the major facilitator superfamily (MFS)8 (TC 2.A.1); (iii) the small multidrug resistance (SMR) family9 (TC 2.A.7); (iv) the resistance-nodulation-cell division (RND) superfamily10 (TC 2.A.6); and (v) the multidrug and toxic compound extrusion (MATE) family11 (TC 2.A.66).
Gram-negative bacteria, including pathogenic E. coli, K. pneumoniae, Pseudomonas aeruginosa, Streptomyces, Salmonella, and Legionella spp., have been found to contain transporters of all five families.12 These organisms are capable of coordinating multiprotein structures that bridge both the inner and outer membranes to expel toxic compounds directly from the cell.13 Composed of an inner membrane transporter, a periplasmic membrane fusion protein (MFP), and an outer membrane factor (OMF) channel protein, tripartite efflux pumps represent the most powerful antibiotic efflux machinery available to bacteria. They are exemplified by the RND superfamily, members of which are essential to the multidrug resistance observed in many pathogens.14
In E. coli, there are seven known RND proteins that can be divided into two subfamilies. The hydrophobic and amphiphilic efflux RND (HAE-RND) protein subfamily10 includes the multidrug efflux pumps AcrB,15–25 AcrD,19,26 AcrF,19,27,28 MdtB,19,29–31 MdtC,19,29–31 and YhiV.19,32,33 The heavy metal efflux RND (HME-RND) subfamily10,34 contains only CusA.19,35–37
The inner membrane copper and silver ion transporter CusA operates in conjunction with the MFP CusB,38 OMF CusC,39,40 and periplasmic metallochaperone protein CusF41,42 to form one of only two known tetrapartite efflux systems. Among all RND family efflux complexes, CusCFBA is one of only three with structural information for all components, including MexAB-OprM43–46 and AcrAB-TolC.47–50
This review will focus on the known structural information for the CusCFBA efflux system, much of which has been solved in our lab. In addition, the proposed transport mechanisms of the pump will be discussed. While the CusCFBA efflux system has been found to have high specificity for Cu+ and Ag+, in contrast to the broad specificity of HAE-RND proteins, both HAE- and HME-RND family pumps are believed to be key components of antimicrobial resistance in Gram-negative pathogens.51
The Cus Determinant
The first HME-RND protein identified was the cadmium and zinc specified CzcA divalent cation efflux pump.52,53 E. coli CusA was later found to be a putative monovalent cation metal transporter,19,54 which is closely related to SilA of Salmonella typhimurium,55–57 The chromosomally encoded cus locus consists of two back-to-back operons, cusCFBA and cusRS. The cusA gene is preceded by cusB, encoding the MFP, cusF, encoding the small periplasmic metallochaperone, and cusC, encoding the OMF protein. Transcribed in the opposite direction, cusR and cusS encode a two component regulatory system.58 Upstream of cusCFBA is a promoter region with high sequence similarity to other known copper and silver resistance genes in Gram-negative bacteria.52,55,59
Early attempts to characterize these genes led to the conclusion that transcription of the cus locus was strongly dependent on both copper and silver. At similar copper and silver concentrations, transcription levels increased by as much as 30- and 5-fold, respectively.59 However, the growth of E. coli EC756 ΔcusA in copper-containing media was unchanged compared with the wild-type strain. In silver-containing media, the growth of E. coli EC756 ΔcusA, compared with the wild-type, was attenuated for only small amounts of AgNO3. Based on these experiments, it was prematurely concluded that the cus genes played no part in copper resistance in E. coli.
While CusCFBA is unique as the only silver resistance machinery in E. coli, there exists one other chromosomally encoded copper resistance determinant. The cue locus encodes both the inner membrane copper transporter CopA and the periplasmic multicopper oxidase CueO, which detoxify Cu+ ions by actively removing them from the cytoplasm or oxidizing them to less dangerous Cu2+, respectively.60–70 While deletion of cusA alone failed to produce any difference in the copper sensitivity of E. coli,59,60 there was a noticeable difference in the minimum inhibitory concentration (MIC) of CuCl2 for E. coli W3110 copA::km ΔcueO::cm (2.25 mM) compared with W3110 copA::km ΔcueO::cm ΔcusCFBA::cm (1.3 mM).60 Additionally, a striking difference was observed between the aerobic and anaerobic growth of E. coli BW25113 ΔcusR.63 In the absence of oxygen, the growth of this strain was attenuated compared with both the ΔcueO strain and the wild type.
Taken together these results indeed support the role of CusCFBA as a Cu+ transporter. In the presence of oxygen the abundant copper species should be Cu2+, and in an anaerobic environment it should be Cu+. The Cu(I) species is much more toxic than Cu(II), due to its ability to participate in harmful Fenton-type reactions, as well as the increased permeability of the bilayer to Cu+ compared with Cu2+.61,71,72 Thus, the importance of CusCFBA in detoxification of copper ions was most apparent in the absence of oxygen. Whereas the lack of one or more copper resistance machines could be compensated for in the aerobic environment, in the anaerobic environment it could not.63,72 The transcription levels seem to fit this scheme as well. While the aerobic half-maximal induction of cusC was measured at 200 µM, the same expression was achieved at anaerobic copper concentrations of less than half (70 µM).64 It is not surprising that CueO was not implicated in silver resistance51 However, some CopA-type transporters do have the capacity to export Ag+ and Cu2+ ions.
Finally, it was found that deletion of each individual Cus protein produced strains that were unable to restore the copper sensitivity of the wild type.73 The results indicated that each component of the CusCFBA tetrapartite efflux system is necessary for the operation of the pump. In the following sections, the roles of CusA, CusB, CusC, and CusF in the transport of copper and silver ions will be discussed, with emphasis on their assembly, interaction, and the relationship between structure and function.
The Inner Membrane Transporter CusA
Crystal structure of CusA
The crystal structure of CusA (PDB ID: 3K07) was first determined to a resolution of 3.52 Å35 (Fig. 1). Approximately 98% of the 1047 residues (residues 5–504 and 516–1040) were included in the final model, which suggests that CusA exists as a homotrimer. Each monomer of CusA is comprised of 12 transmembrane α-helices (TM1–TM12) and a large periplasmic domain formed by two hydrophilic loops between TM1 and TM2 and between TM7 and TM8, characteristic of the RND superfamily of transporter proteins.74 Like HAE-RND family transporters AcrB,16 MexB,46 or MtrD,75 the structure of CusA is organized into six subdomains. The pore domain is comprised of PN1, PN2, PC1, and PC2, while DN and DC form the docking domain for the CusC channel.
Figure 1.
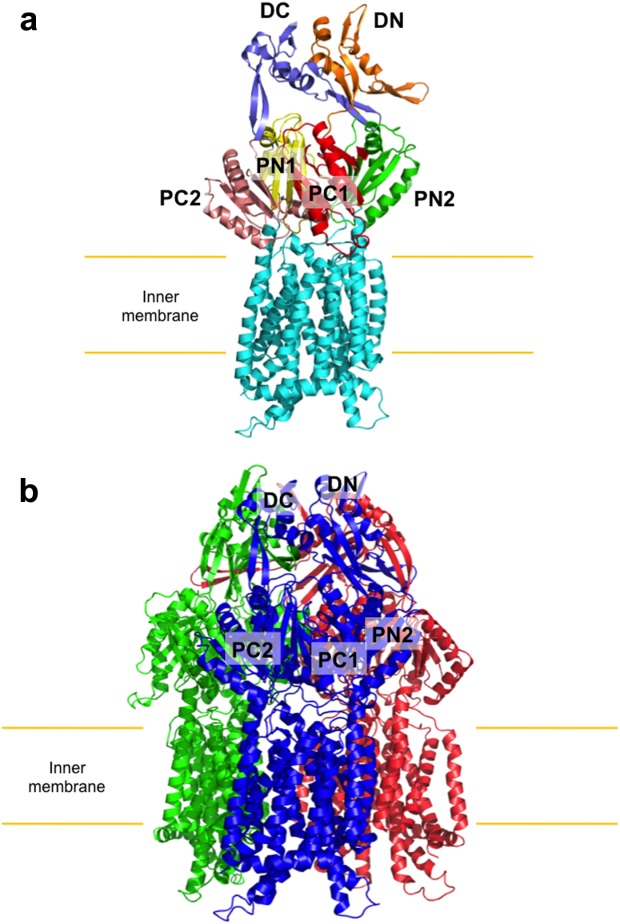
Crystal structure of the CusA transporter. (a) Ribbon diagram of a protomer of CusA. Each domain of CusA is labeled with a different color (cyan, transmembrane helices; yellow, PN1; green, PN2; red, PC1; pink, PC2; orange, DN; slate, DC). (b) Ribbon diagram of the CusA trimer. Each monomer of CusA is labeled with a different color. Subdomains DN, DC, PN2, PC1, and PC2 are labeled for the front subunit (blue), while PN1 is occluded from view.
Interestingly, a cleft in the periplasmic domain, between PC1 and PC2, is covered by one α-helix (residues 690–706) and three β-sheets (residues 681–687, 711–716, and 821–827), which tilt into the cleft to close the opening. At the bottom of this cleft, residues 665–675 form a horizontally oriented α-helix. This structural feature is not seen in the crystal structures of either AcrB, MexB, or MtrD. Sitting atop this helical divider are three adjacent methionine residues (M573, M623, and M672). These methionines were previously identified as conserved residues and appear to cooperate to form a metal ion binding site.66
In addition to the primary three-methionine site, four unique methionine pairs were identified in the crystal structure of CusA. These five methionine pairs/clusters appear to form a relay network to export the bound metal ions. Three of these pairs, M410-M501, M403-M486, and M391-M1009, are found below the primary three-methionine binding site in the transmembrane domain. The fourth, M271-M755, is positioned in the periplasm above the three-methionine site (Fig. 2). Previously, methionine motifs were observed to form metal-binding sites in copper tolerance proteins,76,77 including CusF,42,78 CueR,79 Atx1,80 and CopC.81,82 Thus, these two- and three-methionine clusters in CusA may form a pathway for copper and silver extrusion.
Figure 2.
