

Abstract
Epigenetic silencing of tumor suppressor genes (TSGs) is considered a significant event in the progression of cancer. For example, EPB41L3, a potential biomarker in cervical cancer, is often silenced by cancer-specific promoter methylation. Artificial transcription factors (ATFs) are unique tools to re-express such silenced TSGs to functional levels; however, the induced effects are considered transient. Here, we aimed to improve the efficiency and sustainability of gene re-expression using engineered zinc fingers fused to VP64 (ZF-ATFs) or DNA methylation modifiers (ZF-Tet2 or ZF-TDG) and/or by co-treatment with epigenetic drugs [5-aza-2′-deoxycytidine or Trichostatin A (TSA)]. The EPB41L3-ZF effectively bound its methylated endogenous locus, as also confirmed by ChIP-seq. ZF-ATFs reactivated the epigenetically silenced target gene EPB41L3 (∼10-fold) in breast, ovarian, and cervical cancer cell lines. Prolonged high levels of EPB41L3 (∼150-fold) induction could be achieved by short-term co-treatment with epigenetic drugs. Interestingly, for otherwise ineffective ZF-Tet2 or ZF-TDG treatments, TSA facilitated re-expression of EPB41L3 up to twofold. ATF-mediated re-expression demonstrated a tumor suppressive role for EPB41L3 in cervical cancer cell lines. In conclusion, epigenetic reprogramming provides a novel way to improve sustainability of re-expression of epigenetically silenced promoters.
Keywords: artificial transcription factors, anti-cancer drug response, DNA demethylases, epigenome editing, methylation biomarkers, novel antitumor agents
Introduction
It has become increasingly clear that carcinogenesis is associated with epigenetic alterations, such as DNA hypermethylation of tumor suppressor genes (TSGs), causing aberrant gene expression.1,2 Re-expression of silenced TSGs has emerged as potent anti-cancer tool,3 and epigenetic drugs are successfully exploited to reverse TSG silencing.4 However, these drugs act genome-wide and the disadvantages of these drugs include their lack of gene-specificity and low efficacy in solid tumors.5 A promising alternative is the gene-targeted re-activation of silenced genes using engineered DNA binding domains, including zinc finger proteins (ZFPs) and transcription activator–like (TAL) effectors, fused to gene activators.6 Such fusion proteins strongly exploit the reversibility of epigenetic silencing, which, in contrast to genetic mutations, allow for functional re-expression of the target gene. Nevertheless, to induce long-lasting changes of cell fate in disease, sustained expressional changes are required. Artificial transcription factor (ATF)-mediated expressional changes, however, have shown to be only transient, as targeted promoters are incompletely reprogrammed and genes tend to return to the ‘normal’ state after the ATF is removed.7,8
Recently, a number of TSGs were successfully reactivated using ZFPs linked to VP64, including maspin,7,9,10 CDKN2A,11 and C13ORF18,12 and re-activation of these 3 TSGs was associated with site-specific DNA demethylation, while re-expression of the latter two was also shown to be accompanied with a decreased repressive histone methylation status. Despite these epigenetic changes, ATF-induced effects are transient, and more stable epigenetic reprogramming is required to prolong the effects.13 A possible strategy to achieve this is the co-treatment of ATFs with epigenetic drugs, such as the DNA methylation inhibitor 5-aza-2′-deoxycytidine (5-aza-dC) or the histone deacetylase inhibitor (HDACi) Trichostatin A (TSA). Although epigenetic drugs are often reported as transient actors, they have the potency to improve reprogramming in a number of applications. For example, 5-aza-dC and TSA have previously shown to improve epigenetic reprogramming of somatic cell nuclei,14 and also the reprogramming of somatic cells to become induced pluripotent stem cells15 is increased in the presence of epigenetic drugs.16 Furthermore, TSA synergistically increases 5-aza-dC induced re-expression17 and promoter demethylation,18 and has shown to increase the sustainability of 5-aza-dC induced re-expression.19 Moreover, epigenetic drugs facilitated ATF-mediated activation of epigenetic repressed genes.20,12,21 Such an approach of combined treatment may thus prolong the transient effects of ATFs on gene expression.
An alternative strategy to induce long-lasting transcriptional changes may be through epigenetic editing,22 for example, by targeting DNA demethylases to hypermethylated promoter regions, which can remove DNA methylation at the target site. Targeting members of the ten-eleven translocation (TET) protein family to inactive promoters indeed resulted in gene re-expression.23,24 Also, thymidine DNA glycosylase (TDG), an enzyme involved in the DNA repair pathway, induced similar effects upon targeting.25 Although the reported DNA demethylases re-activated gene expression, the observed effects are sometimes limited and efficiency, as well as sustainability of re-expression, can likely be increased. Methylation levels are quickly recovered by the presence of methyltransferases and other local factors enriched at methylated CpGs sites, resulting in re-methylation and subsequent gene re-silencing.23 TSA, however, is also known to decrease the activity of DNA methyltransferase,26 and may thus inhibit the re-methylation processes. Therefore, the effects of targeting DNA demethylases may be further improved in the presence of TSA.
An important therapeutic target for epigenetic re-expression in cancer is EPB41L3. This gene is part of the 4.1 family of proteins, which actions are implicated in cell adhesion, cell motility and cell growth.27-29 In many cancer types, EPB41L3 is found to be frequently methylated, including breast,30 ovarian,31 and lung cancer.32 As a potential marker for the early detection of (pre)malignant cervical cancer, we previously found that 68% of cervical scrapings of CIN3 are methylated for EPB41L3 vs. 14% of normal cervices.33 This finding was confirmed by others who showed EPB41L3 as the best marker for detecting CIN2/3.34 Such promoter methylation patterns indicate that EPB41L3 is silenced in cervical cancer. A functional consequence of EPB41L3 downregulation in cancer is a disruption of the organization in the cytoskeleton, leading to increased metastasis and invasion of cancer cells. In addition, EPB41L3 overexpression has been linked to strong tumor growth suppression in various cancer types,28,31,35,36 in part through the induction of apoptosis. These properties make EPB41L3 an attractive therapeutic target for re-expression in cancer therapy.
In this study, we aimed to induce sustainable re-expression of EPB41L3 in cancer. For gene-specific re-expression of EPB41L3, we engineered two ATFs (ZFPs + VP64) and confirmed their ability to re-express EPB41L3 in breast, ovarian, and cervical cancer cell lines, displaying various degrees of EPB41L3 methylation. In addition, we analyzed the genome-wide binding of an EPB41L3-targeting ZFP using ChIP-Seq. Furthermore, we tested whether the sustainability of EPB41L3 re-expression by the ATFs could be enhanced by gene-targeting of DNA methylation modifiers (Tet2, TDG) (epigenetic editing) and/or by adding epigenetic drugs. Finally, we studied the functional effects of EPB41L3 re-expression in cervical cancer cell lines, since a tumor suppressive role of this gene has not been studied in this malignancy.
Results
EPB41L3 mRNA expression and epigenetic status
The promoter region for the two main splice variants of EPB41L3 is shown in Figure 1A. The CpG island at the 3′-side of the main transcription start site (TSS) suggests that EPB41L3 expression might be controlled by DNA methylation. We analyzed the mRNA expression of EPB41L3 in breast (MDA-MB-231, SKBR3), ovarian (SKOV3, A2780), and cervical (HeLa, CaSki, C33A, SiHa, and CC-11) cancer cell lines (Fig. 1B): EPB41L3 was silenced in most cell lines, except for SKBR3, C33A and SiHa cells (mRNA relative to GAPDH: 3.8x10–5 (SKBR3), 1.5×10–4 (C33A) and 7.6×10–4 (SiHa)). Examination of the DNA methylation status (region 2, Fig. 1A) showed that EPB41L3 silencing was associated with extensive promoter hypermethylation in SKOV3 (75±9%), HeLa (88 ± 1%) and CaSki (90 ± 1%), but less in MDA-MB-231 (28 ± 1%) and A2780 (58 ± 14%) (Fig. 1C). The cell lines expressing EPB41L3 showed lower degrees of promoter methylation (SKBR3 (6 ± 3%) and C33A (19 ± 5%)).
