

This study examined a direct effect of skin-derived mesenchymal stem cell (S-MSC) treatment in the formation of atherosclerotic plaque in apolipoprotein E-knockout (apoE−/−) mice. Plaque lesion size and the plaque area of the mouse aortic arch were significantly reduced after S-MSC treatment. Results suggest that S-MSCs may have a role in stem cell-based therapy for atherosclerosis.
Keywords: Skin-derived mesenchymal stem cells, Macrophages, Atherosclerosis, Nuclear factor-κB
Abstract
Mesenchymal stem cells (MSCs) exhibit immunosuppressive efficacy and significantly inhibit the formation of the atherosclerosis (AS) plaque in apolipoprotein E-knockout (apoE−/−) mice. Of note, the largest lymphoid organ, the skin, provides a readily accessible and ideal source of tissue for the isolation of MSCs: skin-derived MSCs (S-MSCs). However, the effect and the mechanism of the therapeutic properties of S-MSCs in the progression of AS are unclear. We therefore investigated a direct effect of S-MSC treatment in the formation of atherosclerotic plaque in apoE−/− mice. Fifty apoE−/− mice were divided into four groups: the control group (AS), the S-MSC treatment group (S-MSC treatment), the nuclear factor-κB (NF-κB)−/−-S-MSC treatment group (KO-S-MSC treatment), and the additional S-MSC migration group. Brachiocephalic artery ultrasound biomicroscope (UBM) analysis showed that S-MSC treatment significantly reduced lesion size compared with the control groups (p < .01). Histological studies demonstrated that the plaque area of the mouse aortic arch was significantly decreased after S-MSC treatment. All alterations were dependent on NF-κB activation. After tail-vein injection, S-MSCs were capable of migrating to atherosclerotic plaque and selectively taking up residence near macrophages. S-MSC treatment reduced the release of the proinflammatory cytokine tumor necrosis factor (TNF)-α and increased the expression of the anti-inflammatory factor interleukin (IL)-10 in the atherosclerotic plaque, which was also dependent on NF-κB activation. In vitro, we found lipopolysaccharide (LPS) induced NF-κB-dependent expression of cyclooxygenase-2 (COX-2) in S-MSCs. Prostaglandin E2 (PGE2) expression was markedly increased after LPS-stimulated S-MSCs were cocultured with macrophages. LPS-stimulated macrophages produced less TNF-α/IL-1β and more IL-10 when cultured with S-MSCs, and although both were dependent upon NF-κB, the release of IL-10 was diminished if the S-MSCs were pretreated with a COX-2 inhibitor or an EP2/EP4 antagonist. Our data demonstrated that S-MSCs inhibited the formation of the atherosclerotic plaque in apoE−/− mice by modulating the functionality of macrophages, suggesting that S-MSCs may potentially have a role in stem cell-based therapy for AS.
Significance
A combination of in vitro and in vivo experiments showed that skin-derived mesenchymal stem cells (S-MSCs) can attenuate the plaque size of atherosclerosis. This is probably because S-MSCs beneficially modulate the response of macrophages through an increased release of prostaglandin E2 acting on the EP2 and EP4 receptors of the macrophages, stimulating the production and release of the anti-inflammatory cytokine interleukin-10, and decreasing the production of proinflammatory cytokine tumor necrosis factor-α. S-MSCs inhibited the formation of the atherosclerotic plaque in apolipoprotein E-knockout mice by modulating the functionality of macrophages, and the suppressive property of S-MSCs is dependent on NF-κB signaling. This study provides direct evidence that S-MSCs have a potent immunosuppressive effect in the development of atherosclerosis in mice, suggesting that S-MSCs can easily be cultured and have similar function to bone marrow-derived MSCs, a promising cell source for stem cell-based therapies of atherosclerosis, and possibly also in transplantation.
Introduction
Mesenchymal stem cells (MSCs), also known as multipotent mesenchymal stromal cells, exist in almost all tissues, such as bone marrow, fat, the dermis, arteries, and the umbilical cord [1]. Intravenously injected bone marrow-derived MSCs (BM-MSCs) preferentially home to a variety of organs, with injured or damaged tissues taking priority [2, 3], and they play an important role in regulating the inflammatory response to achieve their immunosuppressive properties [4]. A recent study has characterized a novel population of MSCs derived from skin (S-MSCs), with functional similarities to BM-MSCs, which are a promising cell source for stem cell-based therapies of chronic inflammatory diseases and possibly transplantation, and revealed an important immunomodulatory activity to markedly suppress the development of experimental allergic encephalomyelitis in mice, a mouse model of multiple sclerosis [5].
Inflammation is central to all stages of atherosclerosis (AS). Macrophages produce the majority of the inflammatory cytokines in atherosclerotic plaques and play a critical role in the pathophysiology of AS [6, 7]. Previous studies suggest that MSCs are capable of regulating the function of macrophages and dendritic cells (DCs), either directly through cell-cell contact or indirectly via the production of soluble mediators [8–11]. BM-MSCs (activated by lipopolysaccharide [LPS] or tumor necrosis factor [TNF]-α) reprogram macrophages by releasing prostaglandin E2 (PGE2), which acts on macrophages to release interleukin (IL)-10 through the prostaglandin EP2 and EP4 receptors, and attenuate sepsis [9]. IL-10 is a prototypic anti-inflammatory cytokine; the most recent discoveries suggest that it exerts its antiatherogenic effects on plaque development of AS by influencing the local inflammatory process within the atherosclerotic lesion [7], such as inhibiting macrophage activation as well as matrix metalloproteinase, proinflammatory cytokines, and cyclooxygenase-2 (COX-2) expression in lipid-loaded and activated macrophage foam cells [12]. Moreover, MSCs have been reported to enhance the proportion of regulatory T (Treg) cells in in vitro coculture [8] and to significantly increase the number of Treg cells in different tissues, inhibiting the formation of the atherosclerosis plaque in apolipoprotein E-knockout (apoE−/−) mice [13, 14].
