

Abstract
Resistance to radiation therapy constitutes a significant challenge in the treatment of head and neck squamous cell cancer (HNSCC). Alteration in DNA methylation is thought to play a role in this resistance. Here, we analyzed DNA methylation changes in a matched model of radiation resistance for HNSCC using the Illumina HumanMethylation450 BeadChip. Our results show that compared to radiation-sensitive cells (SCC-61), radiation-resistant cells (rSCC-61) had a significant increase in DNA methylation. After combining these results with microarray gene expression data, we identified 84 differentially methylated and expressed genes between these 2 cell lines. Ingenuity Pathway Analysis revealed ILK signaling, glucocorticoid receptor signaling, fatty acid α-oxidation, and cell cycle regulation as top canonical pathways associated with radiation resistance. Validation studies focused on CCND2, a protein involved in cell cycle regulation, which was identified as hypermethylated in the promoter region and downregulated in rSCC-61 relative to SCC-61 cells. Treatment of rSCC-61 and SCC-61 with the DNA hypomethylating agent 5-aza-2'deoxycitidine increased CCND2 levels only in rSCC-61 cells, while treatment with the control reagent cytosine arabinoside did not influence the expression of this gene. Further analysis of HNSCC data from The Cancer Genome Atlas found increased methylation in radiation-resistant tumors, consistent with the cell culture data. Our findings point to global DNA methylation status as a biomarker of radiation resistance in HNSCC, and suggest a need for targeted manipulation of DNA methylation to increase radiation response in HNSCC.
Keywords: DNA methylation, Gene expression, Head and neck squamous cell cancer (HNSCC), Radiation resistance, The Cancer Genome Atlas (TCGA)
Abbreviations
- 5-Aza
5-aza-2′deoxycitidine
- AKT
Protein kinase B
- AraC
Cytosine arabinoside
- CCNA1
Cyclin A1
- CCND2
Cyclin D2
- CDK4
Cyclin-dependent kinase 4
- CDKN1A
Cyclin-dependent kinase inhibitor 1A (p21, Cip1)
- dmCpG
differentially methylated CpG
- DNMT
DNA methyltransferase
- EIF2AK2
Eukaryotic translation initiation factor 2-αkinase 2
- FASN
Fatty acid synthase
- GSK-3
Glycogen synthase kinase 3
- HM450
HumanMethylation450
- HNSCC
Head and neck squamous cell cancer
- hTERT
human telomerase reverse transcriptase
- IGFBP3
Insulin-like growth factor-binding protein 3
- ILK
Integrin linked kinase
- IPA
Ingenuity pathway analysis
- IRF1
Interferon regulatory factor 1
- KLF4
Kruppel-like factor 4
- KRT19
Keratin 19, LIPG, Endothelial lipase
- LXR
Liver X receptor
- MGMT
O6-methylguanine DNA methyltransferase
- NFATC2
Nuclear factor of activated t-cells cytoplasmic 2
- PCNA
Proliferating cell nuclear antigen
- PTEN
Phosphatase and tensin homolog
- RXR
Retinoid X receptor
- SAM
S-Adenosylmethionine
- SOCS3
Suppressor of cytokine signaling 3
- STAT1
Signal transducers and activators of transcription 1
- TCGA
The Cancer Genome Atlas
- VHL
Von Hippel–Lindau tumor suppressor
Introduction
Head and neck cancer is a collective term for malignancies originating from the oral and nasal cavities, pharynx, and larynx. In the United States, head and neck cancer accounts for 3% of all cancers, with an estimated 42,400 new cases and 8,390 deaths in 2014.1 As with many other cancers, radiation is an important therapeutic approach to treat head and neck cancer, and resistance to this mode of treatment constitutes a significant challenge.2,3 To enable the systematic investigation of the underlying molecular mechanism of radiation resistance, we developed a radiation-resistant head and neck cancer cell line (rSCC-61) from the radiation-sensitive SCC-61 cell line by fractionated radiation and characterized broad phenotypic changes associated with the gain of radiation resistance using proteomics, redox imaging, and complementary methods of analysis.4 To further dissect the mechanisms driving these phenotypic changes, we examined patterns of DNA methylation in the SCC-61/rSCC-61 system and connected changes in DNA methylation with specific gene expression using gene expression microarray data. Recent studies suggest that alterations in DNA methylation pattern have a significant impact on cancer biology and response to therapies.5-8 We found higher levels of DNA methylation in radiation-resistant rSCC-61 cells and identified ILK signaling, glucocorticoid receptor signaling, fatty acid α-oxidation, and cell cycle regulation as top epigenetically regulated canonical pathways associated with radiation resistance. This analysis was extended to tumor data from patients with head and neck squamous cell cancer (HNSCC) in The Cancer Genome Atlas (TCGA). We identified a similar increase in DNA methylation of radiation-resistant tumors, consistent with the cell culture data. Approximately 36% of interrogated CpG methylation sites followed the same methylation trend in the SCC-61/rSCC-61 cell system and TCGA data, further emphasizing the role of genome methylation as regulator of radiation response in HNSCC.
Results
Radiation resistance is accompanied by a significant increase in DNA methylation
To assess DNA methylation changes associated with radiation resistance, we performed epigenome-wide association studies with the HumanMethylation450 (HM450) BeadChip array using radiation-resistant (rSCC-61) and radiation-sensitive (SCC-61) cell lines.4 This high-throughput methylation profiling technology covers 485,577 CpG sites and 99% of RefSeq genes.9 Methylation β values of all CpG sites comparing rSCC-61 and SCC-61 cells are shown in Figure 1A. Most data fell on the diagonal, representing equal methylation levels in the two cell lines. The data above the diagonal represent CpG sites that have higher methylation in rSCC-61 than SCC-61 and the data below the diagonal represent CpG sites that have lower methylation in rSCC-61 than SCC-61. The results indicate that differentially methylated CpG (dmCpG) sites in the rSCC-61 cell line are primarily hypermethylated. This is also illustrated by the pie chart in Figure 1B; roughly 10 times more CpG sites are hypermethylated in rSCC-61 in comparison to SCC-61 cells (140,423 vs. 13,291). The β value distribution for each cell line is shown in Figure 1C and D. In rSCC-61 cells, more than half of the CpG sites have β values greater than 0.8 (Fig. 1C). In contrast, only 31.6% of CpG sites are hypermethylated in SCC-61 cells (Fig. 1D). DNA methylation is catalyzed by DNA methyltransferases (DNMTs). The differences in DNA methylation could be due to differences in DNMT levels or their activities in the two cell lines, as well as other factors. We extracted gene expression data from the HumanHT-12 v4 Expression BeadChip analysis of SCC-61/rSCC-61 system. While the expression of DNMT1 and DNMT3A genes were not changed between the two cell lines, there was a 1.3-fold increase in DNMT3B mRNA level (P = 0 .0015) in rSCC-61.
