

Abstract
Light chain (AL) amyloidosis is an incurable human disease, where the amyloid precursor is a misfolding-prone immunoglobulin light-chain. Here, we identify the role of somatic mutations in the structure, stability and in vitro fibril formation for an amyloidogenic AL-12 protein by restoring four nonconservative mutations to their germline (wild-type) sequence. The single restorative mutations do not affect significantly the native structure, the unfolding pathway, and the reversibility of the protein. However, certain mutations either decrease (H32Y and H70D) or increase (R65S and Q96Y) the protein thermal stability. Interestingly, the most and the least stable mutants, Q96Y and H32Y, do not form amyloid fibrils under physiological conditions. Thus, Q96 and H32 are key residues for AL-12 stability and fibril formation and restoring them to the wild-type residues preclude amyloid formation. The mutants whose equilibrium is shifted to either the native or unfolded states barely sample transient partially folded states, and therefore do not form fibrils. These results agree with previous observations by our laboratory and others that amyloid formation occurs because of the sampling of partially folded states found within the unfolding transition (Blancas-Mejia and Ramirez-Alvarado, Ann Rev Biochem 2013;82:745–774). Here we provide a new insight on the AL amyloidosis mechanism by demonstrating that AL-12 does not follow the established thermodynamic hypothesis of amyloid formation. In this hypothesis, thermodynamically unstable proteins are more prone to amyloid formation. Here we show that within a thermal stability range, the most stable protein in this study is the most amyloidogenic protein.
Keywords: aggregation, protein misfolding, thermodynamics, immunoglobulin fold, light chain amyloidosis, somatic mutations
Introduction
Light chain (AL) amyloidosis is a devastating protein misfolding disease, where the amyloid precursor is a free immunoglobulin light-chain secreted by an aberrant plasma cell population. Light chains (LC) consist of a highly variable N-terminal domain (VL) and a constant C-terminal domain (CL). In normal plasma cells, two light chains associate with two heavy chains in order to form the functional immunoglobulin (Ig)-heterotetramer. However, in AL amyloidosis, there is an excess of free LC or a complete absence of Ig molecules.1 After the free LCs are secreted, they misfold and subsequently aggregate into amyloid fibrils in the extracellular space of many vital organs, resulting in organ failure and death. All immunoglobulin light chains are formed by random recombination of multiple gene segments; and multiple somatic mutations on the VL domain generate the required immunoglobulin diversity and improve the antigen affinity. Because of that, each AL amyloidosis patient possesses a unique amyloidogenic protein sequence with a different set of mutations. These sequence variations may contribute to the differences observed among AL amyloidosis patients, hindering the diagnostic and treatment for AL amyloidosis disease.
Traditionally, the susceptibility to form amyloid fibril formation has been correlated with protein destabilization, assuming that partially folded states promote aggregation.2 In the case of AL proteins, somatic mutations to nonconservative amino acids could result in a reduced thermodynamic stability and higher propensity for amyloid formation.3–7 However, we recently demonstrated that a thermal stability threshold exists for AL fibril formation for some AL proteins.8 In addition, we found that the location of somatic mutations within the LC molecule could alter their dimer interface,3,9 and that the species present at the beginning of the fibril reaction (monomer, canonical or altered dimer) and the reaction conditions (ionic strength, pH, presence of co-factors, etc.) could dramatically affect the kinetics of fibril formation in vitro.10–13
In this study, we characterized the role of somatic mutations in modifying the structure, stability and in vitro fibril formation for AL-12 VL domain (herein referred only as AL-12 for simplicity), an amyloidogenic protein coming from a 64 years-old woman diagnosed with multiple myeloma/cardiac AL amyloidosis. The structure of AL-12 consists of nine β-strands (A, B, C, C′, C″, D, E, F, and G) packed tightly against each other in two antiparallel β-sheets forming the typical Ig β-barrel [Fig. 1(A)].9 AL-12 sequence shares 87% of sequence identity with its germline gene product, κI O18/O8 (also called IGKV 1-33, what would be considered the wild-type) and contains eight somatic mutations, seven of which are nonconservative changes [Fig. 1(B)]. Previous reports have shown that AL-12 maintains the monomeric fold and the dimer interface of the germline κI O18/O8 protein while it is less stable than the germline protein.9,10
Figure 1.
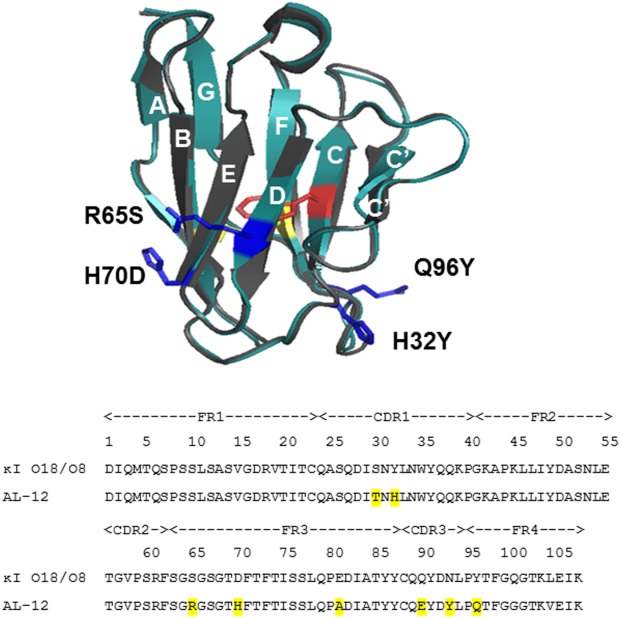
Structural and sequence alignments between AL-12 and germline κI O18/O8. A) Structural alignment of X-ray crystal structures of germline protein κI O18/O8 (gray) and AL-12 (teal) (PDB codes: 2Q20 and 3DVF, respectively). AL-12 VL domain contains a disulfide bond (yellow) and a single Trp-35 (red). The four restorative mutations characterized in this report are shown (blue). B) Sequence alignment showing the 8 somatic mutations (highlighted) present in AL-12.
Here, we have restored four nonconservative mutations to its germline sequence κI O18/O8 corresponding amino acid: H32Y (β-strand C/CDR1), R65S (β-strand D), H70D (β-strand E), and Q96Y (β-strand G). All of these mutations occur at the same level of the β-barrel [Fig. 1(A)], in close proximity with the CDRs. Nonconservative mutations located on the highly variable CDR regions (S30T, Q90E, N93Y) have not been restored.
By restoring these residues, we have to assess the contribution of individual somatic mutations to AL-12 protein stability and amyloidogenicity. We find that somatic mutations can inhibit fibril formation by stabilizing or destabilizing the native state of the protein, in such a way that amyloidogenic species are not sampled enough to trigger fibrillation. Our results confirm that AL proteins access the amyloid formation pathway by sampling partially folded states found within the thermal unfolding transition.
Furthermore, we demonstrate that AL-12 fibril formation does not correlate with protein destabilization because within a thermal stability range, the wild-type AL-12 is at the same time the most stable and the most amyloidogenic protein in this study. Somatic mutations may cause a local effect on the protein structure, affecting the protein amyloidogenicity, independently of its stability.
Together, our results demonstrate that mutations can cause the protein to be too stable or too unstable to form amyloid fibrils under physiological conditions, a finding of significant importance for better understanding of the molecular determinants of amyloid formation.
