

Abstract
Residue types at the interface of protein–protein complexes (PPCs) are known to be reasonably well conserved. However, we show, using a dataset of known 3-D structures of homologous transient PPCs, that the 3-D location of interfacial residues and their interaction patterns are only moderately and poorly conserved, respectively. Another surprising observation is that a residue at the interface that is conserved is not necessarily in the interface in the homolog. Such differences in homologous complexes are manifested by substitution of the residues that are spatially proximal to the conserved residue and structural differences at the interfaces as well as differences in spatial orientations of the interacting proteins. Conservation of interface location and the interaction pattern at the core of the interfaces is higher than at the periphery of the interface patch. Extents of variability of various structural features reported here for homologous transient PPCs are higher than the variation in homologous permanent homomers. Our findings suggest that straightforward extrapolation of interfacial nature and inter-residue interaction patterns from template to target could lead to serious errors in the modeled complex structure. Understanding the evolution of interfaces provides insights to improve comparative modeling of PPC structures.
Keywords: transient protein complexes, structure of protein complexes, interfaces of protein complexes, evolution of protein complexes, protein–protein interactions
Introduction
Diverse biological processes are carried out by physical interactions between proteins to form functional protein–protein complexes (PPCs).1,2 Functional requirements can cause temporary association between proteins to form transient complexes. Proteins involved in the formation of such complexes are also stable in their disassociated forms. However, interactions among protomers in oligomeric proteins are often permanent because protomers of such assemblies are often unstable as a monomer.3–7 Some of the structural features of the protein–protein interfaces in transient complexes studied extensively are size, area, polarity, planarity, shape complementarity, conformational changes upon binding, residue propensities, and residue contacts.3,8–12
It is known that the protein–protein interfacial residues are conserved better than the residues at the rest of the tertiary structural surface.13 Detailed study showed that the interfacial residues in homologous transient PPCs are less well conserved than those in permanent complexes.14 Interfaces in transient PPCs are known to be characterized by faster rate of evolution, thereby leaving weak signals of correlated mutations across the interfaces, while interfaces in permanent complexes tend to co-evolve.14 Nevertheless, high interaction specificity in transient PPCs can be explained by complementarity generally observed in protein surfaces, which is preserved not only in a pair-wise manner but also through clusters of interacting residues.15 de Vries and Bonvin16 have shown improved interface prediction accuracy if pairs of residues are considered as opposed to considering single residues. These studies emphasize the significance of residue–residue contacts and interaction patterns in PPCs.
Two PPCs, say A:B and A′:B′, are said to be homologous if A and A′ are homologous and B and B′ are also homologous. It is generally well known that spatial orientations of interacting proteins in two closely related homologous PPCs are similar.17–19 However, there are also a few studies which have indicated alternate binding modes in homologous PPCs.20–23 To get a broader perspective, we have carried out a comprehensive study specifically for homologous transient PPCs to find out how far the locations of interface and residue–residue interactions across the interacting interfaces are conserved. Further, these structural constraints at the protein interfaces of homologous transient PPCs have been compared with those in homologous permanent homodimers.
Initially we hypothesized high conservation of interface location as well as interaction pattern across the interfaces in homologous transient PPCs. However, in this analysis, we note that this hypothesis is not generally true, thus providing a new perspective on interface evolution. Our observations strongly implicate that during comparative modeling of a PPC, straight forward extrapolation of interface residues and interacting residue pairs from template PPC to the target PPC can be fraught with major errors.
Results
Location of interfacial residues is only moderately conserved in homologous transient protein–protein complexes
Evolution of the interfaces have been studied using a dataset of homologous pairs of transient PPCs of known structures such as A:B complex and A′:B′ complex, where A is homologous to A′ and B is homologous to B′ (refer Materials and Methods section and Table1). Extent of conservation of interface residue locations in homologous proteins has been quantified. Two residues from the homologous proteins are considered to have conserved interfacial location if the residues in question are at the interfaces of the respective complexes and if they are also topologically equivalent. A measure referred henceforth as Conservation of Interface Location (CIL) score has been defined for a pair of homologous proteins (for precise definition, refer Fig. 1 and Materials and Methods section). Essentially, simultaneous satisfaction of following two features adds favorably to the residue location or CIL score:
Table 1.
Homologous Pairs of Transient protein–protein Complexes (PPCs) (PDB Code and the Chain Information Are Provided)
| Homologous protein–protein complex (AB and A′B′) | Seqid AA′ | Seqid BB′ | CIL1 | CIL2 | CIP | Binding affinity 1 (M) | Binding affinity 2 (M) | References |
|---|---|---|---|---|---|---|---|---|
| 1a4y_DE_1dfj_IE | 77 | 34 | 42.86 | 68.18 | 36.7 | 1.00E-15 | 5.90E-14 | 24a,25b |
| 1agr_DH_1fqj_DE | 78 | 37 | 76.19 | 64.00 | 66.7 | 6.70E-08 | 26a,27b | |
| 1agr_DH_3c7k_AB | 73 | 53 | 60.87 | 60.87 | 48.5 | 26a,28 | ||
| 1bxi1mz8_CD | 58 | 69 | 64.29 | 52.17 | 33.3 | –,29 | ||
| 1bxi_AB_7cei_AB | 58 | 68 | 64.29 | 50.00 | 27.9 | –,30b | ||
| 1cse_EI_2sni_EI | 70 | 35 | 64.00 | 76.92 | 64.5 | 2.00E-12 | 31a,32b | |
| 1d5m_AC_1hxy_BD | 34 | 34 | 0.00 | 0.00 | 0 | 5.00E-10 | 33a,34 | |
| 1doa_AB_1ds6_AB | 70 | 72 | 87.50 | 81.63 | 84.5 | 35,36a | ||
| 1euv_AB_1tgz_AB | 33 | 53 | 64.86 | 64.52 | 43.6 | 37a,38 | ||
| 1euv_AB_2iyd_AB | 32 | 47 | 68.29 | 87.50 | 39.3 | 37a,– | ||
| 1evt_BD_1nun_AB | 33 | 73 | 61.54 | 57.89 | 35.1 | 39–40 | ||
| 1ewy_AC_1gaq_AB | 52 | 68 | 37.04 | 43.48 | 5.9 | 3.60E-06 | 41b,42 | |
| 1fqj_DE_2gtp_BC | 78 | 42 | 76.19 | 54.55 | 53.3 | 6.70E-08 | 27b,43 | |
| 1fqj_DE_2ihb_AB | 75 | 43 | 72.73 | 75.00 | 72.7 | 6.70E-08 | 27b,43 | |
| 1fqj_DE_2ik8_CD | 79 | 38 | 63.64 | 58.33 | 54.5 | 6.70E-08 | 27b,43 | |
| 1fqj_DE_2ode_AB | 75 | 39 | 90.91 | 66.67 | 73.3 | 6.70E-08 | 27b,43 | |
| 1fqj_DE_3c7k_AB | 67 | 38 | 83.33 | 84.62 | 70.6 | 6.70E-08 | 27b,28 | |
| 1hxy_BD_1jwm_AD | 33 | 30 | 0.00 | 0.00 | 0 | 5.00E-10 | 34, 44 | |
| 1jiw_PI_1smp_AI | 57 | 37 | 81.25 | 66.67 | 66.7 | 4.00E-12 | 45b,46 | |
| 1kgy3czu_AB | 49 | 30 | 71.11 | 68.42 | 38.2 | 2.50E-08 | 47–48 | |
| 1kgy3hei_OP | 49 | 30 | 72.73 | 70.27 | 36.9 | 2.50E-08 | 47, 49 | |
| 1mda_BM_3c75_BM | 56 | 79 | 50.00 | 30.77 | 31.6 | 50–51 | ||
| 1nb3_BJ_1stf_EI | 41 | 53 | 58.33 | 70.00 | 52.9 | 52–53a | ||
| 1nun_AB_1ry7_AB | 33 | 71 | 52.17 | 52.38 | 23.9 | 2.30E-07 | 40, 54 | |
| 1shw_BA_2hle_AB | 45 | 32 | 45.16 | 48.00 | 25.5 | 4.00E-08 | 55–56b | |
| 1shw_BA_2wo2_AB | 59 | 31 | 55.17 | 43.48 | 35.9 | 1.08E-05 | 55, 57 | |
| 1shw_BA_3czu_AB | 49 | 53 | 48.48 | 44.44 | 40.8 | 55, 48 | ||
| 1shw_BA_3hei_OP | 50 | 53 | 50.00 | 46.15 | 43.5 | 55, 49 | ||
| 1tec_EI_2sni_EI | 43 | 35 | 69.23 | 71.43 | 64.5 | 2.00E-12 | 58, 32b | |
| 1tgz_AB_2iyd_AB | 60 | 53 | 75.00 | 68.97 | 52.2 | 38,– | ||
| 1uea_AB_2e2d_AC | 62 | 42 | 58.54 | 68.57 | 33.3 | 59–60 | ||
| 2e2d_AC_2j0t_BE | 58 | 46 | 60.61 | 46.67 | 29.2 | 4.00E-10 | 60–61b | |
| 2gtp_BC_3c7k_AB | 73 | 52 | 69.57 | 60.00 | 53.3 | 28, 43 | ||
| 2hle_AB_3czu_AB | 40 | 32 | 61.90 | 72.22 | 24.2 | 4.00E-08 | 56b,48 | |
| 2hle_AB_3hei_OP | 39 | 31 | 58.54 | 74.29 | 25.4 | 4.00E-08 | 56b,49 | |
| 2ihb_AB_3c7k_AB | 71 | 41 | 66.67 | 63.64 | 48.5 | 43, 28 | ||
| 2ode_AB_3c7k_AB | 71 | 66 | 66.67 | 81.82 | 66.7 | 43, 28 | ||
| 2wo2_AB_3czu_AB | 55 | 30 | 70.00 | 70.59 | 44.8 | 1.08E-05 | 57, 48 |
Details of sequence identities, CIL scores, conservation of interaction pattern (CIP) scores, binding affinity for AB and A′B′ complexes, wherever available, and references to show their transient nature are listed. The symbol (−) in References column indicates that there is no reference associated with PDB ID.
Crystallized in bound and unbound form (according to Benchmark4 dataset).63
Figure 1.

Extent of conservation of interaction interfaces in homologous transient protein–protein complexes. Homologous pairs of transient protein–protein complexes (PPCs) (A:B and A′:B′) are shown, where A is homologous to A′ and B is homologous to B′. Interface residues in AA′ that are structurally equivalent are circled. Structurally aligned interface residues are said to have conserved interface location (CIL), whereas structurally aligned interactions are said to have conserved interaction pattern (CIP). The interfacial features do not consider the residue type or the nature of interactions.