Figure 1.

Radiation resistance is accompanied by a significant increase in DNA methylation. (A) Scatter plot of β methylation comparing rSCC-61 and SCC-61. Differentially methylated CpG sites in rSCC-61 are primarily hypermethylated, as shown by the data above the diagonal. (B) Pie chart of the methylation change of all CpG sites in rSCC-61 compared with SCC-61. Approximately 10 times more CpG sites are hypermethylated in rSCC-61 in comparison to SCC-61 cells (140,423 vs. 13,291). (C, D) Beta value distribution of rSCC-61 (C) and SCC-61 (D). While over half of the CpG sites in rSCC-61 have β values greater than 0.8, only 31.6% of CpG sites in SCC-61 have β values greater than 0.8. To see this figure in color, please refer to the online version of this article.
Differentially methylated CpG (dmCpG) sites are disproportionally distributed between canonical CpG islands and open sea
To better understand the functional significance of differential DNA methylation between the rSCC-61 and SCC-61 cell lines, we examined the location of the dmCpG sites. The functional composition of the 485,577 CpG sites included on the HM450 BeadChip is shown in Figure 2A (left): promoter (29%), 5′UTR/1st exon (12%), body (31%), 3'UTR (3%), and intergenic (25%). The functional genomic distribution of the dmCpG sites in rSCC-61 cells is shown in Figure 2A (middle and right). In general, the distribution of hyper- and hypomethylated CpG sites reflects their representation on the BeadChip, with most dmCpG sites found in the promoter, gene body, and intergenic regions (Fig. 2A, middle and right).
Figure 2.

Functional genomic distribution (A) and neighborhood location (B) of hypermethylated and hypomethylated CpG sites in rSCC-61 relative to SCC-61. Promoter region is defined as TSS200 and TSS1500 representing sites that are located 200 and 1500 bp, respectively, from a transcription start site. Intergenic regions are defined as the remainder of locations located between genes. Shores and shelves are composed of CpG methylation sites located 0–2 kb and 2–4 kb, respectively, from the nearest CpG island; open sea is defined as CpG methylation sites located >4 kb from a CpG island. (C) Functional genomic distribution of CpG sites located in islands and open sea. To see this figure in color, please refer to the online version of this article.
The neighborhood locations of all CpG sites on the HM450 BeadChip are shown in Figure 2B: 31% of the CpG sites are located in canonical CpG islands, 23% in shores (0–2 kb from the canonical islands), and 10% in shelves (2–4 kb from the canonical islands). The rest of the sequence (36%) is defined as open sea. The annotation of CpG islands was performed following the UCSC Genome Browser guidelines as detailed in the Materials and Methods section. The patterns of the hyper- and hypomethylated CpG sites in rSCC-61 cells deviate from their representation on the BeadChip: only 16% of the hypermethylated CpG sites are located in the canonical CpG islands, while 48% are located in the open sea (Fig. 2B middle). In contrast, 46% of the hypomethylated CpG sites are located in the canonical CpG islands, while only 24% are located in the open sea (Fig. 2B right).
This finding prompted us to perform additional comparisons to determine the functional genomic distribution of dmCpGs located in islands and open sea (Fig. 2C). A comparison among the island and open sea hyper- and hypo-methylated CpGs shows differences in their functional genomic distribution. Hypermethylated CpGs in islands are distributed approximately equally between promoter (25%, 5,872 sites), gene body (29%, 6,743 sites), and intergenic regions (29%, 6,655 sites), and reflect the representation of CpGs on the chip. In contrast, hypomethylated CpGs in islands are more often located in the promoter regions (39%, 2,341 sites). A larger proportion of dmCpGs in the open sea (39% and 44% for hyper- and hypo-methylated sites, respectively) are located in the intergenic region, followed by gene body (29% for both hyper- and hypo-methylated sites), promoter (18% and 15% for hyper- and hypo-methylated sites, respectively), 5'UTR (9% for both hyper- and hypo-methylated sites), and 3'UTR (4% and 3% for hyper- and hypo-methylated sites, respectively). To determine the statistical significance of the association of methylation changes with functional or neighborhood location, we applied the chi-square test to data in Figure 2A (middle and right panels), 2B (middle and right panels) and 2C (all panels). In all cases, we obtained P-values < 2.2E-16, indicating that the methylation changes were strongly associated with both functional and neighborhood locations.
Promoter methylation is negatively correlated with mRNA expression
Since DNA methylation at the promoter region has been more thoroughly established as an epigenetic regulator of gene expression, we next investigated this relationship in the SCC-61/rSCC-61 system by comparing the HM450 DNA methylation data with gene expression data from the HumanHT-12 v4 Expression BeadChip. We analyzed 4,948 genes that had detectable expression values in both rSCC-61 and SCC-61 cell lines. We detected 3,275 genes with statistically significant changes in expression (P-values < 0.05); of these, 162 genes were up- or down-regulated based on the selection criterion |log2 fold change| > 1.5 (Fig. 3A). The DNA methylation data were filtered using the detection P-values and location of probes, resulting in 28,030 CpG sites mapped to the promoter regions of 4,948 expressed genes (Fig. 3B). Further filtering based on the statistical significance of gene expression (P-values < 0.05 and |log2 fold change| > 1.5) and methylation changes (|Δβ| > 0.2) resulted in the identification of 206 dmCpG sites located in the promoter region of 90 genes. Out of these 90 genes, 6 genes had both hyper- and hypo-methylated sites and were thus counted twice in analyses. To correct for this, the 6 genes (MGMT, CCNA1, H19, KRT19, IGFBP3, and LIPG) were manually classified as hyper- or hypo-methylated based on the methylation status of the CpG site in the promoter region showing the largest |Δβ| for each gene. The association between DNA methylation and gene expression changes indicates that genes with hypermethylated promoters tend to be downregulated in the rSCC-61 cell line, with a Pearson correlation of −0.11 (P-value < 2.2E-16, Fig. 3C), which agrees with the previous findings that DNA methylation at the promoter region generally tends to suppress gene expression.