Results
Restorative mutants maintain the immunoglobulin structure
After protein purification, structural features of each mutant were characterized by circular dichroism (CD), fluorescence, and Fourier Transform-infrared (FT-IR) spectroscopy. Native CD spectra were taken at 4°C in order to decrease any conformational fluctuations due to the thermal contribution in the protein stability [see thermal denaturation, Fig. 3(A)].
Figure 3.
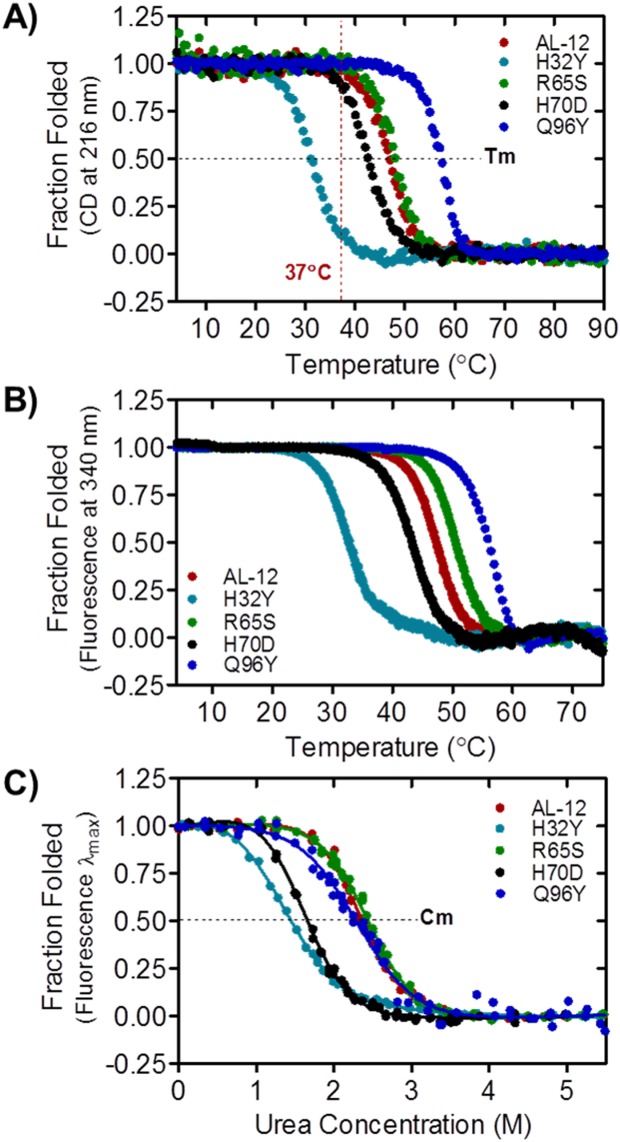
AL-12 and restorative mutants present differences in thermodynamic stability. A) Thermal denaturation followed by CD (ellipticity at 216 nm), from which the Tm and ΔH values were extracted (Table I). B) Thermal denaturation followed by tryptophan fluorescence (emission at 340 nm). C) Chemical denaturation followed by tryptophan fluorescence at 4°C. The Cm and ΔG values (Table I) were extracted by fitting each curve to a two-state model.
By Far UV-CD, all of the restorative mutants exhibit the expected β-sheet secondary structure typical of immunoglobulin light chain variable domains with an ellipticity minimum near 216 nm [Fig. 2(A)]. The native spectra also show a slight minimum around 235 nm characteristic of AL-12,11 likely due to the contribution of aromatic residues that are optically active in the Far UV region.14,15 The H32Y and Q96Y mutants show lower β-sheet content than the rest of the proteins but both of them still adopt the β-sheet structure.
Figure 2.
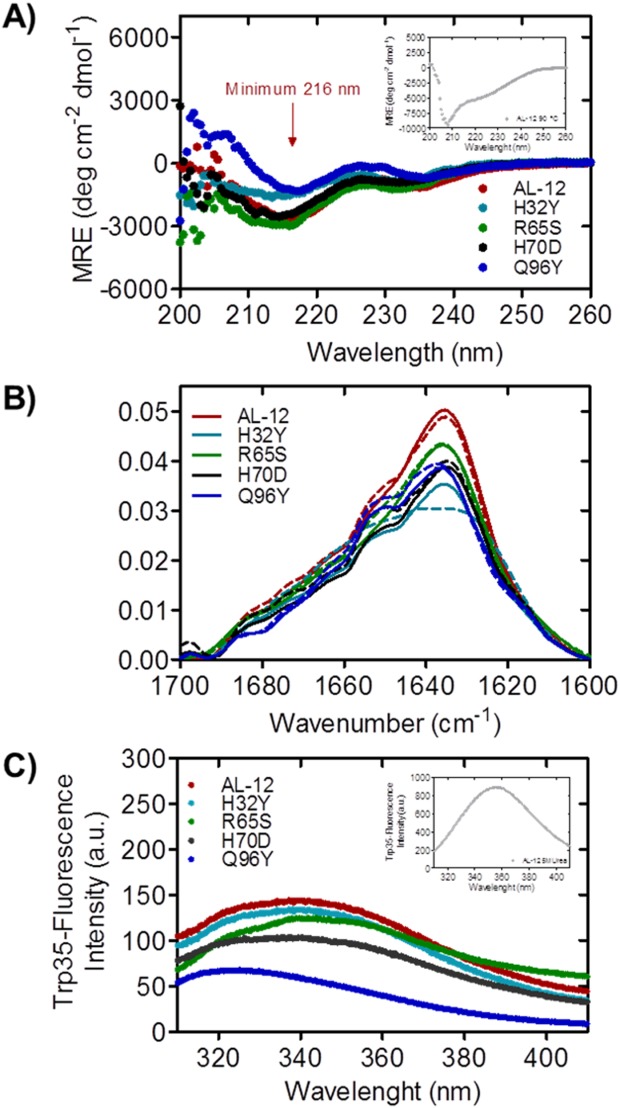
AL-12 and restorative mutants form β-sheets and are fully folded. A) Far UV-CD spectra of AL-12 and restorative mutants. B) FT-IR amide I’ spectra of AL-12 and its restorative mutants, at 4°C (solid line) and 40°C (dashed line). C) Trp35-emission fluorescence spectra of AL-12 and restorative mutants. The spectra of AL-12 denatured state is shown in the insets A and C.
As shown in Figure 2(A) inset, at 90°C, AL-12 adopts a random-coil-like structure with a minimum at 208 nm (see Supporting Information Fig. S1 for the mutants). The spectrum indicates a mixture of random-coil and β-sheet structure, a phenomenon previously described for other AL proteins,16 possibly due to the presence of the disulfide bond between Cys 23 and Cys 88.
By FT-IR [Fig. 2(B)], the amide I’ spectrum at 4°C shows two main maxima: the most representative that corresponds to the native β-sheet component, centered at 1638 cm−1; and a secondary maximum corresponding to the random coil fraction, located at 1650 cm−1. The spectra for the four restorative mutants show the same profile found for AL-12, which is consistent with the crystallographic data previously obtained for the AL-12 protein.9 At 40°C, the spectra remain almost unaltered, with a slightly increase in the random coil component. Notably, we find that the H32Y is the only mutant that shows a more pronounced change in its amide I’ spectrum upon increasing temperature.