Figure 1.

Epigenetic regulation of EPB41L3. (A) Schematic representation of the EPB41L3 gene (two splice variants) and the EPB41L3 promoter, spanning bp −100 to +700. Shown are the TSS, CpGs (indicated as vertical bars), target sites ZFPs (21ab, 22ab) and bisulfite/ChIP regions. (B) EPB41L3 mRNA expression in a panel of breast (MDA-MB-231, SKBR3), ovarian (SKOV3, A2780), and cervical (HeLa, CaSki, C33A, SiHa and CC-11) cancer cell lines. Insert shows the protein expression of an expressing (SiHa) and non-expressing cell line (HeLa) visualized by western blotting using an EPB41L3 specific antibody [4.1B (sc-25965) Santa Cruz]. (C) DNA methylation status of the EPB41L3 promoter for eight cell lines from (B). Three clones per cell line were analyzed by bisulfite sequencing and each third of a circle represents a clone. Also shown are the predicted binding sites of EPB41L3 targeting ATFs (21ab-VP64 (▴), 22ab-VP64 (▾) and the TSS. (D) The DNA methylation status of 15 CpGs in the EPB41L3 promoter of hypomethylated (C33A), intermediately methylated (SKOV3) and high methylated (SiHa) cells as quantified by pyrosequencing. Values represent the mean of at least two independent experiments ± SEM. (The other cell lines from C are shown in supplemental Figure 2) (E) Quantitative ChIP for histone modifications associated with the gene promoter of EPB41L3 [n = 3 or more, (H3K9Ac n = 2)]. (F) mRNA expression of EPB41L3 after treatment with epigenetic drugs {5-aza-dC [500 nM (+)], 5 mM (++) (3 days) and/or TSA (400 nM) (one day)} relative to untreated cells (set as 1). Statistical significance was determined using a student's t-test (*P<0.05).
The cell line with the highest expression (SiHa) was highly methylated (96 ± 4%). Sequencing of the EPB41L3 mRNA of SiHA confirmed expression from the main TSS (isoform 3) (Fig. 1A), and no genetic mutations were observed. Expression of EPB41L3 mRNA in SiHa was confirmed on protein level (Fig. 1B insert). Sequencing of another region in closer proximity to the TSS (region 1) also showed high DNA methylation levels for SiHa (∼89%, Fig. S1). Other cell lines showed comparable methylation levels for EPB41L3 at this more upstream part of the promoter, except for SKOV3 (lower percentage of CpG methylation in this region). To quantify the reduction in methylation levels on single CpGs, we performed pyrosequencing for 15 CpGs in the promoter regions of EPB41L3 (Figs. 1A, 1D, Fig. S2). The methylation patterns showed high DNA methylation levels for the cervical cancer cell lines (except C33A), and intermediate or low levels of DNA methylation for the ovarian and breast cancer cell lines.
Analysis of the histone marks in the hypermethylated cervical cancer cell lines HeLa, CaSki, and SiHa revealed a close association of DNA methylation with the repressive histone mark H3K9me3 (Fig. 1E). Activating histone marks (H3Ac, H4Ac, H3K4me3 and H3K9Ac) were only weakly, and to a similar degree in all three cell lines, associated with the hypermethylated EPB41L3 promoter. Epigenetic drugs (5-aza-dC + TSA) increased re-expression efficacy for EPB41L3: ∼83-fold in HeLa, ∼32-fold in CaSki, and ∼58-fold in C33A. No further increase of EPB41L3 expression in SiHa was observed (Fig. 1F). Based on these findings, EPB41L3 seems a suitable target for gene-targeted interventions by ATFs.
Targeted re-expression of EPB41L3
Next, we investigated whether EPB41L3 induction from the endogenous locus can be achieved by engineering two ATFs (21ab-VP64 and 22ab-VP64) targeted at the EPB41L3 promoter (see Fig. 1A, 2A for the target sites). Efficient expression of EPB41L3-targeting ATFs was demonstrated by qRT-PCR and FACS (Fig. S3). EPB41L3 could be significantly upregulated (Figs. 2B, 2C, 2D) by 21ab-VP64 compared to the effects of an empty vector (pMX) in MDA-MB-231 (11 ± 2.7-fold), SKBR3 (3.6 ± 0.6-fold), A2780 (8.5 ± 2.5-fold), SKOV3 (13 ± 3.3-fold), HeLa (14 ± 4.3-fold), CaSki (13 ± 3.1-fold) (all P<0.01), and in C33A (26 ± 8.0-fold; P<0.05). Also, 22ab-VP64 significantly increased EPB41L3 expression in SKBR3 (11 ± 1.4-fold; P<0.01), CaSki (7.2 ± 2.4-fold; P<0.05), and C33A (6.5 ± 1.1-fold; P<0.01). In CC-11 cells, which are more primary cervical cancer cells,37 EPB41L3 was not significantly upregulated by either ATF. Nevertheless, expressing both ATFs simultaneously resulted in significant gene-induction of ∼32-fold (P<0.05) (Fig. 2E). The overexpression of EPB41L3 cDNA in CC-11 cells yielded levels of ∼8.1×103-fold (P<0.05) compared to the pMX empty vector (Fig. 2E). In Caski cells, EPB41L3 re-expression was also confirmed on the protein level (Fig. 2D insert). Controls, consisting of zinc fingers lacking an effector domain (NoEf) showed comparable EPB41L3 expression as the empty vector control. C33A cells were transduced to express two irrelevant ATFs, which did not influence gene expression of EPB41L3 (data not shown).
Figure 2.

Endogenous re-expression of EPB41L3 by ATFs. (A) Graphical representation of the EPB41L3-targeting ZFPs. Shown are the 18-bp target sequence and the orientation of the VP64 transcriptional activator. EPB41L3 mRNA induction in breast (B), ovarian (C), and cervical (D, E) cancer cell lines by ATF 21ab-VP64 and/or 22ab-VP64 targeted at the EPB41L3 promoter. Quantification of mRNA was performed using qRT-PCR and induction levels were normalized to pMX empty vector (set as 1). ZFP without effector domains (NoEf) were used as extra controls. Each bar represents the mean of at least three independent ± SEM. Statistical significance was determined using a student's t-test (*P<0.05 and **P<0.01). The insert in D show EPB41L3 re-expression in CaSki cells after expressing 21ab-VP64 or EPB41L3 cDNA as visualized by Western blotting. Spice variant one is estimated to be 120 kDa, while the overexpressed splice variant cloned from SiHa cells is estimated to be 100 kDa.
Targeted reversal of histone marks by EPB41L3-ATFs, but moderate binding specificity
Previously, it was shown that ATFs indirectly affect local epigenetic features, such as histone modifications.11,12 Therefore, we also analyzed if the EPB41L3-targeting ATFs changed the histone marks after ATF-mediated re-expression. First, we confirmed successful association of the engineered ZFP 21ab with its target site in the EPB41L3 promoter. Indeed, an enrichment of 10% of input DNA was obtained for 21ab expressing CaSki cells (0.06% for pMX) using ChIP (Fig. 3A). Genome-wide sequencing revealed an enrichment of DNA fragments at the targeted site (area of 20 kb analyzed (Fig. 3B)), while empty vector had little or no reads at this location. Abundant off-targets were identified, but most of them with low coverage per peak (Fig. 3C). The analysis for all peaks with a coverage of 10 or more reads per peak showed that ZFP 21ab did not only bind to the EPB41L3 promoter, but also to other promoter regions (empty vector: 0.43% of all reads mapped to promoter sites, 21ab-NoEf: 7.73%) (Fig. 3D). The number of identified promoter binding sites was 1,397 for the EPB41L3-ZFP, while the number of background binding sites for virus only treated cells (pMX empty) was 204 (Fig. 3E).
Figure 3.