Figure 3.

Analysis of gene expression and DNA methylation levels in rSCC-61 and SCC-61 cells. (A) Volcano plot showing differentially expressed genes between rSCC-61 and SCC-61 cell lines. A total of 4,948 genes have measurable expression values (including black, red and blue dots). 3,275 genes have significantly different expressions (P-value< 0.05) (including red and blue dots). 162 genes have |log2expression change| > 1.5 and P-value < 0.05 (up-/downregulated genes, blue dots only). (B) DNA methylation comparisons between rSCC-61 and SCC-61 cell lines, taking into account CpG site promoter location and changes in gene expression. The compiled plot on top is separated into individual plots for better visualization. Black dots represent 28,030 CpGs on 4,948 gene promoters. Red dots represent 18,233 CpGs on 3,275 gene promoters (gene expressions are significant, P-value < 0.05). Blue dots represent 2,721 CpGs with methylation changes (|Δβ| > 0.2) on 1,336 gene promoters. Green dots represent 206 CpGs with methylation changes located in promoters of genes with regulated expressions. (C) A negative correlation was detected between gene expression changes and methylation changes. 18,233 CpGs on 3,275 gene promoters (gene expressions are significant, P-value < 0.05) yields ∼5.6 CpGs per promoter. CpG-gene pairs were further divided into 4 groups: 1. HYPER DN - 123 hypermethylated CpGs located in 55 downregulated genes (red dots); 2. HYPO DN - 18 hypomethylated CpGs located in 7 downregulated genes (purple dots); 3. HYPO UP - 34 hypomethylated CpGs located in 14 upregulated genes (black dots); and, HYPER UP - 31 hypermethylated CpGs located in 8 upregulated genes (green dots). To see this figure in color, please refer to the online version of this article.
Top pathways regulated by genes with statistically significant dmCpGs and expression patterns
Based on the association between changes of expression and promoter methylation levels, the differentially expressed genes described above (84 genes; comparison of rSCC-61 relative to SCC-61 cells) can be categorized into 4 groups: A) hypermethylated and downregulated genes (HYPER DN); B) hypermethylated and upregulated genes (HYPER UP); C) hypomethylated and downregulated genes (HYPO DN); and D) hypomethylated and upregulated genes (HYPO UP) (see Supplemental File 1). To investigate the functional relevance of the differentially methylated and expressed genes, we applied Ingenuity Pathway Analysis (IPA). The top 20 canonical pathways from the IPA are shown in Table 1, and an extensive list can be found in Supplemental File 2. ILK signaling, glucocorticoid receptor signaling, fatty acid α-oxidation, and cell cycle regulation were among the top pathways represented by this dataset.
Table 1.
Top 20 IPA canonical pathways for differentially methylated and expressed genes in rSCC-61 and SCC-61 cells
| Ingenuity Canonical Pathways | −log(P-value) | Molecules |
|---|---|---|
| ILK Signaling | 3.78E00 | PARVB,FERMT2,VIM,MYL6B,PTGS2,ACTG1 |
| Glucocorticoid Receptor Signaling | 3E00 | SCGB1A1,SGK1,CDKN1A,SMAD4,PTGS2,TSC22D3 |
| Fatty Acid α-oxidation | 2.66E00 | PTGS2,ALDH7A1 |
| Cell Cycle: G1/S Checkpoint Regulation | 2.55E00 | CCND2,CDKN1A,SMAD4 |
| GADD45 Signaling | 2.51E00 | CCND2,CDKN1A |
| Aryl Hydrocarbon Receptor Signaling | 2.48E00 | CCND2,CCNA1,CDKN1A,ALDH7A1 |
| VDR/RXR Activation | 2.31E00 | SERPINB1,CDKN1A,IGFBP3 |
| Cyclins and Cell Cycle Regulation | 2.31E00 | CCND2,CCNA1,CDKN1A |
| Estrogen-mediated S-phase Entry | 2.31E00 | CCNA1,CDKN1A |
| RAR Activation | 2.13E00 | DHRS9,IGFBP3,SMAD4,CRABP2 |
| Choline Degradation I | 2.06E00 | ALDH7A1 |
| Ethanol Degradation II | 2.04E00 | DHRS9,ALDH7A1 |
| Retinol Biosynthesis | 2.01E00 | DHRS9,LIPG |
| Noradrenaline and Adrenaline Degradation | 1.99E00 | DHRS9,ALDH7A1 |
| Interferon Signaling | 1.96E00 | OAS1,IRF1 |
| D-glucuronate Degradation I | 1.88E00 | DCXR |
| LPS/IL-1 Mediated Inhibition of RXR Function | 1.8E00 | IL36G,MGMT,HS6ST2,ALDH7A1 |
| LXR/RXR Activation | 1.79E00 | IL36G,SAA1,PTGS2 |
| Lysine Degradation II | 1.66E00 | ALDH7A1 |
| Lysine Degradation V | 1.66E00 | ALDH7A1 |
Note: Bold indicates upregulated genes. Unbold indicates downregulated genes.
The potential of epigenetic regulation of the differentially methylated and expressed genes was then tested by treating rSCC-61 and SCC-61 cells with 5-aza-2'deoxycitidine (5-Aza) to decrease DNA methylation, and with cytosine arabinoside (AraC) to account for the cytotoxic effects of 5-Aza. We focused first on CCND2 and CDKN1A, two genes involved in cell cycle regulation and characterized by promoter hypermethylation and downregulation of gene expression in rSCC-61 cells (Supplemental File 1, HYPER DN table). Treatment of rSCC-61 and SCC-61 with 5-Aza increased the levels of CCND2 protein in rSCC-61 but not in SCC-61 cells (Fig. 4A, the two bands in SCC-61 cells correspond to unphosphorylated and phosphorylated CCND2 species10). On the other hand, AraC treatment of rSCC-61 or SCC-61 cells resulted in a slight decrease in CCND2 protein. These results were consistent with the analysis of 6 CpG methylation sites in the TSS200 promoter region of CCND2 measured with bisulfite pyrosequencing (Fig. 4B). Treatment with 5-Aza, but not AraC, decreased CCND2 promoter methylation in rSCC-61 cells, confirming that expression of this gene is directly regulated by methylation changes in its promoter region. Western blot analysis of CDKN1A shows a similar overall trend to CCND2 (Fig. S1 in Supplemental File 3). However, since both 5-Aza and AraC (1 μM) induced an increase in CDKN1A protein, we have not followed up on the regulation of the expression of this gene by bisulfite pyrosequencing studies because it would have been difficult to distinguish between cytotoxic and methylation effects.