Thus far, no study has reported the effect of S-MSCs on macrophages or in the treatment of AS. In this study, we demonstrated that S-MSCs inhibited the formation of the atherosclerotic plaque in apoE−/− mice by modulating the functionality of inflammatory macrophages, suggesting that S-MSCs may have therapeutic potential for the treatment of AS.
Materials and Methods
Mice
C57BL/6J, male, apoE−/− mice were purchased from the Animal Center of Peking University, Beijing, China (approval number: SCXK [Jing] 2006-0008). Starting from 8 weeks of age, 50 male apoE−/− mice were randomly divided into 4 groups: AS (control group, n = 15), fed a high-fat diet that contained 21% fat from lard and was supplemented with 0.15% (mass per mass) cholesterol for 24 weeks; S-MSC treatment (wild-type [WT]-S-MSC treatment group, n = 15), fed a high-fat diet for 24 weeks, with the addition of S-MSC treatment by tail-vein injection for the last 5 weeks; KO-S-MSC treatment (nuclear factor-κB [NF-κB]−/−-S-MSC treatment group, n = 10), fed a high-fat diet for 24 weeks, simultaneously receiving KO-S-MSC treatment by tail-vein injection for the last 5 weeks; and the additional S-MSC migration group (n = 10), fed a high-fat diet for 21 weeks, with the addition of S-MSC treatment by tail-vein injection for the last 1 week. Mice with a genetic mutation of genes for NF-кB signaling (NF-кB−/−) have been described previously [15]; the mice included in this study had downregulated NF-кB activity. Enhanced green fluorescent protein (EGFP) transgenic C57BL/6J mice were purchased from Cyagen Biosciences (Guangzhou, China, http://www.cyagen.com). All mice were kept under specific pathogen-free conditions in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals with the approval (SYXK-2003-0026) of the Scientific Investigation Board of Shanghai Jiao Tong University School of Medicine, Shanghai, China.
S-MSC Preparation, Culture, Characterization, and Differentiation
Preparation, culture, characterization, and differentiation of S-MSCs were performed as described previously [5].
Ultrasound Biomicroscopy
Ultrasound biomicroscopy was performed, and measurements of the plaque area in the brachiocephalic arteries and maximal flow velocities were investigated, as described previously [16].
Lipid Analysis
Plasma lipid analyses were performed on the serum samples obtained from blood drawn from the left ventricles of the mice at the end of the studies.
Histology and Immunofluorescence
All animals were killed after UBM investigation for histology examination. The animals were anesthetized, then the left ventricle was perfused with phosphate-buffered saline and 10% neutralized formalin at a constant pressure of 100 mmHg. The aortic arch was removed and embedded in paraffin. Five sections (5 μm) were taken 35 μm apart from the aortic arch of every mouse and were stained with hematoxylin and eosin (H&E) and Masson’s trichrome. The brachiocephalic arteries were removed and cryosectioned; the 8-μm cryosections were fixed in ice-cold acetone for 10 minutes before staining. The following antibodies were used for immunofluorescent staining: anti-mouse CD11b-PE (catalog no. 553311; BD Biosciences, San Jose, CA, http://www.bdbiosciences.com), anti-mouse TNF-α (catalog no. AF-410-NA; R&D Systems, Minneapolis, Minnesota, http://www.rndsystems.com), anti-mouse IL-10 (catalog no. LS-B4913; LifeSpan BioSciences Inc., Seattle, WA, https://www.lsbio.com), Alexa Fluor 555 goat anti-rat IgG (H+L) (catalog no. A21434; Life Technologies, Waltham, MA, http://www.thermofisher.com), and Alexa Fluor 555 donkey anti-goat IgG (H+L) (catalog no. A21432; Life Technologies) as secondary antibodies. 4′,6-Diamidino-2-phenylindole (DAPI) (Fluka, Sigma-Aldrich Co., St. Louis, MO, http://www.sigmaaldrich.com) was used for staining the nuclei.
Gene Expression Analysis
The proteins were analyzed by Western immunoblot and enzyme-linked immunosorbent assay (ELISA). For the Western immunoblot experiments, the following antibodies were used: anti-phos-IκBα (catalog no. 9246; Cell Signaling, Danvers, MA, http://www.cellsignal.com), anti-COX-2 (catalog no. 160106; Cayman Chemical Co., Ann Arbor, MI, http://www.caymanchem.com) and anti-actin antibodies. Chemically pure acetyl-11-keto-b-boswellic acid (AKβBA) was used as an NF-кB inhibitor [17]. Lipopolysaccharide was purchased from Sigma-Aldrich Co. Quantitative analyses of PGE2 and IL-10 protein levels were performed by ELISA using commercially available kits (Biolegend, San Diego, CA, http://www.biolegend.com). The COX-1 inhibitor (indomethacin, 5 µM; catalog no. 70270), the EP2 receptor antagonist (A6809, 10 µM; catalog no. 14050) and the EP4 receptor antagonist (GW627368×, 10 µM; catalog no. 10009162) were purchased from Cayman Chemical Company. The supernatants from the stimulated cell cultures were frozen and analyzed according to the manufacturer’s instructions. Finally, Stop Solution was added, and the optical density of each well was determined within 30 minutes using a microplate reader (450-nm wavelength).