Targeted reversal of histone marks by EPB41L3-ATFs, but moderate binding specificity. Association of 21ab-NoEf with its targeted site as analyzed by an HA tag ChIP (A) and ChIP-Seq (B) for the 21ab-ZFP and an empty vector in EPB41L3 methylated CaSki cells. For the ChIP-Seq, the local coverage is shown for a region of 20 kb as visualized by R using the coverage distribution report obtained from NextGENe. Indicated is the ZFP target site (▴). (C) Representation of the number of identified ChIP-Seq peaks vs. the average coverage per peak [shown for peaks with a coverage of four or more (maximum 100)] determined by the Peak Identification Report obtained from NextGENe. (D) Percentage of ChIP-Seq peaks (with a coverage of 10 or more) bound to promoters, genes or other regions (such as noncoding DNA). (E) Graphical representation of peaks with a coverage of 10 or more bound to promoter regions. (F) Change in histone marks after re-expression of EPB41L3 in methylated CaSki (left) and unmethylated C33A cells (right). A quantitative ChIP for H3Ac and H3K9me3 was performed after treatment with 21ab-VP64 and/or 21ab-NoEf. Values represent the mean percentage of input of three independent experiments ± SEM, (HA tag one experiment).
To determine whether EPB41L3 re-expression by ATFs influenced the histone marks at the ATF target site, H3Ac and H3K9me3 levels were assessed both in methylated (CaSki) or unmethylated (C33A) cells (Fig. 3F) after treatment with a control (21ab-NoEf) and 21ab-VP64. In control cells, CaSki cells showed more association of the repressive H3K9me3 mark with the EPB41L3 promoter than C33A (CaSki 21ab-NoEf 2.4 ± 0.2% of input, C33A 21ab-NoEf 0.5 ± 0.1% of input (P<0.01)), which is consistent with the more silenced/methylated state of EPB41L3 in CaSki. EPB41L3 re-expression by 21ab-VP64 significantly decreased the repressive mark H3K9me3 by 2.3 ± 0.5-fold (P<0.05) in CaSki cells and 3.4 ± 0.6-fold (P = 0.056) in C33A cells. Interestingly, increased EPB41L3 expression in unmethylated C33A cells was associated with a strong increase in the H3Ac mark (23 ± 2.5-fold; P<0.01), while enrichment of H3Ac in the methylated CaSki cells could not be detected upon EPB41L3 re-expression.
Sustained re-expression of EPB41L3
To study sustainability of gene re-expression, we constructed 21ab-VP64 DOX-inducible cell lines (HeLa, SKOV3) and analyzed expression of 21ab-VP64 and EPB41L3 over time, also after removal of DOX. DOX treatment for 2 d resulted in ATF expression (Fig. 4A), which, in turn, upregulated EPB41L3, with highest level reached one day after removal of DOX (HeLa 86 ± 19-fold (Fig. 4A), SKOV3 6.5 ± 1.5-fold (Fig. 4B)). Then, EPB41L3 levels decayed over time, with a delay of 24 h relative to DOX-induced 21ab-VP64 expression levels (as shown in HeLa cells).
Figure 4.
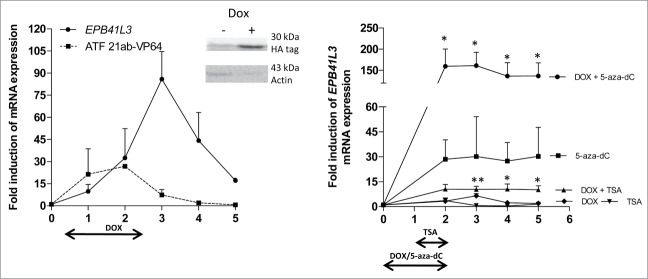
Kinetics of re-expression of EPB41L3 using ATFs and epigenetic drugs. Re-expression of EPB41L3 (and 21ab-VP64) mRNA in HeLa (A) and SKOV3 (B) cells stably transduced with 21ab-VP64 after treatment with DOX. Expression of the ATF was confirmed on protein level after DOX treatment in HeLa cells as visualized with Western blotting (insert A). For SKOV3, DOX-treatment was also administered in the presence of 5-aza-dC (5 μM) or TSA (400 nM) (see treatment schedule at the bottom). Expression of EPB41L3 mRNA was measured during a period of 5 d. Quantification of mRNA was performed using qRT-PCR and induction levels were normalized to untreated cells. Each data point represents the mean of at least three independent experiments measured in triplicate ± SEM. Statistical differences (B) were determined between single treatment (5-aza-dC, TSA) and co-treatment with DOX (5-aza-dC + DOX, TSA + DOX) using a student's t-test (*P<0.05 and **P<0.01).
To explore possibilities to circumvent instable reactivation of the targeted gene, DOX induction was combined with epigenetic drug treatment (5-aza-dC or TSA) in SKOV3 21ab-VP64 stable transfectants (Fig. 4B). In the clinical setting, such “hit-and-run” approaches in which short-term treatment is sufficient to induce sustained effects would be preferred above a long-term drug treatment. Here, we could show that the combination of ATFs and suboptimal doses of epigenetic drugs acts synergistically, as also demonstrated by others.20,21 Interestingly, the improved effects were maintained over time and the reactivation of the targeted gene could be prolonged. Co-treatment strongly increased EPB41L3 re-expression compared to single treatment for 5-aza-dC [at day 2: 5.6 ± 1.4-fold (P<0.05) and TSA (day 2: 2.9 ± 0.8-fold (P = 0.064)]. Moreover, co-treatment of ATFs with 5-aza-dC or TSA resulted in a sustained increase of EPB41L3 expression levels after removal of DOX [day 5: 5-aza-dC 4.5 ± 1.0-fold (P<0.05), TSA 7.0 ± 1.7-fold (P<0.05)]. Such sustained re-expression was also observed for the 5-aza-dC only treated cells (although less prominent), but was not observed for this dose of TSA only.
Targeting demethylating enzymes in the presence of HDACi TSA
Next, we fused the catalytic domain of Tet2 or TDG to the EPB41L3-targeting ZFPs, delivered the construct into CaSki cells and measured the expression of EPB41L3 in the presence or absence of TSA. Without TSA, we failed to significantly re-activate the EPB41L3 promoter after expressing 21ab-Tet2, 22ab-Tet2, 21ab-TDG, or 22ab-TDG (Fig. S4). However, in the presence of TSA, EPB41L3 could be significantly re-activated (Fig. 5A). Co-treatment of 22ab-Tet2 with TSA reached the highest levels of EPB41L3 mRNA [pMX+TSA 1.0 ± 0.1-fold vs. 22ab-Tet2-CD+TSA 2.7 ± 0.8-fold (P<0.05)]. These re-expression levels were approximately similar to the EPB41L3 expression levels reached by the VP64-ATFs (without co-treatment; Fig. 2D). To study if the TSA-induced effects were associated with downregulation of methyltransferases, DNMT3b levels were analyzed after expressing 21ab-Tet2, 22ab-Tet2, the combination of both or irrelevant ZFPs fused to Tet2-CD (GFP expression of cells transduced to express Tet2 fusion proteins was approximately similar as for cells treated with empty vector (Fig. 5B) or VP64-ATFs expressing viruses (Fig. S3D) as monitored by FACS). We found that TSA decreased DNMT3b expression (∼32 ± 7%; P = 0.05) in control cells (Fig. 5C), which was consistent with a previous report.26 Interestingly, we also found that cells transduced to express the Tet-fusion proteins demonstrated a twofold increase in DNMT3b mRNA expression (P<0.05), while no increase in DNMT3b expression was observed in control cells expressing an empty vector (compared to untreated cells). Such increase of DNMT3b mRNA was diminished when Tet-expressing cells were co-treated with TSA.
Figure 5.