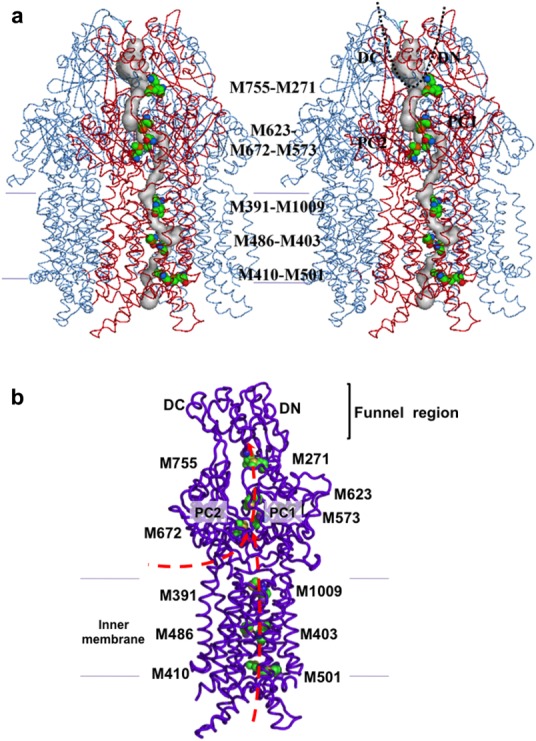
The methionine relay network of CusA. (a) Stereo view of the trimeric CusA pump. Each subunit of CusA independently forms a methionine relay network for exporting metal ions. The methionine relay network is included in the front subunit of the trimer. (b) The methionine cluster (M573, M623, and M672) and four methionine pairs (M271-M755, M401-M501, M403-M486, and M391-M1009) of each monomer of CusA form the metal transport pathway. These residues are depicted as spheres (green, C; orange, O; blue, N; yellow, S). Heavy-metal substrates can enter this pathway through the periplasmic cleft between PC1 and PC2 or through the cytoplasm, illustrated by red arrows.
Additionally, the transmembrane region contains three conserved charged residues, D405 of TM4, E939 of TM10, and K984 of TM11, which are observed to interact with one another. Proteins of the RND superfamily rely on the proton motive force (PMF) for active transport of their substrates.83 These charged residues are expected to be important to the proton relay network of the pump.
In vivo susceptibility assays
To determine the importance of the three-methionine cluster to the operation of the pump, residues M573, M623, and M672 were mutated to isoleucines, individually. When expressed in E. coli BL21(DE3) ΔcueO ΔcusA cells, the single-point mutants, M573I, M623I, and M672I, each produced copper-sensitive strains compared with those expressing wild-type CusA.35,66 Specifically, the MIC of CuSO4 for E. coli cells expressing either M573I, M623I, or M672I (0.50 mM) was found to be much lower compared with those expressing wild-type CusA (2.25 mM). Similar results were observed for susceptibility to silver ions.35
When one methionine in each of the four transmembrane pairs was mutated to isoleucine, individually, the MIC of CuSO4 dropped to 1.25 mM for M391I and 1.75 mM for M410I, M486I, and M755I, compared with 2.25 mM for cells expressing wild-type CusA. Silver tolerance was also clearly attenuated for each mutant strain,35 indicating the importance of these residues in the transport process.
Additionally, the MIC of both CuSO4 and AgNO3 for E. coli carrying the proton relay single-point mutants, D405A, E939A, and K984A, was measured. In each case, the copper and silver tolerance was much less than for cells expressing wild-type CusA and similar to those carrying the empty vector, demonstrating the importance of these charged transmembrane residues.
In vitro transport assays
To investigate the silver transport capabilities of CusA alone, the purified CusA homotrimer was reconstituted into liposomes containing the fluorescent indicator Phen Green SK.35 When Ag+ ions were added into the extravesicular medium, quenching of the fluorescence signal was detected, suggesting the uptake of Ag+ into the intravesicular space.
A stopped-flow transport assay was also employed to determine the importance of the methionine residues, previously found to be essential for copper and silver tolerance.35 Each CusA single-point mutant, M391I, M486I, M573I, M623I, M672I, and M755I, was reconstituted into liposomes encapsulating the same fluorescent indicator. No quenching was detected, and therefore no mutant CusA proteins tested were able to uptake Ag+.
Finally, the importance of the charged transmembrane residues, D405, E939, and K984, for proton translocation was investigated. When reconstituted into liposomes, the mutants, D405A, E939A and K984A, were not observed to uptake Ag+, confirming the importance of these proton relay network residues.35
Crystal structures of CusA-ag+ and CusA-cu+
The crystal structures of copper- and silver-bound E. coli CusA were determined to resolutions of 3.88 Å (PDB ID: 3KSS) and 4.37 Å (PDB ID: 3KSO), respectively.35 The root mean square deviation (RMSD) between the Cu+-and Ag+-bound structures was 1.0 Å, suggesting that these two structures are nearly identical. However, the RMSD between the ligand-bound and apo-CusA structures was 3.9 Å, revealing that metal ion binding triggers a significant conformational change in the pump (Fig. 3).
Figure 3.
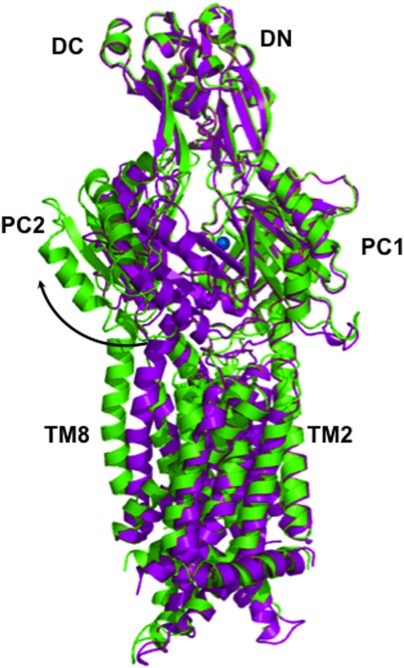
Superposition of apo-CusA (purple) and CusA-Cu(I) (green). The bound Cu(I) is blue. Binding induces a conformational change in the subdomains PC1 and PC2, which creates a 30° opening in the metal-bound structure. The transition from the resting state to the binding state is illustrated by the black arrow.
This deviation is mainly attributed to the cleft between subdomains PC1 and PC2, which creates a 30° opening in both copper- and silver-bound structures, presumably for periplasmic metal ions to enter the pump. This cleft was observed to be closed in the apo-CusA crystal structure, suggesting that the binding site is revealed in the presence of copper or silver and hidden in their absence. Coupled with this 30° swing, the horizontal helix of CusA, residues 665–675, also makes a significant conformational change within the cleft. The C-terminal end of the horizontal helix is found to rotate upwards by approximately 21° in the Cu+-bound structure, compared with the apo-CusA structure. Due to this rotation, residue M672 is moved closer to both residues M573 and M623, which form the transient three-methionine metal-binding site. In addition to exposing this three-methionine binding site to the periplasm, the overall conformational change also more closely coordinates each residue (M573, M623, and M672). Cu+ or Ag+ was found to bind to this same site in each structure (Fig. 4).
Figure 4.
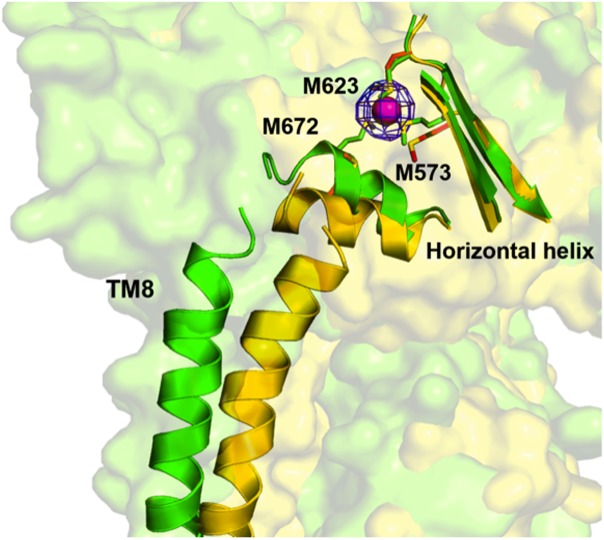
The conformational change induced by binding in the horizontal helix and TM8 of CusA. The apo-CusA structure (yellow) and Cu(I)-bound (green) CusA are superimposed, together with the bound Cu(I) (pink). The blue mesh indicates the anomalous signal of bound Cu(I) (contoured at 8 σ). In the proximity of copper, residues M573, M623, and M672 are closely coordinated and this transient binding site is revealed to the periplasm.
In response to binding, all α-helices in the transmembrane region, except TM2, shift horizontally by as much as 4 Å. Further, TM1, TM3, and TM6 shift toward the periplasm by approximately one turn. Thus, PN1 and the central pore helix also move upward by one turn upon metal binding.
Based on these structures, the dynamics of metal ion transport by CusA were calculated using the elastic network model.35,84 The results suggest that each CusA monomer cycles through three distinct and coupled motions in which the periplasmic binding cleft alternates between the open and closed states, similar to AcrB17,20–22,85,86
The Periplasmic Adaptor CusB
Crystal structure of CusB
The crystal structure of CusB was solved to resolution of 3.40 Å, with 78.1% of the total residues (89–385) included in the final model (PDB ID: 3OOC)38 (Fig. 5). The asymmetric unit contains two molecules of CusB (A and B) that are observed to adopt two distinct conformations, highlighting the flexibility of this protein. Superimposition of molecules A and B results in an overall RMSD of 2.6 Å, which suggests that these structures represent distinct conformational states.
Figure 5.

Crystal structure of the CusB membrane fusion protein. (a) Two distinct conformations of CusB were observed in the crystal structure (gold, purple). (b) Each CusB monomer can be divided into four domains: three β-stranded and one α-helical.
The overall fold of each CusB molecule can be divided into four domains: three β-stranded and one α-helical. The first β-domain (membrane proximal domain) is formed by the N-terminal residues 89 to 102, which comprise one β-strand, and the C-terminal residues 324 to 385, which comprise the remaining five β-strands. Domain 1 in molecules A and B of CusB is almost identical.
The second β-domain (β-barrel domain) of CusB is formed by residues 105 to 115 and 243 to 320 of each molecule. In molecule A, this domain consists of six β-strands and one short α-helix, with the N-terminal residues forming one β-strand and the C-terminal residues forming the remaining β-strand, α-helix, and four-stranded anti-parallel β-sheet.
Domain 3 (lipoyl domain) is another globular β-domain, which consists of residues 121 to 154 and 207 to 239. These residues form an eight β-stranded barrel, which is similar in both molecules A and B.
