

Abstract
Protein secretion is a major contributor to Gram-negative bacterial virulence. Type Vb or two-partner secretion (TPS) pathways utilize a membrane bound β-barrel B component (TpsB) to translocate large and predominantly virulent exoproteins (TpsA) through a nucleotide independent mechanism. We focused our studies on a truncated TpsA member termed hemolysin A (HpmA265), a structurally and functionally characterized TPS domain from Proteus mirabilis. Contrary to the expectation that the TPS domain of HpmA265 would denature in a single cooperative transition, we found that the unfolding follows a sequential model with three distinct transitions linking four states. The solvent inaccessible core of HpmA265 can be divided into two different regions. The C-proximal region contains nonpolar residues and forms a prototypical hydrophobic core as found in globular proteins. The N-proximal region of the solvent inaccessible core, however, contains polar residues. To understand the contributions of the hydrophobic and polar interiors to overall TPS domain stability, we conducted unfolding studies on HpmA265 and site-specific mutants of HpmA265. By correlating the effect of individual site-specific mutations with the sequential unfolding results we were able to divide the HpmA265 TPS domain into polar core, nonpolar core, and C-terminal subdomains. Moreover, the unfolding studies provide quantitative evidence that the folding free energy for the polar core subdomain is more favorable than for the nonpolar core and C-terminal subdomains. This study implicates the hydrogen bonds shared among these conserved internal residues as a primary means for stabilizing the N-proximal polar core subdomain.
Keywords: two-partner secretion, protein folding, beta-helix, domain, subdomain, sequential unfolding
Introduction
Gram-negative bacteria use two-partner secretion (TPS) pathways to transport predominantly virulent proteins across their outer membranes. The TPS pathways are composed of two components, a secreted A exoprotein (TpsA) and an Omp85 related B translocase protein (TpsB). The requisite outer membrane bound B component recognizes and translocates its cognate A partner.1,2 Upon TpsB dependent secretion, TpsA protein family members function as adhesins, cytotoxins, contact growth inhibitors, proteases, and heme binding proteins in pathogenic and non-pathogenic Gram-negative species.3–11 The hemagglutnin TpsB component and its role in translocation has been the subject of many studies (reviewed in Ref.12–13). To better understand the role that TpsA folding plays in translocation, we investigated the structural stability of a hemolysin TPS domain from P. mirabilis, termed HpmA265. We have found that the HpmA265 TPS domain comprises at least three structural subdomains with differing stabilities, and suggest models for TPS translocation based upon the observed sequential unfolding of this domain.
Hemolysin A (HpmA), a prototypical hemolysin TpsA representative from P. mirabilis, is secreted using a TPS pathway.14 Like other A components secreted via the TPS pathway, HpmA is translocated, folded, and activated by its cognate B component (HpmB).15,16 In order to study the folding energetics of the TPS domain, we utilized a cloned fragment of HpmA that encodes methionine 1 through glycine 265 as previously reported.14 We termed this truncated protein HpmA265 in order to indicate its most C-terminal residue based on the full length HpmA sequence. As in the full length protein, HpmA265 expression includes Sec dependent transport across the inner membrane, proteolytic cleavage to remove the Sec signal peptide (Supporting Information Fig. S1), and membrane bound HpmB coupled secretion into the external milieu.6,14,15,17 All of the studies discussed in this work were completed upon fully processed, secreted and purified HpmA265 or variants of HpmA265 harboring site-specific mutations. Therefore, each purified form of HpmA265 under investigation spans residues asparagine 30 through glycine 265. The construct that does not harbor any site-specific mutations is identified as HpmA265 in this study. The naming convention used for the site-specific mutants within this study uses the nomenclature Q125A, where the glutamine residue at position 125 within HpmA265 has been replaced with an alanine.
Structurally, HpmA265 adopts a non-globular three-sided β-helix with parallel and anti-parallel β-sheet segments.14 As described previously, the parallel β-strands form the three-sided HpmA265 β-helix structure, while the anti-parallel β-strands form the N-terminal “cap” and the flanking “flap” regions [Fig. 1(A)].14 The β-helix portion of the HpmA265 structure comprises five full turns or β-circuits, where each circuit comprises three parallel β-strands and three adjoining turns. The β-helix structure also forms a large solvent inaccessible protein core [Fig. 1(B)]. The exposed edges of the β-helix are protected on the N-terminal end by the “cap” structure [Fig. 1(A)], while those on the C-terminal end are protected through dimerization.14
Figure 1.
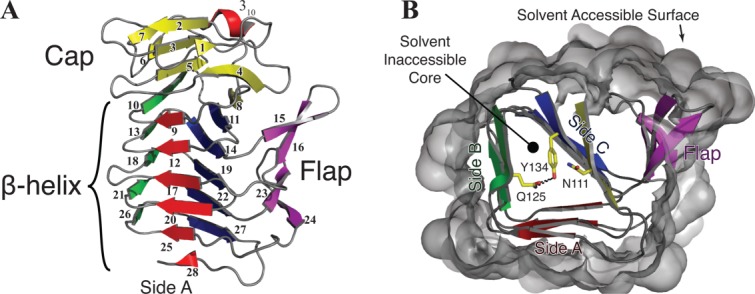
Cartoon representation of the TPS domain from HpmA265 (PDB ID: 4W8Q). A) Side view of structure. PDBsum determined β-strands are numbered starting at the N-terminus. The short 310 helix is labeled. The anti-parallel cap (yellow), flap (purple) and non-globular parallel β-helix (blue, red, green) are indicated. B) Top-down view parallel to helical axis showing the interior of the β-helix, the last β-strand within the cap region and the β-strands associated with β-circuits 1-3 are visible. To clearly show the interior, the remaining β-strands and solvent accessible surface have been removed. Strands are colored as in panel A. The solvent accessible surface is shown in gray. The core of protein is defined as being surrounded by β-strands and inaccessible to solvent within the β-helix. Internal polar residues N111, Q125, and Y134 are shown in stick representation. The hydrogen bonds between Q125 and Y134 are shown as black dotted lines.
The HpmA265 cap structure and the first two β-circuits contain solvent inaccessible polar residues that are highly conserved within the hemolysin family [Fig. 2(A)], but not across the other TpsA family members (data not shown). These conserved residues including N111, Q125 and Y134, are located within the solvent inaccessible core of HpmA265 [Figs. 1(B) and 2(B)]. The hydrogen bond network involving these internal residues terminates at an internal water molecule, which is within hydrogen bonding distance of the side chains of Q125 and C147 and the main chain atoms of G123 and G148. Because these internal polar residues reside between N30 and N151, we have further defined this internal region within the HpmA265 TPS domain as the N-proximal “polar core”. The solvent inaccessible residues within the remaining HpmA265 β-circuits 3 – 5 are nonpolar, giving this segment of the protein a prototypical hydrophobic core. We have defined this internal region of HpmA265 as the C-proximal “hydrophobic core”. Compared to the N-proximal polar residues, the internal residues within the C-proximal hydrophobic core are more poorly conserved within the hemolysin family members. The disparities in internal polarity and conservation suggest that the HpmA265 N-proximal polar and C-proximal hydrophobic cores might have differing stabilities that may be important for TPS domain folding.
Figure 2.

Hemolysin TpsA sequence alignment and structural components. A) TpsA sequence alignment of hemolysin family members. Sequences were aligned using the clustal-omega interface provided by uniprot.org with default parameters. Conservation at each position is noted below the sequences. Residue numbering is in reference to the HpmA full length sequence. Secondary structure based on PDBsum assignments is noted above the sequence in cartoon format. Secondary structure cartoons are colored to indicate polar core (green), hydrophobic core (gray), the junction between these two cores (yellow), or flap (purple). The HpmA sequence is coded to show internal residues as upper-case based on a relative solvent accessibility of 0.2 or less. Highlighted residues indicate a residue that participates in the internal hydrogen bond network: main chain-side chain hydrogen bond (blue), side chain-side chain (red), and disulfide bond (yellow). Assignments of protein segments to cap, flap, and β-circuits are indicated above the secondary structure cartoons. Residues that are mutated as part of this study are indicated with a red x below the alignment. Hemolysins aligned and their uniprot accession numbers are: HpmA, P16466; ShlA, P15320; ChlA, Q7NWR2; PhlA, Q7N9K8; XhlA, D3UYN1; EthA, O32608; HhdA, Q47955; HhhA, F9GTG6. B) HpmA structure showing N-proximal polar core, C-proximal hydrophobic core, extra-helical flap, and the junction between the two cores. Some of the internal polar residues (red highlights in the alignment) are shown as sticks colored by atom type. Internal residues that are part of the hydrophobic core are shown as spheres. Carbon color indicates: yellow – mutated within this study; cyan – trypsin cleavage site; gray – other internal residues. An internal water at the polar/nonpolar core junction is shown as a cyan sphere.