Gene-induction of EPB41L3 with demethylating reagents in the presence of TSA. (A) EPB41L3 mRNA expression after treatment with pMX, 21ab-Tet2-CD, 22ab-Tet2-CD, 21ab-TDG-CD or 22ab-TDG-CD (or combined) in CaSki cells in the presence of TSA (400 nM). (B) Percentage GFP positive cells after retroviral transduction of pMX, 21ab-Tet2-CD, 22ab-Tet2-CD or 21ab/22ab-Tet2-CD. (C) DNMT3b mRNA expression after retroviral transduction with pMX, 21ab-Tet2-CD, 22ab-Tet2-CD, 21ab/22ab-Tet2-CD or expression of control ZFPs carrying the Tet2-CD. Quantification of mRNA was performed using qRT-PCR and induction levels were normalized to an empty vector. Each bar represents the mean of at least three independent ± SEM. Statistical significance was determined using a one-tailed t-test. A P-value of 0.05 or less was considered statistical significant.
EPB41L3 as tumor suppressor gene
Previously, EPB41L3 was identified as a TSG in, among others, breast and ovarian cancer, but in cervical cancer the role of EPB41L3 is currently unknown. To study if ATF-mediated re-activation of EPB41L3 affects cell growth in the EPB41L3 methylated cervical cancer cells, a 5-day MTT assay was performed for HeLa (Fig. 6A) and CaSki cells (Fig. S5). The highest re-expressor of EPB41L3, 21ab-VP64, significantly decreased cell growth compared to controls at day 5 (pMX 100 ± 15%, 21ab-NoEf +125 ± 16%, 21ab-VP64 -41 ± 10% (P<0.05), 22ab-VP64 −50 ± 16% (ns)). For CaSki, growth was even further decreased at day 5 (-80%) compared to pMX (Fig. S5).
Figure 6.

EPB41L3 decreases cell growth and induces apoptosis. (A) Relative cell proliferation was measured with a MTT assay in HeLa cells after transduction with the EPB41L3-targeting constructs. Each data point represents the mean of five independent experiments ± SEM. (B) Percentage of apoptotic cells in methylated MDA-MB-231, A2780, SKOV3, HeLa and CaSki after transduction with the 21ab-VP64 and controls (pMX and 21ab-NoEf) measured by a DilC staining. All bars represent the mean of three independent experiments ± SEM. (C) Visualization of the number of colonies after transduction of the EPB41L3-inducing ATFs and controls in HeLa cells. Picture shows a representative of a triplicate measurement. (D) Percentage of cell death after treatment with EPB41L3 cDNA, 21ab-VP64 + 22ab-VP64 and pMX. (E) Relative cell adhesion of CaSki cells after treatment with EPB41L3-inducing ATFs and a control (F) mRNA expression of the cell cycle regulating genes CDKN2A, p21 and p53 also after transduction of pMX and 21ab-VP64 in CaSki cells. Quantification, statistics, and representation is similar as in Fig. 2.
Next, we studied if the ATF-mediated decrease in growth upon re-expression of EPB41L3 in methylated/silenced cell lines could be partially explained by the induction of apoptosis (Fig. 6B), as demonstrated by others for breast and ovarian cancer cells using EPB41L3 cDNA.31,36 Indeed, apoptosis was induced by 21ab-VP64, the highest re-expressor of EPB41L3, in the methylated breast and ovarian cancer cells (MDA-MB-231 19 ± 5%, SKOV3 11 ± 3%, A2780 66 ± 18 %) compared to pMX (P<0.05). Interestingly, also for the methylated cervical cancer cells, significant apoptosis was induced by 21ab-VP64 (HeLa 13 ± 0.1% (P<0.01); CaSki 21 ± 2% (P<0.05)). ZFPs with no effector domain had similar background levels of apoptosis compared to pMX. The EPB41L3-targeting ATFs also strongly decreased the colony forming potential of the cells compared to NoEf as shown for HeLa cells (Fig. 6). For the more primary CC-11 cells, the re-activation of EPB41L3 by combining 21ab-VP64 and 22ab-VP64 resulted in significant cell death compared to pMX (Fig. 6D). The induced level of cell death was similar to a control condition were EPB41L3 cDNA (isoform 3) was overexpressed (pMX ∼13%, ATFs ∼31%, cDNA ∼29%). As EPB41L3 is known to affect cell adhesion, we analyzed whether targeted re-expression of EPB41L3 affected cell adhesion. Indeed, compared to pMX empty, a significant increase in adhesion was observed for 21ab-VP64 and 22ab-VP64, as shown in Fig. 6E.
To gain a better understanding of how EPB41L3-targeting ATFs induce the effects on cell growth in cervical cancer, we analyzed the mRNA expression of the cell cycle regulators cyclin-dependent kinase inhibitor 1 (CDKN1A, p21), cyclin-dependent kinase inhibitor 2A (CDKN2A, p16), and tumor protein 53 (TP53, p53) in CaSki cells (in which 21ab-VP64 induced the most pronounced apoptotic and growth effects) (Fig. 6F). All three proteins are deregulated in most cancers and play critical roles in cell cycle progression.38,39 However, in cervical cancer, p53 is degraded by the human papillomavirus (HPV) E6 protein, and considered functionally inactive.39 We observed that p21 expression levels were further upregulated by 21ab-VP64 compared to pMX (5.1 ± 1.0-fold; P<0.05), while CDKN2A levels were downregulated (0.28 ± 0.14-fold; P<0.05). P53 expression levels were not changed, as could be expected based on its functional inactive states in cervical cancer. These results suggest that EPB41L3 is related with cell cycle regulators (CDKN2A, p21), which could be associated with the less malignant phenotype of the cells.
Discussion
Here we describe an effective approach to increase sustainability of gene-targeted re-expression by combining effects of ATFs or epigenetic editors with epigenetic drugs. We showed that the DNA methylation inhibitor 5-aza-dC could increase the sustainability of transient ATF-induced re-expression. Furthermore, re-expression of the target gene EPB41L3 was obtained by targeting the demethylation inducers Tet2 or TDG when combined with epigenetic drug treatment (TSA). The combined actions of ATFs/epigenetic editors and epigenetic drugs may induce more complete promoter reprogramming than single treatment19 as shown before for ATFs by others.20 As both epigenetic drugs and DNA targeting approaches are clinically explored, we thus provide a promising way to prolong sustainability of gene re-expression.
The exact mechanism behind the re-enforcing effects between epigenetic drugs and ATFs/epigenetic editors is largely unknown. Several factors may contribute, such as increased binding of the ATF to its endogenous target site,12 acetylation of lysine residues within transcription factors (a mechanism by which cells can overcome gene repression)40, and enhanced expression of the transgenes.41 In addition, TSA-induced repression of DNA methyltransferases may slow down re-methylation/de-reprogramming events.26 DNA methylation has a rapid turnover in human cells,42 and inhibition of the process of re-methylation may therefore increase the reprogramming potential of ATFs. Interestingly, we observed that expression of the Tet2-fusion proteins was associated with increased expression of DNA methyltransferase DNMT3b. A possible explanation for this may be a feedback mechanism in which cells try to neutralize/antagonize decreased methylation levels induced by the demethylating reagents.
Previously, the relation between EPB41L3 promoter methylation and expression silencing has been demonstrated in many cancer types.29-32 In this study, we confirmed the association between promoter hypermethylation and EPB41L3 repression, as we found that, in general, the expressing cell lines showed lower degrees of promoter methylation. However, this relation was not observed for the highest expressing EPB41L3 cell line (SiHa), which turned out to be almost completely methylated in the analyzed part of the CpG island in the EPB41L3 promoter. This observation that TSS-region methylation allows expression is inconsistent with the bulk of literature, as such genes are typically silenced.43
In general, the re-expression of silenced genes by ATFs is considered an advantage compared to traditional cDNA overexpression, as all splice variants can be re-expressed in natural ratios, which may determine the functional outcome of the intervention.44 This may also explain why, despite lower EPB41L3 re-expression capacity, the combination of ATFs were equally efficient in inducing cell death compared to the cDNA overexpression of isoform 3 (EPB41L3 has 38 splice variants). The functional effects we observed, however, may also be partly caused by the aspecific binding of the EPB41L3-targeting ATFs, as also observed for other ZFPs.45 Such aspecific binding may result in modulation of off-target genes, although certain ZF-ATFs might regulate gene expression with single gene resolution.25,46 A possible explanation for such observed gene specificity may be that most of the off-target promoters have an open chromatin structure which is easily accessible by the ATFs; however, the relative impact of ATFs on actively transcribed genes is small. In contrast, the impact of ATFs on repressed regions could be significant; however, as these regions are more difficult to be accessed by ATFs, such off-target events may thus be less frequent. For example, the EPB41L3-ZFP did not show enrichment at the epigenetically repressed C13ORF18-promoter12 in this cell line (data not shown).