Domain 4 (α-helical domain) highlights the uniqueness of CusB. Comprising residues 156 to 205, this domain is all α-helical, with three α-helices folding into an antiparallel three-helix bundle. This structural feature has not been found in any other MFP family protein. The three-helix bundle is approximately 27 Å long, which is at least 20 Å shorter than the two α-helical hairpin domains of MexA44,45,87 and AcrA.48 Both molecules A and B form a similar structure.
While the overall RMSD between molecules A and B was 2.6 Å, each domain in its respective structure formed a similar structure. When domains 1 and 2 of each molecule were superimposed, it resulted in an RMSD of only 0.8 Å. Similarly, the superimposition of domains 3 and 4 of A and B resulted in an RMSD of 0.8 Å. Thus, the difference between each conformation can be summarized as a shift of domains 1 and 2 with respect to 3 and 4 by approximately 20°, about a hinge between domains 2 and 3. This movement is expected to result in a transition from the open conformation of molecule A of CusB to the closed conformation of molecule B.
In vitro binding assays
Extended X-ray absorption fine structure (EXAFS) spectroscopy was used by Bagai et al.88 to identify potential metal ion binding sites of CusB. For copper-loaded CusB, the best fit to the data was obtained for three Cu-S interactions. Because CusB contains no cysteine residues, copper was assumed to be bound by three methionine residues in this experiment, as in CusA.35 Alignment of CusB among similar sequences showed that, of the nine methionines in the mature sequence of CusB, only four (M21, M36, M38, and M283) are conserved.
To determine which three of these four methionines are involved in metal binding, isothermal titration calorimetry (ITC) was employed.88 Each methionine residue was mutated to isoleucine, individually, and the ability of the CusB single-point mutants (M21I, M36I, M38I, and M283I) to bind Ag+ was tested in vitro. In summary, CusB M21I showed a 10-fold reduction in the binding affinity for Ag+, compared with wild-type CusB, mutants M36I and M38I showed no specific binding to Ag+, and the affinity of CusB M283I for Ag+ was the same as the wild-type, with an equilibrium dissociation constant (Kd) of 24.7 nM, indicating that the residues M21, M36, and M38 may comprise a specific three-methionine copper and silver binding site for CusB.88
In vivo susceptibility assays
To determine the importance of these residues to bacterial metal resistance, the CusB mutants M21I, M36I, M38I, and M283I were expressed in the E. coli strain EC950 ΔcueO ΔcusB.88 The MICs of CuCl2 for E. coli expressing the CusB single-point mutants M21I, M36I, M38I, and M283I were approximately 1 mM, compared with 1.5 mM for those cells expressing the wild-type and 0.75 mM for those carrying the empty vector. However, E. coli cells expressing CusB M283I were observed to survive in slightly higher copper concentrations compared with the other three,88 further establishing the importance of residues M21, M36, and M38 for the full operation of the Cus system.
The Adaptor-Transporter Complex CusBA
Crystal structure of CusBA
The first co-crystal structure of an adaptor-transporter complex, CusBA, was solved to a resolution of 2.90 Å (PDB ID: 3NE5), including approximately 91% of the total residues (4–1,043 of CusA, 79–400 of CusB molecule 1, and 79–402 of CusB molecule 2)36 (Fig. 6). Three molecules were present in the asymmetric unit, revealing that each CusA monomer interacts with two monomers of CusB. These two CusB molecules (molecule 1 and molecule 2) were found to represent two distinct conformations of CusB. Surprisingly, these two conformations were also distinct from the previously solved structures of CusB.38
Figure 6.
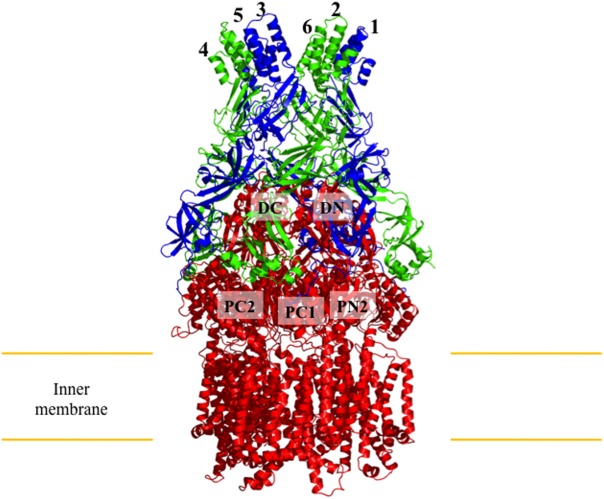
Co-crystal structure of the CusBA adaptor-transporter complex. Three monomers of CusA (red) and six molecules of CusB (blue, molecule A; green, molecule B) form the CusBA efflux complex. The subdomains DC, DN, PC1, PC2, and PN2 of CusA are labeled for the front monomer, while PN1 is occluded from view.
The two CusB molecules are oriented at an angle of approximately 50° to the membrane, and form contacts with the periplasmic domain of CusA via domains 1 and 2. CusB molecule 1 interacts with CusA regions PN2, PC1, and DN, while molecule 2 primarily interacts with PC1, PC2, and DC of the pump. The nature of the interaction between CusB molecule 1 and CusA is electrostatic, with charged residues K95, D386, E388, and R397 of CusB forming salt bridges with charged residues D155, R771, R777, and E584 of CusA, respectively. The interaction between CusB molecule 2 and CusA is dipole-dipole and charge-dipole in nature. Residues Q108, S109, S253, and N312 of CusB form hydrogen bonds with Q785, Q194, D800, and Q198 of CusA, respectively. The interactions between neighboring CusB molecules are primarily in domains 1 to 3, with residues E118, Y119, R186, E252, and R292 of CusB molecule 1 forming hydrogen bonds with T139, D142, T206, N312, and N113 of molecule 2, respectively.
The crystal structure suggests that the trimeric inner membrane CusA pump should interact with six periplasmic CusB molecules, forming a hexameric funnel above the periplasmic domain of CusA. Based on the CusBA complex structure, we have predicted that the complete tripartite efflux assemblage should be in the form of CusC3-CusB6-CusA3 (Fig. 7), which spans the entire bacterial cell envelope to export Cu+/Ag+.36 This assembly is in good agreement with the work of others.50,89–91
Figure 7.
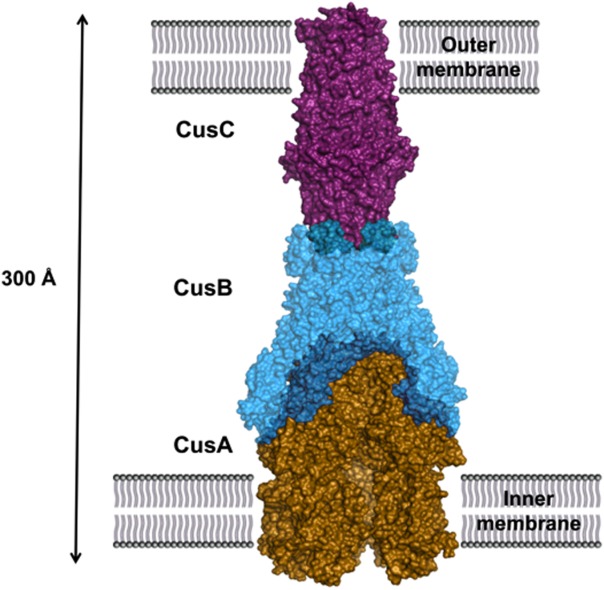
Predicted assembly of the complete CusCBA efflux complex. The pump is shown as a surface rendering of trimeric CusA (gold), hexameric CusB (cyan), and trimeric CusC (purple). The tripartite pump completely spans the inner- and outermembranes of E. coli to expel toxic Cu(I) and Ag(I).
Within the CusBA complex structure, domain 1 and the lower half of domain 2 of each CusB molecule create a cap-like structure, while domain 3, domain 4, and the upper half of domain 2 form the central channel, which is contiguous with that of CusA. Thus, the interior of the channel gives rise to a large elongated cavity with a volume of approximately 65,000 Å.3 The inner surface of this channel is strikingly electronegative, which suggests the capacity to bind positively charged metal ions.
The three CusB methionines (M21, M36, and M38) which were proposed to form a copper and silver binding site cannot be identified in the electron density map of the co-crystal. However, the N-terminal tails of CusB molecules 1 and 2 of CusB can be seen outside the cleft formed between PC1 and PC2 of the CusA pump, which harbors the three-methionine metal-binding site of CusA. It is possible that CusB might help to transfer the metal ions from its N-terminal three-methionine binding site into the CusA pump for extrusion. However, a recent study demonstrated that CusB is not able to transfer metal ions to the CusA pump.92 The role of CusB is more likely to switch the conformation of CusA, allowing this pump to accept ions from the CusF periplasmic chaperone.92 Interestingly, it was also shown that the CopA ATPase is capable of shuttling Cu+ to CusF,93 providing an evidence that both CusA and CopA may work together to clear up Cu+ ions.
Crystal structures of CusBA-cu+
The crystal structures of the Cu+-bound CusBA complex reveal three distinct binding conformations (forms I, II, and III)37 (Fig. 8). The form I structure of Cu+-bound CusBA was solved to a resolution of 3.42 Å (PDB ID: 3T56). In this structure, the Cα chain of residues 664 to 717 and 814 to 888, which forms the horizontal helix TM8 in CusA, displays two distinct conformations (forms Ia and Ib). The occupancy of forms Ia and Ib in the crystal lattice is approximately 0.22:0.78, form Ia:Ib.
Figure 8.