Intrinsic fluorescence gives us some information about the protein tertiary structure. The highly conserved and single Trp35 has its native-maximum emission fluorescence at 340 nm for all the mutants, except for the Q96Y mutant which its maximum is slightly shifted to lower wavelengths [Fig. 2(C)]. This fluorescence shifting indicates a more compact native state upon mutation. At 5 M urea [Fig. 2(C), inset], the tryptophan is fully exposed and the quenching effect of the disulfide bond is blocked. The maximum fluorescence is shifted to 355 nm and the emission fluorescence intensity is significantly increased, indicating that the tertiary structure is completely disrupted upon urea denaturation.
Together, these results demonstrate that, using medium resolution methods (as Far-UV CD and intrinsic fluorescence) there are no significant differences on the characteristic β-sheet protein structure observed in AL-12 and its restorative mutants as well as in all κI light chains previously reported.
Restorative mutants alter the AL-12 thermodynamic stability
The demonstration that AL-12 restorative mutations have no effect on the protein native structure, led us to carry out both thermal and chemical denaturation experiments to determine both the thermodynamic parameters in the unfolding process and the role of each somatic mutation on the protein stability.
As mentioned above, AL-12 is less thermodynamically stable than κI O18/O8 (Table1).12,17 The four restorative mutants display a sigmoidal two-state unfolding transition for both thermal and chemical denaturation experiments (Fig. 3), as previously reported by our laboratory for AL-12.11 In all cases, AL-12 and its restorative mutants follow a reversible unfolding process (Supporting Information Fig. S2). All restorative mutants alter the thermal stability in AL-12, especially those with tyrosine residue located at the canonical dimer interface (H32Y and Q96Y). For H32Y, the change from histidine to tyrosine decrease the Tm value by 15.1°C compared with AL-12 values (from 46.3 to 31.2°C); while the restoration of tyrosine residue in the Q96Y mutant leads to an increase of 9.5°C for the AL-12 thermal stability (from 46.3 to 55.8°C) [Fig. 3(A), Table1]; surpassing the Tm value of the germline κI O18/O8 protein (Tm of 54.7°C). These results suggest that H32 offers a protective effect to AL-12 whereas Q96 is highly detrimental for AL-12 stability. The effects of the restorative mutations R65S and H70D are not quite as dramatic (decreases in the Tm of 2.7°C and 3.8°C, respectively).
Table 1.
Thermodynamic Parameters of AL-12, κI O18/O8, and Restorative Mutants
| Proteina | Tmb(°C) | ΔHvan't Hoffb(Kcal mol−1) | ΔGfoldingc(Kcal mol−1) | mc(Kcal mol−1 M−1) | Cmc(M) |
|---|---|---|---|---|---|
| Q96Y | 55.78 ± 0.97 | −127.80 ± 8.40 | −3.36 ± 1.84 | 1.46 ± 0.73 | 2.30 |
| κI O18/O8 | 54.70 ± 0.30 e | −95.70 ± 2.60d | −6.12 ± 0.23d | 1.53d | 3.98d |
| R65S | 49.04 ± 0.19 | −85.43 ± 3.43 | −4.30 ± 0.91 | 1.76 ± 0.35 | 2.44 |
| AL-12 | 46.31 ± 0.46 | −86.95 ± 3.42 | −4.42 ± 0.84 | 1.90 ± 0.33 | 2.33 |
| H70D | 42.49 ± 0.39 | −73.13 ± 2.03 | −2.97 ± 0.70 | 1.87 ± 0.34 | 1.58 |
| H32Y | 31.19 ± 0.11 | −67.45 ± 4.36 | −1.88 ± 0.36 | 1.48 ± 0.16 | 1.27 |
The thermal unfolding transitions followed by fluorescence [Fig. 3(B)] show the same behavior observed by CD, with the exception of the R65S mutant which shows a slight increase in thermal stability. This result suggests that the restoration of Ser65 affects the global tertiary structure to a higher extent than other mutants.
For a complete thermodynamic characterization, urea-chemical denaturation experiments at 4°C have been carried out, and the thermodynamic parameters have been obtained for all the variants. The experiments were conducted at 4°C due to the fact that H32Y is destabilized and partially unfolded at room temperature.
Figure 3(C) shows AL-12 to be completely unfolded at 3.5M of urea. Both chemical and thermal denaturation experiments reveal a similar trend, although the unfolding cooperativity is lower in the chemical denaturation experiment. The urea concentration at the midpoint of the transition (Cm) for R65S mutant is similar (Cm of 2.44M) to AL-12 (Cm of 2.33M), whereas the H32Y and H70D mutants unfold at lower urea concentrations (Cm of 1.27M and 1.58M, respectively). The most striking results were found for the restorative Q96Y mutant. The chemical denaturation of this thermostable variant is slightly shifted to lower Cm values with respect to AL-12 (Cm of 2.30M), suggesting that under chemical denaturation conditions, Q96Y is less stable than either AL-12 or R65S, and displays the lowest degree of cooperativity, comparable to H32Y (Table1).
In summary, these results show how a single restorative mutation is capable of changing the protein thermodynamic stability. Replacing a single amino acid could offer a protective or deleterious effect in the protein stability.
Restorative mutants display delayed fibril formation
It has been well established that amyloid formation requires an appropriate physicochemical environment. Previous reports in our laboratory showed that AL-12 and others AL proteins derived from the κI O18/O8 sequence, form amyloid fibrils in vitro faster than the germline protein at low pH.12,17 In addition, we found that salt increases the thermodynamic stability of AL proteins, while enhancing amyloid fibril formation under conditions where partially folded states are populated.11,16 The use of denaturants like urea or guanidinium hydrochloride is of great interest for investigating the amyloid fibril formation process. However, is essential to understand the in vivo behavior and the structural fluctuations occurring during the amyloid formation under physiological conditions.2
For that reason, we induced fibril formation of AL-12 and its restorative mutants at physiologic pH, at 37°C. Additionally, we have induced the fibril formation at pH 2.0, at which the fibril kinetics are accelerated for all of our AL proteins and non-amyloidogenic germline control.8,10,12
Figure 4 shows that kinetic traces of AL-12 fibril formation are sigmoidal, suggesting that amyloid fibrils form in a nucleation-dependent manner, similar to other amyloid proteins such as Aβ-peptide or β2-microglobulin.18 This is further supported by the acceleration of fibril formation in the presence of seeds (Blancas-Mejia and Ramirez-Alvarado, in preparation). We find that the time to complete 50% of the fibril reaction for AL-12 (or t50 value) is 253 h at 37°C, pH 7.0 [Fig. 4(A)]. At pH 2.0, AL-12 shows lower fluorescence enhancement compared with pH 7.0 while no notable change is reflected in the t50 value of the reaction [Fig. 4(B), Table2].
Figure 4.