A residue in protein A should be at the interface of A:B complex
The residue in A′ that is topologically equivalent to the residue of A mentioned above should be an interfacial residue in A′:B′ complex
This measure does not consider the residue type (i.e., 20 different types of amino acids). For example unconserved residue type in the topologically equivalent positions in the two homologs with both the residues in the interface will contribute favorably to the CIL score. Two conserved residues (same residue type from among 20 standard types) from the two homologs with the residue in one of the proteins only in the interface while the conserved residue in the other homolog not in the interface will not contribute favorably to the CIL scores.
Two CIL scores are obtained for the two pairs of the homologous subunits (AA′ and BB′) of the homologous transient PPCs. High and low CIL score denotes similar and dissimilar location of interfacial positions, respectively. CIL scores for the entries of homologous pairs of transient PPCs are provided in Tables1 and 2. CIL scores for the entries of homologous pairs of permanent homodimers are provided in Supporting Information Table S1.
Table 2.
List of Entries Corresponding to Same Protein Bound to Two Different Homologous Interacting Partners (PDB Code and the Chain Information Are Provided)
| Homologous protein–protein complex (AB and A′B′) | Seqid AA′ | Seqid BB′ | CIL 1 | CIL 2 | CIP | Binding affinity 1 (M) | Binding affinity 2 (M) | References |
|---|---|---|---|---|---|---|---|---|
| 1agr_DH_2gtp_BC | 100 | 55 | 90.00 | 63.16 | 62.1 | 26a,43 | ||
| 1agr_DH_2ihb_AB | 94 | 47 | 66.67 | 76.19 | 56.3 | 26a,43 | ||
| 1agr_DH_2ik8_CD | 100 | 55 | 85.71 | 57.14 | 62.5 | 26a,43 | ||
| 1agr_DH_2ode_AB | 94 | 57 | 76.19 | 66.67 | 62.1 | 26a,43 | ||
| 1b6c_CD_3h9r_BA | 100 | 69 | 82.76 | 64.00 | 63.4 | 2.80E-07 | 64b,65 | |
| 1b8m_AB_1bnd_AB | 99 | 58 | 84.44 | 93.33 | 78.3 | 66a,67 | ||
| 1bi8_CD_1g3n_AB | 98 | 47 | 86.67 | 81.08 | 62.7 | 68–69 | ||
| 1cmx_AB_1xd3_AB | 33 | 100 | 83.72 | 82.76 | 71.2 | 1.70E-06 | 3.00E-07 | 70–71a |
| 1cse_EI_1mee_AI | 70 | 100 | 66.67 | 66.67 | 64.7 | 31a,72 | ||
| 1cse_EI_1tec_EI | 47 | 98 | 88.89 | 93.33 | 87.5 | 31a,58 | ||
| 1d5m_AC_1jwm_AD | 100 | 69 | 83.33 | 76.92 | 78.3 | 33a,44 | ||
| 1dhk_AB_1viw_AB | 54 | 100 | 85.11 | 72.73 | 71.6 | 3.50E-11 | 73a,74 | |
| 1eui_AC_1udi_EI | 47 | 100 | 84.62 | 90.00 | 78.3 | 75–76b | ||
| 1eui_AC_2j8x_CD | 48 | 100 | 66.67 | 85.00 | 75 | 8.00E-09 | 75, 77 | |
| 1evt_BD_1ry7_AB | 100 | 74 | 64.86 | 75.00 | 67.7 | 2.30E-07 | 39, 54 | |
| 1f8u_AB_1fss_AB | 59 | 100 | 73.33 | 87.50 | 69 | 78–79 | ||
| 1grn_BA_1ow3_AB | 100 | 54 | 90.00 | 84.85 | 86.2 | 2.40E-07 | 80b,81 | |
| 1h1v_AG_3ffk_BA | 100 | 32 | 48.98 | 41.67 | 40.5 | 2.50E-08 | 82b,83 | |
| 1hxy_BD_2g9h_BD | 100 | 33 | 76.92 | 70.59 | 42.1 | 5.00E-10 | 1.00E-07 | 34, 84 |
| 1ibr_CD_2bku_AB | 99 | 41 | 58.18 | 48.48 | 33.7 | 1.00E-09 | 85b,86 | |
| 1kac_AB_2j12_AB | 41 | 99 | 64.00 | 68.97 | 55.8 | 4.00E-10 | 6.90E-11 | 87b,88 |
| 1kgy_CG_1shw_BA | 98 | 32 | 52.94 | 44.44 | 28.6 | 1.50E-08 | 2.00E-08 | 47, 55 |
| 1kgy_CG_2hle_AB | 47 | 99 | 69.77 | 94.44 | 51.5 | 2.50E-08 | 47, 56b | |
| 1kgy_CG_2wo2_AB | 59 | 98 | 82.93 | 88.24 | 69 | 2.50E-08 | 4.00E-08 | 47, 57 |
| 1lqg_BD_1udi_EI | 47 | 100 | 81.48 | 87.18 | 73.9 | 2.50E-08 | 1.08E-05 | 89, 76b |
| 1lqg_BD_2j8x_CD | 47 | 100 | 64.29 | 82.05 | 70.8 | 89, 77 | ||
| 1mda_BM_2gc4_KJ | 90 | 78 | 60.00 | 33.33 | 37.5 | 8.00E-09 | 50,– | |
| 1mee_AI_1tec_EI | 44 | 98 | 71.43 | 62.50 | 64.7 | 72, 58 | ||
| 1qty_RU_1rv6_WX | 53 | 100 | 88.89 | 84.62 | 74.3 | 90–91b | ||
| 1t6g_CA_2b42_BA | 45 | 99 | 73.91 | 66.67 | 60 | 1.00E-11 | 1.70E-10 | 92–93b |
| 1taw_AB_1tpa_EI | 100 | 46 | 81.82 | 94.12 | 64.5 | 1.10E-09 | 94,– | |
| 1taw_AB_1zr0_CD | 100 | 46 | 90.00 | 94.12 | 82.8 | 2.00E-11 | 94–95 | |
| 1taw_AB_2tgp_ZI | 100 | 46 | 86.96 | 94.12 | 77.4 | 2.00E-11 | 1.30E-08 | 94,– |
| 1taw_AB_3d65_EI | 100 | 46 | 85.71 | 100.00 | 71.4 | 2.00E-11 | 2.40E-06 | 94,– |
| 1tpa_EI_1zr0_CD | 100 | 38 | 70.00 | 88.89 | 53.3 | 2.00E-11 | –,95 | |
| 1tpa_EI_3d65_EI | 100 | 46 | 66.67 | 94.12 | 62.1 | 1.30E-08 | –,– | |
| 1udi_EI_2j8x_CD | 41 | 100 | 68.97 | 80.95 | 65.4 | 76–77 | ||
| 1uea_AB_2j0t_BE | 60 | 95 | 52.94 | 68.97 | 36 | 8.00E-09 | 59, 61b | |
| 1ukv_GY_3cph_GA | 100 | 52 | 66.67 | 68.57 | 63.3 | 4.00E-10 | 96–97b | |
| 1zlh_AB_1zli_AB | 47 | 100 | 78.95 | 80.00 | 75.5 | 3.30E-07 | 98b | |
| 1zlh_AB_3d4u_AB | 47 | 100 | 85.00 | 84.62 | 80.7 | 1.30E-09 | 98–99 | |
| 1zr0_CD_2tgp_ZI | 100 | 38 | 76.19 | 88.89 | 66.7 | 95,– | ||
| 1zr0_CD_3d65_EI | 100 | 48 | 94.74 | 94.12 | 74.1 | 1.30E-08 | 2.40E-06 | 95,– |
| 2g81_EI_2iln_BI | 100 | 72 | 72.00 | 0.00 | 0 | 1.30E-08 | 100–101 | |
| 2gc4_KJ_3c75_BM | 63 | 98 | 87.50 | 85.71 | 75.9 | 1.36E-08 | –,51 | |
| 2gtp_BC_2ihb_AB | 94 | 48 | 66.67 | 66.67 | 55.2 | 43, 43 | ||
| 2gtp_BC_2ik8_CD | 100 | 49 | 85.71 | 77.78 | 69 | 43, 43 | ||
| 2gtp_BC_2ode_AB | 94 | 52 | 76.19 | 77.78 | 61.5 | 43, 43 | ||
| 2hle_AB_2wo2_AB | 46 | 98 | 63.16 | 93.75 | 53.6 | 56b,57 | ||
| 2ihb_AB_2ik8_CD | 94 | 41 | 63.64 | 50.00 | 50 | 4.00E-08 | 1.08E-05 | 43, 43 |
| 2ihb_AB_2ode_AB | 100 | 42 | 72.73 | 60.00 | 62.1 | 43, 43 | ||
| 2ik8_CD_2ode_AB | 94 | 68 | 72.73 | 80.00 | 69 | 43, 43 | ||
| 2ik8_CD_3c7k_AB | 74 | 91 | 66.67 | 72.73 | 72.7 | 43, 28 | ||
| 2tgp_ZI_3d65_EI | 100 | 46 | 72.73 | 94.12 | 75.9 | –,– | ||
| 1jtg_AB_3e2l_AC | 40 | 100 | 84.44 | 71.79 | 59.5 | 2.40E-06 | 102b,103 |
Details of sequence identities, CIL scores, conservation of interaction pattern (CIP) scores, binding affinity for AB and A′B′ complexes, wherever available, and references to show their transient nature are listed. The symbol (−) in References column indicates that there is no reference associated with PDB ID.
Crystallized in bound and unbound form (according to Benchmark4 dataset).63
Given the previously established fact that interfacial residue types are reasonably well conserved, we expected that homologous transient PPCs would have high CIL, which implies similar interface location. But surprisingly, high CIL scores (≥70%) are associated with only 45% of the dataset [Fig. 2(a)] corresponding to only 34 homologous chains out of 76. It is intriguing to note that the location of the interface residues in homologous transient PPCs is only moderately conserved. For example, homologous pair of GTPase:GAP (GTPase accelerating proteins)28,43 complex has moderate CIL scores of 67% (for GTPases) and 64% (for GAPs) [Fig. 2(b)]. Interestingly, the extent of CIL is well correlated (r = 0.80, P < 0.0001) in the two pairs of homologous subunits of transient PPCs (AA′ vs. BB′) [Supporting Information Fig. S1(a)].
Figure 2.
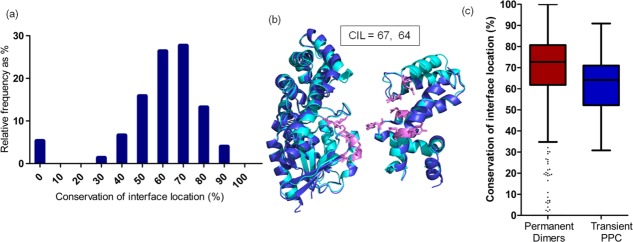
Extent of conservation of interface location (CIL) in homologous transient PPCs. (a) Histogram shows distribution of CIL scores for homologous pairs of transient PPCs. (b) Homologous pair of guanine nucleotide-binding protein G (o) subunit alpha (regulator of G-protein signaling 16) and guanine nucleotide-binding protein G (k) subunit alpha (regulator of G-protein signaling 10) (2ihb, 3c7k) is colored in dark and light blue. Both the homologous subunits show moderate CIL score. Residues with CIL are shown as pink sticks. All the protein structure figures in this work are generated using PyMOL.104 (c) Box plot distribution for CIL in homologous permanent dimers and transient PPC. The lines from bottom specify the minimum, 25th percentile, median, 75th percentile, and maximum for all the box plots in this work. The outliers of the distribution are represented as dots.