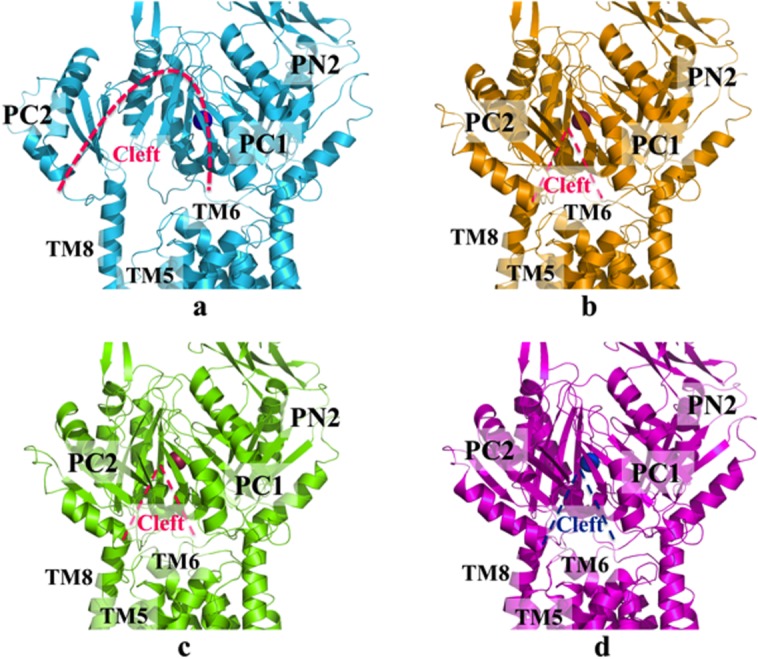
Crystal structures of the CusBA-Cu(I) efflux complex. (a) Form Ia of the crystal structure has been designated as the “pre-extrusion 1” state (cyan), with bound copper (purple). (b) Form Ib of the crystal structure has been designated as the “pre-extrusion 2” state (orange), with bound copper (red). (c) Form II of the crystal structure has been designated as the “pre-extrusion 2” state (green), with bound copper (red). (d) Form III of the crystal structure has been designated as the “extrusion” state (magenta), with bound copper (blue). Molecules of CusB are not shown. In each state, the opening of the periplasmic cleft is depicted by the dashed line.
As in the metal-bound conformation of CusA,35 the periplasmic cleft of CusA is found open in the form Ia CusBA structure. However, the conformation of CusA in form Ib is closer to the apo-CusA structure, in which the periplasmic cleft is closed. In both forms Ia and Ib, a single bound Cu+ is found to coordinate CusA residues M573, M623, and M672, which forms the main three-methionine binding site inside the periplasmic cleft of CusA. In comparison with the structure of the binding state, the horizontal helix (residues 665–675) of form Ib is tilted approximately 10° downward. This rotation shifts M672 away from M573 and M623, partially disassembling the three-sulfur metal coordination site. Further rotation may release the bound metal ion from this site. While it is less obvious in the form Ia structure, this change can be similarly interpreted as a 3° downward tilt. In contrast to the structure of apo-CusBA, no continuous channel is created by either forms Ia and Ib37 as indicated by the program CAVER. Therefore, these conformations have been designated as the pre-extrusion 1 (form Ia) and pre-extrusion 2 (form Ib) states.
The crystal structure of form II of the CusBA-Cu+ complex was solved to a resolution of 3.90 Å (PDB ID: 3T51).37 The overall conformation of this structure is almost identical with the pre-extrusion 2 state. The program CAVER illustrates that the channel formed by the methionine relay network of each protomer of CusA is closed in this conformation. The only cavity that can be identified is the periplasmic cleft, which houses the three-methionine binding site formed by M573, M623, and M672. However, this binding conformation is quite distinct from that of CusA-Ag+ or CusA-Cu+.35 In addition to the changes induced by the pre-extrusion 2 state, a cluster of conserved charged residues, including R83, E567, D617, E625, R669, and K678, is found nearby the bound copper ion. These conserved charged residues are intriguingly positioned along the transmembrane methionine relay network of CusA.
The crystal structure of form III of CusBA-Cu+ was solved to a resolution of 3.30 Å (PDB ID: 3T53).37 The conformation of CusA in this structure is similar to that of apo-CusA. However, a careful inspection of the structures suggests that there are concerted changes in the conformation of CusA that involve several groups of residues. Upon binding to CusA, a short C-terminal helix (residues 391–400) of molecule 1 of CusB seems to press down a helix (residues 582–589) located at the upper half of PC1 of CusA. Consequently, the N-terminal end of the helix tilts downwards by approximately 8° in the form III structure compared with that of apo-CusA. The motion of these helices is concerted with another loop (residues 609–626) that tilts downward, potentially widening the channel for metal transport. Thus, form III has been designated as the extrusion state of the pump.
Superimposition of subdomain PC1 and the horizontal helix of CusA for each state reveals a sequential conformational change during metal transport (Fig. 9). This change can be interpreted as a 10° downward swing of the horizontal helix upon binding copper or silver, from the binding state to the pre-extrusion 1 state.37 This motion shifts M672 away from both M573 and M623, disassembling the three-sulfur metal coordination site and allowing for release of the Cu+/Ag+ into the pump for eventual extrusion.
Figure 9.
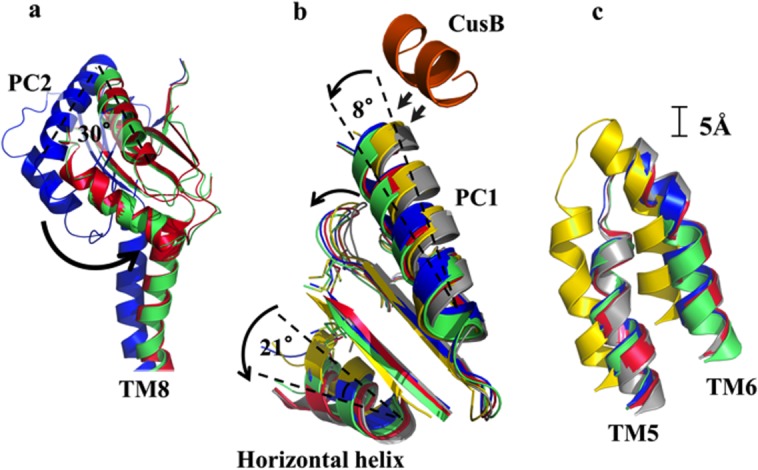
Superimposition of each crystal structure of the CusBA-Cu(I) complex. (a) Superimposition of subdomains PC2 and TM8 of CusA. The 30° rotation of PC2 upon transition from the “pre-extrusion 1” state (blue), through the “pre-extrusion 2” state (green) to the “extrusion” (red) state is depicted by the black arrow. (b) Superimposition of subdomains PC1 and the horizontal helix of CusA. The C-terminal end of the horizontal helix is observed to rotate by 21° upon binding (gray, apo-CusBA; yellow, CusA-Cu(I); blue, form Ia; green, form II; magenta, form III). The C-terminal residues 391 to 400 of molecule 1 of CusB (orange) are also included. (c) Superimposition of TM5 and TM6 of CusA. Upon binding, these subdomains are observed to shift toward the periplasm by approximately one turn (gray, apo-CusBA; yellow, CusA-Cu(I); blue, form Ia; green, form II; red, form III). The structure of form Ib, which is nearly identical to that of form II, has not been included.
In vivo susceptibility assays
Based on the CusBA-Cu+ structures, six charged residues (R83, E567, D617, E625, R669, and K678) were identified in a line along the extrusion pathway of the pump (Fig. 10). To determine whether these residues were important for the operation of the pump, they were mutated to alanines, individually, and expressed in the E. coli strain BL21(DE3) ΔcueO ΔcusA.36 When expressed in a copper environment, each of these CusA single-point mutants was entirely unable to relieve the copper sensitivity of E. coli. The MICs of CuSO4 for E. coli expressing these single-point mutants was similar to those carrying the empty vector (0.5 mM), and much attenuated compared with E. coli which expressed the wild-type pump (2.25 mM). Thus, these residues are necessary to confer full copper resistance.
Figure 10.
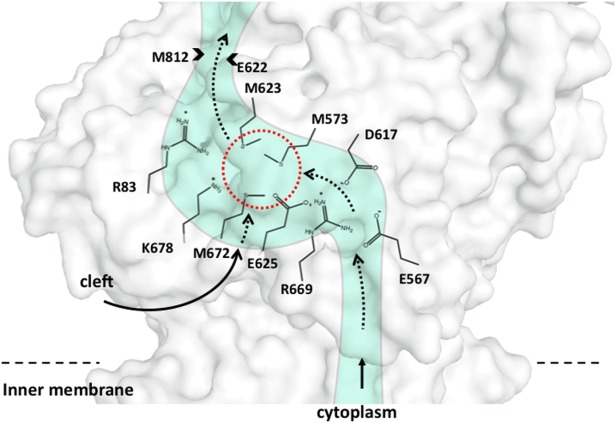
Schematic representation of the metal transport pathway of a subunit of CusA. The conserved charged residues R83, E567, D617, E625, R669, and K678 of a subunit of CusA are labeled. This figure also includes the three-methionine binding site formed by M573, M623, and M672 (dashed red circle) within the subunit. The direction of metal transport is indicated by the black arrows.
In vitro transport assays
To investigate whether the line of charged residues in CusA were essential for metal ion transport, the purified R83A, E567A, D617A, E625A, R669A, and K678A mutant proteins were reconstituted, individually, into liposomes containing the fluorescent indicator Phen Green SK, and a stopped-flow transport assay was used to determine whether the mutant proteins can capture metal ions from the extravesicular medium.36 When Ag+ ions were added into the extravesicular medium, no quenching of the fluorescence signal was detected in each case, in contrast to the results for the wild-type CusA pump. These results suggest that these mutant transporters are unable to uptake Ag+ and that these conserved charged residues are critical for the function of the transporter.
The stopped-flow assay was also used to measure the transport activity of the wild-type CusBA complex.37 The results suggest that the CusBA is more active than CusA alone, as indicated by an approximate 50% attenuation of the fluorescent signal.
Crystal structure of CusB-D405A-cu+
The crystal structure of the mutant complex CusB-D405A-Cu+ was solved to a resolution of 3.10 Å (PDB ID: 4DNT).37 Previously, three conserved charged residues (D405, E939, and K984) of CusA were identified as part of the proton relay network of the pump, and their mutation resulted in loss of transport activity.35,73 Thus, the D405A mutant was chosen to produce the CusB-D405A-Cu(I) co-crystal. The overall structure of CusB-D405A-Cu+ resembles that of apo-CusBA. Superimposition of these two structures resulted in an RMSD of only 0.2 Å. Surprisingly, no copper signal was detected in the crystal, indicating that the D405A mutant is not capable of binding Cu+.
Crystal structure of CusB-R669A-cu+
The crystal structure of the mutant complex CusB-R669A-Cu+ was solved to a resolution of 4.20 Å (PDB ID: 4DOP).37 Residue R669 of CusA was identified as one of six intriguingly positioned residues lining the inner channel of the pump (R83, E567, D617, E625, R669, and K678) and was found to be critical for its function.37 Thus, the CusA mutant R669A was used to produce the co-crystal CusB-R669A-Cu+. Similar to CusB-D405A-Cu+, no copper signal was detected. The overall structure is nearly identical with that of apo-CusBA. Superimposition of the two resulted in an RMSD of only 0.3 Å.