In this study we used HpmA265 and a series of site-specific HpmA265 point mutants to investigate the unfolding and to assign the contributions of the N-proximal polar and C-proximal hydrophobic cores toward TPS domain stability. Here, we present evidence that is consistent with a four-state sequential model for unfolding the HpmA265 TPS domain.18 This multi-state unfolding was surprising since, visually, the TPS functional domain appears to consist of a single structural domain. In order to ascribe the contributions of the N-proximal polar and C-proximal hydrophobic cores toward HpmA265 stability, we designed site-specific mutations targeting the solvent inaccessible residues N66, F80, N111, Q125, Y134, C144, C147, L194, I207, and F241 [Fig. 2(A,B)]. Collectively, the denaturation studies on the HpmA265 mutants have allowed us to assign three subdomains (SD1, SD2, SD3) within the larger functional TPS domain encompassed by HpmA265. Based upon the results presented here, these subdomains are termed the C-terminal, nonpolar core, and polar core subdomains, respectively. The large free energy for folding the polar core subdomain (SD3) suggests that it may play a role in hemolysin TPS domain folding during secretion.
Results
In order to determine the stability of the TPS domain, we undertook guanidine hydrochloride (GdnHCl) denaturation studies of HpmA265 and site-specific mutants of HpmA265. The mutations were selected to test the relative contribution of crystallographically observed internal interactions toward the free energy of folding and to allow assignment of specific unfolding transitions to different subdomains within the HpmA265 TPS domain (PDB ID: 4W8Q). Specifically, the denaturation studies conducted upon HpmA265 have allowed us to divide the hemolysin TPS domain into three structural subdomains, SD1, SD2, and SD3 and quantitatively determine the free energy of folding associated with each subdomain. Denaturation studies of the mutants including N66L, F80L, N111L, Q125A, Q125F, Y134F, C144S-C147S, C144A-C147A, L194N, I207N, and F241K allowed us to describe the regions of HpmA265 associated with each of these newly identified subdomains.
HpmA265 unfolds via three transitions
HpmA265 unfolds via three separate transitions in CD monitored GdnHCl denaturation experiments [Fig. 3(A)]. As with other multi-state unfolding proteins, these multiple transitions indicate that there are multiple structural subdomains within the functional TPS domain.19–22 We propose that the HpmA265 TPS domain comprises three structural subdomains (SD1, SD2, SD3) that unfold sequentially via three distinct transitions, T1, T2, and T3. In this scheme, the unfolding of SD1 during T1 converts the native (N) HpmA265 into intermediate 1 (I1), the unfolding of SD2 during T2 converts I1 to intermediate 2 (I2), and the unfolding of SD3 during T3 converts I2 to the denatured state (D) [Fig. 3(A,B)]. Controls describing complete denaturation (Figure S2), reversibility (Figure S3), and the kinetics of denaturation (Figure S4) are provided in the Supporting Information.
Figure 3.
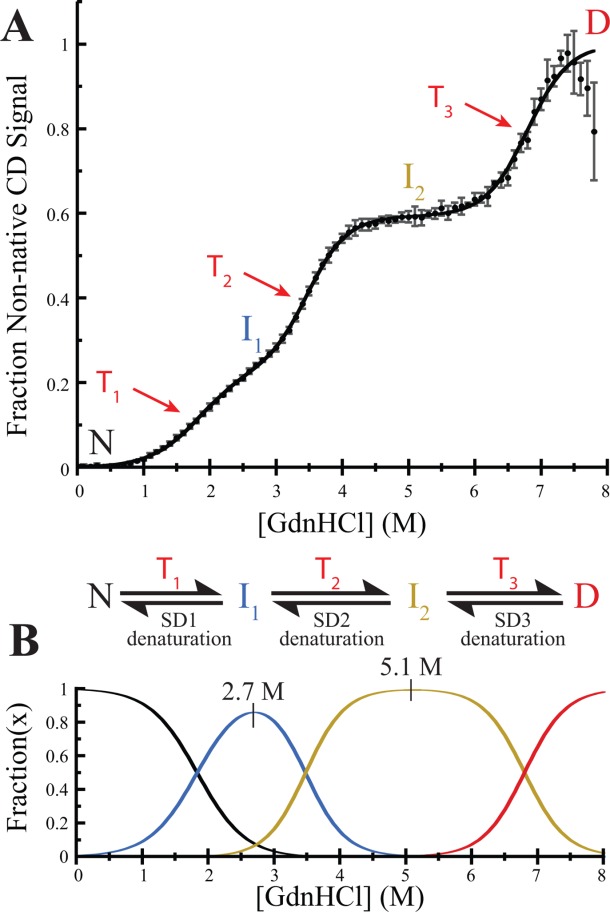
HpmA265 TPS domain unfolding and sequential unfolding model. A) Replicate denaturation profiles presented as average ± standard deviation of three experiments. Native (N), first intermediate (I1), second intermediate (I2), and denatured state (D) are indicated with colors corresponding to panel (B), the transitions between these states (T1, T2, T3) are indicated with red arrows. Transitions 1, 2, and 3 correspond to the denaturation of SD1, SD2, and SD3, respectively. Trend line calculated from average values of the fit parameters as presented in Table I. B) Scheme for sequential unfolding model and fraction of N, I1, I2, and D over a range of GdnHCl concentrations. The approximate GdnHCl concentration necessary to give maximum population of each of the intermediates is indicated above the peak.
Two models for the multi-state subdomain unfolding – sequential and independent unfolding – were considered to fit the denaturation data (Supporting Information, Figure S5). In order to distinguish between the two models, we undertook denaturation studies of N-proximal polar core and the C-terminal hydrophobic core mutants. The results of these denaturation studies with mutant proteins support a sequential model for unfolding [Fig. 3(B)]. Based on this analysis, the values in Table1 and the scheme in Figure 3(B) are for the sequential model only. In a sequential unfolding model, SD1 must always denature prior to denaturation of SD2, which denatures prior to the denaturation of SD3. The mutational analysis below is used to assign approximate boundaries to the subdomains based on both the percentage loss in structure and whether specific mutations affect the stability of the N, I1, or I2 state(s).
Table 1.
Thermodynamic Values for HpmA265 Sequential Folding
| Subdomains |
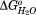 a(kJ/mol) a(kJ/mol) |
m value (kJ/mol/M) | Cm (M) | Fractional CD change | Approximate amount of structureb |
|---|---|---|---|---|---|
| SD1 | −11.7 ± 0.6c | 6.3 ± 0.2 | 1.87 ± 0.04 | 0.19 ± 0.01 | 24% |
| SD2 | −29.9 ± 0.9 | 8.6 ± 0.2 | 3.50 ± 0.01 | 0.27 ± 0.01 | 35% |
| SD3 | −52 ± 3 | 7.7 ± 0.5 | 6.8 ± 0.1 | 0.32 ± 0.02 | 41% |
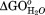 calculated based on a linear model for GdnHCl interactions. Transitions are modeled as sequential transitions.
calculated based on a linear model for GdnHCl interactions. Transitions are modeled as sequential transitions.
Amount of structure in each subdomain based on a total fractional change in CD signal of 0.78 for N→D.
Reported values represent average and standard deviations from at least three trials.
For HpmA265, the three transitions represent unfolding of subdomains within the larger TPS domain. As seen in Table1, the increasing transition midpoints associated with denaturing each of the subdomains is due primarily to large increases in the favorable folding free energies (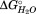 ), and not to large changes in the GdnHCl dependence of the unfolding (m value). The changes in
), and not to large changes in the GdnHCl dependence of the unfolding (m value). The changes in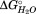 parallel an increasing percentage loss of structure at each transition. The m value for typical globular proteins is correlated with the change in solvent accessible surface area, and thus, the size of the structure that is denatured.23,24 The amount of structure lost in each transition for HpmA265 also correlates with the apparent change in solvent accessible surface area as measured by the m value for each transition. Based upon the HpmA265 structure and the cooperative folding of β-strand25,26 and β-helix27 conformations, a simple assumption for subdomain boundaries would have each subdomain encompass contiguous sets of β-strands. Since any subdomain will consist of contiguous strands, the three HpmA265 subdomains must originate from N-proximal, central, and C-proximal regions of the protein.
parallel an increasing percentage loss of structure at each transition. The m value for typical globular proteins is correlated with the change in solvent accessible surface area, and thus, the size of the structure that is denatured.23,24 The amount of structure lost in each transition for HpmA265 also correlates with the apparent change in solvent accessible surface area as measured by the m value for each transition. Based upon the HpmA265 structure and the cooperative folding of β-strand25,26 and β-helix27 conformations, a simple assumption for subdomain boundaries would have each subdomain encompass contiguous sets of β-strands. Since any subdomain will consist of contiguous strands, the three HpmA265 subdomains must originate from N-proximal, central, and C-proximal regions of the protein.