Location of the interfaces is highly conserved when same protein is bound to two different homologous interacting partners. that is, in the two complexes A:B and A:B′ protein A is common in the two complexes, whereas B is homologous to B′ [Supporting Information Fig. S1(b,c), Table2, and Materials and Methods section]. In such pairs, ≥70% CIL score is represented by 83% of the dataset, which corresponds to 91 chains out of 110.
Comparison of extent of CIL in transient PPCs and permanent homodimeric complexes shows that CIL scores are significantly higher for permanent dimers than transient PPCs (median: transient PPC, 64%; permanent dimers, 73%; medians are significantly different; P < 0.0001) (refer Supporting Information Table S1 and Materials and Methods section). High scores (≥70%) are represented by 63% of the dataset (525 chains out of 834) of homologous permanent homodimers [Fig. 2(c)] and only 45% (34 homologous chains out of 76) of homologous transient PPCs [Fig. 2(a)].
However, one needs to keep in mind that the CIL scores can fluctuate even among identical pairs of transient PPCs (refer Materials and methods section) available in the form of different crystal structures.105,106 (median: transient PPC, 89%; permanent dimers, 92%; medians are significantly different; P < 0.0001) [Supporting Information Fig. S1(d)].
Relationship between the extent of CIL with the overall sequence identity between homologous transient protein subunits was explored. Surprisingly, results show poor correlation between CIL and sequence identity (r = 0.27, P < 0.0001) [Supporting Information Fig. S1(e)]. As the sequence identity increases, the CIL scores are generally high for only permanent homodimers, which is consistent with a previous work.107 But, it is surprising that in transient PPCs even at 100% sequence identity wide range of CIL scores are observed.
Conservation of the type of an interfacial residue in a protein does not imply that the conserved residue in the homolog lie at the interface
It is well known that the residue types (20 amino acids) in the interface of a PPC are generally conserved better than the residue types in the rest of the protein surface.13 This generates an expectation that the residue types conserved in topologically equivalent positions in the homologs are also likely to be in the interface. Based on this expectation, residue type conservation remains as one of the important criteria to predict interface residues in a homolog and model the 3-D structure of the complex. Even though the location of interfaces is only moderately conserved in transient PPCs, we initially believed that the direct extrapolation of conserved residue types to conserved interface location would be valid.
Frequencies of structurally equivalent and conserved residue types that lie at the interfaces in homologous complexes were calculated for all the 20 residue types (refer Materials and Methods section). Interestingly, the distribution of topologically equivalent conserved residue types to be present at the interface is only 54% ± 19% (mean ± SD) and less than 60% for residue types A, C, D, E, G, I, M, R, S, V, and W [Fig. 3(a,b)]. However, this frequency is higher for homologous permanent homodimers than transient PPC [Fig. 3(a)]. Interfaces in homologous pairs of PPCs show moderate CIL with residue types that are only moderately conserved (r = 0.7, P < 0.0001 for transient PPC) [Supporting Information Fig. S2 and Fig. 3(b)]. For example, homologous pairs of GTPase-GAP complex show moderate conserved interface location (CIL = 61%, 61%) and moderate level of conserved residue types with conserved interface location (RC – CIL = 58%, 50%) [Fig. 3(b)].
Figure 3.

Extent of conserved residue types in homologous subunits of protein complexes with conserved interface location. (a) Extent of structurally equivalent conserved residue type to be at interface (residue conservation-conservation of interface location, RC-CIL) for each of the 20 residue types for homologous transient PPC and permanent homodimers are shown. (b) Conserved residue type in a multiple sequence alignment of homologs need not imply that all the conserved residues should lie at the interface. An example of structurally equivalent arginine residues from homologous subunits shows one arginine at the interface (pink) and the other arginine not in the interface (magenta). The extent of structurally equivalent, conserved residue types to be at the interface (RC-CIL = 58.35%, 50%) and conservation of interface location (CIL = 61%, 61%) have been calculated for the homologous subunits of GTPase-GAP complex. (c) In spite of residue types being conserved, interface location is lost in homologous protein complex (dark and light blue) by different ways.
Therefore, conserved residues in two homologs with the residue in one of the homologs at the protein–protein interface need not imply that the conserved residue in the other homolog is also at the interface. This observation seemed counter intuitive to us at the first glance; nevertheless, it is a very important learning from this study.
Circumstances in which conserved residue type may not conserve interfacial feature was explored using the PPC dataset. We made an analysis of homologous PPCs in which an interfacial residue type in a protein is conserved in a homolog, but this residue is not in the interface of the homolog. We note that interfacial location of a conserved residue type is not conserved in a homolog often due to the difference in the nature of residue type, in the interacting proteins, that are proximal to the conserved residue. Residue from the interacting protein can be non-compatible for interaction with the conserved residue types (L-Y and L,K) [Fig. 3(c)], Replacement of longer to smaller residue in the homologous interactor protein can result in loss of interaction with conserved residue type (R-E and R,D) [Fig. 3(c)], or structural differences at the interface of the homologous chains can result in loss of interaction by increased distance with the conserved residue type (D-R and D,K) [Fig. 3(c)].
Therefore, in many pairs of homologous complexes, no residue from the interacting protein interacts with the conserved residue type, which is, therefore, not considered to be in the interface [Fig. 3(c)].
Spatial orientation and structural differences at the interface contribute to interface plasticity in homologous transient protein–protein complexes
It is intriguing to observe only moderate CIL in homologous PPCs, in spite of sharing good sequence similarity between homologs (>30%). The reasons contributing to moderate CIL in homologous proteins could also be due to differences in spatial orientation of interfacial regions (DISO) and/or structural differences at the interface (SDI). DISO describes the differences in the spatial orientation of the homologous subunits with respect to the structurally aligned interactor proteins [Fig. 4(a)]. SDI describes the structural differences between the “equivalent interfaces” in the homologous subunits [Fig. 4(a)]. Analysis shows that when both the homologous subunits of PPCs have low conservation of interfacial location (CIL) (30%–60%), a pronounced difference in DISO and SDI score is observed (median: DISO: 1.5 Å; SDI: 1.7 Å; medians are significantly different between CIL low pair and CIL high pair; P < 0.05) [Fig. 4(b,c)].
Figure 4.

Reasons for non-conservation of interface location in homologous transient PPC. (a) Differences in spatial orientation of the interfacial region (DISO) is calculated by subtracting the RMSD of the interfacial region of homologous subunits (red and orange subunits) in their interfacial region with respect to the structurally aligned proteins (dark and light blue subunits) with RMSD for the interfacial region for the isolated homologous subunits. Structural differences at the interface (SDI) are calculated by calculating the RMSD for the interfacial region in isolated homologous subunits. Distance between the structurally equivalent interface residues are shown as green lines. (b) Box plot distribution for DISO scores for CIL low pair (both the homologous subunits of protein complex have low CIL score (30%–60%)), CIL low–CIL high pair (one homologous subunit has a low CIL score, whereas the other homologous subunit has high CIL score (>60%)), and CIL high pair (both the homologous subunits of protein complex have high CIL score). (c) Box plot distribution for SDI scores for homologous subunits of transient PPC is shown. (d) Homologous pair of ferredoxin: ferredoxin-NADP+ reductase (PDB id: 1ewy, 1gaq) shows drastic DISO. Changes in the spatial orientation are shown by homologous subunits, which are represented as surface. High DISO scores and low CIL scores for both the homologous subunits of the protein complex are observed. (e) Homologous pair of Ephrin-Ephrin receptor complex showing high DISO score for the ephrin receptors and high SDI for the ephrin molecules results in low CIL score (45%, 48%). Changes in the spatial orientation in homologous ephrin receptors are represented as surface. Structure deviation at the interface in the homologous ephrin molecules are highlighted in the circled region.
Homologous pair of ferredoxin (Fd) and ferredoxin-NADP+ (FNR) reductase from cyanobacteria and maize41,42 shows major DISO (21.0 Å, 14.5 Å) resulting in low CIL, (37%, 43%) in spite of sharing good sequence identity of 52% and 68% [Fig. 4(d)]. The difference in spatial orientation did not change the distance of the prosthetic groups between Fd and FNR for the electron transfer to occur. Homologous pair of Ephrin A5-Ephrin receptor B2 and Ephrin B2-Ephrin receptor B455,56 shows high DISO (4.5 Å) between the ephrin receptors and high SDI (4.1 Å) between the ephrin molecules. This additive effect could lead to low CIL (45.2%, 48%) [Fig. 4(e)].
Interaction patterns are poorly conserved in homologous transient protein–protein complexes
Having understood that the location of interfaces is only moderately conserved in homologous proteins in transient PPCs, we wanted to find out how far the pattern of residue–residue interactions across the interfaces is conserved. This pattern is characterized by all pair-wise residue–residue interactions across the interfaces in a PPC. One can compare such a pattern between two homologous PPCs. A residue–residue interaction across the two interacting proteins is considered to be conserved in a homologous complex, if the two residues in the homologous complex that are topologically equivalent to the two interacting residues in the first complex are also interacting across interface. Even if the nature of an interaction is not same (e.g., salt-bridge in a complex and aromatic–aromatic interaction in the homologous complex), the interaction pattern is considered as conserved if the interacting residues are topologically equivalent when 3-D structures of homologous proteins in the two complexes are aligned. “Conservation of Interaction Pattern” (CIP) score has been devised to quantify the extent of residue–residue interactions that are structurally aligned (Fig. 1) (refer Materials and Methods section). High and low CIP scores denote similar and dissimilar location of interactions for a homologous pair of transient PPC. Single CIP score is obtained for a pair of homologous transient PPCs. CIP scores for the entries of homologous pairs of transient PPCs are provided in Tables1 and 2. CIP scores for the entries of homologous pairs of permanent homodimers are provided in Supporting Information Table S1.
High conservation of interaction pattern scores (≥70%) are represented by only 18% (seven homologous PPC pairs out of 38) of the dataset [Fig. 5(a)], which is much lower than the distribution of CIL [Fig. 2(a)]. Homologous pair of GTPase-GAPs shows a moderate CIP score of 49%28,43 [Fig. 5(b)]. Overall, conservation of interaction pattern is poor in homologous PPCs. In other words, homologous complexes are stabilized largely by different patterns of inter-residue interaction network across the interfaces. Non-conservation of an inter-residue interaction results due to the nature of residue type substitution in the interactor protein or structural differences at the interface, as discussed in the previous section [Fig. 3(c)]. Extent of conservation of interfacial location and interaction pattern are well correlated with each other (transient PPC: r = 0.8, P < 0.0001) [Supporting Information Fig. S3(c)]. Nevertheless, interface location seems to be a better conserved structural feature than interaction pattern across the interfaces.