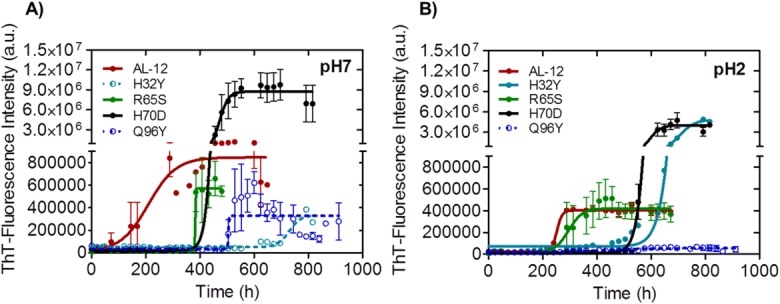
Differences in fibril formation kinetics at different pH values for AL-12 and mutants. Fibril formation traces followed by Thioflavin T (ThT) fluorescence, 37°C, at pH 7 (A) and pH 2 (B). A fourfold increase in ThT fluorescence over time indicates amyloid fibril formation. Continuous lines represent the best fitting. Open circles and dotted line (right panel) indicate no fibril formation, despite the apparent increase in ThT-fluorescence signal; Fibril formation was confirmed with transmission electron microscopy (Fig.5). Absence of an error bar indicates that only one of three replicates had the required fourfold fluorescence enhancement.
Table 2.
Thioflavin T Fluorescence Enhancement as a Function of Time—t50 Values of AL-12 Restorative Mutants
| Proteina | pH 7.0 | pH 2.0 |
|---|---|---|
| H32Y | 420.6 | 703.1 |
| Q96Y | 521.6 ± 17.9 | – |
| H70D | 482.5 ±1.5 | 600.6 ± 12.2 |
| R65S | 374.5 ± 5.9 | 333.3 ± 45.4 |
| κI O18/O8b | – | 258.3 ± 9.8 |
| AL-12 | 237.6 ± 45.2 | 253.1 ± 4.0 |
Error is the SEM from at least three independent experiments (n = 3). Absence of an error indicates that only one of three replicates had the required fourfold fluorescence enhancement. Italic values indicated no fibril formation, despite any ThT-fluorescence signal (t50 values).
Proteins are in order from highest to lowest t50 value.
Values from Blancas-Mejia et al. (12).
The four restorative mutants present the same kinetic profile for amyloid formation compared with AL-12. Interestingly, none of them show accelerated fibril formation kinetics compared with AL-12 [Fig. 4(A,B)]. At pH 7.0, mutants R65S and H70D delay the fibril formation process for several hours, while the H70D mutant shows an increment of ThT-fluorescence enhancement with respect to AL-12 [Fig. 4(A), Table2].
It is noteworthy to mention that the ThT fluorescence final plateau values differ between the AL-12 variants. There could be at least two possible reasons for this difference. First, each protein could form different polymorphic fibril structures that have different ThT binding capabilities.19,20 Second, the fibrils formed by different mutants may differ in stability, reaching different endpoint equilibrium states where different levels of amyloid are populated.21–23
Transmission Electron Microscopy (TEM) images show that the amyloid fibrils formed by R65S are indistinguishable from those formed by AL-12 (Fig. 5). H70D fibrils appear shortened and do not form a mesh, a morphology that concurs with an increase number of fibrils and an enhanced ThT-fluorescence signal. The most interesting results were found with the mutants H32Y and Q96Y as neither of them forms mature amyloid fibrils [even though we observed only a minor enhancement in the ThT-fluorescence emission at pH 7.0 [Fig. 4(A)]. Despite the lack of amyloid fibrils, TEM images show that these two variants are able to form prefibrillar or oligomeric species (Q96Y mutant), and some amorphous aggregates (H32Y mutant), that appear capable of binding ThT (Fig. 5), as it occurs with other amyloidogenic proteins.22,24 Because the kinetics of fibril formation for H32Y and Q96Y are much slower than AL-12, we do not know if only a small portion or the bulk of the low molecular weight aggregates of these mutants will be converted to aggregates after long-term incubations at 37°C.
Figure 5.
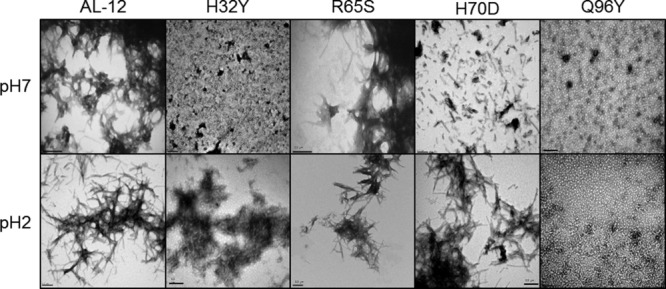
Transmission electron microscopy of amyloid fibrils and other species formed by AL-12 and its variants. TEM images confirm the presence of amyloid fibril structures at the endpoint of the kinetics for AL-12, AL-12 R65S, and AL-12 H70D. Scale bar is 200 nm.
In conclusion, we show that AL-12 forms fibrils faster than any of its restorative mutants, and the most stable and the least stable restorative mutants are unable to form fibrils under the conditions tested.
Discussion
Thermodynamic threshold for amyloid formation—Presence of partially folded species
Figure 6 summarizes the results of the amyloid fibril formation reactions, and compares t50 values as a function of two thermodynamic parameters (Tm and ΔGfolding) to determine a possible correlation between thermodynamic stability and fibrillogenesis for these proteins. Figure 6(B,C) show that proteins with a high or intermediate level of stability (Tm between 40 and 50°C; AL-12, R65S, and H70D) are more prone to form amyloid fibrils. These three proteins have Tm values near 37°C, the temperature where fibril formation occurs both in our in vitro system and certainly in vivo. This observation suggests that these proteins are sampling partially folded states within the time and conditions in which fibril formation occurs. Interestingly, the Tm for AL-12, the fastest amyloid fibril former protein, is the closest to 37°C [Fig. 6(B)]. We previously reported that AL-12 t50 value is significantly decreased at 46°C, the AL-12 Tm value, resulting in an acceleration of amyloid formation (∼60 h versus 238 h at 37°C).11
Figure 6.