Site-specific mutation analysis
To ascribe SD1, SD2, and SD3 to particular regions of the HpmA265 structure, site-specific alterations were constructed within the N-proximal polar and C-proximal hydrophobic cores. Previous structural studies on HpmA265 noted that the side chains of conserved internal polar amino acids N66, Y68, N111, Q125, and Y134 [Fig. 2(B)] participate in a network of hydrogen bonds.14 All of these residues are conserved within the Type Vb TpsA hemolysin members and are located within the N-proximal polar solvent inaccessible interior [Fig. 2(A)]. The nearly conserved C144 and C147 were also targeted as they form an internal disulfide bond at the structural junction between the polar and hydrophobic cores [Fig. 2(B)]. To disrupt the C-proximal hydrophobic core, the internal residues L194, I207, and F241 were mutated to polar residues. In addition, tryptic digests were used to select for stable structures within HpmA265. All of these mutants were analyzed for template assisted hemolytic activity to demonstrate native-like structure (Supporting Information Figure S6). The effects of each site-specific alteration support a sequential unfolding model and allow assignment of regions of HpmA265 to each of the three subdomains, SD1, SD2, and SD3, within the HpmA265 TPS domain. Plots showing average data and fit lines for each of the variants of HpmA265 are provided in the Supporting Information (Figure S7). The residues mutated and their locations within the HpmA265 TPS domain have been provided in Figure 4. This figure serves as a framework for the results presented below in Figure 5 and the logic used to define the boundaries for the three subdomains, SD1, SD2, and SD3. Likewise, Table2 provides the effects of the mutations on the Cm values for each subdomain. In general, large changes in the Cm values due to a mutation are caused by large shifts in the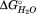 and not to large changes in the m value (Supporting Information Table S1).
and not to large changes in the m value (Supporting Information Table S1).
Figure 4.
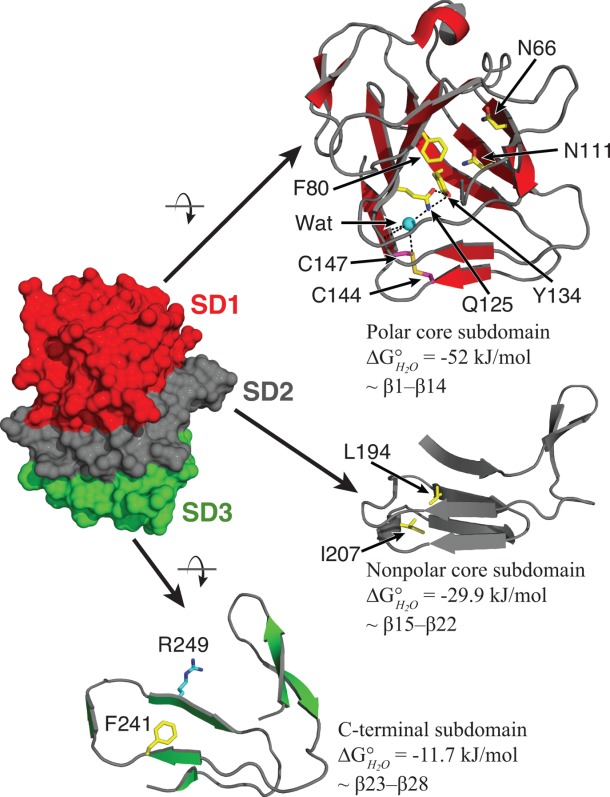
Subdomain divisions and residues mutated within HpmA265. Protein subdomains are as discussed in the text and are colored as polar core subdomain (red), nonpolar core subdomain (gray), C-terminal subdomain (green). The internal amino acid residues mutated in this study are shown as sticks colored by atom type (yellow – carbon; blue – nitrogen; red – oxygen; magenta – sulfur). Carbon atoms on R249, the site of the trypsin cleavage, are colored cyan. Black dashed lines indicate probable hydrogen bonds involving Q125, Y134, and an internal water (Wat, cyan sphere).
Figure 5.

GdnHCl titrations of mutants and resulting sequential model for HpmA265 unfolding. Titration data and trend lines are presented as in Figure 3 with HpmA265 data provided in black in each panel. The effect of each mutation on HpmA265 are noted by a red arrow for each group of mutations. Cartoon insets show site-specifically mutated residues rendered as spheres and colored by atom type. (A) Constructs targeting the C-terminal subdomain resulting in elimination of the N → I1 transition (T1). (B) Constructs targeting the nonpolar subdomain resulting in a shift of the I1 → I2 transition (T2). (C) Representative constructs targeting the polar subdomain resulting in a shift of the I2 → D transition (T3). (D) Sequential unfolding model for HpmA265. The subdomain that is unfolded during each transition is named below the arrow.
Table 2.
Cm Values for HpmA265 Site-Specific Mutants
| Sample | T1 Cm (M) | T2 Cm (M) | T3 Cm (M) | Effect |
|---|---|---|---|---|
| HpmA265 | 1.87 ± 0.04a | 3.50 ± 0.01 | 6.8 ± 0.1 | |
| Carboxy-terminal | ||||
| trHpmA249 | 1.5 ± 0.2b | 3.46 ± 0.05 | 6.6 ± 0.2 | SD1cunstructured |
| F241K | 1.6 ± 0.2b | 3.48 ± 0.07 | WTd | SD1 unstructured |
| Nonpolar core | ||||
| I207N | 0.72 ± 0.03 | 2.02 ± 0.01 | WT | SD1 and SD2 destabilized |
| L194N | 1.29 ± 0.01 | NDe | 6.9 ± 0.1 | SD2 destabilized |
| Polar/Nonpolar core junction | ||||
| C144A-C147A | 2.05 ± 0.10 | 3.44 ± 0.05 | 5.4 ± 0.1 | SD3 destabilized |
| C144S-C147S | 1.99 ± 0.07 | ND | 5.7 ± 0.1 | SD2 and SD3 destabilized, T2 & T1 merge |
| Polar core | ||||
| Y134F | 1.75 ± 0.05 | WT | 5.7 ± 0.1 | SD3 destabilized |
| Q125A | 1.91 ± 0.01 | WT | 4.9 ± 0.1 | SD3 destabilized |
| Q125F | 1.81 ± 0.10 | 3.82 ± 0.05 | ND | SD3 destabilized, T3 & T2 inseparable |
| (1.96 ± 0.06)f | (WT) | (4.2 ± 0.1) | (fixed SD2 to WT separates T2 & T3) | |
| N111L | 1.84 ± 0.09 | 3.82 ± 0.05 | ND | SD3 destabilized, T3 & T2 inseparable |
| (2.26 ± 0.06) | (WT) | (4.1 ± 0.1) | (fixed SD2 to WT separates T2 & T3) | |
| N66L | 1.91 ± 0.07 | WT | 4.6 ± 0.1 | SD3 destabilized |
| F80L | 1.89 ± 0.04 | 3.45 ± 0.08 | 6.5 ± 0.1 | Minimal change from HpmA265 |
Errors represent standard deviations from at least three trials.
Transition corresponding to HpmA265 T1 represents ∼1% of CD signal change.
SD = subdomain.
To constrain the fits these values were held fixed at the values for the unmodified HpmA265 values.
ND = No separate transition was detected for this subdomain.
Estimated values based on using 4-state model.
SD1 is defined as a weakly stable C-terminal subdomain
To assist in delineation of the structural origins of the four-state unfolding, trypsin proteolysis of HpmA265 was used to select for stable structures. Protease sensitivity has been used to define structural domain boundaries in other proteins.28,29 Within 30 min of initiating the trypsin digest, a protease resistant form was generated (Supporting Information Figure S8). Based on an apparent cut site at R249 (Materials and Methods and Supporting Information), the stable trypsin digested species encompassing residues N30-R249 is termed trHpm249. GdnHCl denaturation of the purified trHpmA249 showed complete elimination of unfolding transition T1, with no significant effect on T2 and T3 [Fig. 5(A), Table2]. This means that SD1, the lowest stability subdomain, is nearly completely unstructured in trHpmA249, while the stabilities of SD2 and SD3 are unchanged. So, even though trHpmA249 appears visually and proteolytically to be a single domain, the GdnHCl denaturation studies still provided two distinct transitions. Figure 2(B) provides an illustration of the location for R249 on the HpmA265 structure.
To confirm that destabilization of the C-proximal structure resulted in an unstructured C-terminal subdomain, F241 was site specifically mutated to a lysine (F241K). In the HpmA265 structure, the side chain of F241 resides within the interior and at the end of the C-proximal hydrophobic core where it interacts with internal residues from β-circuits 4 and 5, as well as β-strand 29. Since F241 is found packed within the hydrophobic core [Fig. 2(B)], conversion to a lysine at this position was expected to greatly destabilize the structure in this area of the β-helix. As in trHpmA249, GdnHCl denaturation of F241K showed a virtually complete elimination of T1, with no significant affect on T2 or T3 [Fig. 5(A), Table2]. The effect of these two C-proximal alterations on the denaturation profile allowed us to assign T1 to the denaturation of the C-terminal subdomain, SD1 (Fig. 4).
Based on the change in CD signal during the HpmA265 denaturation, this subdomain contains ∼24% of the residues responsible for the CD signal at 220 nm within the HpmA265 TPS domain. With the assumption that most of the signal at 220 nm is due to the residues in β-strands, we used the loss of the assigned secondary structure as a means of estimating the boundaries of the subdomains in HpmA265. Based upon this ∼24% loss of structure and the requirement for contiguous segments in each subdomain, the boundary of the C-terminal subdomain is approximately residue 216 (see Supporting Information for analysis of alternative groupings of residues into the C-terminal subdomain). Within the error of the CD signal changes this means that the C-terminal subdomain represents β-strands 23 and 24 of the flap region, all of the β-strands associated with β-circuit 5 and β-strand 28 and has an associated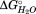 of −11.7 kJ/mol (Fig. 4).
of −11.7 kJ/mol (Fig. 4).