The engineered VP64-ATFs in this study were effective re-activators of EPB41L3, capable of inducing EPB41L3 expression in breast, ovarian, and cervical cancer cell lines, even in hypermethylated cell lines. Recently, TAL effectors and the CRISPR/Cas system have been presented as an alternative for gene targeting, promising higher success rate with regard to specificity.47,48 The advantages of ZFPs may be that they are relative small proteins (6F-ZFP < 200 amino acids, TAL effectors > 800 amino acids), which may have more efficient access to epigenetically silenced regions, and thus might be better suited for gene re-expression. Indeed, the potency of TALEs to induce expression is sometimes hampered, possibly due to their large size.49 However, when comparing the ability to decrease DNA methylation levels at a certain target site when fused to large Tet1 domains, no differences were found between ZFP- or TALE-based fusions.23 Here, we showed that the two engineered VP64-based ATFs targeting a chosen TSS have indeed a high success rate when it comes to gene induction, but no relevant gene inductions were obtained when the ZFPs were fused to Tet domains. Co-treatment with TSA did allow gene induction by these larger constructs. An improvement in the field of TAL effectors was the targeting of combinations of various TAL effectors to a single gene, which resulted in an increase in gene activation compared to a single TAL effector.49 Such synergistic effects were also observed for ZFPs by simultaneously targeting several TDG constructs,25 DNA demethylases (Tet1),23 or H3K9 methylases50 to a single promoter. In addition, here we showed that simultaneously targeting two ZFP-based ATFs to the same promoter facilitates the ATF-effect, when single treatment did not result in gene re-expression.
For cervical cancer, this is the first study demonstrating that EPB41L3 can induce tumor suppressive effects like apoptosis and reduction of cell growth. As was already shown for ovarian and breast cancer, we further validated the role of EPB41L3 as TSG27–29,31,35,36 by the ATFs in cancer. Based on its differential methylation profile in cervical cancer vs. normal tissues33 and the functional effects shown here, EPB41L3 might represent an interesting therapeutic target also for cervical cancer and its high-grade premalignant lesions. Several mechanisms have been described how EPB41L3 may mediate the inhibition of cell growth, for example by increasing activity of caspase-8.36 Additionally, we found that ATF-mediated re-expression of EPB41L3 is related with expression of the cell cycle regulators CDKN2A (down) and p21 (up).
In the future, delivery methods need to be further optimized to reveal the in vivo effectiveness of re-expression of silenced tumor suppressor genes, as currently explored by us and others.9 Attempts are ongoing to make ZFPs, such as our EPB41L3-targeting platform, suitable for clinical use and an ongoing clinical trial (performed by Sangamo Bioscience) already showed promising results. In this trial, ZFPs fused to nucleases (designed to disrupt CDR5 on CD4 cells as "functional cure" for HIV/AIDS) were successfully delivered to target cells and decreased viral load in HIV patients.51 The recent approval for the treatment of lipoprotein lipase deficiency by an adeno-associated viral vector engineered to express lipoprotein lipase52 might facilitate the development of new therapeutic strategies based on gene targeting platforms for future clinical applications.
In conclusion, this study demonstrated that epigenetic drugs can increase the sustainability of selective gene re-expression by ATFs. Furthermore, we showed that the effect of epigenetic editors (Tet2, TDG) on gene-expression could be improved with the HDACi TSA. The synergistic effects of ATFs/epigenetic editors and epigenetic drugs exploit the gene targeted effects of the activators while limiting genome-wide side effects of the epigenetic drug which may be beneficial for future therapeutic applications. For cervical cancer, we showed for the first time the tumor suppressive role of EPB41L3, a gene recently identified as the best marker for detecting early stages of this malignancy.33,34
Materials and Methods
Cell lines
Human breast cancer cell lines (MDA-MB-231, SKBR3), human ovarian cancer cell lines (SKOV3, A2780) and human cervical cancer cell lines (HeLa, SiHa, CaSki, and C33A) were obtained from ATCC (Manassas, VA) and cultured in DMEM (BioWhittaker, Walkersville, MD) supplemented with 10% FBS (BioWhittaker), 2mM L-glutamine and 50 μg/ml gentamycin. All cell lines were confirmed by STR profiling (BaseClear, Leiden, the Netherlands). CC-11 was derived from a cervical squamous cell carcinoma.37
ATF retroviral transduction/development of stable cell lines
Two target regions of ATFs, designated 21ab and 22ab, were selected based on proximity to the TSS and high affinity predictions (www.zincfingertools.org).53 Double stranded DNA oligos (BIO BASIC, Markham, Canada) coding for the two 6-finger ZFPs predicted to bind the target sequences (21ab: GCAACAGGGGGCGGGGGG, 22ab: GGGGAGGAAGCCGCAGCC) were subcloned into the pMX-IRES-GFP containing either the gene activator VP64, no effector domain (NoEf) or the catalytic domain (CD) of Tet224 or TDG.54 Tet2 and TDG DNA fragments were created by PCR (Phusion Hot Start II High-Fidelity DNA polymerase, Thermo Scientific) using construct-specific PCR primers flanked with MluI and PacI restriction sites on pcDNA3-Flag-TET2CD or TDG55 and ligated into the pMX-IRES-GFP by sticky-end ligation with T4 ligase (Thermo Scientific).
ATF-21ab-VP64 (21ab-VP64) was subcloned in the Retro-X Tet-On advanced inducible expression system (Clontech, Mountain View, CA) according to the manufacturer's instructions. For overexpression of EPB41L3, the cDNA fragment of the gene was generated by PCR (Phusion Hot Start II High-Fidelity DNA polymerase) on SiHa cDNA using primers (Table 1) flanked with BamHI and EcoRI restriction sites and ligated into the pMX-IRES-GFP. Transduction of host cells with pMX-IRES-GFP or pRetroX-Tight-Pur was performed as previously described.12 For the expression of two ATFs simultaneously, viral supernatants were mixed (1:1).
Table I.