Figure 5.

Extent of conservation of interaction pattern in homologous transient PPC. (a) Histogram of conservation of interaction pattern (CIP) scores for homologous pairs of transient PPC is colored in blue. (b) Homologous pair of guanine nucleotide-binding protein G (o) subunit alpha (regulator of G-protein signaling 16) and guanine nucleotide-binding protein G (k) subunit alpha (regulator of G-protein signaling 10) (2ihb, 3c7k) is colored in dark and light blue. Residues with CIP are shown as pink sticks, and common interactions are shown as dotted lines. (c) Box plot distribution for CIP in homologous permanent dimers and transient PPC. (d) Residue–residue interaction in a particular transient PPC (K-E) has their residue type conserved in the other homolog. This mostly results in residue–residue interactions being maintained in the other homolog.
However, high CIP scores are observed in complexes when the same protein is bound to two different, but homologous, proteins (≥70% CIP score are represented by 54% of dataset, which corresponds to 30 homologous PPC pairs out of 55) [Supporting Information Fig. S3(a,b)]. Also, homologous permanent dimers show a higher distribution of CIP scores than transient PPCs [Fig. 5(c)] (transient PPC, 42%; permanent dimmers, 55%; medians are significantly different; P < 0.05).
We observed that if the residue–residue interactions in a particular transient PPC have their residue type conserved in a homologous PPC, then the interaction pattern is most often maintained in the homolog [Fig. 5(d); Table3]. On the other hand, 75% (196 of 260) of residues with conserved interaction pattern show conserved or conservatively substituted residue types, 22% (58 of 260) of residues with conserved interaction pattern show conserved/conservatively substituted and drastic substitutions, and 2.3% (six of 260) of residues with conserved interaction pattern show residue types with drastic substitutions in the homologous subunits.
Table 3.
Extent of Conserved Residue Types That Shows Conserved Interaction Pattern
| Conserved residue types that show conserved interaction pattern | Percentage |
|---|---|
| D-T | 100 |
| K-E | 93.1 |
| N-E, N-K, N-Q, N-T | 100, 100, 100, 100 |
| R-D, R-E | 90, 81 |
Following the assessment of CIL and interaction pattern (CIP) in homologous transient PPCs, we investigated the relationship, if any, between the interfacial features and the affinity of interaction. Even though the transient complexes dissociate and associate to carry out biological function, their binding affinities are variable—they can be strong transient interactions or weak transient interactions. The stronger interactions typically require a trigger for association/dissociation, whereas the weaker interactions show a fast bound–unbound equilibrium. The evidence for transient nature and the binding affinity values for transient PPCs used in this study are provided in Tables1 and 2.
We found no distinct trend for CIL and CIP scores in strong transient binders versus weak transient binders (for details refer Supporting Information text and Supporting Information Fig. S4).
Interaction type is poorly conserved in residues with conserved interaction pattern
Even though the conservation of inter-residue interaction pattern is generally poor in homologous transient PPCs, we wanted to analyze the nature of interaction type where the interaction patterns are conserved. Non-conservation of interaction type is quite common [Fig. 6(a)]. Only 53% (370 of 698) of the residues with conserved interaction pattern showed conserved interaction type. If the interacting residues are hydrophobic in nature, then such hydrophobic interactions are conserved better (111 of 147, 76%). This shows the crucial role of hydrophobic residues in the core of the interface.108 Ionic interaction is more often substituted by side chain–side chain hydrogen bond interaction than being conserved as shown in Figure 6(a). These findings are consistent with a previous study on good conservation of non-polar interface contacts and poor conservation of polar contacts in homologous protein complexes.20 An interesting feature is the substitution of cation–pi interaction to hydrophobic interaction (four of 14, 14.8%) next to the conservation of cation–pi interaction (five of 14, 18.5%). This could account for the drastic substitution of R↔L [Fig. 6(b)].
Figure 6.
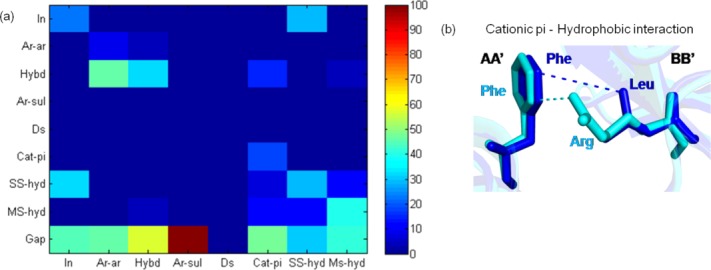
Interaction type substitutions for residues with conserved interaction pattern in homologous transient protein–protein complexes. (a) Interaction type substitution for residues with CIP in homologous pairs of transient PPC is shown. Various interaction types include hydrophobic (hybd), disulfide (Ds), side chain–side chain hydrogen bonding (SS-hyd), main chain–side chain hydrogen bonding (MS-hyd), ionic (In), aromatic–aromatic (Ar-Ar), aromatic–sulfur (Ar-sul), and cation–pi (Cat-pi). Gap denotes non-conservation of interaction pattern. (b) Cation–pi (Phe-Arg) interaction is getting substituted to hydrophobic interaction (Phe-Leu) in residues with CIP in homologous pairs of transient PPC. Residues with CIP are shown as sticks, and the common interactions are shown as dotted lines.
Core interface residues show higher CIL and interaction pattern than rim interface residues
Interface residues can be subdivided into core and rim depending on whether the residues occur in the middle or the periphery of the interaction interface patch. It has been shown in previous studies that the residue type at the core of the interface is conserved better than at rim of the interface.3,109 We have extended this idea by investigating the extent of CIL and CIP for core and rim interface residues in the dataset. Results showed that the core interface residues have their interface location better conserved than rim interface residues [Fig. 7(a)] (medians are significantly different; P < 0.0001). The frequency of residues with conserved residue type and interface location are higher for core interface residues 64% ± 21% (mean ± SD) than rim interface residues 42% ± 13% for different residue types [Fig. 7(b)]. Similarly, interaction pairs involving only core interface residues (C-C: core–core) have their interaction patterns conserved better than those involving only rim interface residues or core–rim residues (R-R: rim–rim, C-R: core–rim) [Fig. 7(c)] (medians are significantly different; P < 0.05). Therefore, the interface and interactions involving core residues are evolutionarily more constrained, making their predictions more reliable than the interface residues in the rim. Similar analysis carried out on same protein bound to homologous proteins showed higher CIL and CIP for core interface residues than homologous transient PPCs (Supporting Information Fig. S5).
Figure 7.
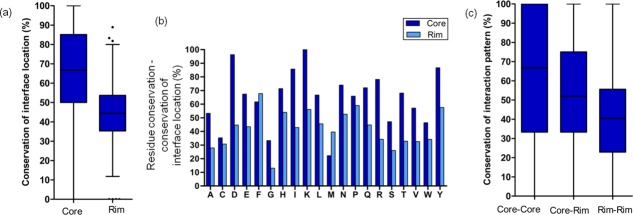
Extent of conservation of interface location and interaction pattern in core and rim interface residues. (a) Extent of core and rim interface residues to show conserved interface location. (b) Extent of core and rim residues that have conserved residue type and conserved interface location have been shown for all the 20 residue types. (c) Extent of core–core, core–rim, and rim–rim interactions to show conserved interaction pattern.
Discussion
We have carried out a comprehensive analysis that focuses on the evolution of interfaces for a dataset of transient homologous PPCs of known 3-D structures. It is well known that generally structures of homologous PPCs are similar.18,19,110 Therefore, one might expect that the interface location and the interaction pattern would be highly conserved in homologous transient PPCs. The knowledge gained from learning these structural features could aid in improving the comparative modeling of PPCs.
The foremost finding of this study, which has been shown for the first time, is the observation that conservation of an interface residue type12 among homologous members need not always guarantee that all the conserved residues in a family would lie at the interface. Therefore, the popular approach of directly extrapolating conserved residues as interface residues during interface residue prediction and comparative modeling of PPC111 is not always valid. Ability of a potentially interacting residue to interact seems to depends on the residue type in the interacting partner (longer to smaller residue substitution, non-compatible interaction) or structural differences at the interfaces resulting in absence of interaction(s).
This study has provided a new perspective on interface evolution in transient PPCs. We observed only modest CIL scores (Fig. 1) between homologous pairs of PPCs. Low CIL is possible in hub proteins, which are capable of transiently binding to many interacting partners by alternate binding modes.23 Ephrin-ephrin receptor shows that ephrin receptor B2 recognizes ephrin molecule B2 and A5 by different interfaces exhibiting interaction promiscuity and low CIL (53%, 44%) from the dataset.47,55 An exceptional case of CIL being zero between HLA class II histocompatibility antigen–staphylococcal enterotoxin H and HLA class II histocompatibility–staphylococcal enterotoxin B from the dataset shows completely different binding modes.33,34
In fact, interface location differs even between two crystal structures corresponding to the identical PPCs [Supporting Information Fig. S1(d)]. Pronounced interface plasticity in the homologous chains of homologous transient PPC is due to subtle DISO and structure deviation at the interface. The extent of conservation of interfacial location is not correlated well with the sequence identity between the homologous proteins. Even though sequence identity between the homologous pair is an important indicator, using sequence identity as the only descriptor to model protein interactions might not be reliable.
We also observed that the conservation of interaction pattern (Fig. 1) in homologous transient PPCs is poor. This means that there are different ways to stabilize homologous PPCs. Residues with conserved interaction pattern mostly have their residue types conserved and also the vice versa is true. It is very interesting to note that in spite of interaction patterns being conserved, the interaction type is usually different. For example, substitution of ionic interaction to side chain mediated hydrogen bonds is observed more often than conservation of ionic interaction [Fig. 6(a)]. This study highlights the inherent interface and interaction plasticity observed in transient PPCs.
We noticed that the interfacial location and interaction pattern are conserved better in permanent homodimers than transient heteromeric PPCs. Therefore, there is less selection pressure on protein interfaces in transient PPC than permanent proteins at both sequence14 and structural level. This is consistent with the recent finding of heteromeric protein complexes being more flexible than the homomeric proteins.112 The general notion of homologous protein complexes binding in similar way holds true mainly for permanent protein complex which had been shown in previous analysis.107 Even though homologous transient PPCs might bind grossly in a similar way,18,19,110 detailed analysis in this work reveals interface and interaction plasticity.
Encouragingly, we noticed that the core interface residues and interactions involving them show higher conservation of interfacial location and interaction pattern than interface residues in the rim. This makes the predictions of core interface residues more reliable than rim interface residues. Unlike a dataset of homologous pairs of transient PPCs, a dataset of same protein bound to homologous proteins shows a high CIL and CIP. Therefore, prediction of interface residues and interactions using comparative modeling of these complexes becomes straightforward.