Given the fact that they are incapable of binding copper, it is likely that the CusA mutants D405A and R669A are trapped in one of the transient states of the pump and unable to change conformation to adopt the binding state. Based on their similarity to the apo-CusBA structure, these CusB-mutant CusA structures should correspond to the resting state of the pump. Taken together, these experiments support our hypothesis that the Cus system undergoes sequential transitions during the extrusion process, from the resting state to the binding, pre-extrusion 1, pre-extrusion 2, and extrusion states37 (Fig. 1).
Figure 11.
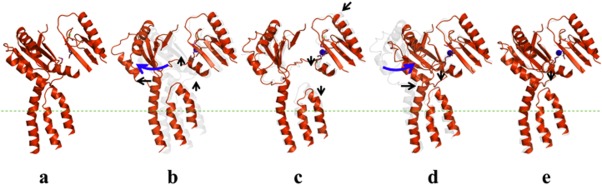
Sequential conformational changes of the CusBA complex. To transport metal ions, this pump transitions from the “resting” state through the “extrusion” state (a, “resting” state; b, “binding” state; c, “pre-extrusion 1” state; d, “pre-extrusion 2” state; e, “extrusion” state). Subdomains PC1, PC2 and the helices TM5, TM6, and TM8 of CusA are shown for each state with the bound copper ion (blue). The change in conformation from the previous state is depicted by the black and blue arrows.
The Outer Membrane Channel CusC
Crystal structure of CusC
The crystal structure of wild-type CusC was determined to a resolution of 2.09 Å (PDB ID: 4K7R)39 (Fig. 2). Previously, the structure had been solved by Kulathila et al. 40 to a resolution of 2.30 Å (PDB ID: 3PIK). These two structures are nearly identical. Superimposition of the two results in an RMSD of 0.28 Å over 429 Cα atoms.39
Figure 12.
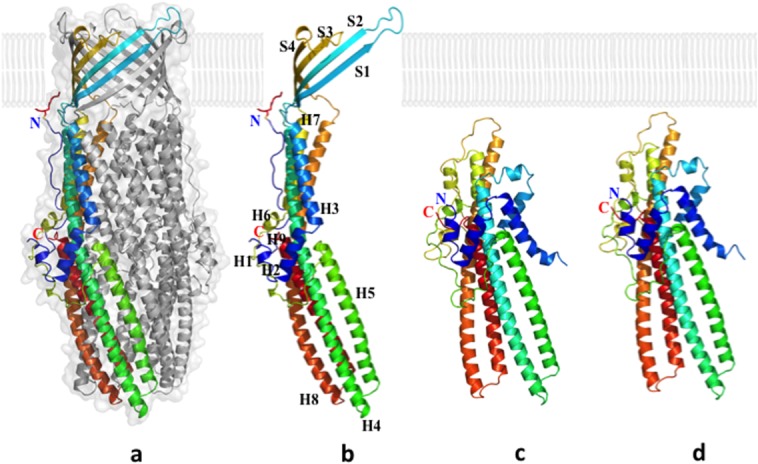
Crystal structures of the CusC outer membrane channel. (a) One monomer of the CusC trimer is shown in rainbow colors, while the other two monomers are depicted in gray. (b) Each CusC monomer (rainbow) can be divided into four β-strands and nine α-helices. The CusC protomer is acylated (red) through the residue C1 to anchor to the outer membrane. (c) Structure of the monomeric ΔC1 mutant. (d) Structure of the monomeric C1S mutant. The mutant structures are seen to adopt a dramatically different, partially folded conformation, compared with the wild-type CusC.
The overall structure of CusC suggests that it forms a homotrimeric α/β barrel, approximately 130 Å long, with a large cylindrical internal cavity, approximately 28,000 Å3 in volume. Like TolC,47 each CusC monomer can be divided into four β-strands: S1 (residues 84–94), S2 (104–116), S3 (290–301), and S4 (313–322). In the periplasmic domain of CusC, nine α-helices from each protomer contribute to form an elongated α-barrel: H1 (residues 36–41), H2 (43–56), H3 (58–80), H4 (121–188), H5 (193–230), H6 (242–246), H7 (256–286), H8 (329–397), and H9 (403–437).
The cannon shape of the CusC trimer is similar to other proteins of the OMF family, including TolC,47 OprM,43 MtrE,94 and CmeC.95 Many outer membrane channels appear to be interchangeable. For example, the E. coli genome contains over 30 inner membrane transporters of the RND, MFS, and ABC superfamilies, but only four OMF family proteins, including TolC.96 However, TolC was unable to replace CusC in the CusCBA complex.73 Thus, CusC is believed to work only within the Cus efflux system. Its irreplaceability may be due to the unique overall structure of CusB compared with other members of the MFP family.38
Although CusC is highly specific to the CusCFBA system, there is no evidence of copper or silver specificity in the channel. When the apo-CusC crystals were prepared in copper or silver concentrations exceeding 10 mM, no bound metal ions were detected.40 Whereas CusA has been shown to bind copper and silver using a three-methionine motif, the five methionines in CusC are far remote from one another in the crystal structure of apo-CusC. Thus, it is unlikely that CusC possesses any specificity for these metal ions. As seen in the electrostatic charge distribution, the interior surface of the trimeric CusC channel is strikingly electronegative, similar to that of the hexameric CusB channel within the CusBA complex.36,37 The electrostatic gradient created by the internal surfaces of the CusB hexamer and CusC trimer may provide a possible mechanism for metal ion transport.14,51,97,98
Interestingly, these CusC structures also reveal that the first N-terminal cysteine residue (C1) is covalently bonded to an outer membrane lipid via a thioester bond,39,40 securing the anchoring of the trimeric CusC channel to the outer membrane.
Crystal structure of ΔC1 CusC
As we suspected that the C1 residue of CusC might play an important role in the protein-membrane interaction, we decided to remove this residue to create the ΔC1 mutant channel. The crystal structure of the ΔC1 CusC mutant was determined to a resolution of 2.53 Å (PDB ID: 4K7K) (Fig. 2), with all residues included in the final model except 21 to 31 and 99 to 129, which formed disordered regions.39 The overall structure of ΔC1 CusC was quite distinct from that of wild-type CusC. Superimposition of these two structures resulted in an RMSD of 22.49 Å over 378 Cα atoms (Fig. 3). The most significant differences can be seen in the amino acids that form the four transmembrane β-strands (S1–S4) in the wild-type structure. These residues adopt a dramatically different conformation in ΔC1 CusC. The majority of these residues in the ΔC1 channel appear to be unstructured and form two large random loops. Residues 290 to 326, which form the transmembrane β-strands S3 and S4 of wild-type CusC, are found to flip down to the outermost surface of the periplasmic α-helical tunnel. Additionally, the top portion of the periplasmic α-helices H7 and H8 are found to bend downward to accommodate for the change. Residues N276 of H7 and N334 of H8 appear to form hinges for the bending.
Figure 13.
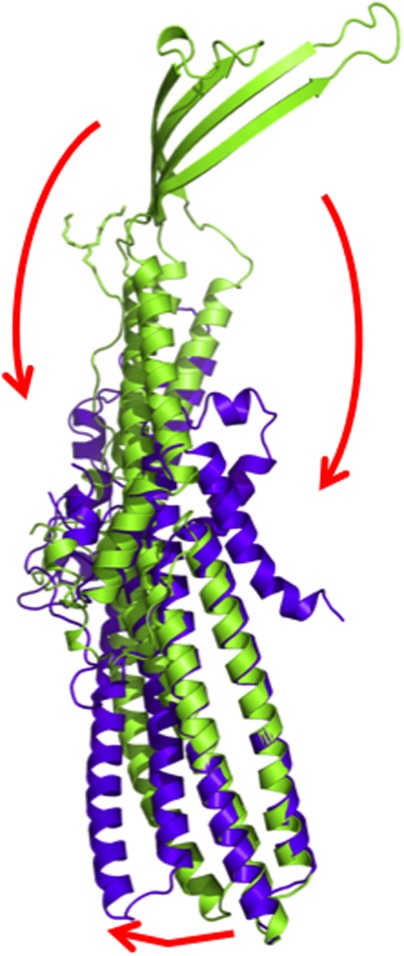
Superimposition of a monomer of wild-type CusC onto that of the ΔC1 mutant. The structures of the wild-type CusC and ΔC1 protomers are colored green and purple, respectively. The arrows indicate the drastic changes in positions and secondary structures when comparing the conformations of the wild-type and ΔC1 CusC.
Interestingly, the drastic conformational changes as described above are accompanied by the structural changes of residues 84 to 116. These residues form the transmembrane β-strands S1 and S2 of wild-type CusC. In the ΔC1 mutant, the electron density map in this region is very unclear, suggesting that the majority of the secondary structure of this area is disordered.39 Thus, this region also creates a large random loop in the ΔC1 mutant. Similar to H7 and H8, the top portion of the periplasmic α-helices H3 and H4 also bend downward about a hinge created by residues N76 and T137.
The periplasmic domain of the ΔC1 CusC mutant is largely unchanged compared with that of the wild-type protein. However, the symmetry of the ΔC1 CusC crystal suggests that ΔC1 CusC is monomeric, compared with the trimeric wild-type CusC channel. This observation was confirmed by gel filtration assays.39
Crystal structure of C1S CusC
We also mutated this C1 residue to a serine to produce a C1S mutant channel. The crystal structure of the single-point mutant C1S CusC was determined to a resolution of 2.69 Å (PDB ID: 4K34)39 (Fig. 2). This structure was almost identical to that of the ΔC1 CusC mutant. Superimposition of these two structures resulted in an RMSD of only 0.37 Å over 381 Cα atoms. Taken together, the structures of ΔC1, C1S, and wild-type CusC provide molecular details of the assembly and folding of the CusC outer membrane channel, similar to the OMF family protein OmpA.99–102
In the aqueous phase, before encountering the outer membrane, the β-barrel domain of OmpA is unstructured. Interaction between the aqueous phase and the outer membrane leaflet produces the molten intermediate IM1.101 However, similar to ΔC1 and C1S CusC, the protein is still highly disordered. After membrane association, two more sequential intermediates are formed, molten disk (IM2) and molten globule (IM3), before the native state. In the IM2 state, there is some β-structure, but its formation is localized to the membrane surface. Then, in the IM3 state, these β-hairpin loops are partially inserted into the bilayer. Finally, extensive rearrangement of side chains and formation of backbone hydrogen bonds is necessary to achieve the final transmembrane β-channel structure.