Primer sequences
| mRNA Primer | Sequence |
|---|---|
| EPB41L3 Fw | AGGAGGAGCAGCAGCAGGCC |
| EPB41L3 Rv | GCTGTTTTGCAGCCCTGGCA |
| EPB41L3 probe | TGCGGAGGGAGGTCACTGACAAG |
| GAPDH Fw | CCACATCGCTCAGACACCAT |
| GAPDH Rv | GCGCCCAATACGACCAAAT |
| GAPDH probe | GTTGACTCCGACCTTCACCTTCCC |
| VP64 Fw | AAGCGACGCATTGGATGAC |
| VP64 Rv | GGAACGTCGTACGGGTAGTTAATT |
| VP64 probe | TCGGCTCCGATGCT |
| CDKN2A Fw | CACCGAATAGTTACGGTCGGA |
| CDKN2A Rv | GATGTAGAGCGGGCCTTTGA |
| p21 Fw | GGCAGACCAGCATGACAGAT |
| p21 Rv | GATGTAGAGCGGGCCTTTGA |
| p53 Fw | GGTTGGCTCTGACTGTACCA |
| p53 Rv | CAAAGCTGTTCCGTCCCAGT |
| DNMT3b Fw | CGTGAAGCACGAGGGGAATA |
| DNMT3b Rv | TTCCGCCAATCACCAAGTCA |
| cDNA EPB41L3 Fw | GAGGGATCCGCCACCATGACGACCGAATCT |
| cDNA EPB41L3 Rv | CTCGAATTCCTACCGATTCAATCCTCTCCATCTTCTG |
| Bs seq. region 1 Fw | ATTATTTAAGTGGGAATAAAGGGTTAA |
| Bs seq. region 1 Rv | CCCCCTATTACAAAAAACACC |
| Bs seq region 2Fw | GTAATAGGGGGYGGGGGGAATAG61 |
| Bs seq. region 2Rv | AACCCCCTCGCAATCCCCCACTC |
| ChIP Fw | CCCGGGCTCCCTGCTGATCC |
| ChIP Rv | CCTCGGGCTCTTCCTCCGCA |
| Pyrosequence Fw | GTTGGGAGGGTAGGT |
| Pyosequence Rev | Btn AACCCCCTCCCAATCCCCCACT |
| Pyrosequence seq. | TTTTTAGTAAGGTTTTTGG |
To obtain stable RetroX-Tet-On double transfectants, cells transduced with pRetroX-Tet-On/pRetroX-Tight-Pur (ratio 1:3) were placed under selection with G418 sulfate (InvitroGen, San Diego, CA) (600 μg/ml) and puromycin (Invitrogen) (1 μg/ml) for 2 weeks. To express 21ab-VP64 in double transfectants, cells were treated with doxycycline (DOX) (Clontech) (500 ng/ml) for 2 d DOX-treated cells were also co-treated with 5-aza-dC (Sigma, St Louis, MO) (5 μM) or TSA (Sigma) (400 nM).
Quantitative real-time PCR
RNA was extracted using the RNeasyPlus Mini Kit (Qiagen, Hilden, Germany) and converted into cDNA (Fermentas, Leon-Rot, Germany). cDNA (20 ng) was used for qRT-PCR for the quantification of EPB41L3, ZFP-VP64 constructs, CDKN2A, p21, p53, and GAPDH, as previously described.12 Samples without amplification curves were assigned a Ct value of 40. Sequences of primers and probes are listed in Table 1. RNA levels were determined by the following formula 2ΔCt(relative to GAPDH) or 2-ΔΔCt(relative to empty vector).
Western blotting
Protein detection of EPB41L3, Actin and the ATFs was performed using standard Western blot techniques with antibodies detecting EPB41L3 [Santa Cruz, sc-100641 (1:250)], Actin (Millipore clone C4 (1:5000)) or HA tag [Abcam ChIP grade, Ab9110 (1:5000)]. Bands were visualized with the Pierce ECL chemiluminescence detection kit (Thermo Scientific, Rockford, USA). Protein mass was calculated using http://www.sciencegateway.org/tools/proteinmw.htm.
Bisulfite/pyrosequencing
DNA of untreated cells was bisulfite converted (EZ DNA Methylation-GoldTM Kit, Zymo research, Irvine, CA) and amplified with primers (Table 1) specific for two regions in the EPB41L3 promoter (Fig. 1A). PCR products were cloned into the pCR 2.1-TOPO Vector (Invitrogen, Carlsbad, CA) and sequenced subsequently.
For pyrosequencing, bisulfite-treated DNA of the EPB41L3 promoter was amplified with the Pyromark PCR kit (Qiagen, Hilden, Germany) using biotin-labeled primers (Table 1), and subsequently sequenced using the Pyromark Q24 MD pyrosequencer (Qiagen). Pyromark Q24 Software (Qiagen) was used to determine the methylation levels of single CpGs.
Chromatin Immunoprecipitation (ChIP)
A ChIP for the detection of the ZFP (HA tag), acetylation of histone 3 (H3Ac) and trimethylation of Lys9 of histone H3 (H3K9me3) was performed 72 h after transduction. ChIP was performed as previously described12 with the following antibodies: normal rabbit IgG (ab46540) (Abcam, Cambridge, UK), HA tag (101P-200) (Covance, Uden, the Netherlands), H3Ac (06–599) and H3K9me3 (07–442) (Millipore, Billerica, MA). DNA specific for the EPB41L3 promoter (Fig. 1A) was amplified with primers listed in Table 1.
ChIP-Seq was performed as previously described56 using the HA-tag antibody on a HiSeq2000 (Illumina). Reads were analyzed using NextGENe (SoftGenetics, LLC, USA). Alignment was done excluding reads mapping to multiple locations. Peak regions were identified by setting the coverage to 4 reads, gap to 110 bp, and setting the noise to zero. The coverage per peak was calculated as the 75th percentile of the number of aligned basepairs across the region. The plot of the EPB41L3 region was made using the “coverage distribution rapport” obtained from NextGENe and plotted with R software and is shown as the amount and location of reads at the EPB41L3 locus.
Apoptosis, cell death, cell growth, and colony forming assays
To quantify the fraction of apoptotic cells, cells were incubated in 1,1′,3,3,3′,3′;-Hexamethylindodicarbocyanine iodide (DilC) (Enzo Life Sciences, Farmingdale, NY) containing medium (50 nM, 20 min), and analyzed by flow cytometry using a FACS Calibur cytometer and CellQuest software (BD Biosciences, San Jose, CA). The fraction of living cells with decreased DiLC signal was considered apoptotic, as exemplified in Supplementary Fig. 6. A MTT assay (Sigma) was performed to examine metabolic activity representing cell growth of transduced cells as previously described.12 PI staining to measure late apoptosis was performed using a standard PI staining protocol, as previously described.57 For colony forming assay, transduced cells were seeded at a density of 750 cells per well, incubated at 37°C for 3 weeks and subsequently stained with Coomassie brilliant blue.
Adhesion assay
To measure cell adhesion, transduced cells were seeded on laminin coated dishes and incubated for 1 h at 37°C. Loose cells were detached by shaking the plate at 2000 rpm for 15 s. After washing, cells were fixed with paraformaldehyde and stained with crystal violet (0.05%) in distilled water for 30 min. After washing, the dye was dissolved in methanol and OD was measured at 540 nM.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We acknowledge Marcel Ruiters for helpful discussion.
Funding
This work was financially supported by the Netherlands Organization for Scientific Research (VIDI grant number 91786373 to MGR).