Results from this work clearly shows that thorough investigations of conservation of structural features at the interfaces is a necessary input in building models of PPCs of improved accuracies. Even though, this analysis shows that direct extrapolation of interface residues and interactions may not provide accurate PPC models, we propose that residues with conserved interaction pattern that show high residue type conservation/conservative substitution could serve as anchor to guide protein–protein docking.
In a previous work prediction of protein interface residues has been carried out using sequence comparison methods.113 In this work, we have shown that, even though the extent of conservation of interaction pattern is generally low, a strategic way to increase the number of residues with conserved interaction pattern would be to get information from multiple homologous PPC templates. Moreover, core-core interactions in the homologous pairs of protein complexes would more likely show conservation of interaction pattern. These pointers could help improved knowledge guided protein–protein docking.
In fact we found that information about residues with conserved interaction pattern from multiple homologous templates of GTPase-GAP complexes served as anchor residues to dock PPC with improved accuracy. This method could predict higher number of interface and interacting residues in the PPC correctly than the traditional protein–protein docking where the interface residues in the PPCs are blindly extrapolated to predict that the equivalent residues in the homolog are in the interface (for details refer Supporting Information text and Supporting Information Fig. S6). Even though the modeling of transient PPCs could be challenging, this study provides important leads to improve the accuracy of homology models of PPCs.
Materials and Methods
Datasets of homologous pairs of transient protein–protein complexes and homologous pairs of permanent homodimers
The primary dataset comprises of 3-D structures of homologous pairs of transient PPCs A:B and A′:B′, where A is homologous to A′ and B is homologous to B′ (Fig. 1; Table1). We had set-up a high-quality dataset, which satisfies following important conditions:
Dataset must be made of entries with each entry corresponding to two homologous PPCs. The two pairs of homologous proteins should preferably have a sequence identity greater than 30%.
However, we did not want to use a PPC structure in more than one entry (i.e., every complex structure has been used in only one pairwise comparison). This condition has been imposed to avoid the use of homologous complexes between entries, which will cause bias in the dataset. Imposition of this condition resulted in elimination of large number of complex structures, such as those corresponding to proteinase-inhibitor complexes.
All the complex structures in the dataset should have a clear prior evidence for their transient nature of association.
We did not want to use crystal structures of complexes with missing electron densities
We did not want to use complex structures involving mutant protein(s).
Chain lengths of the interacting partners should be greater than 50 residues.
Antigen–antibody complexes were not considered due to permanent interaction upon binding.
Interacting partners involving only a single interface patch have been included in the data set.
Although subjecting several conditions would compromise on the size of the dataset, the quality of the dataset is quite high.
Transient nature of PPCs was inferred from various studies, including information from literature, SWISSPROT, and availability of 3-D structures in both complexed and uncomplexed forms.8,13,63 This dataset has been further filtered so that mutant entries are avoided and protein complex structures with a crystallographic resolution better than 3 Å are only considered but after ensuring that electron density is not missing in any of the structures. Bias in the dataset was avoided by using the family information available in the SCOP114 (Structure Classification of Proteins) code, resulting in a pairs of homologous complex structures with no complex used in more than one pairwise comparison. Homologs of both the interacting proteins of the nonredundant transient PPCs were identified by searching the PDB115 using BLAST116 (length coverage ≥ 70%, sequence identity ≥ 30%, and E-value cutoff: 10−4)
We have considered only close homologs (≥30% sequence identity) because it is observed that distant homologs tend to have non-equivalent interfaces in their tertiary structures and different interacting partners.117 The dataset of homologous transient PPC were clustered at 80% sequence identity using BLASTCLUST116 to ensure that identical protein complexes or protein complexes with point mutations were excluded from the analysis. Further, identical protein bound to two different homologous proteins was considered as a separate dataset for analysis (refer Supporting Information Table S1).
Another dataset of homologous pairs of permanent homodimer AA and A′A′ (where A and A′ are homologous) were generated (Supporting Information Table S1). List of permanent homodimers were derived from previous studies, which comprised of manually curated dataset of dimers in solution.118
Homologs for permanent homodimers were picked as discussed previously. The dataset were made by clustering them at 25% sequence identity using BLASTCLUST. All the homomers correspond to the biological assembly unit, which are biologically functional in their dimeric form. This information is obtained from PDB.119
The entries in the dataset generated were searched in PDB using BLAST for identical transient protein complexes and permanent protein complexes solved in different crystalline conditions. Different crystalline conditions refer to different space groups in which protein structures have been solved.
Number of permanent protein complexes solved has surpassed the number of transient PPC structures. In this analysis, even though the dataset size is modest in transient PPC as compared with permanent dimers, experimental information of the transient nature is available for all the entries of the dataset, making it robust and reliable. Moreover, it was made sure that the transient PPC dataset is not biased by multiple entries of complex structures of same kind but includes different enzyme-inhibitors, hormone-receptors, signaling proteins, immune proteins, and so forth. Owing to the differences in the dataset size of homologous permanent and transient PPCs, we had to use appropriate cut offs for dataset preparation of homologous pairs of PPCs.
Identification of interface residues in protein–protein complex structures
Protein Interaction Calculator (PIC) program120 was used to identify the interface residues and types of interactions between interfacial residues (hydrophobic, disulfide, hydrogen bonding, ionic, aromatic–aromatic, aromatic–sulfur, and cation–pi) using 3D structure of protein complex as an input. Interface residues are identified based on standard distance criteria that were published previously.120 Only side chain mediated interactions were considered.
CIL score
The extent of CIL in homologous subunits is calculated using CIL score. CIL score quantifies the extent of interfacial residues that are structurally equivalent in homologous pairs of proteins of PPC (Fig. 1). Structural equivalence of a residue pair is identified by using the structure-based alignment program DALI.121 Two CIL scores (between A and A′ and between B and B′) are computed for the two homologous subunits independently. It must be noted that CIL score is a structural feature of the interface, which does not consider the residue type.
A relaxed structural equivalence criteria has been considered to identify topologically equivalent Cα atoms (<3 Å). Only structure alignments with low root mean square deviation (RMSD) and high Z-score were chosen for the analysis (Supporting Information Table S2). Z-score provides the measure of quality of structure alignment. It was made sure that there are no missing residues at the interaction interfaces in the 3-D structures used for the analysis.
Extent of conservation of residue type and interface location in homologous protein–protein complexes
The extent of conservation of residue types and interface location (AA′ and BB′) were calculated for each of the 20 residue types for the dataset of homologous pairs of protein complexes
 |
DISO score
DISO quantifies the differences in the spatial orientation of the homologous proteins in their interfacial region with respect to the structurally aligned interactor proteins [Fig. 4(a)]. Two DISO scores were calculated for the homologous subunits (AA′ and BB′) of transient PPC. Local RMSDs between the interfacial regions of the homologous subunits are based on the structurally aligned residues.121
SDI score
SDI quantifies the structural differences between the “equivalent” interfaces [Fig. 4(a)]. Two SDI scores are obtained for both the homologous subunits (AA′ and BB′) of the protein complex.
CIP score
If P:Q and P′:Q′ are interacting residues in a pair of homologous transient PPC where P and P′, Q and Q′ are structurally equivalent, they are said to have a conserved interaction pattern. The nature of interaction type or the residue type involved in the interaction of the homologous PPC is not considered. A single CIP score is obtained for a homologous pair of protein complexes.
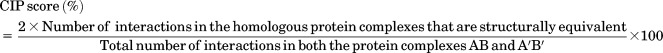 |
Interaction type substitution matrix
Interaction types such as hydrophobic, disulfide, hydrogen bonding, ionic, aromatic–aromatic, aromatic–sulfur, and cation–pi are considered. Percentage of occurrence of conserved and non-conserved interaction type but with conserved interaction pattern is used for generation of the matrix (Fig. 6). Non-conservation of interaction pattern is labeled as gap in the matrix.
Extent of conservation of residue type and interaction pattern in homologous transient protein–protein complexes
This measure is defined by,
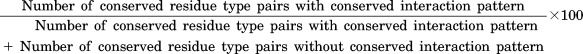 |
This value is calculated for all possible residue type pairs (20 × 20). Only residue type pairs interacting pairs with more than 10 interactions are shown in Table3.
Residues with conserved interaction pattern in which the homologous subunits (AA′ and BB′) are conserved/conservatively substituted/drastically substituted were counted. This value is normalized with the total number of residues with conserved interaction pattern.
Analysis of core/rim interface residues with conserved interface location and interaction pattern
Core interface residues are defined by the relative solvent accessibility ≤7% in complexed form and ≥10% in unbound form.122–124 The remaining interface residues are termed as rim interface residues.
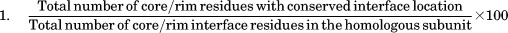
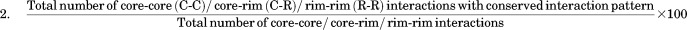
Extent of conserved residue types in homologous subunits (AA′ and BB′) with CIL was calculated for each of the 20 residue type for core and rim interface residues for the dataset.
Statistical analysis
Wilcoxon's matched pairs is a non-parametric test to compare two paired groups. It identifies whether the populations have different medians. Spearmen's rank correlation, a non-parametric measure of statistical dependence between two values was used to calculate correlation. Statistical analyses were carried out using Graphpad prism.125
Acknowledgments
The authors thank members of their group for discussions and suggestions. They declare no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
Supporting Information Table 1.
Supporting Information Table 2.