In this respect, the crystal structures of both ΔC1 and C1S CusC should correspond to the unstructured IM1 intermediate conformation of the CusC channel. As both mutant proteins were purified from the outer membrane for crystallizations, they must be at least partly associated with the bilayer. However, as in the IM1 state of OmpA, they have no β-structure and are not yet inserted into the membrane.102 Thus, the next conformational steps for CusC may be the formation of a monomeric molten disk (mono-IM2) intermediate, containing partial β-strands, but no β-barrel motif. At this stage, as in ΔC1 and C1S CusC, mono-IM2 is still monomeric. Upon oligomerization, the trimeric molten disk (tri-IM2) intermediate can transition to the trimeric molten globule intermediate, IM3, which is now partially inserted to the inner leaflet of the outer membrane. The native trimeric CusC structure is finally achieved upon folding of the 12-stranded β-barrel.39,40
The Periplasmic Metallochaperone CusF
Crystal structure of CusF
The crystal structure of CusF was determined by Loftin et al.41 to a resolution of 1.50 Å (PDB ID: 1ZEQ), including residues 6 to 88. The overall structure of CusF is a small five-stranded β-barrel, formed by two antiparallel β-sheets. The first β-sheet is composed of strands β1-β3, while the second strand comprises β4, β5, and the shared β1. The secondary structure of CusF is unique among small copper-binding proteins, including Atx180 or CopC.81,82 However, alignment of CusF with the top 35 homologous sequences reveals that there are three totally conserved residues (H36, M47, and M49) located between β2 and β3.
In vitro binding assay
To determine whether the three conserved residues of CusF are important for metal binding, a heteronuclear single quantum coherence (HSQC) spectrum was collected from CusF in the presence of Cu+ or Cu2+.41 In the case of Cu2+, 28 residues corresponding to the N-terminal and C-terminal ends were observed to participate in binding. However, the authors note that the C-terminal residues include both cloning artifacts and an affinity tag for purification. Thus, these residues are probably not involved in specific binding of the copper substrate. Accordingly, when a twofold molar excess of Cu2+ ions were added to the sample no change in the spectrum was observed. In the case of Cu+, analysis shows that the residues most affected by the addition of copper, as measured by chemical shift, are T48, M47, M49, R50, D37, H36, E46, Q74, W44, and Q75, respectively. Most of these residues are located in β2 and β3, including the highly conserved M47, M49, and H36. Thus, these residues should participate in the specific binding of Cu+ by CusF.
Crystal structure of CusF-ag+ and CusF-cu+
The crystal structure of Cu+-bound CusF was determined by Xue et al.42 to a resolution of 1.70 Å (PDB ID: 2VB2), including 88 residues of the mature protein. The crystal structure of Ag+-bound CusF was similarly solved to a resolution of 2.33 Å. Both structures of CusF-Cu+ and CusF-Ag+ are almost identical to that of apo-CusF. Interestingly, both bound copper and silver ions were found to be coordinated by residues H36, M47, and M49. This metal-binding region (residues 36–49) of the CusF-Cu+ structure is observed to change conformation slightly, compared with that of apo-CusF, with the M47 and M49 side chains reorienting to make favorable Cu-S bonds. Residues H36 and T44 also shift slightly in this conformation. Space-filling representations reveal that the metal-binding residues M47 and M49 are exposed to substantially more solvent in the apo-structure, compared with the Cu+- and Ag+-bound structures, possibly facilitating substrate capture from the periplasm.
The CusCFBA Transport Pathway
The divalent cation efflux pump CzcA83 and the aminoglycoside efflux pump AcrD26 were previously shown to be capable of capturing their substrates in vitro from both the periplasm and cytoplasm. We hypothesize that CusA is also able to extrude copper and silver ions from both sides of the inner membrane.103 From the cytoplasm, the methionine ladder created by transmembrane pairs M410-M501, M403-M486, and M391-M1009 should shuttle Cu+ and Ag+ ions to the primary three-methionine binding site. The periplasmic cleft of CusA containing these residues (M573, M623, and M672) presumably remains closed in the resting state. However, in the presence of Cu+ or Ag+, the periplasmic cleft opens, closely coordinating the three-methionine binding site and revealing it to the cytoplasm. Metal ions should be able to enter the CusA pump directly through this cleft or via the cytoplasmic tunnel. However, the major path for metal ion export should be via the periplasmic cleft. Adjacent to the periplasmic metal-binding site of CusA is the N-terminal tail of CusB.38 There is a possibility that this region of CusB, which contains the methionine triad (M21, M36, and M38) may engage to shuttle Cu+ and Ag+ ions to the periplasmic metal-ion binding site of CusA. However, it has been found that CusB is not capable of transferring ions to the CusA efflux pump.92 Thus, it is most likely that the metallochaperone CusF is responsible for scavenging Cu+/Ag+ and shuttling these metal ions to CusA.92 Transport of these metal ions in CusA is likely to involve a stepwise process that shuttles the metal ions from one cluster of methionines to another.104 Upon entering the central channel, the electrostatic gradient formed by the inner surfaces of the hexameric CusB and trimeric CusC channels may draw the ions across the outer membrane for final extrusion.
Conclusion
The availability of the three-dimensional structures of these efflux transporters and their accessory proteins should allow us to block their function. Potentially, by the rational design of inhibitors, as demonstrated by AcrB and DARPin.22 In this respect, the work is still in its infancy. To date, there is no high-resolution three dimensional structure of a completely assembled tripartite efflux system. It is our hope that this invaluable structural information may one day provide a platform for producing new drugs that heighten bacterial sensitivity to the available antibiotics.
References
- World Health Organization. Antimicrobial resistance: global report on surveillance. Geneva: WHO; 2014. . Available at: http://www.who.int/drugresistance/documents/surveillancereport/en/ [Google Scholar]
- Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007;446:749–757. doi: 10.1038/nature05630. [DOI] [PubMed] [Google Scholar]
- Routh MD, Zalucki Y, Su CC, Long F, Zhang Q, Shafer WM, Yu EW. Efflux pumps of the resistance-nodulation-division family: a perspective of their structure, function, and regulation in gram-negative bacteria. Adv Enzymol Relat Areas Mol Biol. 2011;77:109–146. doi: 10.1002/9780470920541.ch3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver S. Bacterial resistances to toxic metal ions division. Gene. 1996;179:9–19. doi: 10.1016/s0378-1119(96)00323-x. [DOI] [PubMed] [Google Scholar]
- Saier MH., Jr Molecular phylogeny as a basis for the classification of transport proteins from bacteria, archaea, and eukarya. Adv Microb Physiol. 1998;40:81–136. doi: 10.1016/s0065-2911(08)60130-7. [DOI] [PubMed] [Google Scholar]
- Saier MH, Jr, Beatty JT, Goffeau A, Harley KT, Heijne WH, Huang SC, Jack DL, Jähn PS, Lew K, Liu J, Pao SS, Paulsen IT, Tseng TT, Virk PS. The major facilitator superfamily. J Mol Microbiol Biotechnol. 1999;1:257–279. [PubMed] [Google Scholar]
- Higgins CF. ABC transporters: from microorganism to man. Annu Rev Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- Pao SS, Paulsen IT, Saier MH., Jr Major facilitator superfamily. Microbiol Mol Biol Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen IT, Skurray RA, Tam R, Saier MH, Jr, Turner RJ, Weiner JH, Goldberg EB, Grinius LL. The SMR family: a novel family of multidrug efflux proteins involved with the efflux of lipophilic drugs. Mol Microbiol. 1996;19:1167–1175. doi: 10.1111/j.1365-2958.1996.tb02462.x. [DOI] [PubMed] [Google Scholar]
- Tseng TT, Gratwick KS, Kollman J, Park D, Nies DH, Goffeau A, Saier MH., Jr The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J Mol Microbiol Biotechnol. 1999;1:107–125. [PubMed] [Google Scholar]
- Brown MH, Paulsen IT, Skurray RA. The multidrug efflux protein NorM is a prototype of a new family of transporters. Mol Microbiol. 1999;31:394–395. doi: 10.1046/j.1365-2958.1999.01162.x. [DOI] [PubMed] [Google Scholar]
- Nikaido H. Structure and mechanism of RND-type multidrug efflux pumps. Adv Enzymol Relat Areas Mol Biol. 2011;77:1–60. doi: 10.1002/9780470920541.ch1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgurskaya HI, Nikaido H. Multidrug resistance mechanisms: drug efflux across two membranes. Mol Microbiol. 2000;37:219–225. doi: 10.1046/j.1365-2958.2000.01926.x. [DOI] [PubMed] [Google Scholar]
- Delmar JA, Su CC, Yu EW. Bacterial multidrug efflux transporters. Annu Rev Biophys. 2014;43:93–117. doi: 10.1146/annurev-biophys-051013-022855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D, Xu QS, Lee JY, Ankoudinova I, Huang C, Lou Y, DeGiovanni A, Kim R, Kim SH. Crystal structure of the multidrug efflux transporter AcrB at 3.1 Å resolution reveals the N-terminal region with conserved amino acids. J Struct Biol. 2007;158:494–502. doi: 10.1016/j.jsb.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature. 2006;443:173–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- Nikaido H, Takatsuka Y. Mechanisms of RND multidrug efflux pumps. Biochim Biophys Acta. 2009;1794:769–781. doi: 10.1016/j.bbapap.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino K, Yamaguchi A. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J Bacteriol. 2001;183:5803–5812. doi: 10.1128/JB.183.20.5803-5812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pos KM. Drug transport mechanism of the AcrB efflux pump. Biochim Biophys Acta. 2009;1794:782–793. doi: 10.1016/j.bbapap.2008.12.015. [DOI] [PubMed] [Google Scholar]