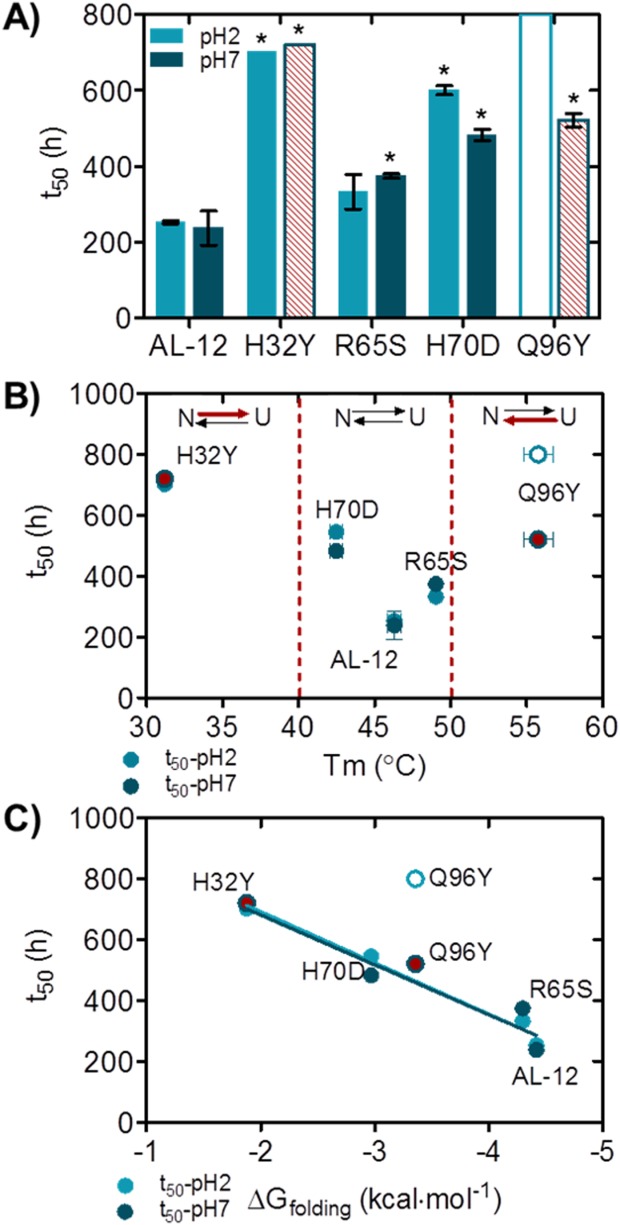
Differences in amyloid formation kinetics for AL-12 and mutants; correlation between protein stability and amyloid formation kinetics. A) Comparison of t50 values at pH 7 (dark blue) and pH 2 (light blue). B) Relationship between Tm and t50 values. Red arrows indicate unfolding reaction is shifted to the native or unfolded states. C) Relationship between t50 and ΔGfolding values. pH 7 and pH 2 data points were fitted separately into a linear regression model. Red stripped bars (A) and red circles (B, C) indicate no fibril formation, despite any ThT-fluorescence signal (t50 values). Opened stripped bar (A) and opened circle (B, C) denotes no fibril formation (t50 > 800 h). Absence of an error indicates that only one of three replicates had the required fourfold fluorescence enhancement. *P value <0.05 with respect to AL-12.
Together, these results suggest that partially folded states populated during the thermal unfolding transition become the key species in amyloidogenesis and thus incubation under conditions where these species are populated accelerates the process.25–27
Following this rationale, mutants that present Tm values above (shifting to native state) or below (shifting to unfolded state) the unfolding transition for AL-12 will not be able to form amyloid fibrils. H32Y and Q96Y Tm values are outside the fibrillation threshold/unfolding transition at pH 7.0 (40–50°C) and are the proteins that show no fibril formation in this study. Because the presence of some oligomeric species or amorphous aggregates, it is possible that these two mutants could eventually form fibrils after a long-term incubation. However, their apparent inability to form amyloid fibrils confirms our previous observation related to the thermodynamic threshold for fibril formation in which we proposed that proteins can be too unstable to form amyloid fibrils under certain conditions.8
By demonstrating that the folded and unfolded states are unable to form fibrils, we confirm that the amyloid formation reaction proceeds due to the sampling of partially folded states that are favored during the unfolding transition of these proteins.
Incongruency with the low thermodynamic stability hypothesis suggests new mechanism for AL fibril formation
Our results present an unusual case on the generally accepted model for how amyloidogenicity is achieved in globular proteins. Considering the thermodynamics parameters [Fig. 6(C)], AL-12 is one of the most stable proteins analyzed in this study, yet AL-12 is the most amyloidogenic. AL-12 forms amyloid significantly faster than the less stable R65S and H70D mutants. At the temperature of the aggregation reaction (37°C), the three variants populate partial unfolded species [Fig. 3(A)]. While most of it is populated by the H70D mutant (at 37°C is 11% unfolded), the protein nonetheless aggregates later than AL-12, presumably because the H70D mutant not only globally destabilizes the protein but also locally makes some non-native state less amyloidogenic.
This special behavior provides new insights on the AL fibril formation mechanism redefining the current thermodynamic hypothesis (low thermodynamic stability, high amyloidogenic tendency).
Different amyloid potential observed by different AL proteins showcases the difficulty in diagnosis and treatment for AL amyloidosis patients and justifies the study of different amyloidogenic protein sequences.
Several studies using AL proteins have shown that mutations that reduce the thermodynamic stability are more prone to form amyloid fibrils. Among these, Hurle et al. postulated that certain mutations at particular position enhance partially unfolded states and promote AL fibril formation.5 Wall et al. provided further evidence for a correlation between LC thermodynamic stability and tertiary structural features, with the propensity of these molecules to form amyloid.6,7 Also, Kim et al. demonstrated that an amyloidogenic light chain protein was inherently less thermodynamically stable and more prone to form fibrils than its nonpathological counterpart.4 Also, these authors proposed an approach to reduce fibril formation by stabilizing the native protein by reducing the levels of partially folded molecules using specific ligands.4 In our laboratory, Baden et al. compared the germline κI O18/O8 with the AL-09 protein and found that restorative and reciprocal mutations in AL-09 display a clear correlation between protein stability and amyloid fibril formation, with less stable AL-09 mutants forming fibrils at a faster rate than more stable ones.3,17 Later, Poshusta et al. worked with AL-T03, the most unstable AL protein found to date (Tm of 29.8°C).8 At 37°C, AL-T03 is 70% unfolded and does not form amyloid fibrils. Based on these results, Poshusta et al. proposed a thermodynamic threshold hypothesis for fibril formation indicating that some LC proteins are too highly unstable to form amyloid fibrils under certain conditions.
Notably, in contrast with these earlier reports, our results reported here demonstrate that thermodynamic stability and tendency to aggregation are not always directly correlated in some AL proteins. This new knowledge should be considered when designing therapeutic strategies, assuming that increased stability could be not enough to avoid fibril formation as our results demonstrate.
Location of restorative mutants affects the AL-12 stability and amyloidogenesis
Above all, our data confirm that the thermodynamic contribution is not the only factor that modulates amyloid aggregation. Most likely, other properties intrinsically linked to the primary structure of the protein (isoelectric point, β-sheet propensity, hydropathy, net charge, etc.) significantly influence the propensity of proteins to form amyloid fibrils. In the case of AL proteins, the location and amino acid properties of somatic mutations may be an important amyloidogenic determinant.28
The idea of globular protein amyloidogenicity is determined by a composite of global stability and local sequence amyloidogenicity has been well established. Interestingly, all of the four restorative mutations studied here are exposed to the solvent [Fig. 1(A)], thus in principle, they should neither impact on the hydrophobic collapse nor the global β-sheet structure of the protein. However, a single mutation could facilitate the accumulation of a non-native state that is prone to aggregation, or increase the intrinsic tendency of such a state to aggregate.29 Nowadays, the amyloidogenicity of globular proteins is frequently understood to derive from some combination of these two effects.
Furthermore, restoration of residues H32Y and H70D have a detrimental effect on the AL-12 stability. Comparing with other AL proteins with histidine mutations, the restorative mutation AL-103 H92D also destabilizes the protein,12 suggesting that histidine residues introduced by somatic mutations around the CDR regions and the top of the β-hairpin DE may confer a protective effect on the stability of the light chain. In addition, restoration of residue Q96 to Y significantly increases the protein stability, which is similar to our previous report on the AL-09 H87Y mutant.3 The fact that both residues are located at the dimer interface, leads us to infer that tyrosine residues in this area contributes favorably to the AL stabilization, probably by enhancing dimerization.30
Interestingly, restoration of tyrosine in the mutants H32Y and Q96Y rendered the highest differences in the thermal stability but, strikingly, in the opposite direction (decreasing and increasing, respectively). Takano et al. have demonstrated that –OH groups in side chains (such as the tyrosine phenol group) can contribute favourably to thermal stability, even if they are not hydrogen-bonded, when the specific amino acid is located within a favourable emvironment. Conversely, -OH groups can be detrimental if found in an unfavourable environment.31 This argument could explain the opposite effect of two tyrosine residues. Notably, this antagonistic effect is also shown by FT-IR, where H32Y significantly increases its random coil content at higher temperatures, whereas Q96Y remains unaltered.