Figure 4.
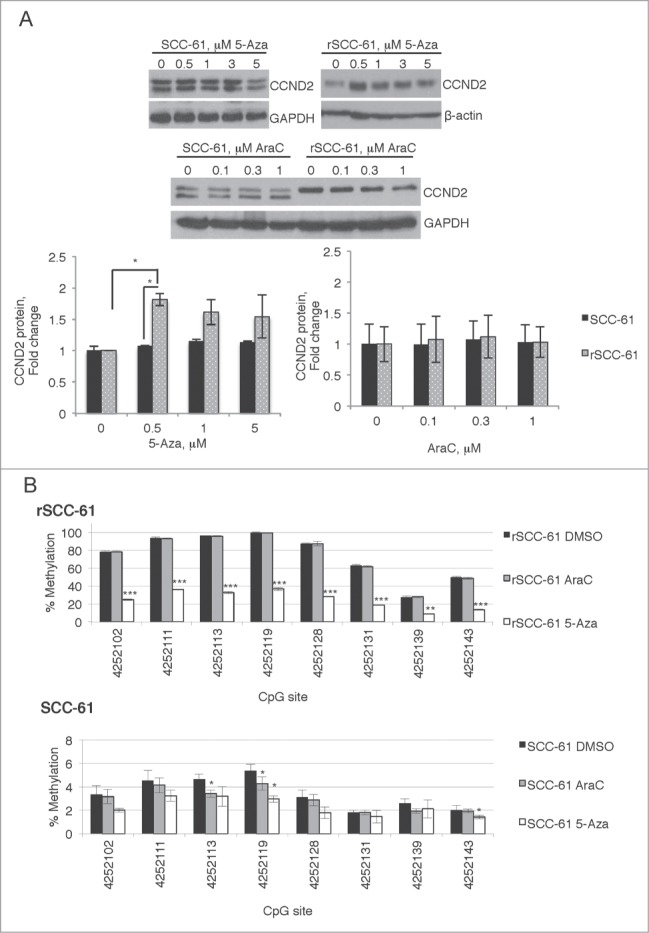
Targeted analysis of CCND2 regulation by promoter methylation. SCC-61 and rSCC-61 cells were treated with 0–5 µM 5-Aza or 0–1 µM AraC for 4 days. After this, the cells were either lysed for Western blot analysis (A) or processed for bisulfite pyrosequencing experiments (B). (A) Quantification of the Western blots is shown from 3 independent experiments. Only rSCC-61 cells treated with 5-Aza showed a statistically significant increase in CCND2 protein. (B) Similar to Western blot data, only rSCC-61 cells treated with 5-Aza (1 μM) produced a decrease in promoter methylation at all sites investigated, clearly independent of the cytotoxic effects of AraC (1 μM) in control experiments. In panels (A) and (B), asterisks indicate statistically significant changes [α = 0.05, P-values of 0.01–0.05 (*), 0.001–0.01 (**), or <0.001 (***)]. In (B), the statistical significance of 5-Aza and AraC changes was calculated relative to the DMSO control at each methylation site.
To gain insight into potential upstream regulatory mechanisms driving the expression of the 84 genes other than promoter methylation changes, we analyzed this data set using the Regulator Effects feature in IPA. The analysis produced a network of regulators that are predicted to affect the expression of these genes (Fig. 5A shows a subset of genes of interest; original network is included as Fig. S2 in Supplemental File 3). We sought validation of the prediction of upstream regulators shown in Figure 5A using the HumanHT-12 v4 Expression data. Interestingly, STAT1 and KLF4, which are known to promote expression of CCND2, are both predicted to be downregulated in rSCC-61 (Fig. 5A) and confirmed to be downregulated by mRNA expression analysis (Fig. 5B). Other predicted upstream regulators showed either a similar trend to STAT1 and KLF4 (e.g., IRF1 and EIF2AK2), no change in mRNA expression (e.g., AKT, SOCS3) or were contradictory (e.g., NFATC2, VHL) (Fig. 5B), suggesting alternate modes of regulation such as by posttranslational modifications. For example, AKT had equal expression based on mRNA levels but was predicted by this analysis to be downregulated in rSCC-61. Indeed, we have previously reported data showing decreased phosphorylation and thus activation of this signaling protein in rSCC-61, consistent with the predictions of the Regulator Effects described here.4
Figure 5.