Cytokine mRNA was quantified by real-time polymerase chain reaction (PCR). Total RNA was extracted from cultured cells with TRIzol Reagent (Invitrogen/Thermo Fisher Scientific, Waltham, MA, http://www.thermofisher.com). Reverse transcription was performed using the PrimeScript RT Reagent kit (TaKaRa, Shiga, Japan, http://www.clontech.com). Quantitative PCR was carried out using the SYBR Green PCR Master Mix (TaKaRa) in the ABI Prism 7900 (Applied Biosystems/Thermo Fisher Scientific, Waltham, MA, http://www.appliedbiosystems.com). All gene expression results were normalized to the expression of the housekeeping gene GAPDH. Primer sequences were as follows: TNF-α (forward: GAACTGGCAGAAGAGGCACT; reverse: AGGGTCTGGGCCATAGAACT); IL-1β (forward: ACCTTCCAGGAT GAGGACATGA; reverse: CTAATGGGAACGTCACACACCA); IL-10 (forward: CAGCCGGGAAGACAATAACT; reverse: GCATTAAGGAGTCGGTTAGCA); GAPDH (forward: TGTGTCCGTCGTGGATCTGA; reverse: CCTGCTTCACCACCTTCTTGA).
Statistical Analysis
Values shown represent the mean ± SEM, where applicable. Statistical significances were calculated with the Newman-Keuls test. Differences were considered significant at p < .05.
Results
Effect of S-MSC Treatment on the Progression of Atherosclerotic Lesions
Cells with phenotypic and functional characteristics of MSCs, besides bone marrow MSCs, have been identified [18]. The latest study has characterized a novel population of MSCs, skin-derived MSCs, which reveal in vivo immunosuppressive properties in autoimmunity [5]. This study was the basis for our investigation of the role of S-MSCs in the treatment of AS. We isolated the cells from the dissociated dermis of 1- to 3-day-old wild-type or NF-κB−/− mice and characterized them by either cell surface makers or differential abilities in specific culture medium conditions, as published recently [5].
We examined the effects of S-MSCs on the formation of atherosclerotic plaques by using a mouse model for AS in apoE−/− mice with 24 weeks of fat feeding or with 24 weeks of fat feeding with the addition of WT-S-MSCs or NF-κB−/−-S-MSC treatment for the last 5 weeks. At the end of experiment, all mice were assessed by UBM, and the analysis images revealed significant differences in plaque sizes in the brachiocephalic arteries. Additionally, the images of the pulse-wave Doppler measurements of maximal flow velocities were also markedly decreased in the S-MSC treatment but not in NF-κB−/−-S-MSC treatment, compared with the AS groups (Table 1; Fig. 1A, 1B). Histological analysis of sections taken from the aortic roots demonstrated that S-MSCs could specifically prevent atherosclerosis formation, but not for NF-κB−/−-S-MSCs (Fig. 1C, 1D). Plasma lipid analyses were performed on the terminal blood samples obtained by cardiac puncture. There were no significant differences among AS groups, S-MSC treatment groups, and NF-κB−/−-S-MSC treatment groups (supplemental online Table 1). Treatment with S-MSCs did not affect the plasma lipid concentration levels. Our results suggest that S-MSCs derived from skin harbor strong immunosuppressive properties for inhibiting the development of AS in mice, which is dependent on NF-κB activation.
Table 1.
Effects of S-MSC treatment assessed by UBM on established plaque progression in brachiocephalic artery
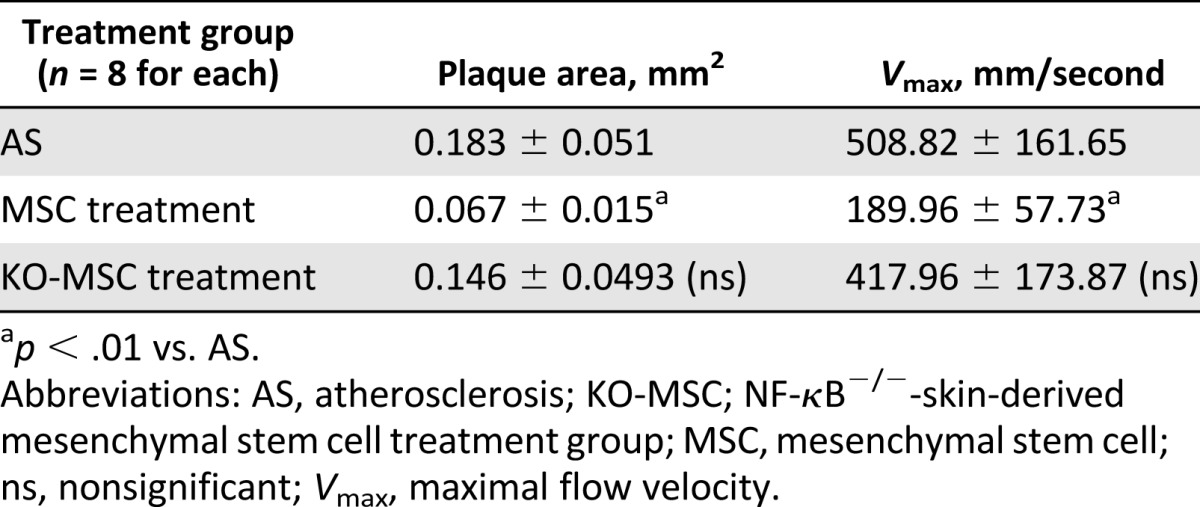
Figure 1.