β-circuit 4 lies at the junction of SD1 and SD2
To further refine the boundary between SD1 and SD2, I207 was mutated to an asparagine (I207N). In the HpmA265 structure, I207 makes contacts with internal residues from both β-circuit 3 and 5. The placement of I207 within the HpmA265 hydrophobic core is illustrated in Figure 2(B). The I207N mutation results in a shift in the Cm for both T1 and T2, resulting from a destabilization to both SD1 and SD2 [Fig. 5(A), Table2]. The fact that I207N destabilizes both of these subdomains suggests that β-circuit 4 lies at the interface between SD1 and SD2 and supports the assignment of β23 - β28 as the C-terminal subdomain.
Disruption of the hydrophobic core defines SD2 as a nonpolar core subdomain
The HpmA265 solvent inaccessible interior spanning approximately β15 (starting at T152) through the C-terminal residues after β28 (ending at G265) is filled with nonpolar residues [Fig. 2(B)]. Even after accounting for the C-terminal subdomain, the β-strands in this segment could account for the CD change that accompanies either T2 or T3, and thus, this region of the protein is a major component of either of the remaining two subdomains. In order to ascribe the contribution of this contiguous section of the protein to a specific subdomain, we targeted the internal residue L194, which is part of β-circuit 3 and buried within the HpmA265 hydrophobic core [Fig. 2(B)]. The GdnHCl denaturation of L194N is well fit by a three-state unfolding model [Fig. 5(B), Table2] with an increase in both the m value and fractional change in CD (0.38 ± 0.01) and a slight increase in the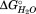 associated transition 1, T1, in comparison to HpmA265 (Supporting Information Table S1). These changes suggest an increased loss of structure during T1, suggesting that SD2 now denatures in conjunction with the C-terminal subdomain, SD1 (Supporting Information Figure S5). This effect is different than the I207N mutation suggesting that β-circuit 3 does not form an interface with the C-terminal subdomain. Therefore, we propose that T2 represents the denaturation of the subdomain that encompasses much of the hydrophobic core, SD2, now termed the nonpolar core subdomain. Based on the loss of CD signal for HpmA265, denaturation of this nonpolar core subdomain is accompanied by loss of approximately 35% of the structure. Given the boundaries on the C-terminal subdomain, the nonpolar subdomain corresponds to approximately β-strands 12 through 22 and has an associated
associated transition 1, T1, in comparison to HpmA265 (Supporting Information Table S1). These changes suggest an increased loss of structure during T1, suggesting that SD2 now denatures in conjunction with the C-terminal subdomain, SD1 (Supporting Information Figure S5). This effect is different than the I207N mutation suggesting that β-circuit 3 does not form an interface with the C-terminal subdomain. Therefore, we propose that T2 represents the denaturation of the subdomain that encompasses much of the hydrophobic core, SD2, now termed the nonpolar core subdomain. Based on the loss of CD signal for HpmA265, denaturation of this nonpolar core subdomain is accompanied by loss of approximately 35% of the structure. Given the boundaries on the C-terminal subdomain, the nonpolar subdomain corresponds to approximately β-strands 12 through 22 and has an associated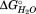 of −29.9 kJ/mol (Fig. 4). The final subdomain, SD3, then represents the remaining N-proximal structure, which is now termed the polar core subdomain. This subdomain contains the cap structure, as well as β-circuit 1 and has an associated
of −29.9 kJ/mol (Fig. 4). The final subdomain, SD3, then represents the remaining N-proximal structure, which is now termed the polar core subdomain. This subdomain contains the cap structure, as well as β-circuit 1 and has an associated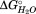 of −52 kJ/mol (Fig. 4). As suggested structurally, these results implicate β-circuit 2 as an interface between the nonpolar and polar subdomains, SD2 and SD3. The boundaries between the nonpolar (SD2) and polar (SD3) subdomains will be refined based on the effects of the mutations below.
of −52 kJ/mol (Fig. 4). As suggested structurally, these results implicate β-circuit 2 as an interface between the nonpolar and polar subdomains, SD2 and SD3. The boundaries between the nonpolar (SD2) and polar (SD3) subdomains will be refined based on the effects of the mutations below.
Disulfide bond disruption at the polar-nonpolar junction alters the stability of SD2 and SD3
The C144-C147 disulfide bond provides both a hydrogen bond acceptor for the internal water in the polar core and van der Waals interactions with residues in the nonpolar core [(Fig. 2(B)]. This bridging between the two internal environments suggests that removal of the hydrogen bonding acceptors will destabilize the polar core subdomain, but inclusion of more polar residues will destabilize the nonpolar core subdomain. In addition, removal of the structure nucleating properties of the disulfide bond30 will result in destabilization of its encompassing subdomain.
To help delineate the effect of the disulfide bond and refine the subdomain boundaries, two sets of site-specific mutations of the cysteine pair were studied. In the first, the two cysteine residues were replaced with alanine residues. This tests both the removal of the disulfide bond and the absence of a hydrogen bond acceptor. Elimination of the disulfide bond and the hydrogen bonding capabilities at positions 144 and 147 via double mutation of the cysteine residues to alanines results in virtually no change in the Cm for denaturation of the nonpolar core transition, but does affect the polar core transition [Table2]. This loss in polar core stability and the absence of effects on the nonpolar core stability suggests that the disulfide bond is part of the polar core subdomain. In the second, the two cysteine residues were replaced with the more polar, but nearly isosteric serine residues. The C144S-C147S double replacement affects both SD2 and SD3 [Fig. 5(B)]. SD1 for this mutant denatures with a similar transition midpoint (Cm) as HpmA265 (Table2), yet this transition is now accompanied by increases in the fractional change in CD (0.54 ± 0.01),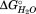 , and m value (Supporting Information Table S1). As in L194N, these results suggest an increased cooperative loss of structure associated with T1. Additionally, the double replacement destabilizes SD3 and reduces its Cm. By increasing the polarity at the junction between the polar and nonpolar core subdomains, the C144S-C147S double substitution destabilizes the interactions that β-circuit 2 makes with β-circuits 1 and 3 (Supporting Information). The results from the two sets of cysteine replacements are consistent with β-circuit 2 acting as a junction between the nonpolar core subdomain, SD2, and the final subdomain, SD3. These results also suggest that the sequence boundary between the polar core and nonpolar core subdomains resides after the disulfide bond, placing β15 as the approximate start to the nonpolar core subdomain (Fig. 4).
, and m value (Supporting Information Table S1). As in L194N, these results suggest an increased cooperative loss of structure associated with T1. Additionally, the double replacement destabilizes SD3 and reduces its Cm. By increasing the polarity at the junction between the polar and nonpolar core subdomains, the C144S-C147S double substitution destabilizes the interactions that β-circuit 2 makes with β-circuits 1 and 3 (Supporting Information). The results from the two sets of cysteine replacements are consistent with β-circuit 2 acting as a junction between the nonpolar core subdomain, SD2, and the final subdomain, SD3. These results also suggest that the sequence boundary between the polar core and nonpolar core subdomains resides after the disulfide bond, placing β15 as the approximate start to the nonpolar core subdomain (Fig. 4).
Substitution of polar residues in N-proximal polar core confirms SD3 as the polar core subdomain
The conserved internal residues N66, N111, Q125, and Y134 all lie within the N-proximal polar core spanning β4 through β12 [Figs. 2(A) and 2(B)]. Mutations at these positions result in a destabilization of the polar core subdomain and shifts to the Cm for T3 (Figure 5C, Table2). The amount of the destabilization correlates roughly with the potential number of hydrogen bonds lost in the order Y134F < N66L < Q125A < N111L ≈ Q125F (see Supporting Information for detailed discussion, Table S1). The final two mutations in that series, Q125F and N111L, each destabilize the polar core subdomain to the point that denaturation of the nonpolar and polar core subdomains is nearly indistinguishable. Practically, the broad second transition for these mutants has a larger fractional CD change which encompasses the HpmA265 CD changes for T2 and T3. However, both have lower m values for the second transition (Supprting Information Table S1) compared to the HpmA265 values for either T2 or T3. This suggests that T2 for these mutants is comprised of multiple transitions. Based on this analysis, a fit to a four-state model was attempted with the added constraints that the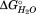 and m value for SD1 and SD2 were unchanged from HpmA265. With these constraints, an estimate of the stability and Cm of the polar core subdomain, SD3 is possible (Table2 and Supporting Information Table S1). The mutation F80L was used to test the effect of changes in packing on the stability of the polar core. This semi-conservative mutation resulted in little change to T3 [Fig. 5(C), Table2]. This set of mutations confirmed that SD3 represents the polar core subdomain of HpmA265 and spans approximately β-strands 1-14 (Fig. 4).
and m value for SD1 and SD2 were unchanged from HpmA265. With these constraints, an estimate of the stability and Cm of the polar core subdomain, SD3 is possible (Table2 and Supporting Information Table S1). The mutation F80L was used to test the effect of changes in packing on the stability of the polar core. This semi-conservative mutation resulted in little change to T3 [Fig. 5(C), Table2]. This set of mutations confirmed that SD3 represents the polar core subdomain of HpmA265 and spans approximately β-strands 1-14 (Fig. 4).