- Seeger MA, Diederichs K, Eicher T, Brandstchsor L, Schiefner A, Verrey F, Pos KM. The AcrB efflux pump: conformational cycling and peristalsis lead to multidrug resistance. Curr Drug Targets. 2008;9:729–749. doi: 10.2174/138945008785747789. [DOI] [PubMed] [Google Scholar]
- Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grorche MG. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS One. 2007;5:e7. doi: 10.1371/journal.pbio.0050007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, Yu EW. Conformation of the AcrB multidrug efflux pump in mutants of the putative proton relay pathway. J Bacteriol. 2006;188:7290–7296. doi: 10.1128/JB.00684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu EW, Aires JR, McDermott G, Nikaido H. A periplasmic drug-binding site of the AcrB multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J Bacteriol. 2005;187:6804–6815. doi: 10.1128/JB.187.19.6804-6815.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE., Jr Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science. 2003;300:976–980. doi: 10.1126/science.1083137. [DOI] [PubMed] [Google Scholar]
- Aires JR, Nikaido H. Aminoglycosides are captured from both periplasm and cytoplasm by the AcrD multidrug efflux transporter of Escherichia coli. J Bacteriol. 2005;187:1923–1929. doi: 10.1128/JB.187.6.1923-1929.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau SY, Zgurskaya HI. Cell division defects in Escherichia coli deficient in the multidrug efflux transporter AcrEF-TolC. J Bacteriol. 2005;187:7815–7825. doi: 10.1128/JB.187.22.7815-7825.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. Molecular cloning and characterization of acrA and acrE genes of Escherichia coli. J Bacteriol. 1993;175:6299–6313. doi: 10.1128/jb.175.19.6299-6313.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranova N, Nikaido H. The baeSR two-component regulatory system activates transcription of the yegMNOBmdtABCD) transporter gene cluster in Escherichia coli and increases its resistance to novobiocin and deoxycholate. J Bacteriol. 2002;184:4168–4176. doi: 10.1128/JB.184.15.4168-4176.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Nikaido H. Different function ofMdtB and MdtC subunits in the heterotrimeric efflux transporter MdtB2C complex of Escherichia coli. Biochemistry. 2012;51:4188–4197. doi: 10.1021/bi300379y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagakubo S, Nishino K, Hirata T, Yamaguchi A. The putative response regulator BaeR stimulates multidrug resistance of Escherichia coli via a novel multidrug exporter system, MdtABC. J Bacteriol. 2002;184:4161–4167. doi: 10.1128/JB.184.15.4161-4167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert JA, Schuster S, Fchuster E, Trittler R, Kern WV. Altered spectrumofmultidrug resistance associated with a single point mutation in the Escherichia coli RND-type MDR efflux pump YhiV (MdtF) J Antimicrob Chemother. 2007;59:1216–1222. doi: 10.1093/jac/dkl426. [DOI] [PubMed] [Google Scholar]
- Kobayashi N, Nishino K, Yamaguchi A. Novel macrolide-specific ABC-type efflux transporter in Escherichia coli. J Bacteriol. 2001;183:5639–5644. doi: 10.1128/JB.183.19.5639-5644.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nies DH. Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol Rev. 2003;27:313–339. doi: 10.1016/S0168-6445(03)00048-2. [DOI] [PubMed] [Google Scholar]
- Long F, Su CC, Zimmermann MT, Boyken SE, Rajashankar KR, Jernigan RL, Yu EW. Crystal structures of the CusA efflux pump suggest methionine-mediated metal transport. Nature. 2010;467:484–488. doi: 10.1038/nature09395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Long F, Zimmermann MT, Rajashankar KR, Jernigan RL, Yu EW. Crystal structure of the CusBA heavy-metal efflux complex of Escherichia coli. Nature. 2011;470:558–563. doi: 10.1038/nature09743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Long F, Lei HT, Bolla JR, Do SV, Rajashankar KR, Yu EW. Charged amino acids (R83, E567, D617, E625, R669, and K678) of CusA are required for metal ion transport in the Cus efflux system. J Mol Biol. 2012;422:429–441. doi: 10.1016/j.jmb.2012.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Yang F, Long F, Reyon D, Routh MD, Kuo DW, Mokhtari AK, Van Ornam JD, Rabe KL, Hoy JA, Lee YJ, Rajashankar KR, Yu EW. Crystal structure of the membrane fusion protein CusB from Escherichia coli. J Mol Biol. 2009;393:342–355. doi: 10.1016/j.jmb.2009.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei HT, Bolla JR, Bishop NR, Su CC, Yu EW. Crystal structures of CusC review conformational changes accompanying folding and transmembrane channel formation. J Mol Biol. 2014;426:403–411. doi: 10.1016/j.jmb.2013.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulathila R, Kulathila R, Indic M, van den Berg B. Crystal structure of Escherichia coli CusC, the outer membrane component of a heavy metal efflux pump. PLoS One. 2011;6:e15610. doi: 10.1371/journal.pone.0015610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftin IR, Franke S, Roberts SA, Weichsel A, Heichs A, Montfort WR, Rensing C, McEvoy MM. A novel copper-binding fold for the periplasmic copper resistance protein CusF. Biochemistry. 2005;44:10533–10540. doi: 10.1021/bi050827b. [DOI] [PubMed] [Google Scholar]
- Xue Y, Davis AV, Balakrishnan G, Stasser JP, Staehlin BM, Focia P, Spiro TG, Penner-Hahn JE, periplas OTV. Cu(I) recognition via cation-JE, O periplasmic copper resistance prNat. Chem Biol. 2008;4:107–109. doi: 10.1038/nchembio.2007.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akama H, Kanemaki M, Yoshimura M, Tsukihara T, Kashiwagi T, Yoneyama H, Narita S, Nakagawa A, Nakae T. Crystal structure of the drug discharge outer membrane protein, OprM, of Pseudomonas aeruginosa: dual modes of membrane anchoring and occluded cavity end. J Biol Chem. 2004;279:52816–52819. doi: 10.1074/jbc.C400445200. [DOI] [PubMed] [Google Scholar]
- Akama H, Matsuura T, Kashiwagi S, Yoneyama H, Narita S, Tsukihara T, Nakagawa A, Nakae T. Crystal structure of the membrane fusion protein, MexA, of the multidrug transporter in Pseudomonas aeruginosa. J Biol Chem. 2004;279:25939–25942. doi: 10.1074/jbc.C400164200. [DOI] [PubMed] [Google Scholar]
- Higgins MK, Bokma E, Koronakis E, Hughes C, Koronakis V. Structure of the periplasmic component of a bacterial drug efflux pump. Proc Natl Acad Sci USA. 2004;101:9995–9999. doi: 10.1073/pnas.0400375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennhauser G, Bukowksa MA, Briand C, Griands MG. Crystal structure of the multidrug exporter MexB from Pseudomonas aeruginosa. J Mol Biol. 2009;389:134–145. doi: 10.1016/j.jmb.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405:914–919. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- Mikolosko J, Bobyk K, Zgurskaya HI, Ghosh P. Conformational flexibility in the multidrug efflux system protein AcrA. Structure. 2006;14:577–587. doi: 10.1016/j.str.2005.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science. 2006;313:1295–1298. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- Du D, Wang Z, James NR, Voss JE, Klimont E, Ohene-Agyei T, Venter H, Chiu W, Luisi BF. Structure of the AcrAB-TolC multidrug efflux pump. Nature. 2014;509:512–515. doi: 10.1038/nature13205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmar JA, Su CC, Yu EW. Structural mechanisms of heavy-metal extrusion by the Cus efflux system. Biometals. 2013;26:593–607. doi: 10.1007/s10534-013-9628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nies DH, Nies A, Chu L, Silver S. Expression and nucleotide sequence of a plasmid-determined divalent cation efflux system from Alcaligenes eutrophus. Proc Natl Acad Sci USA. 1989;86:7351–7355. doi: 10.1073/pnas.86.19.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier MH, Tam R, Reizer A, Reizer J. Two novel families of bacterial membrane proteins concerned with nodulation, cell division and transport. Mol Microbiol. 1994;11:841–847. doi: 10.1111/j.1365-2958.1994.tb00362.x. [DOI] [PubMed] [Google Scholar]
- Silver S, Phung LT, Silver G. Silver as biocides in burn and wound dressings and bacterial resistance to silver compounds. J Ind Microbiol Biotechnol. 2006;33:627–634. doi: 10.1007/s10295-006-0139-7. [DOI] [PubMed] [Google Scholar]
- Gupta A, Matsui K, Lo JF, Silver S. Molecular basis for resistance to silver cations in Salmonella. Nat Med. 1999;5:183–188. doi: 10.1038/5545. [DOI] [PubMed] [Google Scholar]
- Silver S. Bacterial silver resistance: molecular biology and uses and misuses of silver compounds. FEMS Microbiol Rev. 2003;27:341–353. doi: 10.1016/S0168-6445(03)00047-0. [DOI] [PubMed] [Google Scholar]
- Paulsen IT, Nguyen L, Sliwinski MK, Rabus R, Saier MH., Jr Microbial genome analyses: comparative transport capabilities in eighteen prokaryotes. J Mol Biol. 2000;301:75–100. doi: 10.1006/jmbi.2000.3961. [DOI] [PubMed] [Google Scholar]
- Munson GP, Lam DL, Outten FW, Halloran O′TV. Identification of a copper-responsive two-component system on the chromosome of Escherichia coli K-12. J Bacteriol. 2000;182:5864–5871. doi: 10.1128/jb.182.20.5864-5871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke S, Grass G, Nies DH. The product of the ybdE gene of the Escherichia coli chromosome is involved in detoxification of silver ions. Microbiology. 2001;147:965–972. doi: 10.1099/00221287-147-4-965. [DOI] [PubMed] [Google Scholar]
- Grass G, Rensing C. Genes involved in copper homeostasis in Escherichia coli. J Bacteriol. 2001;183:2145–2147. doi: 10.1128/JB.183.6.2145-2147.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macomber L, Imlay JA. The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc Natl Acad Sci USA. 2009;106:8344–8349. doi: 10.1073/pnas.0812808106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nies DH. Microbial heavy-metal resistance. Appl Microbiol Biotechnol. 1999;51:730–750. doi: 10.1007/s002530051457. [DOI] [PubMed] [Google Scholar]