Effects of S-MSC treatment on established atherosclerotic plaque progression in brachiocephalic arteries of apolipoprotein E-knockout (apoE−/−) mice. We used a mouse model for AS in apoE−/− mice and constructed three groups of mice: an AS (control group) comprising apoE−/− mice fed a high-fat diet for 24 weeks, and another two groups of apoE−/− mice that were fed a high-fat diet for 24 weeks and received 0.5 million wild-type S-MSCs or KO-S-MSCs weekly injections for the final 5 weeks. Ultrasound biomicroscopy (was performed after 24 weeks of treatment. Representative ultrasound biomicroscope (UBM) images showed the brachiocephalic arteries of the three groups of mice (AS, S-MSC treatment, KO-S-MSC treatment). (A): S-MSC treatment reduced the plaque area in brachiocephalic arteries of apoE−/− mice. White bar for UBM images = 1 mm. (B): Images of pulse-wave Doppler measurements of maximal flow velocities in the corresponding groups. (C, D): H&E (C) and Masson trichrome staining (D) show histopathologic changes of the mouse aortic arch. (E): Quantitative analysis of lesion size in the three groups. All results shown are representative of the arithmetic mean ± SEM of eight independent experiments. Scale bar for histological images = 200 µm. ∗∗, p < .01 versus AS. Abbreviations: AS, atherosclerosis; KO-S-MSCs, nuclear factor-κB−/− skin-derived mesenchymal stem cell treatment group; ns, nonsignificant; S-MSC, skin-derived mesenchymal stem cell.
S-MSC Migration
We next determined the fate of injected S-MSCs in vivo in AS mice. The mice were fed a high-fat diet for 21 weeks and were also injected i.v. with S-MSCs derived from enhanced green fluorescent protein (EGFP) transgenic mice (Fig. 2A) in the last week. Immunofluorescence staining revealed that the injected S-MSCs labeled with EGFP preferentially trafficked to the plaque (Fig. 2B), and seemed to migrate toward macrophages (Fig. 2C). These data indicate that the injected S-MSCs have the ability to migrate toward inflamed tissues and possibly to interact with activated macrophages within the atherosclerotic plaque.
Figure 2.

Determination of the fate of intravenously injected S-MSCs in a mouse model of atherosclerosis. Apolipoprotein E-knockout mice fed a high-fat diet for 21 weeks also received S-MSC-labeled enhanced green fluorescent protein (EGFP) injections during the last week. (A): Immunofluorescent staining showed S-MSCs labeled with EGFP. (B): Immunohistochemical staining showing that S-MSCs traveled to the plaque (green punctate staining). (C): S-MSCs take up residence near macrophages (CD11b, a marker of the macrophage). Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; GFP, green fluorescent protein; S-MSC, skin-derived mesenchymal stem cell.
S-MSCs Modulated Cytokine Expression in AS Mouse Model
To assess the inhibitory function of S-MSCs in vivo, immunofluorescence microscopy of sections from atherosclerotic arteries of three groups mice (AS groups, S-MSC treatment group, and NF-κB−/−-S-MSC treatment group) were stained for the proinflammatory cytokine TNF-α and anti-inflammatory cytokine IL-10. In contrast to the AS groups, TNF-α expression was markedly decreased (Fig. 3A), whereas IL-10 expression was increased in the S-MSC treatment group but not in the NF-κB−/−-S-MSC treatment group (Fig. 3B). Because TNF-α and IL-10 play essential roles in the pathogenesis of AS, our results reveal that S-MSCs could specifically inhibit proinflammatory cytokines and increase the anti-inflammatory cytokine expression in vivo.
Figure 3.

Effects of S-MSC treatment on cytokine release in the plaque of brachiocephalic arteries of apolipoprotein E-knockout mice. Representative sections from eight mice are shown. (A, B): Immunofluorescent staining of cryosections of the three groups (Fig. 1) of mouse atherosclerotic plaques with either goat anti-mouse TNF-α (primary antibody)/Alexa Fluor 555 donkey anti-goat IgG (H+L) (secondary antibody) (A), or rat anti-mouse IL-10 (primary antibody)/Alexa Fluor 555 goat anti-rat IgG (H+L) (secondary antibody) (B). Cell nuclei were counterstained with DAPI (blue). The accumulation of TNF-α was significantly decreased and IL-10 was increased in the plaques in mice treated with wild-type S-MSCs compared with AS mice, but not for KO-S-MSCs. Graphs show quantification of TNF-α and IL-10 in the plaque area of the three groups. All results are representative of the arithmetic mean ± SEM of eight independent experiments. ∗∗, p < .01 versus AS. Scale bars = 100 µm. Original magnifications, ×100 (main panels) and ×400 (insets). Abbreviations: AS, atherosclerosis; DAPI, 4′,6-diamidino-2-phenylindole; IL-10, interleukin 10; KO-S-MSCs, nuclear factor-κB−/− skin-derived mesenchymal stem cell treatment group; ns, nonsignificant; S-MSC, skin-derived mesenchymal stem cell; TNF-α, tumor necrosis factor-α.