References
- Janin J, Bahadur RP, Chakrabarti P. Protein-protein interaction and quaternary structure. Q Rev Biophys. 2008;41:133–180. doi: 10.1017/S0033583508004708. [DOI] [PubMed] [Google Scholar]
- Keskin O, Gursoy A, Ma B, Nussinov R. Principles of protein–protein interactions: what are the preferred ways for proteins to interact? Chem Rev. 2008;108:1225–1244. doi: 10.1021/cr040409x. [DOI] [PubMed] [Google Scholar]
- Bahadur RP, Chakrabarti P, Rodier F, Janin J. Dissecting subunit interfaces in homodimeric proteins. Proteins. 2003;53:708–719. doi: 10.1002/prot.10461. [DOI] [PubMed] [Google Scholar]
- Nooren IM, Thornton JM. Diversity of protein-protein interactions. EMBO J. 2003;22:3486–3492. doi: 10.1093/emboj/cdg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbabacan SE, Engin HB, Gursoy A, Keskin O. Transient protein-protein interactions. Protein Eng Des Sel. 2011;24:635–648. doi: 10.1093/protein/gzr025. [DOI] [PubMed] [Google Scholar]
- Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C. Transient protein-protein interactions: structural, functional, and network properties. Structure. 2010;18:1233–1243. doi: 10.1016/j.str.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Nishi H, Bryant S, Panchenko AR. Caught in self-interaction: evolutionary and functional mechanisms of protein homooligomerization. Phys Biol. 2011;8:035007. doi: 10.1088/1478-3975/8/3/035007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari S, Helms V. Statistical analysis of predominantly transient protein-protein interfaces. Proteins. 2005;61:344–355. doi: 10.1002/prot.20593. [DOI] [PubMed] [Google Scholar]
- De S, Krishnadev O, Srinivasan N, Rekha N. Interaction preferences across protein-protein interfaces of obligatory and non-obligatory components are different. BMC Struct Biol. 2005;5:15. doi: 10.1186/1472-6807-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci U S A. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nooren IM, Thornton JM. Structural characterisation and functional significance of transient protein-protein interactions. J Mol Biol. 2003;325:991–1018. doi: 10.1016/s0022-2836(02)01281-0. [DOI] [PubMed] [Google Scholar]
- Valdar WS, Thornton JM. Protein-protein interfaces: analysis of amino acid conservation in homodimers. Proteins. 2001;42:108–124. [PubMed] [Google Scholar]
- Choi YS, Yang JS, Choi Y, Ryu SH, Kim S. Evolutionary conservation in multiple faces of protein interaction. Proteins. 2009;77:14–25. doi: 10.1002/prot.22410. [DOI] [PubMed] [Google Scholar]
- Mintseris J, Weng Z. Structure, function, and evolution of transient and obligate protein-protein interactions. Proc Natl Acad Sci U S A. 2005;102:10930–10935. doi: 10.1073/pnas.0502667102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madaoui H, Guerois R. Coevolution at protein complex interfaces can be detected by the complementarity trace with important impact for predictive docking. Proc Natl Acad Sci U S A. 2007;105:7708–7713. doi: 10.1073/pnas.0707032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries SJ, Bonvin AM. Intramolecular surface contacts contain information about protein-protein interface regions. Bioinformatics. 2006;22:2094–2098. doi: 10.1093/bioinformatics/btl275. [DOI] [PubMed] [Google Scholar]
- Aloy P, Ceulemans H, Stark A, Russell RB. The relationship between sequence and interaction divergence in proteins. J Mol Biol. 2003;332:989–998. doi: 10.1016/j.jmb.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Teichmann SA. The constraints protein-protein interactions place on sequence divergence. J Mol Biol. 2002;324:399–407. doi: 10.1016/s0022-2836(02)01144-0. [DOI] [PubMed] [Google Scholar]
- Zhang QC, Petrey D, Norel R, Honig BH. Protein interface conservation across structure space. Proc Natl Acad Sci U S A. 2010;107:10896–10901. doi: 10.1073/pnas.1005894107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreani J, Faure G, Guerois R. Versatility and invariance in the evolution of homologous heteromeric interfaces. PLoS Comput Biol. 2012;8:e1002677. doi: 10.1371/journal.pcbi.1002677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamp T, Rost B. Alternative protein-protein interfaces are frequent exceptions. PLoS Comput Biol. 2012;8:e1002623. doi: 10.1371/journal.pcbi.1002623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundrotas PJ, Vakser IA. Protein-protein alternative binding modes do not overlap. Protein Sci. 2013;22:1141–1145. doi: 10.1002/pro.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WK, Henschel A, Winter C, Schroeder M. The many faces of protein-protein interactions: a compendium of interface geometry. PLoS Comput Biol. 2006;2:e124. doi: 10.1371/journal.pcbi.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papageorgiou AC, Shapiro R, Acharya KR. Molecular recognition of human angiogenin by placental ribonuclease inhibitor–an X-ray crystallographic study at 2.0 A resolution. EMBO J. 1997;16:5162–5177. doi: 10.1093/emboj/16.17.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobe B, Deisenhofer J. A structural basis of the interactions between leucine-rich repeats and protein ligands. Nature. 1995;374:183–186. doi: 10.1038/374183a0. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4–activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- Slep KC, Kercher MA, He W, Cowan CW, Wensel TG, Sigler PB. Structural determinants for regulation of phosphodiesterase by a G protein at 2.0 A. Nature. 2001;409:1071–1077. doi: 10.1038/35059138. [DOI] [PubMed] [Google Scholar]
- Slep KC, Kercher MA, Wieland T, Chen CK, Simon MI, Sigler PB. Molecular architecture of Galphao and the structural basis for RGS16-mediated deactivation. Proc Natl Acad Sci U S A. 2008;105:6243–6248. doi: 10.1073/pnas.0801569105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui MJ, Tsai LC, Hsia KC, Doudeva LG, Ku WY, Han GW, Yuan HS. Metal ions and phosphate binding in the H-N-H motif: crystal structures of the nuclease domain of ColE7/Im7 in complex with a phosphate ion and different divalent metal ions. Protein Sci. 2002;11:2947–2957. doi: 10.1110/ps.0220602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko TP, Liao CC, Ku WY, Chak KF, Yuan HS. The crystal structure of the DNase domain of colicin E7 in complex with its inhibitor Im7 protein. Structure. 1999;7:91–102. doi: 10.1016/s0969-2126(99)80012-4. [DOI] [PubMed] [Google Scholar]
- Bode W, Papamokos E, Musil D. The high-resolution X-ray crystal structure of the complex formed between subtilisin Carlsberg and eglin c, an elastase inhibitor from the leech Hirudo medicinalis. Structural analysis, subtilisin structure and interface geometry. Eur J Biochem. 1987;166:673–692. doi: 10.1111/j.1432-1033.1987.tb13566.x. [DOI] [PubMed] [Google Scholar]
- McPhalen CA, James MN. Structural comparison of two serine proteinase-protein inhibitor complexes: eglin-c-subtilisin Carlsberg and CI-2-subtilisin Novo. Biochemistry. 1988;27:6582–6598. [PubMed] [Google Scholar]
- Bolin DR, Swain AL, Sarabu R, Berthel SJ, Gillespie P, Huby NJ, Makofske R, Orzechowski L, Perrotta A, Toth K, Cooper JP, Jiang N, Falcioni F, Campbell R, Cox D, Gaizband D, Belunis CJ, Vidovic D, Ito K, Crowther R, Kammlott U, Zhang X, Palermo R, Weber D, Guenot J, Nagy Z, Olson GL. Peptide and peptide mimetic inhibitors of antigen presentation by HLA-DR class II MHC molecules. Design, structure-activity relationships, and X-ray crystal structures. J Med Chem. 2000;43:2135–2148. doi: 10.1021/jm000034h. [DOI] [PubMed] [Google Scholar]
- Petersson K, Hakansson M, Nilsson H, Forsberg G, Svensson LA, Liljas A, Walse B. Crystal structure of a superantigen bound to MHC class II displays zinc and peptide dependence. EMBO J. 2001;20:3306–3312. doi: 10.1093/emboj/20.13.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman GR, Nassar N, Cerione RA. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell. 2000;100:345–356. doi: 10.1016/s0092-8674(00)80670-4. [DOI] [PubMed] [Google Scholar]
- Scheffzek K, Stephan I, Jensen ON, Illenberger D, Gierschik P. The Rac-RhoGDI complex and the structural basis for the regulation of Rho proteins by RhoGDI. Nat Struct Biol. 2000;7:122–126. doi: 10.1038/72392. [DOI] [PubMed] [Google Scholar]
- Mossessova E, Lima CD. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell. 2000;5:865–876. doi: 10.1016/s1097-2765(00)80326-3. [DOI] [PubMed] [Google Scholar]
- Reverter D, Lima CD. A basis for SUMO protease specificity provided by analysis of human Senp2 and a Senp2-SUMO complex. Structure. 2004;12:1519–1531. doi: 10.1016/j.str.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Plotnikov AN, Hubbard SR, Schlessinger J, Mohammadi M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell. 2000;101:413–424. doi: 10.1016/s0092-8674(00)80851-x. [DOI] [PubMed] [Google Scholar]
- Yeh BK, Igarashi M, Eliseenkova AV, Plotnikov AN, Sher I, Ron D, Aaronson SA, Mohammadi M. Structural basis by which alternative splicing confers specificity in fibroblast growth factor receptors. Proc Natl Acad Sci U S A. 2003;100:2266–2271. doi: 10.1073/pnas.0436500100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R, Kachalova G, Vellieux F, Charon MH, Frey M. Crystallographic studies of the interaction between the ferredoxin-NADP+ reductase and ferredoxin from the cyanobacterium Anabaena: looking for the elusive ferredoxin molecule. Acta Crystallogr D Biol Crystallogr. 2000;56:1408–1412. doi: 10.1107/s0907444900010052. [DOI] [PubMed] [Google Scholar]
- Kurisu G, Kusunoki M, Katoh E, Yamazaki T, Teshima K, Onda Y, Kimata-Ariga Y, Hase T. Structure of the electron transfer complex between ferredoxin and ferredoxin-NADP(+) reductase. Nat Struct Biol. 2001;8:117–121. doi: 10.1038/84097. [DOI] [PubMed] [Google Scholar]
- Soundararajan M, Willard FS, Kimple AJ, Turnbull AP, Ball LJ, Schoch GA, Gileadi C, Fedorov OY, Dowler EF, Higman VA, Hutsell SQ, Sundström M, Doyle DA, Siderovski DP. Structural diversity in the RGS domain and its interaction with heterotrimeric G protein alpha-subunits. Proc Natl Acad Sci U S A. 2008;105:6457–6462. doi: 10.1073/pnas.0801508105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg EJ, Andersen PS, Schlievert PM, Karjalainen K, Mariuzza RA. Structural, energetic, and functional analysis of a protein-protein interface at distinct stages of affinity maturation. Structure. 2003;11:1151–1161. doi: 10.1016/s0969-2126(03)00187-4. [DOI] [PubMed] [Google Scholar]