Since residues more N-proximal than the C144-C147 disulfide bond only destabilize SD3, these results confirm that the final transition represents unfolding the polar core subdomain. Elimination of the interactions involving internal polar residues destabilizes the polar core subdomain. The GdnHCl denaturation studies undertaken on HpmA265 and various site-specific mutants of HpmA265 support a sequential model for TPS domain unfolding, where the C-terminal subdomain, SD1, unfolds first, followed by the nonpolar core subdomain, SD2, and finally the polar core subdomain, SD3 [Fig. 5(D)].
Discussion
The right-handed β-helix structure of HpmA265 appears visually as a single domain.31 Thus, the four-state transition was an unexpected result from the denaturation studies. However, multi-transitional denaturation curves have been shown for other apparent single domain structures and the absence of clear structural boundaries for domains is well documented.28,32,33 In line with other multi-state unfolding proteins, we have assigned structural subdomain boundaries through unfolding studies of the HpmA265 TPS domain. Most importantly, we implicate the HpmA265 sequential unfolding mechanism and the high stability within the N-terminal polar core subdomain, SD3, as a means for the TPS pathway to couple translocation and folding.
Sequential unfolding
The multi-state denaturation data for HpmA265 is fit equally well by independent and sequential schemes for unfolding, characterized by parallel or linear unfolding respectively; therefore, additional information is required to distinguish between these two schemes. To start, the structural fold of HpmA265 favors a sequential unfolding model. Unlike typical globular protein folds, the placement of β-strands onto the β-helix depends upon an in-register and progressive framework formed from contiguous segments of protein.29,34 This means that the individual subdomains are packed against each other and gain stability through their inter-subdomain interactions.27,35 These interactions suggest that perturbations of the interface should destabilize both subdomains. Independently folding subdomains would be insensitive to changes in stability of the other subdomains, thus mutations that affect both subdomains support a sequential model of unfolding. The C144S-C147S mutations affect both the polar core (SD3) and nonpolar core (SD2) subdomains [Fig. 5(B)], suggesting that these residues lie at the interface of the two subdomains and that the stabilities of the two subdomains are not independent of each other. Likewise, the I207N mutation also affects two subdomains, the nonpolar core (SD2) and C-terminal (SD1) subdomains [Fig. 5(B)].
A second hallmark of the sequential unfolding model is that merged transitions result in a larger m value for denaturation. Since the m value is proportional to the amount of structure in each cooperative unit denatured, two independent subdomains with indistinguishable transition midpoints (Cm) would report a low m value relative to the total amount of structure denatured. In this scenario, the CD change would monitor the total loss of structure but the fit to real experimental data would provide an m value that is an average of the m values for the two independent subdomains. Conversely, a sequential model will result in a larger m value for the cooperative denaturation of the two subdomains (Supporting Information). This means destabilization of the nonpolar or polar core subdomains (SD2 and SD3) resulting in a merged transition (T2 → T1, or T3 → T2), should result in an increase in the m value if the sequential model is correct. This is exactly what occurs for the L194N mutation, which merges T2 with T1 with both a higher m value and a higher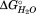 . In addition, the midpoint of the T1 transition has shifted down to 1.3 M (Table2), suggesting that the nonpolar subdomain (SD2) has become so destabilized that once the C-terminal subdomain (SD1) denatures, the nonpolar subdomain denatures. The fact that the C-terminal domain did not maintain its same transition midpoint implies that either the L194N substitution destabilized both subdomains, or that the C-terminal subdomain is dependent on the nonpolar subdomain being folded. The increase in m value implies the sequential unfolding model.
. In addition, the midpoint of the T1 transition has shifted down to 1.3 M (Table2), suggesting that the nonpolar subdomain (SD2) has become so destabilized that once the C-terminal subdomain (SD1) denatures, the nonpolar subdomain denatures. The fact that the C-terminal domain did not maintain its same transition midpoint implies that either the L194N substitution destabilized both subdomains, or that the C-terminal subdomain is dependent on the nonpolar subdomain being folded. The increase in m value implies the sequential unfolding model.
The sequential model for unfolding is further supported by the merger of T2 and T1 by the double mutant C144S-C147S, which also has a larger m value associated with the first transition. Destabilization of the interface between the nonpolar core (SD2) and C-terminal (SD1) subdomains is not an explanation for this effect, as C144 and C147 do not share an interface with the C-terminal subdomain. The lack of a similar effect with mutations targeting the polar subdomain (SD3) is explained by the large destabilization of the polar subdomain necessary to merge its stability with that of the nonpolar subdomain. Even Q125F, the most destabilizing of the polar core mutants, does not destabilize the polar core subdomain enough to merge its denaturation with the nonpolar subdomain. This is supported by the fact that a small, 5°C, decrease in temperature is enough to separate the T2 and T3 transitions for the Q125F mutation (MW, DG, TW, unpublished observation). Amino acid sequence conservation also suggests subdomain divisions within the larger TPS domain (data not shown). Collectively, these data suggest a specific, sequentially ordered folding for HpmA265 wherein the N-proximal polar core subdomain (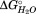 of −52 kJ/mol) must fold first followed by the nonpolar subdomain (
of −52 kJ/mol) must fold first followed by the nonpolar subdomain (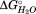 of −29.9 kJ/mol) and finally the C-terminal subdomain (
of −29.9 kJ/mol) and finally the C-terminal subdomain (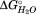 of −11.7 kJ/mol) [Fig. 5(D)]. This sequential folding model is supported by AFM studies on the hemagglutinin TpsA member FHA, where a mechanically resistant N-proximal subdomain was identified.18
of −11.7 kJ/mol) [Fig. 5(D)]. This sequential folding model is supported by AFM studies on the hemagglutinin TpsA member FHA, where a mechanically resistant N-proximal subdomain was identified.18
Polar core subdomain provides selectivity and stability
Based on our results, the folding of the highly stable N-proximal polar core subdomain provides the initial template for folding the nonpolar and C-terminal subdomains. The polar core subdomain stabilization is enhanced via placement of internal polar residues, including N66, N111, Q125, and Y134 (Fig. 4) near hydrogen bonding partners. As evidenced in the GdnHCl denaturation studies, replacement of N66, N111, Q125, and Y134 destabilizes the polar core subdomain as associated with the final TPS domain transition, T3 [Fig. 5(C)]. Furthermore, the effect of each site directed mutation correlates with the degree that the HpmA265 interactions are reduced or removed. For example, replacement of polar with nonpolar residues is especially destabilizing (e.g., Q125F or N66L) [Fig. 5(C), Table2], while the F80L alteration does little to affect the HpmA265 stability [Fig. 5(A), Table2]. The results of the mutations within the polar core point to the importance of the internal hydrogen bond network in stabilizing the polar core subdomain (Fig. 4).
An open question is why these hydrogen bonds are effectively stronger than side chain-water hydrogen bonds, and thus capable of stabilizing the subdomain. The structure of HpmA265 leads to the hypothesis that neighboring nonpolar side chains around this polar core provide an anhydrous and insulating sheath, which would lower the local dielectric constant and enhance the electrostatic interactions (the nonpolar residues are not illustrated in Fig. 4 for clarity). The thermodynamic contribution of the local microenvironment surrounding hydrogen-bonding partners has been investigated by others,29 who have found that increasing the hydrophobic microenvironment around a hydrogen bond pair leads to an average 2.9 kJ/mol increase in stabilization energy.36 A high conservation of internal polar residues has been found when they formed hydrogen bonds with neighboring atoms, where polar residues sharing two hydrogen bonds with two main chain atoms were the most highly conserved within this group.16,37 In HpmA265, this situation occurs for N111 and N66, supporting the destabilization observed in the leucine replacements. Conversely, the lesser destabilization of Y134F points to the aromatic ring being able to accept and donate hydrogen bonds,38–40 albeit more weakly than the phenolic hydroxyl group of Y134. The hydrogen bond network within the polar core subdomain provides much of the stability, enhanced by the insulation of these interactions from the high dielectric solvent by the nonpolar residues that surround it. The high conservation across all of the hemolysin family members of the internal polar residues within the polar core subdomain supports the importance of this hydrogen bond network (Supporting Information).