Molecular Basis of S-MSC and Macrophage Interaction
In vivo, it remains unclear which factor(s) attracts S-MSCs to migrate toward infiltrating macrophages and whether these two different types of cells interact with each other when they meet. To understand the molecular basis of the interaction between S-MSCs and macrophages, we performed a series of experiments in vitro. We stimulated WT-S-MSCs and NF-κB−/−-S-MSCs with LPS and found that NF-κB signaling is activated only in WT-S-MSCs 15 minutes after LPS stimulation (Fig. 4A). In a number of cell types, NF-κB has been shown to induce prostaglandin production and release through a pathway involving COX-2 [9, 19]. Herein, we found a significant increase in the expression of COX-2 in S-MSCs after LPS stimulation for 3 hours and 5 hours (Fig. 4B); however, when the S-MSCs were collected from NF-κB−/− mice (Fig. 4B) or pretreated with NF-κB inhibitor AKβBA (10 µM) for 30 minutes, there was no change in COX-2 expression when S-MSCs were stimulated with LPS (Fig. 4C). Estimation of P-IKBα or COX-2 protein band intensity normalized to actin (Fig. 4A–4C). Thus, these data suggest that the COX-2 expression was dependent on the activation of the NF-κB signaling pathway in S-MSCs, and COX-2 could produce the substrate for prostaglandin synthase enzymes [9]. For this reason, using ELISA, we measured the amount of PGE2 in the supernatants of cultures of S-MSCs isolated from wild-type or NF-κB−/− mice in the presence of macrophages for an additional period of 3 or 5 hours with LPS stimulation (100 ng/ml). We observed a significant increase of PGE2 release in the supernatants of cultures with wild-type S-MSCs but not NF-κB−/− S-MSCs, suggesting that the PGE2 release was dependent on NF-κB signaling (Fig. 4D). Interestingly, a recent study has determined that PGE2 produced from BM-MSCs could affect other immune cells via EP1 and EP4 receptors [20]. Furthermore, macrophages expressing PGE2 receptors EP2 and EP4 were identified [21]. Together, our findings indicate that PGE2 released from S-MSCs might, indeed, be responsible for reprogramming the infiltrated macrophages in atherosclerotic lesions.
Figure 4.

Examination of molecular alterations underlying the effect of S-MSCs on macrophages. (A, B): Wild-type S-MSCs or KO-S-MSCs (1 × 106 per assay) were stimulated with LPS (100 ng/ml) for the indicated time. Whole-cell lysates were separated and analyzed by Western immunoblots with antibodies phosphor-IκBα (A) and COX-2 (B). (C): Effects of NF-κB on the LPS-induced COX-2 expression. S-MSCs pretreated with 10 µmol/l AKβBA for 30 minutes were stimulated with LPS (100 ng/ml) for the indicated time on the immunoblot with staining of actin-loading controls. Densitometric scanning of three Western blots is quantified in the bar graphs. The results shown are representative of at least three independent experiments. (D): S-MSCs isolated from wild-type or NF-κB−/− mice were grown to confluence and cultured or cocultured with macrophages for an additional period of 3 or 5 hours with LPS (100 ng/ml) stimulation. Supernatants were collected, and the levels of PGE2 were determined by enzyme-linked immunosorbent assay (n = 4). Error bars represent mean ± SEM. ∗∗, p < .01. Abbreviations: AKβBA, acetyl-11-keto-β-boswellic acid; COX-2, cyclooxygenase-2; KO-S-MSCs, nuclear factor-κB−/− skin-derived mesenchymal stem cell treatment group; LPS, lipopolysaccharide; PGE2, prostaglandin E2; S-MSC, skin-derived mesenchymal stem cell.
Effect of S-MSCs on Cytokine Production in LPS-Induced Macrophages
To further examine the modulatory effect of S-MSCs on macrophages in detail, we cultured macrophages alone or with S-MSCs isolated from either wild-type or NF-κB−/− mice and stimulated them for an additional 6 hours with LPS. We then measured TNF-α, IL-1β, and IL-10 expression by real-time-polymerase chain reaction (RT-PCR). We consistently found that TNF-α and IL-1β expression were decreased and IL-10 expression was increased after coculture, and both alterations for TNF-α and IL-10 were dependent on NF-κB activation of S-MSCs (Fig. 5A). These results suggested that S-MSCs could regulate the profile of cytokines produced by activated macrophages in vitro.
Figure 5.