References
- 1. Black JC, Whetstine JR. Chromatin landscape: methylation beyond transcription. Epigenetics 2011; 6:9-15; PMID:20855937; http://dx.doi.org/ 10.4161/epi.6.1.13331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lendvai A, Johannes F, Grimm C, Eijsink JJ, Wardenaar R, Volders HH, Klip HG, Hollema H, Jansen RC, Schuuring E, et al. Genome-wide methylation profiling identifies hypermethylated biomarkers in high-grade cervical intraepithelial neoplasia. Epigenetics 2012; 7:1268-78; PMID:23018867; http://dx.doi.org/ 10.4161/epi.22301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Esteller M. Epigenetics provides a new generation of oncogenes and tumour-suppressor genes. Br J Cancer 2007; 96:R26-30; PMID:17393582 [PubMed] [Google Scholar]
- 4. Kim TY, Bang YJ, Robertson KD. Histone deacetylase inhibitors for cancer therapy. Epigenetics 2006; 1:14-23; PMID:17998811; http://dx.doi.org/ 10.4161/epi.1.1.2644 [DOI] [PubMed] [Google Scholar]
- 5. Al-Salihi M, Yu M, Burnett DM, Alexander A, Samlowski WE, Fitzpatrick FA. The depletion of DNA methyltransferase-1 and the epigenetic effects of 5-aza-2'deoxycytidine (decitabine) are differentially regulated by cell cycle progression. Epigenetics 2011; 6:1021-8; PMID:21725200; http://dx.doi.org/ 10.4161/epi.6.8.16064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gersbach CA, Perez-Pinera P. Activating human genes with zinc finger proteins, transcription activator-like effectors and CRISPR/Cas9 for gene therapy and regenerative medicine. Expert Opin Ther Targets 2014; 18:835-839; PMID:24917359; http://dx.doi.org/ 10.1517/14728222.2014.913572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beltran AS, Russo A, Lara H, Fan C, Lizardi PM, Blancafort P. Suppression of breast tumor growth and metastasis by an engineered transcription factor. PLoS One 2011; 6:e24595; PMID:21931769; http://dx.doi.org/ 10.1371/journal.pone.0024595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tan S, Guschin D, Davalos A, Lee YL, Snowden AW, Jouvenot Y, Zhang HS, Howes K, McNamara AR, Lai A, et al. Zinc-finger protein-targeted gene regulation: genomewide single-gene specificity. Proc Natl Acad Sci U S A 2003; 100:11997-2002; PMID:14514889; http://dx.doi.org/ 10.1073/pnas.2035056100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lara H, Wang Y, Beltran AS, Juarez-Moreno K, Yuan X, Kato S, Leisewitz AV, Cuello Fredes M, Licea AF, Connolly DC, et al. Targeting serous epithelial ovarian cancer with designer zinc finger transcription factors. J Biol Chem 2012; 287:29873-86; PMID:22782891; http://dx.doi.org/ 10.1074/jbc.M112.360768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beltran AS, Blancafort P. Reactivation of MASPIN in non-small cell lung carcinoma (NSCLC) cells by artificial transcription factors (ATFs). Epigenetics 2011; 6:224-35; PMID:20948306; http://dx.doi.org/ 10.4161/epi.6.2.13700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang B, Xiang S, Zhong Q, Yin Y, Gu L, Deng D. The p16-specific reactivation and inhibition of cell migration through demethylation of CpG islands by engineered transcription factors. Hum Gene Ther 2012; 23:1071-81; PMID:22738793; http://dx.doi.org/ 10.1089/hum.2012.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huisman C, Wisman GB, Kazemier HG, van Vugt MA, van der Zee AG, Schuuring E, Rots MG. Functional validation of putative tumor suppressor gene C13ORF18 in cervical cancer by artificial transcription factors. Mol Oncol 2013; 7:669-79; PMID:23522960; http://dx.doi.org/ 10.1016/j.molonc.2013.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rivenbark AG, Stolzenburg S, Beltran AS, Yuan X, Rots MG, Strahl BD, Blancafort P. Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics 2012; 7:350-60; PMID:22419067; http://dx.doi.org/ 10.4161/epi.19507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y, Su J, Wang L, Xu W, Quan F, Liu J, Zhang Y. The effects of 5-aza-2'- deoxycytidine and trichostatin A on gene expression and DNA methylation status in cloned bovine blastocysts. Cell Reprogram 2011; 13:297-306; PMID:21486115; http://dx.doi.org/ 10.1089/cell.2010.0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Czepiel M, Balasubramaniyan V, Schaafsma W, Stancic M, Mikkers H, Huisman C, Boddeke E, Copray S. Differentiation of induced pluripotent stem cells into functional oligodendrocytes. Glia 2011; 59:882-92; PMID:21438010; http://dx.doi.org/ 10.1002/glia.21159 [DOI] [PubMed] [Google Scholar]
- 16. Han J, Sachdev PS, Sidhu KS. A combined epigenetic and non-genetic approach for reprogramming human somatic cells. PLoS One 2010; 5:e12297; PMID:20808872; http://dx.doi.org/ 10.1371/journal.pone.0012297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet 1999; 21:103-7; PMID:9916800; http://dx.doi.org/ 10.1038/5047 [DOI] [PubMed] [Google Scholar]
- 18. Sato N, Fukushima N, Matsubayashi H, Goggins M. Identification of maspin and S100P as novel hypomethylation targets in pancreatic cancer using global gene expression profiling. Oncogene 2004; 23:1531-8; PMID:14716296; http://dx.doi.org/ 10.1038/sj.onc.1207269 [DOI] [PubMed] [Google Scholar]
- 19. Mossman D, Kim KT, Scott RJ. Demethylation by 5-aza-2'-deoxycytidine in colorectal cancer cells targets genomic DNA whilst promoter CpG island methylation persists. BMC Cancer 2010; 10:366-2407-10-366; PMID:20618997; http://dx.doi.org/ 10.1186/1471-2407-10-366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bultmann S, Morbitzer R, Schmidt CS, Thanisch K, Spada F, Elsaesser J, Lahaye T, Leonhardt H. Targeted transcriptional activation of silent oct4 pluripotency gene by combining designer TALEs and inhibition of epigenetic modifiers. Nucleic Acids Res 2012; 40:5368-77; PMID:22387464; http://dx.doi.org/ 10.1093/nar/gks199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beltran AS, Sun X, Lizardi PM, Blancafort P. Reprogramming epigenetic silencing: artificial transcription factors synergize with chromatin remodeling drugs to reactivate the tumor suppressor mammary serine protease inhibitor. Mol Cancer Ther 2008; 7:1080-90; PMID:18483297; http://dx.doi.org/ 10.1158/1535-7163.MCT-07-0526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Groote ML, Verschure PJ, Rots MG. Epigenetic editing: targeted rewriting of epigenetic marks to modulate expression of selected target genes. Nucleic Acids Res 2012; 40:10596-613; PMID:23002135; http://dx.doi.org/ 10.1093/nar/gks863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, Ho QH, Sander JD, Reyon D, Bernstein BE, et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol 2013; 31:1137-42; PMID:24108092; http://dx.doi.org/ 10.1038/nbt.2726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen H, Kazemier HG, de Groote ML, Ruiters MH, Xu GL, Rots MG. Induced DNA demethylation by targeting ten-eleven translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res 2014; 42:1563-74; PMID:24194590; http://dx.doi.org/ 10.1093/nar/gkt1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gregory DJ, Zhang Y, Kobzik L, Fedulov AV. Specific transcriptional enhancement of inducible nitric oxide synthase by targeted promoter demethylation. Epigenetics 2013; 8:1205-12; PMID:24008769; http://dx.doi.org/ 10.4161/epi.26267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiong Y, Dowdy SC, Podratz KC, Jin F, Attewell JR, Eberhardt NL, Jiang SW. Histone deacetylase inhibitors decrease DNA methyltransferase-3B messenger RNA stability and down-regulate de novo DNA methyltransferase activity in human endometrial cells. Cancer Res 2005; 65:2684-9; PMID:15805266; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-2843 [DOI] [PubMed] [Google Scholar]
- 27. Tran YK, Bogler O, Gorse KM, Wieland I, Green MR, Newsham IF. A novel member of the NF2/ERM/4.1 superfamily with growth suppressing properties in lung cancer. Cancer Res 1999; 59:35-43; PMID:989218012115567 [PubMed] [Google Scholar]