- Hege T, Feltzer RE, Gray RD, Baumann U. Crystal structure of a complex between Pseudomonas aeruginosa alkaline protease and its cognate inhibitor: inhibition by a zinc-NH2 coordinative bond. J Biol Chem. 2001;276:35087–35092. doi: 10.1074/jbc.M104020200. [DOI] [PubMed] [Google Scholar]
- Baumann U, Bauer M, Letoffe S, Delepelaire P, Wandersman C. Crystal structure of a complex between Serratia marcescens metallo-protease and an inhibitor from Erwinia chrysanthemi. J Mol Biol. 1995;248:653–661. doi: 10.1006/jmbi.1995.0249. [DOI] [PubMed] [Google Scholar]
- Himanen JP, Rajashankar KR, Lackmann M, Cowan CA, Henkemeyer M, Nikolov DB. Crystal structure of an Eph receptor-ephrin complex. Nature. 2001;414:933–938. doi: 10.1038/414933a. [DOI] [PubMed] [Google Scholar]
- Himanen JP, Yermekbayeva L, Janes PW, Walker JR, Xu K, Atapattu L, Rajashankar KR, Mensinga A, Lackmann M, Nikolov DB, Dhe-Paganon S. Architecture of Eph receptor clusters. Proc Natl Acad Sci U S A. 2010;107:10860–10865. doi: 10.1073/pnas.1004148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himanen JP, Goldgur Y, Miao H, Myshkin E, Guo H, Buck M, Nguyen M, Rajashankar KR, Wang B, Nikolov DB. Ligand recognition by A-class Eph receptors: crystal structures of the EphA2 ligand-binding domain and the EphA2/ephrin-A1 complex. EMBO Rep. 2009;10:722–728. doi: 10.1038/embor.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Durley R, Poliks BJ, Hamada K, Chen Z, Mathews FS, Davidson VL, Satow Y, Huizinga E, Vellieux FM, Hol WG. Crystal structure of an electron-transfer complex between methylamine dehydrogenase and amicyanin. Biochemistry. 1992;31:4959–4964. doi: 10.1021/bi00136a006. [DOI] [PubMed] [Google Scholar]
- Cavalieri C, Biermann N, Vlasie MD, Einsle O, Merli A, Ferrari D, Rossi GL, Ubbink M. Structural comparison of crystal and solution states of the 138 kDa complex of methylamine dehydrogenase and amicyanin from Paracoccus versutus. Biochemistry. 2008;47:6560–6570. doi: 10.1021/bi7023749. [DOI] [PubMed] [Google Scholar]
- Jenko S, Dolenc I, Guncar G, Dobersek A, Podobnik M, Turk D. Crystal structure of stefin A in complex with cathepsin H: N-terminal residues of inhibitors can adapt to the active sites of endo- and exopeptidases. J Mol Biol. 2003;326:875–885. doi: 10.1016/s0022-2836(02)01432-8. [DOI] [PubMed] [Google Scholar]
- Stubbs MT, Laber B, Bode W, Huber R, Jerala R, Lenarcic B, Turk V. The refined 2.4 A X-ray crystal structure of recombinant human stefin B in complex with the cysteine proteinase papain: a novel type of proteinase inhibitor interaction. EMBO J. 1990;9:1939–1947. doi: 10.1002/j.1460-2075.1990.tb08321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen SK, Ibrahimi OA, Raucci A, Zhang F, Eliseenkova AV, Yayon A, Basilico C, Linhardt RJ, Schlessinger J, Mohammadi M. Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proc Natl Acad Sci U S A. 2004;101:935–940. doi: 10.1073/pnas.0307287101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himanen JP, Chumley MJ, Lackmann M, Li C, Barton WA, Jeffrey PD, Vearing C, Geleick D, Feldheim DA, Boyd AW, Henkemeyer M, Nikolov DB. Repelling class discrimination: ephrin-A5 binds to and activates EphB2 receptor signaling. Nat Neurosci. 2004;7:501–509. doi: 10.1038/nn1237. [DOI] [PubMed] [Google Scholar]
- Chrencik JE, Brooun A, Kraus ML, Recht MI, Kolatkar AR, Han GW, Seifert JM, Widmer H, Auer M, Kuhn P. Structural and biophysical characterization of the EphB4*ephrinB2 protein-protein interaction and receptor specificity. J Biol Chem. 2006;281:28185–28192. doi: 10.1074/jbc.M605766200. [DOI] [PubMed] [Google Scholar]
- Bowden TA, Aricescu AR, Nettleship JE, Siebold C, Rahman-Huq N, Owens RJ, Stuart DI, Jones EY. Structural plasticity of eph receptor A4 facilitates cross-class ephrin signaling. Structure. 2009;17:1386–1397. doi: 10.1016/j.str.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros P, Fujinaga M, Dijkstra BW, Kalk KH, Hol WG. Crystallographic refinement by incorporation of molecular dynamics: thermostable serine protease thermitase complexed with eglin c. Acta Cryst. 1989;B45:488–499. doi: 10.1107/s0108768189006038. [DOI] [PubMed] [Google Scholar]
- Gomis-Ruth FX, Maskos K, Betz M, Bergner A, Huber R, Suzuki K, Yoshida N, Nagase H, Brew K, Bourenkov GP, Bartunik H, Bode W. Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature. 1997;389:77–81. doi: 10.1038/37995. [DOI] [PubMed] [Google Scholar]
- Maskos K, Lang R, Tschesche H, Bode W. Flexibility and variability of TIMP binding: X-ray structure of the complex between collagenase-3/MMP-13 and TIMP-2. J Mol Biol. 2007;366:1222–1231. doi: 10.1016/j.jmb.2006.11.072. [DOI] [PubMed] [Google Scholar]
- Iyer S, Wei S, Brew K, Acharya KR. Crystal structure of the catalytic domain of matrix metalloproteinase-1 in complex with the inhibitory domain of tissue inhibitor of metalloproteinase-1. J Biol Chem. 2007;282:364–371. doi: 10.1074/jbc.M607625200. [DOI] [PubMed] [Google Scholar]
- Mintseris J, Weng Z. Atomic contact vectors in protein-protein recognition. Proteins. 2003;53:629–639. doi: 10.1002/prot.10432. [DOI] [PubMed] [Google Scholar]
- Hwang H, Vreven T, Janin J, Weng Z. Protein-protein docking benchmark version 4.0. Proteins. 2010;78:3111–3114. doi: 10.1002/prot.22830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M, Chen YG, Massague J, Kuriyan J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell. 1999;96:425–436. doi: 10.1016/s0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- Chaikuad A, Alfano I, Kerr G, Sanvitale CE, Boergermann JH, Triffitt JT, von Delft F, Knapp S, Knaus P, Bullock AN. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J Biol Chem. 2012;287:36990–36998. doi: 10.1074/jbc.M112.365932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RC, Radziejewski C, Spraggon G, Greenwald J, Kostura MR, Burtnick LD, Stuart DI, Choe S, Jones EY. The structures of the neurotrophin 4 homodimer and the brain-derived neurotrophic factor/neurotrophin 4 heterodimer reveal a common Trk-binding site. Protein Sci. 1999;8:2589–2597. doi: 10.1110/ps.8.12.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RC, Radziejewski C, Stuart DI, Jones EY. Structure of the brain-derived neurotrophic factor/neurotrophin 3 heterodimer. Biochemistry. 1995;34:4139–4146. doi: 10.1021/bi00013a001. [DOI] [PubMed] [Google Scholar]
- Russo AA, Tong L, Lee JO, Jeffrey PD, Pavletich NP. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature. 1998;395:237–243. doi: 10.1038/26155. [DOI] [PubMed] [Google Scholar]
- Jeffrey PD, Tong L, Pavletich NP. Structural basis of inhibition of CDK-cyclin complexes by INK4 inhibitors. Genes Dev. 2000;14:3115–3125. doi: 10.1101/gad.851100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SC, Riddle SM, Cohen RE, Hill CP. Structural basis for the specificity of ubiquitin C-terminal hydrolases. EMBO J. 1999;18:3877–3887. doi: 10.1093/emboj/18.14.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misaghi S, Galardy PJ, Meester WJ, Ovaa H, Ploegh HL, Gaudet R. Structure of the ubiquitin hydrolase UCH-L3 complexed with a suicide substrate. J Biol Chem. 2005;280:1512–1520. doi: 10.1074/jbc.M410770200. [DOI] [PubMed] [Google Scholar]
- Dauter Z, Betzel C, Genov N, Pipon N, Wilson KS. Complex between the subtilisin from a mesophilic bacterium and the leech inhibitor eglin-C. Acta Cryst. 1991;B47:707–730. doi: 10.1107/s0108768191004202. [DOI] [PubMed] [Google Scholar]
- Bompard-Gilles C, Rousseau P, Rouge P, Payan F. Substrate mimicry in the active center of a mammalian alpha-amylase: structural analysis of an enzyme-inhibitor complex. Structure. 1996;4:1441–1452. doi: 10.1016/s0969-2126(96)00151-7. [DOI] [PubMed] [Google Scholar]
- Nahoum V, Farisei F, Le-Berre-Anton V, Egloff MP, Rouge P, Poerio E, Payan F. A plant-seed inhibitor of two classes of alpha-amylases: X-ray analysis of Tenebrio molitor larvae alpha-amylase in complex with the bean Phaseolus vulgaris inhibitor. Acta Crystallogr D Biol Crystallogr. 1999;55:360–362. doi: 10.1107/S0907444998010701. [DOI] [PubMed] [Google Scholar]
- Ravishankar R, Bidya Sagar M, Roy S, Purnapatre K, Handa P, Varshney U, Vijayan M. X-ray analysis of a complex of Escherichia coli uracil DNA glycosylase (EcUDG) with a proteinaceous inhibitor. The structure elucidation of a prokaryotic UDG. Nucleic Acids Res. 1998;26:4880–4887. doi: 10.1093/nar/26.21.4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savva R, Pearl LH. Nucleotide mimicry in the crystal structure of the uracil-DNA glycosylase-uracil glycosylase inhibitor protein complex. Nat Struct Biol. 1995;2:752–757. doi: 10.1038/nsb0995-752. [DOI] [PubMed] [Google Scholar]
- Geoui T, Buisson M, Tarbouriech N, Burmeister WP. New insights on the role of the gamma-herpesvirus uracil-DNA glycosylase leucine loop revealed by the structure of the Epstein-Barr virus enzyme in complex with an inhibitor protein. J Mol Biol. 2007;366:117–131. doi: 10.1016/j.jmb.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Kryger G, Harel M, Giles K, Toker L, Velan B, Lazar A, Kronman C, Barak D, Ariel N, Shafferman A, Silman I, Sussman JL. Structures of recombinant native and E202Q mutant human acetylcholinesterase complexed with the snake-venom toxin fasciculin-II. Acta Crystallogr D Biol Crystallogr. 2000;56:1385–1394. doi: 10.1107/s0907444900010659. [DOI] [PubMed] [Google Scholar]
- Harel M, Kleywegt GJ, Ravelli RB, Silman I, Sussman JL. Crystal structure of an acetylcholinesterase-fasciculin complex: interaction of a three-fingered toxin from snake venom with its target. Structure. 1995;3:1355–1366. doi: 10.1016/s0969-2126(01)00273-8. [DOI] [PubMed] [Google Scholar]
- Nassar N, Hoffman GR, Manor D, Clardy JC, Cerione RA. Structures of Cdc42 bound to the active and catalytically compromised forms of Cdc42GAP. Nat Struct Biol. 1998;5:1047–1052. doi: 10.1038/4156. [DOI] [PubMed] [Google Scholar]
- Graham DL, Lowe PN, Grime GW, Marsh M, Rittinger K, Smerdon SJ, Gamblin SJ, Eccleston JF. MgF(3)(-) as a transition state analog of phosphoryl transfer. Chem Biol. 2002;9:375–381. doi: 10.1016/s1074-5521(02)00112-6. [DOI] [PubMed] [Google Scholar]
- Choe H, Burtnick LD, Mejillano M, Yin HL, Robinson RC, Choe S. The calcium activation of gelsolin: insights from the 3A structure of the G4-G6/actin complex. J Mol Biol. 2002;324:691–702. doi: 10.1016/s0022-2836(02)01131-2. [DOI] [PubMed] [Google Scholar]