S-MSCs inhibit the production of inflammatory cytokines and enhance the production of IL-10 by LPS-induced MACs via PGE2. MACs were cultured overnight in the absence or presence of S-MSCs or KO-MSCs (MACs: S-MSCs or KO-MSCs ratio = 10:1). (A): Cells were then washed and incubated for 6 hours with or without LPS (100 ng/ml), and cytokines were analyzed in cells by real-time polymerase chain reaction (RT-PCR). Effect of the indomethacin (COX-1/2 inhibitor; 5 µM), A6809 (EP2 receptor antagonist; 10 µM), and GW627368X (EP4 receptor antagonist; 10 µM) on the LPS-induced IL-10 release in MAC and S-MSC cocultures. Cell cocultures pretreated with COX-2 inhibitor or EP2/EP4 antagonist or medium (none) for 30 minutes were stimulated with or without LPS (100 ng/ml) for an additional 12 hours. (B): The production of IL-10 was analyzed in supernatants of cocultures by enzyme-linked immunosorbent assay. All results shown are representative of the arithmetic mean ± SEM of six independent experiments. ∗, p < .05, ∗∗, p < .01, versus MACs plus LPS. Abbreviations: COX-2, cyclooxygenase-2; Ctrl, control; IL-1β, interleukin 1-β; IL-10, interleukin-10; KO-MSC, NF-κB−/−-skin-derived mesenchymal stem cell treatment group; LPS, lipopolysaccharide; MAC, macrophage; MSC, skin-derived mesenchymal stem cell; ns, nonsignificant; TNF-α, tumor necrosis factor-α.
In addition, we pretreated the S-MSCs and macrophages with EP2 and EP4 receptor antagonists or a COX-2 inhibitor for 30 minutes before LPS stimulation for 12 hours, and the IL-10 release in coculture supernatants was investigated by ELISA. The result displayed blockades of EP2 and EP4, or that COX-2 prevented the increase of IL-10 secretion in LPS-stimulated S-MSCs cocultured with macrophages (Fig. 5B). The data suggested that S-MSCs regulated the profile of cytokines produced by activated macrophages in vitro, which may be due to the COX-2 or PGE2 released from S-MSCs.
The combination of our in vivo and in vitro experiments showed that the interaction between S-MSCs and macrophages was likely due to an increased release of COX-2 or PGE2 from the S-MSCs acting on the EP2 and EP4 receptors of the macrophages. In turn, this stimulated the release of anti-inflammatory cytokine IL-10 and decreased release of inflammatory cytokines TNF-α and IL-1β to perform their immunosuppressive effects that eventually led to reduced atherosclerotic lesions in apoE−/− mice (Fig. 6).
Figure 6.

Scheme for the S-MSCs suppressing the progression of AS via regulating macrophage activation. Abbreviations: AS, atherosclerosis; COX-2, cyclooxygenase-2; IL-1β, interleukin 1-β; IL-10, interleukin-10; LPS, lipopolysaccharide; S-MSC, skin-derived mesenchymal stem cell; TNF-α, tumor necrosis factor-α.
Discussion
The development of atherosclerotic plaques is a multistep inflammatory process [22]. Macrophages are ubiquitous in all stages of AS and possess crucial effector functions in the process of AS [6, 23, 24]. Specifically, macrophages are the main source of cytokines in atherosclerotic plaques [25].
Proinflammatory cytokines such as TNF-α may act as autocrine or paracrine molecules on the cells to promote the development of AS [26]. Consistently, apoE/TNF-α double-knockout mice show a significant decrease in relative atherosclerotic lesion size [27]. Elevated levels of TNF-α are associated with an elevated risk for recurrent myocardial infarction and cardiovascular death after a first myocardial infarction [25]. A prototypic anti-inflammatory cytokine, IL-10, produced primarily by the macrophages and Th2 subtype T lymphocytes [28], has been shown to attenuate atherogenesis by inhibition of the release of several proinflammatory cytokines (including IL-1β and TNF-α) and matrix metalloproteinase [12], and alter lipid metabolism in macrophages in AS [7]. A recent publication showed that IL-10 induces macrophage polarization toward the M2 phenotype, which reduces murine AS [29, 30]. The athero-protective role of IL-10 was confirmed by IL-10-deficient mice on the C57BL/6J background and confirmed in IL-10 and apoE double-knockout mice, as demonstrated by Caligiuri et al. [31, 32]. Previous results suggest that BM-MSCs (activated by LPS or TNF-α) reprogram macrophages to increase their IL-10 production and attenuate sepsis [9]. BM-MSCs regulate the number of Treg cells and significantly inhibit the formation of atherosclerotic plaques in apoE−/− mice [13].
BM-MSCs have been successfully used for the treatment of human diseases, and they are a good alternative for cell-based therapy in inflammatory disorders [33, 34]. However, skin-derived MSCs, which have similar functions to BM-MSCs, express major histocompatibility complex class I, which also offers them an advantage in transplantation by partially bypassing rejection responses [5].
S-MSCs are unique, multipotent stem cells and have inherent host compatibility, immunosuppressive ability, and susceptibility to gene modification, and they are readily expandable in vitro and display a greater growth potential in culture, similar to BM-MSCs [5, 35–37]. Thus, the skin is considered an ideal source for the isolation of MSCs and perhaps has an even more remarkable feature [5]: the present study is the first to demonstrate that S-MSCs are able to markedly attenuate the progression of atherosclerotic plaques in apoE−/− mice due to reprogramming macrophages for their IL-10 production.