- 28. Charboneau AL, Singh V, Yu T, Newsham IF. Suppression of growth and increased cellular attachment after expression of DAL-1 in MCF-7 breast cancer cells. Int J Cancer 2002; 100:181-8; PMID:12115567; http://dx.doi.org/ 10.1002/ijc.10470 [DOI] [PubMed] [Google Scholar]
- 29. Bernkopf DB, Williams ED. Potential role of EPB41L3 (protein 4.1B/Dal-1) as a target for treatment of advanced prostate cancer. Expert Opin Ther Targets 2008; 12:845-853; PMID:18554153; http://dx.doi.org/ 10.1517/14728222.12.7.845 [DOI] [PubMed] [Google Scholar]
- 30. Heller G, Geradts J, Ziegler B, Newsham I, Filipits M, Markis-Ritzinger EM, Kandioler D, Berger W, Stiglbauer W, Depisch D, et al. Downregulation of TSLC1 and DAL-1 expression occurs frequently in breast cancer. Breast Cancer Res Treat 2007; 103:283-91; PMID:17260099; http://dx.doi.org/ 10.1007/s10549-006-9377-7 [DOI] [PubMed] [Google Scholar]
- 31. Dafou D, Grun B, Sinclair J, Lawrenson K, Benjamin EC, Hogdall E, Kruger-Kjaer S, Christensen L, Sowter HM, Al-Attar A, et al. Microcell-mediated chromosome transfer identifies EPB41L3 as a functional suppressor of epithelial ovarian cancers. Neoplasia 2010; 12:579-89; PMID:2065198722782504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y, Xu R, Li G, Xie X, Long J, Wang H. Loss of expression of the differentially expressed in adenocarcinoma of the lung (DAL-1) protein is associated with metastasis of non-small cell lung carcinoma cells. Tumour Biol 2012; 33:1915-25; PMID:22782504; http://dx.doi.org/ 10.1007/s13277-012-0452-x [DOI] [PubMed] [Google Scholar]
- 33. Eijsink JJ, Lendvai A, Deregowski V, Klip HG, Verpooten G, Dehaspe L, de Bock GH, Hollema H, van Criekinge W, Schuuring E, et al. A four-gene methylation marker panel as triage test in high-risk human papillomavirus positive patients. Int J Cancer 2012; 130:1861-9; PMID:21796628; http://dx.doi.org/ 10.1002/ijc.26326 [DOI] [PubMed] [Google Scholar]
- 34. Vasiljevic N, Scibior-Bentkowska D, Brentnall AR, Cuzick J, Lorincz AT. Credentialing of DNA methylation assays for human genes as diagnostic biomarkers of cervical intraepithelial neoplasia in high-risk HPV positive women. Gynecol Oncol 2014; 132:709-14; PMID:24508839; http://dx.doi.org/ 10.1016/j.ygyno.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wong SY, Haack H, Kissil JL, Barry M, Bronson RT, Shen SS, Whittaker CA, Crowley D, Hynes RO. Protein 4.1B suppresses prostate cancer progression and metastasis. Proc Natl Acad Sci U S A 2007; 104:12784-9; PMID:17640904; http://dx.doi.org/ 10.1073/pnas.0705499104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jiang W, Newsham IF. The tumor suppressor DAL-1/4.1B and protein methylation cooperate in inducing apoptosis in MCF-7 breast cancer cells. Mol Cancer 2006; 5:4; PMID:16420693; http://dx.doi.org/ 10.1186/1476-4598-5-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Koopman LA, Szuhai K, van Eendenburg JD, Bezrookove V, Kenter GG, Schuuring E, Tanke H, Fleuren GJ. Recurrent integration of human papillomaviruses 16, 45, and 67 near translocation breakpoints in new cervical cancer cell lines. Cancer Res 1999; 59:5615-24; PMID:1055404319440234 [PubMed] [Google Scholar]
- 38. Abbas T, Dutta A. P21 in cancer: Intricate networks and multiple activities. Nat Rev Cancer 2009; 9:400-14; PMID:19440234; http://dx.doi.org/ 10.1038/nrc2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sano T, Oyama T, Kashiwabara K, Fukuda T, Nakajima T. Expression status of p16 protein is associated with human papillomavirus oncogenic potential in cervical and genital lesions. Am J Pathol 1998; 153:1741-8; PMID:9846965; http://dx.doi.org/ 10.1016/S0002-9440(10)65689-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bannister AJ, Miska EA. Regulation of gene expression by transcription factor acetylation. Cell Mol Life Sci 2000; 57:1184-92; PMID:11028911; http://dx.doi.org/ 10.1007/PL00000758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Condreay JP, Witherspoon SM, Clay WC, Kost TA. Transient and stable gene expression in mammalian cells transduced with a recombinant baculovirus vector. Proc Natl Acad Sci U S A 1999; 96:127-32; PMID:9874783; http://dx.doi.org/ 10.1073/pnas.96.1.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yamagata Y, Szabo P, Szuts D, Bacquet C, Aranyi T, Paldi A. Rapid turnover of DNA methylation in human cells. Epigenetics 2012; 7:141-5; PMID:22395463; http://dx.doi.org/ 10.4161/epi.7.2.18906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Razin A, Cedar H. DNA methylation and gene expression. Microbiol Rev 1991; 55:451-8; PMID:1943996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rebar EJ, Huang Y, Hickey R, Nath AK, Meoli D, Nath S, Chen B, Xu L, Liang Y, Jamieson AC, et al. Induction of angiogenesis in a mouse model using engineered transcription factors. Nat Med 2002; 8:1427-32; PMID:12415262; http://dx.doi.org/ 10.1038/nm1202-795 [DOI] [PubMed] [Google Scholar]
- 45. Grimmer MR, Stolzenburg S, Ford E, Lister R, Blancafort P, Farnham PJ. Analysis of an artificial zinc finger epigenetic modulator: widespread binding but limited regulation. Nucleic Acids Res 2014; 42:10856-68; PMID:25122745; http://dx.doi.org/ 10.1093/nar/gku708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang HS, Liu D, Huang Y, Schmidt S, Hickey R, Guschin D, Su H, Jovin IS, Kunis M, Hinkley S, et al. A designed zinc-finger transcriptional repressor of phospholamban improves function of the failing heart. Mol Ther 2012; 20:1508-15; PMID:22828502; http://dx.doi.org/ 10.1038/mt.2012.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science 2011; 333:1843-46; PMID:21960622; http://dx.doi.org/ 10.1126/science.1204094 [DOI] [PubMed] [Google Scholar]
- 48. Munoz Bodnar A, Bernal A, Szurek B, Lopez CE. Tell me a tale of TALEs. Mol Biotechnol 2013; 53:228-35; PMID:23114874; http://dx.doi.org/ 10.1007/s12033-012-9619-3 [DOI] [PubMed] [Google Scholar]
- 49. Perez-Pinera P, Ousterout DG, Brunger JM, Farin AM, Glass KA, Guilak F, Crawford GE, Hartemink AJ, Gersbach CA. Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat Methods 2013; 10:239-42; PMID:23377379; http://dx.doi.org/ 10.1038/nmeth.2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Snowden AW, Gregory PD, Case CC, Pabo CO. Gene-specific targeting of H3K9 methylation is sufficient for initiating repression in vivo. Curr Biol 2002; 12:2159-66; PMID:12498693; http://dx.doi.org/ 10.1016/S0960-9822(02)01391-X [DOI] [PubMed] [Google Scholar]
- 51. Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 2014; 370:901-10; PMID:24597865; http://dx.doi.org/ 10.1056/NEJMoa1300662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yla-Herttuala S. Endgame: Glybera finally recommended for approval as the first gene therapy drug in the european union. Mol Ther 2012; 20:1831-2; PMID:23023051; http://dx.doi.org/ 10.1038/mt.2012.194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mandell JG, Barbas CF, 3rd. Zinc finger tools: custom DNA-binding domains for transcription factors and nucleases. Nucleic Acids Res 2006; 34:W516-23; PMID:16845061; http://dx.doi.org/ 10.1093/nar/gkl209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011; 333:1303-7; PMID:21817016; http://dx.doi.org/ 10.1126/science.1210944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Muller U, Bauer C, Siegl M, Rottach A, Leonhardt H. TET-mediated oxidation of methylcytosine causes TDG or NEIL glycosylase dependent gene reactivation. Nucleic Acids Res 2014; 42:8592-604; PMID:24948610; http://dx.doi.org/ 10.1093/nar/gku552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Falahi F, Huisman C, Kazemier HG, van der Vlies P, Kok K, Hospers GA, Rots MG. Towards sustained silencing of HER2/neu in cancer by epigenetic editing. Mol Cancer Res 2013; 11:1029-39; PMID:23814024; http://dx.doi.org/ 10.1158/1541-7786.MCR-12-0567 [DOI] [PubMed] [Google Scholar]
- 57. Li Q, van der Wijst MG, Kazemier HG, Rots MG, Roelfes G. Efficient nuclear DNA cleavage in human cancer cells by synthetic bleomycin mimics. ACS Chem Biol 2014; 9(4):1044–51; PMID:24527883; http://dx.doi.org/ 10.1021/cb500057n [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.