- Nies DH. Bacterial transition metal homeostasis. In: Nies DH, Silver S, editors. Molecular microbiology of heavy metals. Berlin: Springer; 2007. pp. 117–142. . In: Eds. ( [Google Scholar]
- Outten FW, Huffman DL, Hale JA, Halloran O′TV. The independent cue and cus systems confer copper tolerance during aerobic and anaerobic growth in Escherichia coli. J Biol Chem. 2001;276:30670–30677. doi: 10.1074/jbc.M104122200. [DOI] [PubMed] [Google Scholar]
- Rensing C, Fan B, Sharma R, Mitra B, Rosen BP. CopA: an Escherichia coli Cu(I)-translocating P-type ATPase. Proc Natl Acad Sci USA. 2000;97:652–656. doi: 10.1073/pnas.97.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensing C, Grass G. Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol Rev. 2003;27:197–213. doi: 10.1016/S0168-6445(03)00049-4. [DOI] [PubMed] [Google Scholar]
- Roberts SA, Weichsel A, Grass G, Thakali K, Hazzard JT, Tollin G, Rensing C, Montfort WR. Crystal structure and electron transfer kinetics of CueO, a multicopper oxidase required for copper homeostasis in Escherichia coli. Proc Natl Acad Sci USA. 2002;99:2766–2771. doi: 10.1073/pnas.052710499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Grass G, Rensing C, Montfort WR. Cuprous oxidase activity of CueO from Escherichia coli. J Bacteriol. 2004;186:7815–7817. doi: 10.1128/JB.186.22.7815-7817.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Roberts SA, McDevitt SF, Weichsel A, Wildner GF, Grass GB, Rensing C, Montfort WR. Crystal structures of multicopper oxidase CueO bound to copper(I) and silver(I): functional role of a methionine-rich sequence. J Biol Chem. 2011;286:37849–37857. doi: 10.1074/jbc.M111.293589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Ishihama A. Transcriptional response of Escherichia coli to external copper. Mol Microbiol. 2005;56:215–227. doi: 10.1111/j.1365-2958.2005.04532.x. [DOI] [PubMed] [Google Scholar]
- Haber F, Weiss J. On the catalysis of hydroperoxides. Naturwissenschaften. 1932;20:948–950. [Google Scholar]
- Nies DH. RND efflux pumps for metal cations. In: Yu EW, Zhang Q, Brown MH, editors. Microbial efflux pumps: current research. Norfolk: Caister Academic; 2013. pp. 79–121. . In: Eds. ( [Google Scholar]
- Franke S, Grass G, Rensing C, Nies DH. Molecular analysis of the copper-transporting efflux system CusCFBA of Escherichia coli. J Bacteriol. 2003;185:3804–3812. doi: 10.1128/JB.185.13.3804-3812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson PJ. The 12-transmembrane helix transporters. Curr Opin Cell Biol. 1993;5:708–721. doi: 10.1016/0955-0674(93)90144-f. [DOI] [PubMed] [Google Scholar]
- Bolla JR, Su CC, Do SV, Radhakrishnan A, Kumar N, Long F, Chou TH, Delmar JA, Lei HT, Rajashankar KR, Shafer WM, Yu EW. Crystal structure of the Neisseria gonorrhoeae MtrD inner membrane multidrug efflux pump. PLoS One. 2014;9:e97903. doi: 10.1371/journal.pone.0097903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Thiele DJ. Identification of a novel high affinity copper transport complex in the fission yeast Schizosaccharomyces pombe. J Biol Chem. 2001;276:20529–20535. doi: 10.1074/jbc.M102004200. [DOI] [PubMed] [Google Scholar]
- Jiang J, Nadas IA, Kim MA, Franz KJ. A Mets motif peptide found in copper transport proteins selectively bind Cu(I) with methionine-only coordination. Inorg Chem. 2005;44:9787–9794. doi: 10.1021/ic051180m. [DOI] [PubMed] [Google Scholar]
- Loftin IR, Franke S, Blackburn NJ, McEvoy MM. Unusual Cu(I)/Ag(I) coordination of Escherichia coli CusF as revealed by atomic resolution crystallography and X-ray absorption spectroscopy. Prot Sci. 2007;16:2287–2293. doi: 10.1110/ps.073021307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changela A, Chen K, Xue Y, Holschen J, Outten CE, Outtenenevealed by atomi O. Molecular basis of metal-ion selectivity and zeptomolar sensitivity by CueR. Science. 2003;301:1383–1387. doi: 10.1126/science.1085950. [DOI] [PubMed] [Google Scholar]
- Arnesano F, Banci L, Bertini I, Huffman DL, Ouffman B TV. Solution structure of the Cu(I) and apo forms of the yeast metallochaperone, Atx1. Biochemistry. 2001;40:1528–1539. doi: 10.1021/bi0014711. [DOI] [PubMed] [Google Scholar]
- Arnesano F, Banci L, Bertini I, Mangani S, Thompsett AR. A redox switch in CopC: an intriguing copper trafficking protein that binds copper(I) and copper(II) at different sites. Proc Natl Acad Sci USA. 2003;100:3814–3819. doi: 10.1073/pnas.0636904100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Koay M, Maher MJ, Xiao Z, Wedd AG. Intermolecular transfer of copper ions from the CopC protein of Pseudomonas syringae. Crystal structures of fully loaded CuICuII forms. J Am Chem Soc. 2006;128:5834–5850. doi: 10.1021/ja058528x. [DOI] [PubMed] [Google Scholar]
- Goldberg M, Pribyl T, Juhnke S, Nies DH. Energetics and topology of CzcA, a cation/proton antiporter of the resistance-nodulation-cell division protein family. J Biol Chem. 1999;273:26065–26070. doi: 10.1074/jbc.274.37.26065. [DOI] [PubMed] [Google Scholar]
- Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher T, Cha HJ, Seeger MA, Brandstropy L, El-Delik J, Bohnert JA, Kern WV, Verrey F, Grctter MG, Diederichs K, Pos KM. Transport of drugs by themultidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc Natl Acad Sci USA. 2012;109:5687–5692. doi: 10.1073/pnas.1114944109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S. Multidrug efflux transporter, AcrB transporter AcrB involvCurr. Opin Struct Biol. 2008;18:459–465. doi: 10.1016/j.sbi.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Symmons M, Bokma E, Koronakis E, Hughes C, Koronakis V. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc Natl Acad Sci USA. 2009;106:7173–7178. doi: 10.1073/pnas.0900693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagai I, Liu W, Rensing C, Blackburn NJ, McEvoy MM. Substrate-linked conformational change in the periplasmic component of a Cu(I)/Ag(I) efflux system. J Biol Chem. 2007;282:35695–35702. doi: 10.1074/jbc.M703937200. [DOI] [PubMed] [Google Scholar]
- Janganan TK, Bavro VN, Zhang L, Matak-Vinkovic D, Barrera NP, Robinson CV, Borges-Walmsley MI, Walmsley AR. Evidence for the assembly of a bacterial tripartite multidrug pump with a stoichiometry of 3:6:3. J Biol Chem. 2011;286:26900–26912. doi: 10.1074/jbc.M111.246595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensing C, Pribyl T, Nies DH. New functions for the three subunits of the CzcCBA cation-proton antiporter. J Bacteriol. 1997;179:6871–6879. doi: 10.1128/jb.179.22.6871-6879.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier JF, Polleichtner G, Brandes N, Hotz C, Andersen C. Importance of the adaptor (membrane fusion) protein hairpin domain for the functionality of multidrug efflux pumps. Biochemistry. 2006;45:10303–10312. doi: 10.1021/bi060320g. [DOI] [PubMed] [Google Scholar]
- Chacón KN, Mealman TD, McEvoy MM, Blackburn NJ. Tracking metal ions through a Cu/Ag efflux pump assigns the functional roles of the periplasmic proteins. Proc Natl Acad Sci USA. 2014;111:15373–15378. doi: 10.1073/pnas.1411475111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla-Benavides T, Thompson AMG, McEvoy MM, Argüello JM. Mechanism of ATPase-mediated Cu+ export and delivery to periplasmic chaperones. J Biol Chem. 2014;289:20492–20501. doi: 10.1074/jbc.M114.577668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei HT, Chou TH, Su CC, Bolla JR, Kumar N, Radhakrishnan A, Long F, Delmar JA, Do SV, Rajashankar KR, Shafer WM, Yu EW. Crystal structure of the open state of the Neisseria gonorrhoeae MtrE outer membrane channel. PLoS One. 2014;9:e97475. doi: 10.1371/journal.pone.0097475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Radhakrishnan A, Kumar N, Long F, Bolla JR, Lei HT, Delmar JA, Do SV, Chou TH, Rajashankar KR, Zhang Q, Yu EW. Crystal structure of the Campylobacter jejuni CmeC outer membrane channel. Prot Sci. 2014;23:954–961. doi: 10.1002/pro.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronakis V, Eswaran J, Hughes C. Structure and function of TolC: the bacterial exit duct for proteins and drugs. Annu Rev Biochem. 2004;73:467–489. doi: 10.1146/annurev.biochem.73.011303.074104. [DOI] [PubMed] [Google Scholar]
- Hinchliffe P, Symmons MF, Hughes C, Koronakis V. Structure and operation of bacterial tripartite pumps. Annu Rev Microbiol. 2013;67:221–242. doi: 10.1146/annurev-micro-092412-155718. [DOI] [PubMed] [Google Scholar]
- Long F, Su CC, Lei HT, Bolla JR, Do SV, Yu EW. Structure and mechanism of the tripartite CusCBA heavy-metal efflux complex. Phil Trans R Soc B. 2012;367:1047–1058. doi: 10.1098/rstb.2011.0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pautsch A, Schulz GE. Structure of the outer membrane protein A transmembrane domain. Nat Struct Biol. 1998;5:1013–1017. doi: 10.1038/2983. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt JH, Tamm LK. Folding intermediates of a beta-barrel membrane protein. Kinetic evidence for a multi-step membrane insertion mechanism. Biochemistry. 1996;35:12993–13000. doi: 10.1021/bi961478b. [DOI] [PubMed] [Google Scholar]
- Tamm LK, Arora A, Kleinschmidt JH. Structure and assembly of beta-barrel membrane proteins. J Biol Chem. 2001;276:32399–32402. doi: 10.1074/jbc.R100021200. [DOI] [PubMed] [Google Scholar]
- Tamm LK, Hong H, Liang B. Folding and assembly of beta-barrel membrane proteins. Biochim Biophys Acta. 2004;1666:250–263. doi: 10.1016/j.bbamem.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Su CC, Long F, Yu EW. CusBA heavy-metal efflux complex of Escherichia coli. In: Messerschmidt A, editor. Encyclopedia of Inorganic and Bioinorganic Chemistry, Online. NJ: Wiley; 2012. . In: Ed. (. DOI: 10.1002/9781119951438.eibc2054. [Google Scholar]
- Su CC, Long F, Yu EW. The Cus efflux system removes toxic ions via a methionine shuttle. Prot Sci. 2011;20:6–18. doi: 10.1002/pro.532. [DOI] [PMC free article] [PubMed] [Google Scholar]