S-MSCs seem to be potent immunomodulators, but their mechanism of action is largely unknown. S-MSCs are reported to inhibit the polarization of pathogenic T-helper type 1 (Th1) and Th17 cells and markedly suppress the development of experimental allergic encephalomyelitis in mice [5]. In the present study, we show that S-MSCs can be used to improve AS in mice. We demonstrate that, in S-MSCs, NF-κB signaling is required to induce prostaglandin production through a pathway involving COX-2, which produces the substrate for prostaglandin synthase enzymes [19]. BM-MSCs have been shown to produce PGE2 and possibly affect other immune cells via EP1-EP4 receptors [20]. In peritoneal macrophages, PGE2 was shown to modulate inflammatory reactions via the EP2 and EP4 receptors [21]. In agreement with the previous studies described in this article, we reveal a significant increase in the expression of COX-2 in S-MSCs and PGE2 in cocultures of S-MSCs and macrophages, and these actions occur in an NF-κB-dependent manner after LPS stimulation. Additionally, we note that the increased IL-10 release from the cocultures is dependent on the COX-2 or PGE2.
In conclusion, we provide direct evidence that S-MSCs have a potent immunosuppressive effect in the development of AS in mice. The combination of our in vitro and in vivo experiments shows that S-MSCs can attenuate the plaque size of AS, which is likely because S-MSCs beneficially modulate the response of macrophages through an increased release of PGE2 acting on the EP2 and EP4 receptors of the macrophages, stimulating the production and release of the anti-inflammatory cytokine IL-10 and decreasing the production of proinflammatory cytokine TNF-α. Our results suggest that S-MSCs can easily be cultured and have similar function to BM-MSCs, and they are a promising cell source for stem cell-based therapies of AS and, possibly, transplantation.
Supplementary Material
Acknowledgments
This research was supported by grants from the New Scholars Foundation of Ministry of Education of China (Grant 20110073120103), the National Natural Science Foundation of China (Grants 81202298 and 81230071), and the General Program of the Shanghai Board of Health (Grant 20124239).
Author Contributions
Q.L.: study design, collection and/or assembly of data, data analysis, manuscript writing; W.S.: collection and/or assembly of data, manuscript writing; X.W., K.Z., and W.X.: collection and/or assembly of data; P.G.: manuscript writing.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
References
- 1.Bianco P, Robey PG, Simmons PJ. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell. 2008;2:313–319. doi: 10.1016/j.stem.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Devine SM, Cobbs C, Jennings M, et al. Mesenchymal stem cells distribute to a wide range of tissues following systemic infusion into nonhuman primates. Blood. 2003;101:2999–3001. doi: 10.1182/blood-2002-06-1830. [DOI] [PubMed] [Google Scholar]
- 3.Kawada H, Fujita J, Kinjo K, et al. Nonhematopoietic mesenchymal stem cells can be mobilized and differentiate into cardiomyocytes after myocardial infarction. Blood. 2004;104:3581–3587. doi: 10.1182/blood-2004-04-1488. [DOI] [PubMed] [Google Scholar]
- 4.Shi Y, Hu G, Su J, et al. Mesenchymal stem cells: A new strategy for immunosuppression and tissue repair. Cell Res. 2010;20:510–518. doi: 10.1038/cr.2010.44. [DOI] [PubMed] [Google Scholar]
- 5.Ke F, Zhang L, Liu Z, et al. Autocrine interleukin-6 drives skin-derived mesenchymal stem cell trafficking via regulating voltage-gated Ca(2+) channels. Stem Cells. 2014;32:2799–2810. doi: 10.1002/stem.1763. [DOI] [PubMed] [Google Scholar]
- 6.Li Q, Laumonnier Y, Syrovets T, et al. Plasmin triggers cytokine induction in human monocyte-derived macrophages. Arterioscler Thromb Vasc Biol. 2007;27:1383–1389. doi: 10.1161/ATVBAHA.107.142901. [DOI] [PubMed] [Google Scholar]
- 7.Han X, Boisvert WA. Interleukin-10 protects against atherosclerosis by modulating multiple atherogenic macrophage function. Thromb Haemost. 2015;113:505–512. doi: 10.1160/TH14-06-0509. [DOI] [PubMed] [Google Scholar]
- 8.Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815–1822. doi: 10.1182/blood-2004-04-1559. [DOI] [PubMed] [Google Scholar]
- 9.Németh K, Leelahavanichkul A, Yuen PS, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15:42–49. doi: 10.1038/nm.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spaggiari GM, Capobianco A, Abdelrazik H, et al. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: Role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood. 2008;111:1327–1333. doi: 10.1182/blood-2007-02-074997. [DOI] [PubMed] [Google Scholar]
- 11.Maggini J, Mirkin G, Bognanni I, et al. Mouse bone marrow-derived mesenchymal stromal cells turn activated macrophages into a regulatory-like profile. PLoS One. 2010;5:e9252. doi: 10.1371/journal.pone.0009252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hansson GK. Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc Biol. 2001;21:1876–1890. doi: 10.1161/hq1201.100220. [DOI] [PubMed] [Google Scholar]
- 13.Wang ZX, Mao S, Li Y, et al. [Effects of MSCs on the progression of atherosclerosis plaque in ApoE-knock out mice] Xibao Yu Fenzi Mianyixue Zazhi. 2012;28:244–246. [PubMed] [Google Scholar]