- Nag S, Ma Q, Wang H, Chumnarnsilpa S, Lee WL, Larsson M, Kannan B, Hernandez-Valladares M, Burtnick LD, Robinson RC. Ca2+ binding by domain 2 plays a critical role in the activation and stabilization of gelsolin. Proc Natl Acad Sci U S A. 2009;106:13713–13718. doi: 10.1073/pnas.0812374106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez MM, Guan R, Swaminathan CP, Malchiodi EL, Mariuzza RA. Crystal structure of staphylococcal enterotoxin I (SEI) in complex with a human major histocompatibility complex class II molecule. J Biol Chem. 2006;281:25356–25364. doi: 10.1074/jbc.M603969200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter IR, Arndt A, Kutay U, Gorlich D, Wittinghofer A. Structural view of the Ran-Importin beta interaction at 2.3 A resolution. Cell. 1999;97:635–646. doi: 10.1016/s0092-8674(00)80774-6. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Matsuura Y, Liu SM, Stewart M. Structural basis for nuclear import complex dissociation by RanGTP. Nature. 2005;435:693–696. doi: 10.1038/nature03578. [DOI] [PubMed] [Google Scholar]
- Bewley MC, Springer K, Zhang YB, Freimuth P, Flanagan JM. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science. 1999;286:1579–1583. doi: 10.1126/science.286.5444.1579. [DOI] [PubMed] [Google Scholar]
- Seiradake E, Lortat-Jacob H, Billet O, Kremer EJ, Cusack S. Structural and mutational analysis of human Ad37 and canine adenovirus 2 fiber heads in complex with the D1 domain of coxsackie and adenovirus receptor. J Biol Chem. 2006;281:33704–33716. doi: 10.1074/jbc.M605316200. [DOI] [PubMed] [Google Scholar]
- Saikrishnan K, Bidya Sagar M, Ravishankar R, Roy S, Purnapatre K, Handa P, Varshney U, Vijayan M. Domain closure and action of uracil DNA glycosylase (UDG): structures of new crystal forms containing the Escherichia coli enzyme and a comparative study of the known structures involving UDG. Acta Crystallogr D Biol Crystallogr. 2002;58:1269–1276. doi: 10.1107/s0907444902009599. [DOI] [PubMed] [Google Scholar]
- Starovasnik MA, Christinger HW, Wiesmann C, Champe MA, de Vos AM, Skelton NJ. Solution structure of the VEGF-binding domain of Flt-1: comparison of its free and bound states. J Mol Biol. 1999;293:531–544. doi: 10.1006/jmbi.1999.3134. [DOI] [PubMed] [Google Scholar]
- Christinger HW, Fuh G, de Vos AM, Wiesmann C. The crystal structure of placental growth factor in complex with domain 2 of vascular endothelial growth factor receptor-1. J Biol Chem. 2004;279:10382–10388. doi: 10.1074/jbc.M313237200. [DOI] [PubMed] [Google Scholar]
- Sansen S, De Ranter CJ, Gebruers K, Brijs K, Courtin CM, Delcour JA, Rabijns A. Structural basis for inhibition of Aspergillus niger xylanase by Triticum aestivum xylanase inhibitor-I. J Biol Chem. 2004;279:36022–36028. doi: 10.1074/jbc.M404212200. [DOI] [PubMed] [Google Scholar]
- Pollet A, Sansen S, Raedschelders G, Gebruers K, Rabijns A, Delcour JA, Courtin CM. Identification of structural determinants for inhibition strength and specificity of wheat xylanase inhibitors TAXI-IA and TAXI-IIA. FEBS J. 2009;276:3916–3927. doi: 10.1111/j.1742-4658.2009.07105.x. [DOI] [PubMed] [Google Scholar]
- Scheidig AJ, Hynes TR, Pelletier LA, Wells JA, Kossiakoff AA. Crystal structures of bovine chymotrypsin and trypsin complexed to the inhibitor domain of Alzheimer's amyloid beta-protein precursor (APPI) and basic pancreatic trypsin inhibitor (BPTI): engineering of inhibitors with altered specificities. Protein Sci. 1997;6:1806–1824. doi: 10.1002/pro.5560060902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt AE, Chand HS, Cascio D, Kisiel W, Bajaj SP. Crystal structure of Kunitz domain 1 (KD1) of tissue factor pathway inhibitor-2 in complex with trypsin. Implications for KD1 specificity of inhibition. J Biol Chem. 2005;280:27832–27838. doi: 10.1074/jbc.M504105200. [DOI] [PubMed] [Google Scholar]
- Rak A, Pylypenko O, Durek T, Watzke A, Kushnir S, Brunsveld L, Waldmann H, Goody RS, Alexandrov K. Structure of Rab GDP-dissociation inhibitor in complex with prenylated YPT1 GTPase. Science. 2003;302:646–650. doi: 10.1126/science.1087761. [DOI] [PubMed] [Google Scholar]
- Ignatev A, Kravchenko S, Rak A, Goody RS, Pylypenko O. A structural model of the GDP dissociation inhibitor rab membrane extraction mechanism. J Biol Chem. 2008;283:18377–18384. doi: 10.1074/jbc.M709718200. [DOI] [PubMed] [Google Scholar]
- Arolas JL, Popowicz GM, Lorenzo J, Sommerhoff CP, Huber R, Aviles FX, Holak TA. The three-dimensional structures of tick carboxypeptidase inhibitor in complex with A/B carboxypeptidases reveal a novel double-headed binding mode. J Mol Biol. 2005;350:489–498. doi: 10.1016/j.jmb.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Sanglas L, Valnickova Z, Arolas JL, Pallares I, Guevara T, Sola M, Kristensen T, Enghild JJ, Aviles FX, Gomis-Ruth FX. Structure of activated thrombin-activatable fibrinolysis inhibitor, a molecular link between coagulation and fibrinolysis. Mol Cell. 2008;31:598–606. doi: 10.1016/j.molcel.2008.05.031. [DOI] [PubMed] [Google Scholar]
- Barbosa JA, Silva LP, Teles RC, Esteves GF, Azevedo RB, Ventura MM, de Freitas SM. Crystal structure of the Bowman-Birk Inhibitor from Vigna unguiculata seeds in complex with beta-trypsin at 1.55 A resolution and its structural properties in association with proteinases. Biophys J. 2007;92:1638–1650. doi: 10.1529/biophysj.106.090555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capaldi S, Perduca M, Faggion B, Carrizo ME, Tava A, Ragona L, Monaco HL. Crystal structure of the anticarcinogenic Bowman-Birk inhibitor from snail medic (Medicago scutellata) seeds complexed with bovine trypsin. J Struct Biol. 2007;158:71–79. doi: 10.1016/j.jsb.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Lim D, Park HU, De Castro L, Kang SG, Lee HS, Jensen S, Lee KJ, Strynadka NC. Crystal structure and kinetic analysis of beta-lactamase inhibitor protein-II in complex with TEM-1 beta-lactamase. Nat Struct Biol. 2001;8:848–852. doi: 10.1038/nsb1001-848. [DOI] [PubMed] [Google Scholar]
- Hanes MS, Jude KM, Berger JM, Bonomo RA, Handel TM. Structural and biochemical characterization of the interaction between KPC-2 beta-lactamase and beta-lactamase inhibitor protein. Biochemistry. 2009;48:9185–9193. doi: 10.1021/bi9007963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. . Available at: http://www.pymol.org. [Google Scholar]
- Kondrashov DA, Zhang W, Aranda RT, Stec B, Phillips GNJ. Sampling of the native conformational ensemble of myoglobin via structures in different crystalline environments. Proteins. 2008;70:353–362. doi: 10.1002/prot.21499. [DOI] [PubMed] [Google Scholar]
- van Aalten DM, Crielaard W, Hellingwerf KJ, Joshua-Tor L. Conformational substates in different crystal forms of the photoactive yellow protein–correlation with theoretical and experimental flexibility. Protein Sci. 2000;9:64–72. doi: 10.1110/ps.9.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayhoff JE, Shoemaker BA, Bryant SH, Panchenko AR. Evolution of protein binding modes in homooligomers. J Mol Biol. 2010;395:860–870. doi: 10.1016/j.jmb.2009.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Huang Y, Swaminathan CP, Smith-Gill SJ, Mariuzza RA. Magnitude of the hydrophobic effect at central versus peripheral sites in protein-protein interfaces. Structure. 2005;13:297–307. doi: 10.1016/j.str.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Guharoy M, Chakrabarti P. Conservation and relative importance of residues across protein-protein interfaces. Proc Natl Acad Sci U S A. 2005;102:15447–15452. doi: 10.1073/pnas.0505425102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloy P, Russell RB. Interrogating protein interaction networks through structural biology. Proc Natl Acad Sci U S A. 2002;99:5896–5901. doi: 10.1073/pnas.092147999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FP, Barkan DT, Eswar N, McKerrow JH, Sali A. Host pathogen protein interactions predicted by comparative modeling. Protein Sci. 2007;16:2585–2596. doi: 10.1110/ps.073228407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh JA, Teichmann SA. Protein flexibility facilitates quaternary structure assembly and evolution. PLoS Biol. 2014;12:e1001870. doi: 10.1371/journal.pbio.1001870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue LC, Dobbs D, Honavar V. HomPPI: a class of sequence homology based protein-protein interface prediction methods. BMC Bioinform. 2011;12:244. doi: 10.1186/1471-2105-12-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Rekha N, Machado SM, Narayanan C, Krupa A, Srinivasan N. Interaction interfaces of protein domains are not topologically equivalent across families within superfamilies: implications for metabolic and signaling pathways. Proteins. 2005;58:339–353. doi: 10.1002/prot.20319. [DOI] [PubMed] [Google Scholar]
- Dey S, Pal A, Chakrabarti P, Janin J. The subunit interfaces of weakly associated homodimeric proteins. J Mol Biol. 2010;398:146–160. doi: 10.1016/j.jmb.2010.02.020. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Tina KG, Bhadra R, Srinivasan N. PIC: protein interactions calculator. Nucleic Acids Res. 2007;35:W473–W476. doi: 10.1093/nar/gkm423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Park J. DaliLite workbench for protein structure comparison. Bioinformatics. 2000;16:566–567. doi: 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- Aiello D, Caffrey DR. Evolution of specific protein-protein interaction sites following gene duplication. J Mol Biol. 2012;423:257–272. doi: 10.1016/j.jmb.2012.06.039. [DOI] [PubMed] [Google Scholar]
- Swapna LS, Bhaskara RM, Sharma J, Srinivasan N. Roles of residues in the interface of transient protein-protein complexes before complexation. Sci Rep. 2012;2:334. doi: 10.1038/srep00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard S. NACCESS: program for calculating accessibilities. London: Department of Biochemistry and Molecular Biology, University College of London; 1992. . Available at: http://www.bioinf.manchester.ac.uk/naccess/ [Google Scholar]
- Motulsky H. Analyzing data with GraphPad prism. San Diego, GA: GraphPad Software Inc; 1999. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information Table 1.
Supporting Information Table 2.