Biological significance of sequential unfolding
Our sequential unfolding model has led us to propose a mechanism for hemolysin TPS domain directed secretion across the Gram-negative bacterial outer membrane. Upon Sec pathway mediated secretion of unfolded HpmA265 across the inner membrane,37 disulfide bond formation between Cys144 and Cys147 (the only two cysteine residues within the 1,577 full length HpmA amino acid sequence) would be catalyzed by the Dsb system within the periplasm of the host organism, P. mirabilis.41 The Dsb system has been shown to introduce disulfide bonds into a variety of exported proteins,42 including exotoxins,43 via a vectorial mechanism.44 Disulfide bond formation is a critical stabilizing step45,46 during the protein folding process and has been shown to be very slow unless catalyzed by an enzyme. Thus, the Dsb system would facilitate rapid disulfide bond formation within the periplasmic space47 between Cys144 and Cys147 at the junction between the polar and nonpolar subdomains. The disulfide bond would prevent backsliding into the periplasmic space, while providing a nucleation point for vectorial β-strand addition within the polar core subdomain via a β-augmentation process as proposed by others.48
HpmB POTRA domains would then recognize the unfolded TPS domain36 and allow the transmembrane pore of HpmB to translocate an HpmA265 TPS domain hairpin structure across the outer membrane. This would allow N-terminal and C-terminal contact on the periplasmic side of the outer membrane as others have suggested.12,13 As in the FhaC/FHA system,36,49 the POTRA domains would remain critical for continued recognition of unfolded HpmA265 during the coupled vectorial folding and secretion process. The polar core subdomain appears to require TpsB to fold (Supporting Information). However, the presence of a folded polar core subdomain allows rapid folding of the nonpolar core and C-terminal subdomains. This leads to a model where translocation of the TPS domain hairpin by TpsB allows polar core subdomain folding. The polar core subdomain folding would be facilitated by the internal hydrogen bonds shared between the conserved residues N66, Y68, N111, Q125, and Y134. The stability of the folded N-terminal polar core subdomain would further prevent transport back into the periplasmic space, while enabling sequential folding and secretion of the entire TPS domain to the external environment. This model is similar to the Brownian ratchet mechanism proposed for the secretion of other proteins across membranes, where translocation is coupled to sequential folding events.50–52
Three possible Brownian ratchet models for TPS domain translocation across the outer membrane are presented in Figure 6. Our unfolding studies conducted upon HpmA265 and various HpmA site-specific mutants demonstrate sequential folding starting with the N-proximal polar core subdomain that is inconsistent with a model that folds the C-proximal portion of the TPS domain first [Fig. 6(A)]. Furthermore, our studies suggest that the N-proximal polar core subdomain stability may provide the free energy to initiate β-augmentation driven protein folding at the cell surface concomitant with translocation across the outer membrane. Thus, our studies are most consistent with models where the inherent stability of the polar subdomain facilitates vectorial β-strand addition during nucleotide independent secretion across the outer membrane [Fig. 6(B,C)]. Interestingly, recent studies have shown that the TPS domain stays associated to the POTRA domain only at the beginning of the translocation process and the N-terminus of the TPS domain is released first prior to the complete secretion of the entire TpsA polypeptide.15 Thus, the secretion models illustrated in Figure 6(B,C) would appear most consistent with this recent study. Our proposed N → C terminal TPS secretion model, complete with differentiated subdomain stability, is an inverted form of the proposed secretion method for the autotransporter protein family (Type Va), the other Type V family. In the autotransporter family the stability within the C-terminus facilitates the secretion in a C → N-terminal direction.50 More detailed studies are necessary to distinguish between our two models for polar core subdomain driven secretion [Fig. 6(B,C)].
Figure 6.
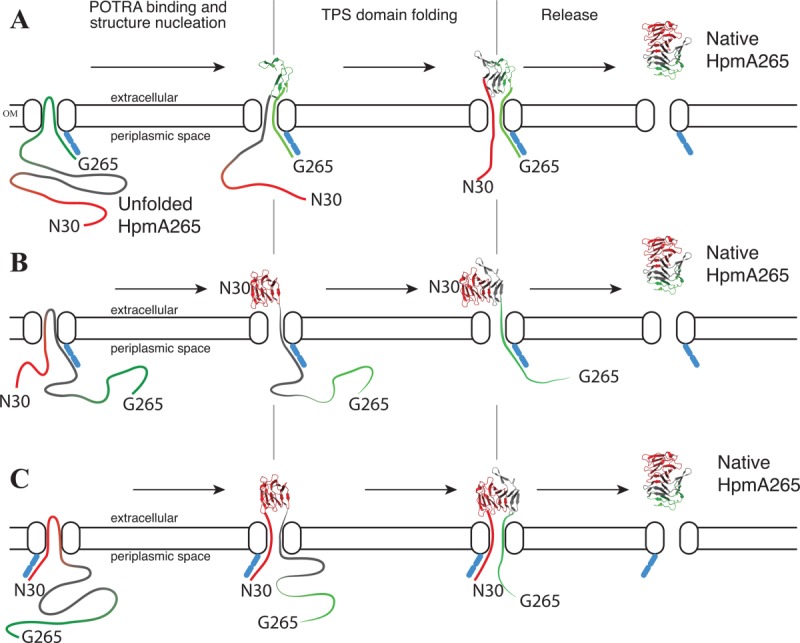
TPS domain driven secretion schemes for HpmA265. Unfolded HpmA265 is shown as a ribbon that is colored by subdomain: polar core (red), nonpolar core (gray), C-terminal (green). The N- and C-terminus are labeled. The outer membrane is shown with an embedded TpsB component. Secretion and folding proceed from left to right in each panel. The POTRA domains are found on the periplasmic side and are represented by blue ovals. Three possible transport schemes are shown. (A) POTRA domains bind C-proximally on HpmA265 and remain bound until translocation is (nearly) complete. (B) POTRA domains binds initially near the middle of HpmA265. As secretion progresses, the POTRA domains are bound in progressively more C-proximal locations. (C) POTRA domains bind N-proximally on HpmA265 and remain bound until translocation is (nearly) complete.
Some of the TpsA family members are anchored to the outer membrane via their N-terminal domains,53 some via their C-terminal domains,54,55 while others are secreted into the external environment.6,7,14 Therefore, the exact mechanism of outer membrane translocation may differ across the TpsA family members. Supporting this analysis are studies that have shown the TpsA families are incapable of catalyzing the transport of TpsA proteins with different functions.56 For example, the TpsB associated with the hemolysin group does not transport TpsA components from the adhesin group. TpsB components can, however, transport proteins within the same functional family, but from a different organism.56 Furthermore, this divergence in transport mechanisms suggests that the details of TPS domain directed secretion might differ between various TpsA members. Regardless, TpsA folding at the extracellular cell surface is thought to provide the energy necessary for vectorial secretion, as active transport is not an option for crossing the outer membrane.16,27,37
Conclusions
Denaturation studies of HpmA265 have lead us to propose that the TPS domain is composed of three structural subdomains termed the C-terminal (SD1), nonpolar core (SD2), and polar core (SD3) subdomains that unfold via a sequential model. By studying the effects of site-specific substitutions on the denaturation of HpmA265, we have mapped the approximate subdomain boundaries (Fig. 4). Intriguingly, the polar core subdomain has the largest favorable folding free energy of the three subdomains. Mutations to the polar residues destabilize this subdomain, suggesting that the polar residues themselves help maintain this high stability. Based on our results, we have proposed a model that utilizes the N-proximal polar core subdomain to provide the structural and energetic framework for translocation of hemolysin TPS domains [Fig. 6(B,C)]. This model may be applicable to the transport of TPS domains from other TpsA families.
Materials and Methods
Cell strains, culturing and site directed mutagenesis
Cell strains used in this study were C41 (DE3) (Lucigen, Madison, WI) and RAU126.14 All cells were cultured at 37°C via rotary shaking in Luria-Broth supplemented with appropriate antibiotics. The site directed mutants were generated using the QuikChange Kit (Stratagene, La Jolla, CA), pHpmA265 plasmid, and sets of oligonucleotide primers designed to replace the codons for N66, F80, N111, Q125, Y134, C144, C147, L194, and F241 (GenScript Inc., Piscataway, NJ). All site directed mutants were verified by DNA sequencing (Eton Bioscience Inc., San Diego, CA or GenScript Inc., Piscataway, NJ).
Protein isolation, molecular weight determination and electrophoresis
All HpmA proteins were purified via metal-chelate chromatography as previously described.14 Samples were desalted via gel filtration (Life Technologies, Madison, WI). Denaturing protein gel electrophoresis was conducted as described by Laemmli.45 A representative purified HpmA265 sample has been illustrated (Supporting Information). Absolute molecular weights of selected samples were determined via coupled SEC-LS (size-exclusion chromatography and light scattering (Keck Biotechnology Resource Laboratory at Yale University, New Haven, CT). Mass analysis was determined via MALDI-TOF MS (Biotechnology Center at the University Wisconsin-Madison, Madison, WI).
Tryptic digestion
Trypsin digests were conducted in PBS at a trypsin:HpmA265 ratio of 1:100 (w:w) (Promega Corporation, Madison, WI). MALDI-TOF of this stable fragment reported a mass of 23,050 Da and amino acid sequencing verified an intact SEC pathway processed N-terminus starting at N30.
Circular dichroism (CD) spectroscopy
CD monitored wavelength scans and unfolding experiments were performed on an AVIV Model 420 Circular Dichroism Spectrometer (AVIV Biomedical Inc., Lakewood, NJ) equipped with a stepper-motor driven syringe system for performing titrations (Hamilton, Reno, NV). HpmA265, equilibrated in 10 mM Na2HPO4, pH 7.4, was subjected to constant volume guanidine·HCl (GdnHCl) denaturation experiments at 25°C. GdnHCl stocks were prepared in 10 mM Na2PO4, pH 7.4 and the concentration was confirmed by measuring the refractive index.23 GdnHCl titrations were conducted using either 0.1 M or 0.2 M GdnHCl step sizes and 1 min stir times from 0 to 5 M GdnHCl followed by 2 min stir times from 5 to 7.8 M GdnHCl (Supporting Information).