- 14.Wang ZX, Wang QW, Li XY, et al. Mesenchymal stem cells alleviate atherosclerosis by elevating number and function of CD4CD25 FOXP3 regulatory T-cells and inhibiting macrophage foam cell formation. Mol Cell Biochem. 2015;400:163–172. doi: 10.1007/s11010-014-2272-3. [DOI] [PubMed] [Google Scholar]
- 15.Jimi E, Strickland I, Voll RE, et al. Differential role of the transcription factor NF-kappaB in selection and survival of CD4+ and CD8+ thymocytes. Immunity. 2008;29:523–537. doi: 10.1016/j.immuni.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu DJ, Xu JZ, Wu YJ, et al. Effects of fasudil on early atherosclerotic plaque formation and established lesion progression in apolipoprotein E-knockout mice. Atherosclerosis. 2009;207:68–73. doi: 10.1016/j.atherosclerosis.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 17.Li Q, Syrovets T, Simmet T, et al. Plasmin induces intercellular adhesion molecule 1 expression in human endothelial cells via nuclear factor-κB/mitogen-activated protein kinases-dependent pathways. Exp Biol Med (Maywood) 2013;238:176–186. doi: 10.1177/1535370212473700. [DOI] [PubMed] [Google Scholar]
- 18.da Silva Meirelles L, Chagastelles PC, Nardi NB. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J Cell Sci. 2006;119:2204–2213. doi: 10.1242/jcs.02932. [DOI] [PubMed] [Google Scholar]
- 19.Nakao S, Ogtata Y, Shimizu E, et al. Tumor necrosis factor alpha (TNF-alpha)-induced prostaglandin E2 release is mediated by the activation of cyclooxygenase-2 (COX-2) transcription via NFkappaB in human gingival fibroblasts. Mol Cell Biochem. 2002;238:11–18. doi: 10.1023/a:1019927616000. [DOI] [PubMed] [Google Scholar]
- 20.Sotiropoulou PA, Perez SA, Gritzapis AD, et al. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells. 2006;24:74–85. doi: 10.1634/stemcells.2004-0359. [DOI] [PubMed] [Google Scholar]
- 21.Shinomiya S, Naraba H, Ueno A, et al. Regulation of TNFalpha and interleukin-10 production by prostaglandins I(2) and E(2): Studies with prostaglandin receptor-deficient mice and prostaglandin E-receptor subtype-selective synthetic agonists. Biochem Pharmacol. 2001;61:1153–1160. doi: 10.1016/s0006-2952(01)00586-x. [DOI] [PubMed] [Google Scholar]
- 22.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 24.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tedgui A, Mallat Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–581. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 26.Little PJ, Chait A, Bobik A. Cellular and cytokine-based inflammatory processes as novel therapeutic targets for the prevention and treatment of atherosclerosis. Pharmacol Ther. 2011;131:255–268. doi: 10.1016/j.pharmthera.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 27.Brånén L, Hovgaard L, Nitulescu M, et al. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2004;24:2137–2142. doi: 10.1161/01.ATV.0000143933.20616.1b. [DOI] [PubMed] [Google Scholar]
- 28.Mallat Z, Heymes C, Ohan J, et al. Expression of interleukin-10 in advanced human atherosclerotic plaques: Relation to inducible nitric oxide synthase expression and cell death. Arterioscler Thromb Vasc Biol. 1999;19:611–616. doi: 10.1161/01.atv.19.3.611. [DOI] [PubMed] [Google Scholar]
- 29.Boehler RM, Kuo R, Shin S, et al. Lentivirus delivery of IL-10 to promote and sustain macrophage polarization towards an anti-inflammatory phenotype. Biotechnol Bioeng. 2014;111:1210–1221. doi: 10.1002/bit.25175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolfs IM, Stöger JL, Goossens P, et al. Reprogramming macrophages to an anti-inflammatory phenotype by helminth antigens reduces murine atherosclerosis. FASEB J. 2014;28:288–299. doi: 10.1096/fj.13-235911. [DOI] [PubMed] [Google Scholar]
- 31.Mallat Z, Besnard S, Duriez M, et al. Protective role of interleukin-10 in atherosclerosis. Circ Res. 1999;85:e17–e24. doi: 10.1161/01.res.85.8.e17. [DOI] [PubMed] [Google Scholar]
- 32.Caligiuri G, Rudling M, Ollivier V, et al. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med. 2003;9:10–17. [PMC free article] [PubMed] [Google Scholar]
- 33.Le Blanc K, Rasmusson I, Sundberg B, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363:1439–1441. doi: 10.1016/S0140-6736(04)16104-7. [DOI] [PubMed] [Google Scholar]
- 34.Yang J, Jiang Z, Fitzgerald DC, et al. Adult neural stem cells expressing IL-10 confer potent immunomodulation and remyelination in experimental autoimmune encephalitis. J Clin Invest. 2009;119:3678–3691. doi: 10.1172/JCI37914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haniffa MA, Wang XN, Holtick U, et al. Adult human fibroblasts are potent immunoregulatory cells and functionally equivalent to mesenchymal stem cells. J Immunol. 2007;179:1595–1604. doi: 10.4049/jimmunol.179.3.1595. [DOI] [PubMed] [Google Scholar]
- 36.Riekstina U, Muceniece R, Cakstina I, et al. Characterization of human skin-derived mesenchymal stem cell proliferation rate in different growth conditions. Cytotechnology. 2008;58:153–162. doi: 10.1007/s10616-009-9183-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salvolini E, Orciani M, Vignini A, et al. Skin-derived mesenchymal stem cells (S-MSCs) induce endothelial cell activation by paracrine mechanisms. Exp Dermatol. 2010;19:848–850. doi: 10.1111/j.1600-0625.2010.01104.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.