Previously, Blancas-Mejia et al. showed with another AL protein that incubation at lower pH accelerates the fibril formation process.12 AL-12 fibril formation seems to be highly independent of the pH [Fig. 6(A)], as it was shown with AL-0910 because the t50 values are not significantly different between pH 2.0 and pH 7.0 for AL-12 and the four restorative mutants.
Q96Y mutant deserves special attention because its increment in stability followed by thermal denaturation is not reflected as an increment in stability followed by chemical denaturation. This suggests that thermal and urea denaturation follow different unfolding mechanisms.32,33 This is further complicated by the fact that we had to perform our chemical denaturation experiments at 4°C due to the evidence that one of the mutants is very unstable at room temperature and therefore, the comparison is more difficult to make between these two experiments. Further on, the thermodynamic discrepancy between the thermal and chemical denaturation—Tm and Cm value—could be described as a paradox where the two parameters are not equivalent, and the reason relays on the protein cooperativity (m value), or the change of its accessible surface area upon chemical unfolding.34
In summary, in the current study we demonstrate for the first time that somatic mutations can dramatically modulate the protein stability to prevent amyloid formation. In addition, we show that low thermodynamic stability does not always imply more amyloidogenicity, as is generally accepted. Also, the thermodynamic paradox opens doors to further studies on thermodynamic characterization in AL proteins, as could be the effect of temperature on the chemical denaturation.
Our new finding is significantly important for elucidate other factors implied on fibril formation. Apart from the impact of this new idea on the AL amyloidosis molecular mechanism, we believe these results may help to better understand other amyloid diseases.
Experimental Procedures
Site-directed mutagenesis
AL-12 shows 7 nonconservative mutations. From these, we have chosen to restore the following: AL-12 H32Y and AL-12 Q96Y, because they are located on the dimer interface; and AL-12 R65S and AL-12 H70D, because both of them are located in the β-hairpin DE; a region of the protein that we have not characterized before, but that has been shown to bind to the endoplasmic reticulum chaperone BiP and an inhibitor peptide.35,36 Restorative mutants were generated using the QuikChange Multi Site-directed Mutagenesis kit (Stratagene). The Mayo Clinic DNA Sequencing Core facility confirmed the mutated sequences. To make it easier, the AL-12 variable domain is referred as AL-12 protein; also, the AL-12 restorative mutants are referred as H32Y, R65S, H70D and Q96Y throughout the text.
Cloning, expression, extraction, and purification of AL-12 protein and mutants
The AL-12 VL sequence came from a patient diagnosed with cardiac AL amyloidosis as previously described.11 Briefly, the AL-12 sequence (Gen Bank accession number AF490912) was cloned, sequenced, and then expressed using the pET12a expression system in Escherichia coli BL21 (DE3) Gold-competent cells. The protein expression was induced with 0.8 mM IPTG for 16 to 18 h; after that, the cells were harvested and frozen at 20°C. Restorative AL-12 mutants were obtained following the same expression protocol.
AL-12, and the mutants R65S and H70D, were extracted from periplasmic space by breaking the cells through one freeze-thaw cycle using PBS buffer (pH 7.0). The periplasmic fraction was then dialyzed into 10 mM Tris-HCl (pH 7.0). Q96Y and H32Y mutants were extracted from solubilized inclusion bodies using 5M urea and refolded by dialysis against 10 mM Tris-HCl (pH 7.0). The proteins were purified using size exclusion chromatography (HiLoad 16/60 Superdex 75 column) on an AKTA FPLC (GE Healthcare, Piscataway, NJ).
Pure fractions were checked by SDS-PAGE and their protein concentration was determined by UV absorption at 280 nm using an extinction coefficient (ɛ) calculated from the amino acid sequence (13,610M−1 cm−1, for AL-12, R65S, and H70D; 14,890M−1 cm−1, for H32Y and Q96Y mutants). Pure fractions were combined, aliquoted at >100 μM, flash frozen, and stored at −80°C. Proteins were thawed at 4°C, filtered, and/or ultracentrifuged before they were used for each study.
Protein structure characterization
Secondary and tertiary structures were monitored by far UV-CD spectroscopy (Jasco spectropolarimeter 810), and by fluorescence (Varian Cary Eclypse spectrofluorometer) and infrared (Variant Resolutions Pro spectrometer) spectroscopies.
Circular Dichroism spectroscopy
Secondary structure of all proteins was monitored by far UV-CD, scanning from 260–200 nm at 4°C; temperature where all proteins are found in their native state. Samples contained 20 μM of protein (10 mM Tris buffer, pH 7.4) in a 0.2 cm quartz-cuvette. Five accumulations were recorded and the measurements were taken every 0.5 nm with a scan rate of 50 nm/min at 4°C, as previously reported.12
Fluorescence spectroscopy
Single-tryptophan residue was excited at 290 nm (slit 5 nm) and the fluorescence emission spectrum was recorded from 310 to 410 nm (slit 5 nm). Samples contained 20 μM of protein (10 mM Tris buffer, pH 7.4) in a 1 cm quartz-cuvette. Five accumulations were recorded and measurements were taken every 0.5 nm with a scan rate of 50 nm/min at 4°C.
Fourier-transform infrared spectroscopy (FT-IR)
Samples at 200 μM (2.4 mg/mL) were dialyzed at 4°C against deuterated-10 mM Tris buffer pH 7.0, using 15000 MWCO MINI Dialysis Units (G-Biosciences). Spectra were acquired at 4°C and 40°C using an excavated cell with a 50 μm path (Reflex Analytical) and the series software licensed under OMNIC (Thermo Scientific). Thousand spectra were recorded at a scan rate of 95 cm−1/min and averaged. FT-IR results are shown as the original amide I’ band.
Thermal denaturation
Thermal denaturation experiments were carried out by far-UV CD spectroscopy and intrinsic tryptophan fluorescence of 20 μM protein samples (10 mM Tris buffer, pH 7.4), from 4 to 90°C. Protein refolding was also measured from 90 to 4°C immediately after the denaturation experiment. CD and fluorescence spectra were recorded initially at 4°C, at 90°C, and again after cooling the samples to 4°C. Samples were done in triplicate.
Circular dichroism spectroscopy
The unfolding transition was followed by the change of ellipticity at 216 nm, the minimum of β-sheet structure. The temperature was increased by 0.5°C/min with a response time of 32 s, and measurements were taken every 0.5°C. The two-state unfolding curves were analyzed as described previously to determine the fraction folded and the Tm, or temperature where 50% of the protein is unfolded.16
Fluorescence spectroscopy
The thermal unfolding was also followed with the change of fluorescence at 340 nm (the maximum emission corresponding to the native single-Trp35) as a function of temperature. Samples were excited at 290 nm (5 nm slit), measurements were taken every 0.1°C, and the temperature was increased by 1°C/min with a response time of 2 s.