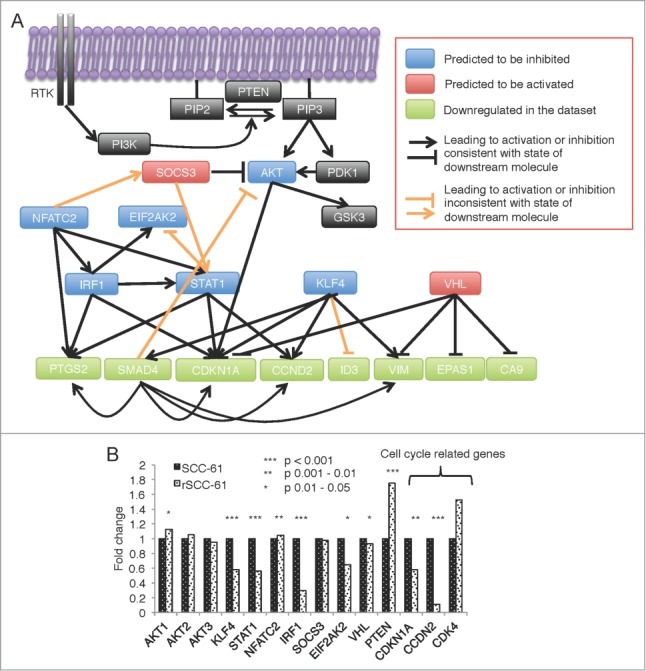
IPA predicted upstream regulator network for differentially methylated and expressed genes in rSCC-61 cells. (A) The complete network obtained from performing the Regulator Effects analysis of the differentially methylated and expressed genes is shown in Figure S2 in Supplemental File 3. The AKT-connected subnetwork was extracted and enriched with other relevant components of the AKT pathway (black boxes). From bottom up in this panel, the molecules in light green are a subset of genes with downregulated gene expression. Based on these findings, the molecules shown in blue boxes are predicted by the Regulator Effects analysis to be inhibited, and molecules shown in red are predicted to be activated. Black lines represent relationship effects (activating or inhibitory) consistent between our experimental data and IPA literature knowledge, and orange lines represent findings inconsistent with state of downstream molecule. (B) mRNA expression levels of the predicted genes and cell cycle-related genes extracted from the HumanHT-12 v4 Expression BeadChip data. Asterisks indicate statistically significant changes in gene expression in rSCC-61 relative to SCC-61 cells [α = 0.05, P-values of 0.01–0.05 (*), 0.001–0.01 (**), or <0.001 (***)].
Given the potential connection between AKT activation and cell cycle regulation,11,12 we next investigated whether DNA hypermethylation may also contribute to downregulation of AKT phosphorylation. Using the same treatment conditions as described for CCND2 studies in Figure 4A, we monitored PTEN expression and AKT phosphorylation by Western blot analysis. Consistent with our previous report and the prediction from IPA, AKT phosphorylation was lower in untreated rSCC-61 cells compared with SCC-61 cells. When treated with increasing concentrations of 5-Aza, rSCC-61 cells displayed a gradual increase in AKT phosphorylation (Fig. 6A). This increase in AKT phosphorylation was not evident in AraC-treated cells, confirming regulation at the level of DNA methylation. Because PTEN is a known negative regulator of the AKT pathway, and since DNA methylation does not directly impact protein posttranslational modifications, we proposed that increased AKT phosphorylation by 5-Aza treatment was due to decreased PTEN expression. As shown in Figure 6B, treatment of rSCC-61 cells with 5-Aza resulted in a gradual decrease in PTEN expression, in congruence with the changes in AKT phosphorylation. Future studies will address whether the expression of PTEN gene is controlled directly by methylation in its promoter region or other potential mechanisms described in the Discussion.
Figure 6.

Treatment of rSCC-61 cells with 5-Aza decreases PTEN levels and upregulates downstream AKT signaling. SCC-61 and rSCC-61 cells were treated with 0–5 µM 5-Aza or 0–1 µM AraC for 4 days. Cells were then lysed for Western blot analysis using indicated antibodies for monitoring AKT phosphorylation (A) and PTEN expression (B). Treatment of rSCC-61 cells with 5-Aza induced an increase in AKT phosphorylation and a decrease in PTEN expression independent of its cytotoxic effects seen in AraC control experiments. Quantification of the Western blots is shown from 3 independent experiments. Asterisks indicate statistically significant changes in pAKT or PTEN at each 5-Aza or AraC concentration relative to the untreated rSCC-61 and SCC-61 cells, respectively [α = 0.05, P-values of 0.01–0.05 (*), 0.001–0.01 (**), or <0.001 (***)].
Analysis of HNSCC patient data extracted from The Cancer Genome Atlas (TCGA)
To determine whether our findings in the SCC-61/rSCC-61 cell model have broader relevance to radiation resistance in HNSCC patients, we performed a similar series of analyses using publicly available data from the NIH-funded TCGA project. We screened the TCGA database and selected HNSCC patients who had both DNA methylation and gene expression data available using the following criteria: HPV status “negative,” perspective collection “yes,” and primary tumors “yes.” Based on radiation treatment outcomes, we categorized samples into sensitive tumors (“complete remission/ response”) and resistant tumors (“stable disease,” “partial remission/response,” or “progressive disease”). This screening yielded data from 27 patients (Supplemental File 4), 4 with radiation-resistant tumors and 23 with radiation-sensitive tumors. The level-3 data for both DNA methylation (Illumina HumanMethylation450 BeadChip, β values) and gene expression (IlluminaHiseq RNAseq V2, RSEM values) were downloaded from TCGA for the 27 samples. [Note: The TCGA level-3 data contains NULL entries, representing probes that overlap with known SNPs or other genomic variations, or probes with non-detection probability (P-value) greater than 0.05.]
DNA methylation. Analysis of the 27 patient samples identified 396,064 CpG sites, with 118,556 CpG sites located in the promoters of 20,891 genes. For each CpG site, we calculated the average β value for each group (radiation-resistant and -sensitive) and identified 2,182 CpG sites with |Δβ| > 0.2 located in the promoter region of 1,400 genes. Consistent with the SCC-61/rSCC-61 results, more CpG sites were hypermethylated in the radiation-resistant compared with radiation-sensitive tumors (1,322 versus 860, respectively) (Fig. 7A and B). Out of 4,383 CpG methylation sites that overlapped between the SCC-61/rSCC-61 and TCGA data, 1,605 (36.6%) had the same hyper- or hypo-methylated trend, based on filtering by delta β values (Fig. 7C). While approximately 25% of these methylation sites were located in promoter regions, ∼50% were intergenic with as yet unknown function.
Figure 7.