Assignment of secondary structure
The standard bioinformatics programs DSSP,46 STRIDE,57 and PDBsum,58 as well as a manual tabulation of residues with β-strand like dihedrals were used to assign secondary structure (Supporting Information). Programs were run with their default parameters based on the 4W8Q crystal structure. To be consistent with the PDB default values, all figures are prepared using the results from PDBsum using the 4W8Q.pdb file as input.
Data analysis
Depending on the site directed mutant protein being analyzed, the GdnHCl titration data were fit to a three- or four-state unfolding model (see Results). Good fits to the data were obtained using both an independent and a sequential model for the multiple observed transitions. A full description of the models and the equations used to fit the data as well as analysis of different data processing procedures are found in the Supporting Information. For both models, each transition is modeled as depending linearly on the concentration of GdnHCl using the relationship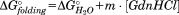 . Initially, estimates of the individual fractional CD change,
. Initially, estimates of the individual fractional CD change,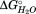 , and m values were obtained by fitting each transition separately using a two-state model. Little change was observed in the fit parameters for each transition between fits to the four–state model and initial estimates of the fit values for each transition. Replicate data sets were fit using OriginPro v.8 (OriginLab Corp, Northampton, MA).
, and m values were obtained by fitting each transition separately using a two-state model. Little change was observed in the fit parameters for each transition between fits to the four–state model and initial estimates of the fit values for each transition. Replicate data sets were fit using OriginPro v.8 (OriginLab Corp, Northampton, MA).
Acknowledgments
The authors also would like to thank Gregory Barrett-Wilt, Ph.D. and Grzegorz Sabat, M.S. from the Mass Spectrometry Facility in the Biotechnology Center at UW-Madison for the MALDI-TOF results and Ewa Folta-Stogniew from the Keck Biophysics Resource Center at Yale University for the SEC-LS/UV/RI results. NIH Award 1S10RR023748-01 to Ewa Folta-Stogniew supported the SEC-LS/UV/RI instrumentation. The authors wish to thank the fall 2014 BIO 436 class within the Biology Department at UW-L and the instructors, S. Cooper and J. Klein, for generating the N66L, F80L, and N111L site-specific mutants discussed within this study. The authors are grateful for critical comments from A. Sanderfoot, S. Cooper, A. Opdahl, and C. Maeder. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Glossary
- CD
circular dichroism
- Cm
guanidine hydrochloride concentration at transition mid-point
- FHA
Bordetella pertussis filamentous hemagglutinin
- D
denatured HpmA265 TPS domain
- GdnHCl
guanidine•HCl
- HpmA
Proteus mirabilis full length hemolysin A
- HpmA265
truncation fragment of P. mirabilis hemolysin A processed to start at asparagine 30 and cloned to end at glycine 265
- I1 and I2
HpmA265 unfolding intermediates 1 and 2
- MALDI-TOF MS
matrix-assisted laser desorption/ionization time of flight mass spectrometry
- N
native HpmA265 TPS domain
- PBS
phosphate buffered saline
- POTRA domain
polypeptide-transport associated domain
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SEC-LS
size-exclusion chromatography light scattering
- SD1
SD2, SD3, structural subdomains within the HpmA265 TPS domain
- T1
T2, T3, transitions 1, 2, and 3 within the HpmA265 TPS domain four-state unfolding model
- TPS
two-partner secretion
- TpsA
two-partner secretion pathway A component
- TpsB
two-partner secretion pathway B component
- Vmax
maximum velocity.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- Jacob-Dubuisson F, Locht C, Antoine R. Two-partner secretion in Gram-negative bacteria: a thrifty, specific pathway for large virulence proteins. Mol Microbiol. 2001;40:306–313. doi: 10.1046/j.1365-2958.2001.02278.x. [DOI] [PubMed] [Google Scholar]
- Walker G, Hertle R, Braun V. Activation of Serratia marcescens hemolysin through a conformational change. Infect Immun. 2004;72:611–614. doi: 10.1128/IAI.72.1.611-614.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locht C, Bertin P, Menozzi FD, Renauld G. The filamentous haemagglutinin, a multifaceted adhesion produced by virulent Bordetella spp. Mol Microbiol. 1993;9:653–660. doi: 10.1111/j.1365-2958.1993.tb01725.x. [DOI] [PubMed] [Google Scholar]
- Geme JWS, Thanassi DG, Yeo H-J, Stathopoulos C, Karkal A, Li H. A prototype two-partner secretion pathway: the Haemophilus influenzae HMW1 and HMW2 adhesin systems. Trends Microbiol. 2009;17:355–360. doi: 10.1016/j.tim.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Swihart KG, Welch RA. Cytotoxic activity of the Proteus hemolysin HpmA. Infect Immun. 1990;58:1861–1869. doi: 10.1128/iai.58.6.1861-1869.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uphoff TS, Welch RA. Nucleotide sequencing of the Proteus mirabilis calcium-independent hemolysin genes (hpmA and hpmB) reveals sequence similarity with the Serratia marcescens hemolysin genes (shlA and shlB) J Bacteriol. 1990;172:1206–1216. doi: 10.1128/jb.172.3.1206-1216.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun V, Hobbie S, Ondraczek R. Serratia marcescens forms a new type of cytolysin. FEMS Microbiol Lett. 1992;100:299–305. doi: 10.1111/j.1574-6968.1992.tb14056.x. [DOI] [PubMed] [Google Scholar]
- Aoki SK, Pamma R, Hernday AD, Bickham JE, Braaten BA, Low DA. Contact-dependent inhibition of growth in Escherichia coli. Science. 2005;309:1245–1248. doi: 10.1126/science.1115109. [DOI] [PubMed] [Google Scholar]
- Kida Y, Higashimoto Y, Inoue H, Shimizu T, Kuwano K. A novel secreted protease from Pseudomonas aeruginosa activates NF-kappaB through protease-activated receptors. Cell Microbiol. 2008;10:1491–1504. doi: 10.1111/j.1462-5822.2008.01142.x. [DOI] [PubMed] [Google Scholar]
- Fournier C, Smith A, Delepelaire P. Haem release from haemopexin by HxuA allows Haemophilus influenzae to escape host nutritional immunity. Mol Microbiol. 2011;80:133–148. doi: 10.1111/j.1365-2958.2011.07562.x. [DOI] [PubMed] [Google Scholar]
- Cope LD, Thomas SE, Hrkal Z, Hansen EJ. Binding of heme-hemopexin complexes by soluble HxuA protein allows utilization of this complexed heme by Haemophilus influenzae. Infect Immun. 1998;66:4511–4516. doi: 10.1128/iai.66.9.4511-4516.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazar J, Cotter PA. New insight into the molecular mechanisms of two-partner secretion. Trends Microbiol. 2007;15:508–515. doi: 10.1016/j.tim.2007.10.005. [DOI] [PubMed] [Google Scholar]
- van Ulsen P, Rahman SU, Jong WSP, Daleke-Schermerhorn MH, Luirink J. Type V secretion: from biogenesis to biotechnology. Biochim Biophys Acta. 2014;1843:1592–1611. doi: 10.1016/j.bbamcr.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Weaver TM, Smith JA, Hocking JM, Bailey LJ, Wawrzyn GT, Howard DR, Sikkink LA, Ramirez-Alvarado M, Thompson JR. Structural and functional studies of truncated hemolysin A from Proteus mirabilis. J Biol Chem. 2009;284:22297–22309. doi: 10.1074/jbc.M109.014431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guérin J, Baud C, Touati N, Saint N, Willery E, Locht C, Vezin H, Jacob-Dubuisson F. Conformational dynamics of protein transporter FhaC: large-scale motions of plug helix. Mol Microbiol. 2014;92:1164–1176. doi: 10.1111/mmi.12585. [DOI] [PubMed] [Google Scholar]
- Jacob-Dubuisson F, Fernandez R, Coutte L. Protein secretion through autotransporter and two-partner pathways. Biochim BiophysActa. 2004;1694:235–257. doi: 10.1016/j.bbamcr.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Maier T, Clantin B, Gruss F, Dewitte F, Delattre A-S, Jacob-Dubuisson F, Hiller S, Villeret V. Conserved Omp85 lid-lock structure and substrate recognition in FhaC. Nat Commun. 2015;6:7452. doi: 10.1038/ncomms8452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsteens D, Martinez N, Jamin M, Jacob-Dubuisson F. Sequential unfolding of beta helical protein by single-molecule atomic force microscopy. PLOS One. 2013;8:e73572. doi: 10.1371/journal.pone.0073572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JS. The anatomy and taxonomy of protein structure. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- Hughson FM, Wright PE, Baldwin RL. Structural characterization of a partly folded apomyoglobin intermediate. Science. 1990;249:1544–1548. doi: 10.1126/science.2218495. [DOI] [PubMed] [Google Scholar]