Chemical denaturation
Chemical denaturation was performed as previously described.37 Briefly, 2 μM protein samples containing progressive urea concentration (0–6M) were equilibrated in 10 mM Tris-HCl (pH 7.0) overnight at 4°C. That temperature was selected to ensure that all proteins were fully folded at the beginning of the reaction and also to reduce the contribution of the temperature during the determination of the thermodynamic parameters by chemical denaturation. Final urea concentrations were calculated with a hand refractometer.38 The denaturation experiment was followed by tryptophan fluorescence, and the emission spectrum of each sample was recorded as described above. The unfolding transitions were analyzed by following the red-shift in the maximum emission spectra; maxima were obtained by fitting each spectrum to a four-order polynomial equation [Eq. 1]:39
| 1 |
The equilibrium parameters from spectroscopic data were calculated for each urea concentration using Eq. 2, a nonlinear least-squares fitting to a two-state model:40
| 2 |
where, FN = a + (b × x); FU = c + (d × x); K = exp(−ΔG/(R × T); ΔG = G − m × x; in which the dependence of the intrinsic fluorescence in the native (FN) and unfolded (FU) states upon increasing denaturant concentration (x) is taken into account by the terms b × x and d × x, respectively (linear approximation). ΔG is the free energy of unfolding in the absence of denaturant; m is ΔG/Cm; R is the ideal gas constant; and T is temperature. Cm, where 50% of the protein is unfolded, was calculated as the ΔG/m ratio. The proteins are considered monomeric at the concentration used for this experiment based on the Kd value found for AL-12 (∼400 μM).
Fibril formation assay
Because of the presence of preformed aggregates may accelerate the fibril formation kinetics,3,16 protein samples were ultracentrifuged before the fibril formation assay following the protocol described by DiCostanzo et al.10 Fibril formation assays were performed in triplicate using black 96-well polystyrene plates and shaken continuously at 300 rpm at 37°C in a New Brunswick Scientific Innova40 incubator shaker. Each well contained 260 μL of 20 μM protein, 150 mM NaCl, 10 μM thioflavin T (ThT), 0.02% NaN3 in 10 mM sodium acetate, boric acid, and sodium citrate (ABC) buffer at pH 2.0 or 7.0. ThT-fluorescence was used to follow the fibril formation kinetics,21,41 and was monitored daily on a plate reader (Analyst AD, Molecular Devices) with an excitation wavelength of 440 nm and an emission wavelength of 480 nm, until the reaction reached the plateau (∼600–800 h).
A fibril formation reaction was considered completed when ThT fluorescence enhancement reached a plateau; and was considered positive when ThT-fluorescence readings reached at least four times the lowest ThT reading of that particular reaction (usually 200,000 counts/s).
The t50 value, or the time at which the fibril formation reaction is 50% complete, was obtained by fitting each kinetic trace to a nonlinear Boltzmann sigmoidal equation as defined by the Prism software.12 The comparison of fibril formation between AL-12 and the restorative mutants was based on paired Student's t-test. Significance was reported at the 95% (P < 0.05) confidence level.
Transmission electron microscopy
The presence of amyloid fibril structures at the endpoint of kinetic reactions was confirmed by TEM. A 5 μL fibril sample was placed on a 300 mesh copper formvar/carbon grid (Electron Microscopy Science, Hatfield, PA), and excess liquid was removed. The samples were negatively stained with 4% uranyl acetate, washed once with sterile H2O, and air-dried. Grids were analyzed on a Philips Tecnai T12 transmission electron microscope at 80 kV (FEI, Hillsboro, OR).
Acknowledgments
The authors thank Professor Raul Urrutia and Dr. Alexander Tischer for helpful discussion and critical reading of this manuscript. The authors also thank the generosity of amyloidosis patients and their families. S.V., M.M.A., and M.R.A. designed the experiments, M.M.A. and J.G.B. performed the experiments, M.M.A., S.V., and L.B.M. analyzed the data, M.M.A., S.V., M.R.A. wrote the article.
Glossary
- AL amyloidosis
light chain amyloidosis
- CDR
complementary determining region
- CL
light chain constant domain
- Ig
immunoglobulin
- LC
light chain
- t50 value
the time to complete 50% of the fibril reaction
- ThT
Thioflavin T
- VL
light chain variable domain.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
Supporting Information
References
- Baden EM, Sikkink LA, Ramirez-Alvarado M. Light chain amyloidosis - Current findings and future prospects. Curr Protein Pept Sci. 2009;10:500–508. doi: 10.2174/138920309789351949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat Chem Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- Baden EM, Randles EG, Aboagye AK, Thompson JR, Ramirez-Alvarado M. Structural insights into the role of mutations in amyloidogenesis. J Biol Chem. 2008;283:30950–30956. doi: 10.1074/jbc.M804822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Wall JS, Meyer J, Murphy C, Randolph TW, Manning MC, Solomon A, Carpenter JF. Thermodynamic modulation of light chain amyloid fibril formation. J Biol Chem. 2000;275:1570–1574. doi: 10.1074/jbc.275.3.1570. [DOI] [PubMed] [Google Scholar]
- Hurle MR, Helms LR, Li L, Chan W, Wetzel R. A role for destabilizing amino acid replacements in light-chain amyloidosis. Proc Natl Acad Sci USA. 1994;91:5446–5450. doi: 10.1073/pnas.91.12.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall J, Schell M, Murphy C, Hrncic R, Stevens FJ, Solomon A. Thermodynamic instability of human lambda 6 light chains: correlation with fibrillogenicity. Biochemistry. 1999;38:14101–14108. doi: 10.1021/bi991131j. [DOI] [PubMed] [Google Scholar]
- Wall JS, Gupta V, Wilkerson M, Schell M, Loris R, Adams P, Solomon A, Stevens F, Dealwis C. Structural basis of light chain amyloidogenicity: comparison of the thermodynamic properties, fibrillogenic potential and tertiary structural features of four Vlambda6 proteins. J Mol Recognit. 2004;17:323–331. doi: 10.1002/jmr.681. [DOI] [PubMed] [Google Scholar]
- Poshusta TL, Katoh N, Gertz MA, Dispenzieri A, Ramirez-Alvarado M. Thermal stability threshold for amyloid formation in light chain amyloidosis. Int J Mol Med. 2013;14:22604–22617. doi: 10.3390/ijms141122604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randles EG, Thompson JR, Martin DJ, Ramirez-Alvarado M. Structural alterations within native amyloidogenic immunoglobulin light chains. J Mol Biol. 2009;389:199–210. doi: 10.1016/j.jmb.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCostanzo AC, Thompson JR, Peterson FC, Volkman BF, Ramirez-Alvarado M. Tyrosine residues mediate fibril formation in a dynamic light chain dimer interface. J Biol Chem. 2012;287:27997–28006. doi: 10.1074/jbc.M112.362921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikkink LA, Ramirez-Alvarado M. Salts enhance both protein stability and amyloid formation of an immunoglobulin light chain. Biophys Chem. 2008;135:25–31. doi: 10.1016/j.bpc.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancas-Mejia LM, Tischer A, Thompson JR, Tai J, Wang L, Auton M, Ramirez-Alvarado M. Kinetic control in protein folding for light chain amyloidosis and the differential effects of somatic mutations. J Mol Biol. 2014;426:347–361. doi: 10.1016/j.jmb.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancas-Mejia LM, Hammernik J, Marin-Argany M, Ramirez-Alvarado M. Differential effects on light chain amyloid formation depend on mutations and type of glycosaminoglycans. J Biol Chem. 2015;290:4953–4965. doi: 10.1074/jbc.M114.615401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albinsson B, Norden B. Excited-state properties of the indole chromophore: electronic transition moment directions from linear dichroism measurements: Effect of methyl and methoxy substituents. J Phys Chem. 1992;96:6204–6212. [Google Scholar]