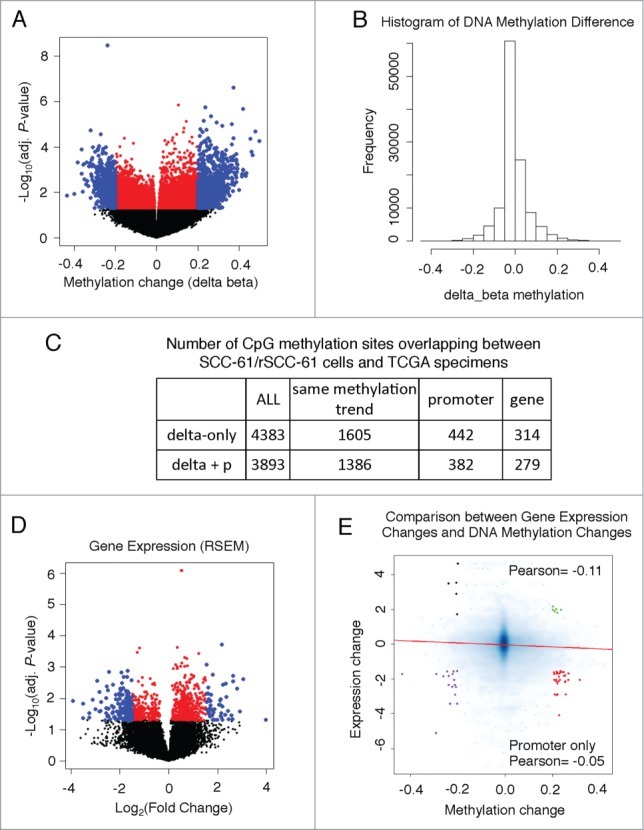
Gene expression and DNA methylation levels in TCGA samples. (A) Volcano plot showing differences in DNA methylation between radiation-resistant and radiation-sensitive HNSCC tumors. 2,182 CpG sites, located in 1,400 gene promoters, had methylation changes (|Δβ| > 0.2), including 1,322 hyper- and 860 hypo-methylated ones (blue dots). (B) Histogram of promoter CpG methylation changes (resistant tumors – sensitive tumors). (C) Summary table of CpG methylation sites overlapping between the SCC-61/rSCC-61 cells and radiation-sensitive or radiation-resistant HNSCC tumors. (D) Gene expression comparison between radiation-sensitive or radiation-resistant HNSCC tumors using a volcano plot. To reduce noise, we filtered out the genes with RSEM< 5 for both groups (RSEM of one gene for each group is the average RSEM over all samples). This decreased the number of genes to 12,985. Out of these, 531 genes had a significantly changed expression (|log2fold-change| > 1.5) (blue dots). (E) A negative correlation was detected between gene expression changes and DNA methylation level changes. Further analysis divided the CpG-gene pairs into 4 groups: 1. HYPER DN - 34 hypermethylated CpGs located in 21 downregulated genes (red dots); 2. HYPO DN - 19 hypomethylated CpGs located in 14 downregulated genes (purple dots); 3. HYPO UP - 5 hypomethylated CpGs located in 5 upregulated genes (black dots); and, HYPER UP - 6 hypermethylated CpGs located in 5 upregulated genes (green dots).
Gene expression. TCGA level-3 data available for the 27 patient samples provided gene expression values (RSEM) for 13,265 genes. After removing the genes with RSEM < 5 across samples, we calculated the average gene expression in each group and selected 531 genes as differentially expressed genes with |log2fold change| > 1.5 (Fig. 7D).
Relationships between promoter methylation and gene expression. We analyzed the correlation between gene expression changes and DNA methylation changes in the promoter regions. The negative correlation between changes in promoter methylation and changes in gene expression supports the observation that DNA methylation at the promoter region generally tends to suppress gene expression (Fig. 7E). In addition, similar to the studies using the SCC-61/rSCC-61 system, we categorized the differentially expressed genes, also containing hyper-/hypo-methylated CpGs, into 4 groups (Fig. 7E). Then we performed IPA analysis on all 45 genes passing the filtering criteria for both changes in promoter methylation and gene expression (Table 2). The LXR/RXR pathway appears in both top 20 canonical pathways from IPA using cell lines and patient data. The breakdown of the 45 genes into the 4 groups: (21) HYPER DN, (14) HYPO DN, (5) HYPO UP, and (5) HYPER UP is shown in Supplemental File 5; the complete list of IPA canonical pathways can be found in Supplemental File 6.
Table 2.
Top 20 IPA canonical pathways for differentially methylated and expressed genes in radiation-resistant and -sensitive HNSCC tumors
| Ingenuity Canonical Pathways | −log(P-value) | Molecules |
|---|---|---|
| PAK Signaling | 3.04E00 | PIK3CG,EPHA3,TNF |
| IL-6 Signaling | 2.71E00 | IL1R2,PIK3CG,TNF |
| LXR/RXR Activation | 2.66E00 | IL1R2,LPL,TNF |
| IL-9 Signaling | 2.62E00 | PIK3CG,TNF |
| IL-12 Signaling and Production in Macrophages | 2.52E00 | ALOX15,PIK3CG,TNF |
| Docosahexaenoic Acid (DHA) Signaling | 2.5E00 | ALOX15,PIK3CG |
| 3-phosphoinositide Biosynthesis | 2.36E00 | PPP1R1B,PIK3CG,PTPRN |
| MSP-RON Signaling Pathway | 2.36E00 | PIK3CG,TNF |
| Tec Kinase Signaling | 2.33E00 | PIK3CG,TNF,FGR |
| Ephrin A Signaling | 2.32E00 | PIK3CG,EPHA3 |
| NF-κB Signaling | 2.22E00 | IL1R2,PIK3CG,TNF |
| Superpathway of Inositol Phosphate Compounds | 2.1E00 | PPP1R1B,PIK3CG,PTPRN |
| Eicosanoid Signaling | 2.1E00 | ALOX15,FPR2 |
| IL-15 Signaling | 2.06E00 | PIK3CG,TNF |
| IL-10 Signaling | 2.03E00 | IL1R2,TNF |
| Ceramide Signaling | 1.9E00 | PIK3CG,TNF |
| PPAR Signaling | 1.76E00 | IL1R2,TNF |
| Glucocorticoid Receptor Signaling | 1.74E00 | IL1R2,PIK3CG,TNF |
| HIF1α Signaling | 1.69E00 | PIK3CG,MMP10 |
| Renin-Angiotensin Signaling | 1.64E00 | PIK3CG,TNF |
Note: Bold indicates upregulated genes. Unbold indicates downregulated genes.