- Tsytlonok M, Sormanni P, Rowling PJE, Vendruscolo M, Itzhaki LS. Subdomain architecture and stability of a giant repeat protein. J Phys Chem B. 2013;117:13029–13037. doi: 10.1021/jp402360x. [DOI] [PubMed] [Google Scholar]
- Barrick D, Ferreiro DU, Komives EA. Folding landscapes of ankyrin repeat proteins: experiments meet theory. Curr Opin Struct Biol. 2008;18:27–34. doi: 10.1016/j.sbi.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street TO, Courtemanche N, Barrick D. Protein folding and stability using denaturants. Methods Cell Biol. 2008;84:295–325. doi: 10.1016/S0091-679X(07)84011-8. [DOI] [PubMed] [Google Scholar]
- Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharman GJ, Searle MS. Cooperative interaction between the three strands of a designed antiparallel β-sheet. J Am Chem Soc. 1998;120:5291–5300. [Google Scholar]
- Schenck HL, Gellman SH. Use of a designed triple-stranded antiparallel β-sheet to probe β-sheet cooperativity in aqueous solution. J Am Chem Soc. 1998;120:4869–4870. [Google Scholar]
- Tsemekhman K, Goldschmidt L, Eisenberg D, Baker D. Cooperative hydrogen bonding in amyloid formation. Protein Sci. 2007;16:761–764. doi: 10.1110/ps.062609607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker M, Schuster CC, McDonnell AV, Sorg KA, Finn MC, Berger B, Clark PL. Pertactin beta-helix folding mechanism suggests common themes for the secretion and folding of autotransporter proteins. Proc Natl Acad Sci USA. 2006;103:4918–4923. doi: 10.1073/pnas.0507923103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace CN, Fu H, Lee Fryar K, Landua J, Trevino SR, Schell D, Thurlkill RL, Imura S, Scholtz JM, Gajiwala K, Sevcik J, Urbanikova L, Myers JK, Takano K, Herbert EJ, Shirley BA, Grimsley GR. Contribution of hydrogen bonds to protein stability. Protein Sci. 2014;23:652–661. doi: 10.1002/pro.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier CS, Kaiser CA. Formation and transfer of disulphide bonds in living cells. Nat Rev Mol Cell Biol. 2002;3:836–847. doi: 10.1038/nrm954. [DOI] [PubMed] [Google Scholar]
- Yoder MD, Yoder MD, Keen NT, Keen NT, Jurnak F, Jurnak F. New domain motif: the structure of pectate lyase C, a secreted plant virulence factor. Science. 1993;260:1503–1507. doi: 10.1126/science.8502994. [DOI] [PubMed] [Google Scholar]
- Porter LL, Rose GD. A thermodynamic definition of protein domains. Proc Natl Acad Sci USA. 2012;109:9420–9425. doi: 10.1073/pnas.1202604109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamen DE, Kamen DE, Griko Y, Griko Y, Woody RW, Woody RW. The stability, structural organization, and denaturation of pectate lyase C, a parallel beta-helix protein. Biochemistry. 2000;39:15932–15943. doi: 10.1021/bi001900v. [DOI] [PubMed] [Google Scholar]
- Kobe B, Kajava AV. When protein folding is simplified to protein coiling: the continuum of solenoid protein structures. Trends Biochem Sci. 2000;25:509–515. doi: 10.1016/s0968-0004(00)01667-4. [DOI] [PubMed] [Google Scholar]
- Nelson R, Sawaya MR, Balbirnie M, Madsen AØ, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodak H, Clantin B, Willery E, Villeret V, Locht C, Jacob-Dubuisson F. Secretion signal of the filamentous haemagglutinin, a model two-partner secretion substrate. Mol Microbiol. 2006;61:368–382. doi: 10.1111/j.1365-2958.2006.05242.x. [DOI] [PubMed] [Google Scholar]
- Thanassi DG, Stathopoulos C, Karkal A, Li H. Protein secretion in the absence of ATP: the autotransporter, two-partner secretion and chaperone/usher pathways of gram-negative bacteria. Mol Membr Biol. 2005;22:63–72. doi: 10.1080/09687860500063290. [DOI] [PubMed] [Google Scholar]
- Scheiner S, Kar T, Pattanayak J. Comparison of various types of hydrogen bonds involving aromatic amino acids. J Am Chem Soc. 2002;124:13257–13264. doi: 10.1021/ja027200q. [DOI] [PubMed] [Google Scholar]
- Steiner T, Koellner G. Hydrogen bonds with pi-acceptors in proteins: frequencies and role in stabilizing local 3D structures. J Mol Biol. 2001;305:535–557. doi: 10.1006/jmbi.2000.4301. [DOI] [PubMed] [Google Scholar]
- Saggu M, Levinson NM, Boxer SG. Experimental quantification of electrostatics in X-H···π hydrogen bonds. J Am Chem Soc. 2012;134:18986–18997. doi: 10.1021/ja305575t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson MM, Sebaihia M, Churcher C, Quail MA, Seshasayee AS, Luscombe NM, Abdellah Z, Arrosmith C, Atkin B, Chillingworth T, Hauser H, Jagels K, Moule S, Mungall K, Norbertczak H, Rabbinowitsch E, Walker D, Whithead S, Thomson NR, Rather PN, Parkhill J, Mobley HLT. Complete genome sequence of uropathogenic Proteus mirabilis, a master of both adherence and motility. J Bacteriol. 2008;190:4027–4037. doi: 10.1128/JB.01981-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto H, Bardwell JCA. Catalysis of disulfide bond formation and isomerization in the Escherichia coli periplasm. Biochim Biophys Acta. 2004;1694:111–119. doi: 10.1016/j.bbamcr.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, Martin JL. DSB proteins and bacterial pathogenicity. Nat Rev Micro. 2009;7:215–225. doi: 10.1038/nrmicro2087. [DOI] [PubMed] [Google Scholar]
- Kadokura H, Beckwith J. Detecting folding intermediates of a protein as it passes through the bacterial translocation channel. Cell. 2009;138:1164–1173. doi: 10.1016/j.cell.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Touw WG, Baakman C, Black J, Beek te TAH, Krieger E, Joosten RP, Vriend G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015;43:D364–D368. doi: 10.1093/nar/gku1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Łasica AM, Jagusztyn-Krynicka EK. The role of Dsb proteins of gram-negative bacteria in the process of pathogenesis. FEMS Microbiol Rev. 2007;31:626–636. doi: 10.1111/j.1574-6976.2007.00081.x. [DOI] [PubMed] [Google Scholar]
- Delattre A-S, Saint N, Clantin B, Willery E, Lippens G, Locht C, Villeret V, Jacob-Dubuisson F. Substrate recognition by the POTRA domains of TpsB transporter FhaC. Mol Microbiol. 2011;81:99–112. doi: 10.1111/j.1365-2958.2011.07680.x. [DOI] [PubMed] [Google Scholar]
- Clantin B, Delattre A-S, Rucktooa P, Saint N, Méli AC, Locht C, Jacob-Dubuisson F, Villeret V. Structure of the membrane protein FhaC: a member of the Omp85-TpsB transporter superfamily. Science. 2007;317:957–961. doi: 10.1126/science.1143860. [DOI] [PubMed] [Google Scholar]
- Renn JP, Junker M, Besingi RN, Braselmann E, Clark PL. ATP-independent control of autotransporter virulence protein transport via the folding properties of the secreted protein. Chem Biol. 2012;19:287–296. doi: 10.1016/j.chembiol.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depperschmidt A, Ketterer N, Pfaffelhuber P. A Brownian ratchet for protein translocation including dissociation of ratcheting sites. J Math Biol. 2013;66:505–534. doi: 10.1007/s00285-012-0519-8. [DOI] [PubMed] [Google Scholar]
- Simon SM, Peskin CS, Oster GF. What drives the translocation of proteins? Proc Natl Acad Sci USA. 1992;89:3770–3774. doi: 10.1073/pnas.89.9.3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renauld-Mongénie G, Cornette J, Mielcarek N, Menozzi FD, Locht C. Distinct roles of the N-terminal and C-terminal precursor domains in the biogenesis of the Bordetella pertussis filamentous hemagglutinin. J Bacteriol. 1996;178:1053–1060. doi: 10.1128/jb.178.4.1053-1060.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noël CR, Mazar J, Melvin JA, Sexton JA, Cotter PA. The prodomain of the Bordetella two-partner secretion pathway protein FhaB remains intracellular yet affects the conformation of the mature C-terminal domain. Mol Microbiol. 2012;86:988–1006. doi: 10.1111/mmi.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscher AZ, Grass S, Heuser J, Roth R, St Geme JW. Surface anchoring of a bacterial adhesin secreted by the two-partner secretion pathway. Mol Microbiol. 2006;61:470–483. doi: 10.1111/j.1365-2958.2006.05236.x. [DOI] [PubMed] [Google Scholar]
- Jacob-Dubuisson F, Buisine C, Willery E, Renauld-Mongénie G, Locht C. Lack of functional complementation between Bordetella pertussis filamentous hemagglutinin and Proteus mirabilis HpmA hemolysin secretion machineries. J Bacteriol. 1997;179:775–783. doi: 10.1128/jb.179.3.775-783.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- de Beer TAP, Berka K, Thornton JM, Laskowski RA. PDBsum additions. Nucleic Acids Res. 2014;42:D292–D296. doi: 10.1093/nar/gkt940. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information