- Sreerama N, Manning MC, Powers ME, Zhang JX, Goldenberg DP, Woody RW. Tyrosine, phenylalanine, and disulfide contributions to the circular dichroism of proteins: Circular dichroism spectra of wild-type and mutant bovine pancreatic trypsin inhibitor. Biochemistry. 1999;38:10814–10822. doi: 10.1021/bi990516z. [DOI] [PubMed] [Google Scholar]
- McLaughlin RW, De Stigter JK, Sikkink LA, Baden EM, Ramirez-Alvarado M. The effects of sodium sulfate, glycosaminoglycans, and Congo red on the structure, stability, and amyloid formation of an immunoglobulin light-chain protein. Protein Sci. 2006;15:1710–1722. doi: 10.1110/ps.051997606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baden EM, Owen BA, Peterson FC, Volkman BF, Ramirez-Alvarado M, Thompson JR. Altered dimer interface decreases stability in an amyloidogenic protein. J Biol Chem. 2008;283:15853–15860. doi: 10.1074/jbc.M705347200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MR, Yerbury JJ, Poon S. Potential roles of abundant extracellular chaperones in the control of amyloid formation and toxicity. Mol Biosyst. 2008;4:42–52. doi: 10.1039/b712728f. [DOI] [PubMed] [Google Scholar]
- Lindberg DJ, Wranne MS, Gilbert Gatty M, Westerlund F, Esbjorner EK. Steady-state and time-resolved Thioflavin-T fluorescence can report on morphological differences in amyloid fibrils formed by Abeta(1-40) and Abeta(1-42) Biochem Biophys Res Commun. 2015;458:418–423. doi: 10.1016/j.bbrc.2015.01.132. [DOI] [PubMed] [Google Scholar]
- Qiang W, Yau WM, Tycko R. Structural evolution of Iowa mutant beta-amyloid fibrils from polymorphic to homogeneous states under repeated seeded growth. J Am Chem Soc. 2011;133:4018–4029. doi: 10.1021/ja109679q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana R, Coleman C, Ionescu-Zanetti C, Carter SA, Krishna V, Grover RK, Roy R, Singh S. Mechanism of thioflavin T binding to amyloid fibrils. J Struct Biol. 2005;151:229–238. doi: 10.1016/j.jsb.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Lindgren M, Sorgjerd K, Hammarstrom P. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys J. 2005;88:4200–4212. doi: 10.1529/biophysj.104.049700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AD, Shivaprasad S, Wetzel R. Alanine scanning mutagenesis of Abeta(1-40) amyloid fibril stability. J Mol Biol. 2006;357:1283–1294. doi: 10.1016/j.jmb.2006.01.041. [DOI] [PubMed] [Google Scholar]
- Marin-Argany M, Rivera-Hernandez G, Marti J, Villegas S. An anti-Abeta (amyloid beta) single-chain variable fragment prevents amyloid fibril formation and cytotoxicity by withdrawing Abeta oligomers from the amyloid pathway. Biochem J. 2011;437:25–34. doi: 10.1042/BJ20101712. [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Ann Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Ramirez-Alvarado M, Merkel JS, Regan L. A systematic exploration of the influence of the protein stability on amyloid fibril formation in vitro. Proc Natl Acad Sci USA. 2000;97:8979–8984. doi: 10.1073/pnas.150091797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancas-Mejia LM, Ramirez-Alvarado M. Systemic amyloidoses. Ann Rev Biochem. 2013;82:745–774. doi: 10.1146/annurev-biochem-072611-130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poshusta TL, Sikkink LA, Leung N, Clark RJ, Dispenzieri A, Ramirez-Alvarado M. Mutations in specific structural regions of immunoglobulin light chains are associated with free light chain levels in patients with AL amyloidosis. PLoS One. 2009;4:e5169. doi: 10.1371/journal.pone.0005169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel R. Mutations and off-pathway aggregation of proteins. Trends Biotechnol. 1994;12:193–198. doi: 10.1016/0167-7799(94)90082-5. [DOI] [PubMed] [Google Scholar]
- Brumshtein B, Esswein SR, Landau M, Ryan CM, Whitelegge JP, Phillips ML, Cascio D, Sawaya MR, Eisenberg DS. Formation of amyloid fibers by monomeric light chain variable domains. J Biol Chem. 2014;289:27513–27525. doi: 10.1074/jbc.M114.585638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano K, Scholtz JM, Sacchettini JC, Pace CN. The contribution of polar group burial to protein stability is strongly context-dependent. J Biol Chem. 2003;278:31790–31795. doi: 10.1074/jbc.M304177200. [DOI] [PubMed] [Google Scholar]
- Vogt G, Woell S, Argos P. Protein thermal stability, hydrogen bonds, and ion pairs. J Mol Biol. 1997;269:631–643. doi: 10.1006/jmbi.1997.1042. [DOI] [PubMed] [Google Scholar]
- Bennion BJ, Daggett V. The molecular basis for the chemical denaturation of proteins by urea. Proc Natl Acad Sci USA. 2003;100:5142–5147. doi: 10.1073/pnas.0930122100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskakov IV, Bolen DW. The paradox between m values and deltaCp's for denaturation of ribonuclease T1 with disulfide bonds intact and broken. Protein Sci. 1999;8:1314–1319. doi: 10.1110/ps.8.6.1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DP, Gallo G, Vogen SM, Dul JL, Sciarretta KL, Kumar A, Raffen R, Stevens FJ, Argon Y. Both the environment and somatic mutations govern the aggregation pathway of pathogenic immunoglobulin light chain. J Mol Biol. 2001;313:1021–1034. doi: 10.1006/jmbi.2001.5092. [DOI] [PubMed] [Google Scholar]
- Dul JL, Davis DP, Williamson EK, Stevens FJ, Argon Y. Hsp70 and antifibrillogenic peptides promote degradation and inhibit intracellular aggregation of amyloidogenic light chains. J Cell Biol. 2001;152:705–716. doi: 10.1083/jcb.152.4.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Hernandez G, Marin-Argany M, Blasco-Moreno B, Bonet J, Oliva B, Villegas S. Elongation of the C-terminal domain of an anti-amyloid beta single-chain variable fragment increases its thermodynamic stability and decreases its aggregation tendency. mAbs. 2013;5:678–689. doi: 10.4161/mabs.25382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace CN, Scholtz M. Protein structure: A practical approach. Vol. 1. Oxford University Press; 1997. p. 383. , Vol.. San Diego, California. [Google Scholar]
- Knappik A, Pluckthun A. Engineered turns of a recombinant antibody improve its in vivo folding. Protein Eng. 1995;8:81–89. doi: 10.1093/protein/8.1.81. [DOI] [PubMed] [Google Scholar]
- Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal Biochem. 1989;177:244–249. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information