5-Aza treatment does not increase DNA damage induced by ionizing radiation
The global increase in DNA methylation identified in radiation-resistant cell lines and patient samples suggests that this epigenetic modification is involved in radiation resistance. Other reports have also indicated that treatment of cancer cell lines with 5-Aza decreases DNA methylation and results in increased sensitivity to radiation treatment.13-18 To determine if global demethylation of rSCC-61 would promote DNA damage and cell death induced by ionizing radiation, we treated rSCC-61 cells with 5-Aza and measured the response to radiation using cell viability assays under two experimental conditions (Fig. 8A and 8C, top schemes). Methylation data (Fig. 8D) show that 4 days of treatment with 5-Aza (1–3 μM) resulted in a 66% decrease in DNA methylation in rSCC-61 cells. Based on this analysis, a minimum of 15% decrease in methylation is expected with 24 h treatment. Treatment with 5-Aza alone in rSCC-61 cells was cytotoxic to cells and caused 60% or 50% cell death (Fig. 8A and B, respectively, bottom graphs), similar or lower than the cytotoxicity caused by the control reagent cytosine arabinoside (AraC) and higher than radiation alone. Thus, we conclude that 5-Aza did not cause increased response to radiation in rSCC-61 cells beyond the cytotoxic effects of AraC.
Figure 8 (See previous page).

Treatment with 5-Aza does not enhance radiation-induced DNA damage and cell death. (A) SCC-61 and rSCC-61 cells were incubated with 2 µM of 5-Aza or AraC overnight and then irradiated with a single dose of 2, 4, or 8 Gy. Cell viability was determined at 72 h post-irradiation using an MTT assay. (B) rSCC-61 cells were incubated with 2 µM 5-Aza for 4 days and then irradiated with a single dose of 2 Gy. Cell lysates were collected at 15 min and 3 h post-irradiation and subjected to Western blot analysis using antibodies against p-γ-H2AX and GAPDH. (C) SCC-61 and rSCC-61 cells were incubated with 2 µM 5-Aza, 2 µM AraC, or 100 µM SAM for 4 days (refreshing the medium every other day), and treated with a single dose of 2 or 4 Gy irradiation starting on the second day. Cell viability was determined at 72 h post-irradiation using an MTT assay. In both (A) and (C), asterisks indicate statistically significant changes in cell viability for 5-Aza- and AraC-treated cells at each radiation treatment condition relative to the DMSO control [α = 0.05, P-values of 0.01–0.05 (*), 0.001–0.01 (**), or <0.001 (***)]. (D) rSCC-61 cells were incubated with 0–10 µM 5-Aza for 4 days and 5-methylcytosine levels were measured using MethylFlash Methylated DNA Quantification Kit. Increasing concentrations of 5-Aza decreased global DNA methylation. Data are from 2 independent experiments. Asterisks indicate statistically significant changes [α = 0.05, P-values of 0.01–0.05 (*), 0.001–0.01 (**), or <0.001 (***)].
Since similar or stronger effects were noted with AraC treatment, we reasoned that the methylation changes induced by 5-Aza are more likely to protect against radiation-induced cell death (see Fig. 8C, bottom graph). To further support these findings, we monitored radiation-induced DNA damage in rSCC-61 cells treated with 5-Aza by Western blot analysis of phosphorylated γH2AX, a known marker for DNA damage. As shown in Figure 8B, there was a strong increase in phosphorylated γH2AX in response to radiation, which was attenuated in cells treated with 5-Aza. These results are consistent with the cell viability assays in Figure 8A and C (bottom graphs). We also performed similar cell viability assays using SCC-61 cells (Fig. 8A and C, upper graphs). As expected and reported earlier, ionizing radiation alone caused more cell death in SCC-61 than rSCC-61.4 Similar to the results in rSCC-61 cells, 5-Aza treatment did not increase cell death in irradiated SCC-61 cells. Moreover, when combined with radiation, 5-Aza treatment caused less cell death compared with the control reagent AraC.
We then attempted to increase DNA methylation in SCC-61 cells using S-adenosylmethionine (SAM), and measured the response to radiation to determine whether the increased DNA methylation would increase radiation resistance. Under conditions that allowed us to observe increased DNA methylation (1 mM SAM, 5mC% from 0.5 to 0.7%), SAM was highly cytotoxic, impairing our ability to measure the effects of increased methylation on radiation response in this cell line. Lowering the SAM concentration to 100 μM, as reported in other studies, allowed us to perform cell viability experiments.19 As shown in Fig. 8C (middle graph), SAM treatment had a minimal effect on the response to radiation at 2 Gy, and virtually no effect at 4 Gy.
Discussion
Epigenetic mechanisms such as DNA methylation are essential for the regulation of gene expression in normal mammalian development; aberrant epigenetic alterations have been linked to many pathological conditions, including cancer.20 Development of technologies that allow for genome-wide investigation of DNA methylation has significantly increased the number of investigations into the function of methylation changes in cancer development and response to therapies. Since radiation is an important modality of cancer treatment, its effects on DNA methylation and the role of DNA methylation in radiation resistance have been investigated by a number of groups. These studies indicate that radiation induces substantial changes in DNA methylation. Although these changes are dynamic and depend on many factors (e.g., radiation dose, sex, tissue), the analysis of differentially methylated genes shows a common enrichment in cell cycle, DNA repair, and apoptosis pathways.21-25 For example, Kim et al. analyzed the DNA methylation profile of two non-small cell lung cancer cell lines, the radiation-sensitive H460 and radiation-resistant H1299 cell lines, and found a higher proportion of hypermethylation in radiation-resistant cells.26 This work used cell lines derived from patients with different genetic backgrounds, and lacked in vivo validation of the findings. To overcome these limitations, in the present study we paralleled the analyses in matched HNSCC cells with those using tumor data from HNSCC patients extracted from TCGA. The goals of our studies were two-fold: first, to establish global DNA hypermethylation as a biomarker of radiation resistance in HNSCC, and second, to identify pathways regulated by DNA promoter methylation that may contribute to the radiation resistance phenotype. The results are supported by a series of validation experiments showing epigenetic regulation of genes involved in cell cycle control and other relevant pathways.
Consistent with the findings of Kim et al., we observed similar patterns of increased DNA methylation in both radiation-resistant cells and radiation-resistant HNSCC tumors. The mechanisms driving hypermethylation and the implications of hypermethylation in radiation resistance are mostly unknown. Mechanistically, global DNA methylation is regulated by many factors, including the level and activity of DNMTs. Expression of the DNMT3B gene was increased in rSCC-61 cells, providing a partial explanation for the differences in DNA methylation.
Several studies have investigated the complex relationship between DNMTs activities, redox microenvironment and DNA methylation status.27 One of these studies demonstrated DNA dehydroxymethylase activity of DNMT3A and DNMT3B under oxidative conditions converting 5-hydroxymethylcytosine (5hmC) to cytosine.28 In an earlier study, Turk et al. reported that DNA adduct 8-hydroxyguanine, which is also formed by oxidation, impairs the methylation of cytosines at CpG sites.29 We previously reported increased levels of reactive oxygen species in SCC-61 compared with rSCC-61 cells, and increased labeling with the redox-sensitive probe BP1 in radiation-sensitive vs. -resistant HNSCC tumors.4 Thus, it is feasible that differences in DNA methylation observed in both cell culture and tumor tissues reported here have common redox-regulated mechanisms, which will be the topic of future studies.
To begin to address the function of dmCpGs in radiation resistance, we first determined their distribution across the genome. The most striking finding was in the pattern of neighborhood location, with the hypermethylated sites in radiation-resistant cells being primarily located in the open sea and the hypomethylated sites primarily located in CpG islands. Although a significant proportion of the hypermethylated CpGs in the open sea were assigned to sites in the intergenic regions, more than 50% of sites were located in regions with known function in the regulation of gene expression (e.g., enhancers). Previous studies have shown that methylation at CpG sites other than CpG islands is more dynamic than methylation in CpG islands and occurs in tissue-specific and cancer-specific ways.30,31 However, regardless of the neighborhood location of promoter methylation sites, DNA methylation in the promoter region is most often associated with downregulation of gene expression.32 Consistent with this, our data examining the relationship between gene expression and promoter methylation using cell lines and patient samples revealed a negative correlation.
Genes that were characterized by both significant changes in the promoter methylation and gene expression were further analyzed using IPA and the Regulator Effects tool within the IPA. We found the “Cyclins and Cell Cycle Regulation” pathway to be among the top pathways identified by this analysis and AKT as top upstream regulator of proteins involved in these pathways. These results are not surprising, given the importance of cell cycle regulation in radiation resistance and the known relationship between AKT activity, CDKN1A, and Proliferating Cell Nuclear Antigen (PCNA), involved in DNA replication and repair.11,12,33 IPA analysis also identified LXR/RXR activation in the top 20 canonical pathways using both cell culture and tumor tissue data. Although this pathway has not been directly linked to radiation resistance, its target genes are involved in cholesterol and lipid metabolism,34 including fatty acid synthase (FASN). We have shown that regulation of cholesterol and lipid metabolism is important for radiation response and FASN protein is highly upregulated in rSCC-61.4 Further investigations into the LXR/RXR pathway and the regulation of cholesterol and lipid metabolism may reveal the mechanism by which this pathway contributes to radiation response.
To further validate DNA methylation as a regulator of proteins controlling cell cycle, we used 5-Aza, a deoxycytidine analog that induces DNA hypomethylation.35,36 As a demethylating agent, 5-Aza is generally thought to increase gene expression by decreasing methylation at CpG islands in the promoter region. However, the effect of 5-Aza on gene expression depended on the cell type and the specific gene tested, sometimes inducing a decrease in gene expression.37,38 Another confounding factor in studies using 5-Aza is the need to distinguish between its demethylating and cytotoxic effects, which both could induce changes in protein levels. For example, the targeted analysis included here shows that treatment of rSCC-61 with low doses of 5-Aza increased the protein levels of both CCND2 and CDKN1A. However, while the AraC treatment also increased the CDKN1A protein, it did not affect CCND2. Further analysis of CCND2 promoter methylation confirmed the regulation of this gene's expression by methylation. Our studies also revealed the presence of both unphosphorylated and phosphorylated CCND2 species in SCC-61, suggesting posttranslational mechanisms of CCND2 regulation independent of DNA methylation.
Since AKT was predicted by the IPA Regulator Effects analysis to be an upstream positive regulator of CDKN1A, we investigated AKT phosphorylation in the presence of 5-Aza and AraC. AKT phosphorylation was increased by 5-Aza but not AraC, leading to the conclusion that DNA methylation is involved in the regulation of AKT phosphorylation.
We then investigated whether PTEN expression, a known upstream regulator of AKT phosphorylation, is controlled (directly or indirectly) by DNA methylation. PTEN expression showed dose-dependent changes, decreasing with 5-Aza treatment and showing virtually no change with AraC treatment in rSCC-61 cells. Although the mechanism of PTEN repression by demethylation is unclear and remains to be further investigated, several studies have shown decreased gene expression after treatment of 5-Aza or 5-azacytidine and provided possible explanations.39-41 Ando et al. reported that 5-Aza decreased P-glycoprotein expression in leukemia cells as a result of demethylation at the repressor binding sites.39 Kitagawa et al. demonstrated that 5-Aza repressed hTERT transcription in prostate cancer cells, possibly due to reactivation of upstream inhibitor p16.40 Both mechanisms could be relevant for regulation of PTEN levels in the system described here. In one mechanism, hypomethylation at CpG sites in the promoter region of PTEN caused by 5-Aza treatment would allow repressor access and inhibition of expression. In the other mechanism, DNA hypomethylation would upregulate a PTEN transcriptional repressor, thus decreasing the expression of the PTEN gene and PTEN protein content. The epigenetic regulation of PTEN and its upstream effectors remain to be studied.
Activation of the AKT pathway has been associated with radiation resistance in vivo and in vitro.42-45 In patients with oral squamous cell carcinoma treated with radiation therapy after surgery, activity of the Ras/PI3K/AKT pathway was negatively correlated with disease-free survival.46 In our SCC-61/rSCC-61 system, rSCC-61 cells were characterized by lower AKT phosphorylation and higher PTEN expression, demonstrating that activation of the AKT pathway is not always the driving force for radiation resistance and alternative mechanisms for radiation resistance might exist. Our results suggest that concerted regulation of cell cycle proteins and AKT could be the driving force for cell proliferation and radiation response in the SCC-61/rSCC-61 system. We recently reported increased proliferation of rSCC-61 compared with SCC-61 cells and G2/M arrest in SCC-61 cells exposed to radiation.4,47 Neither one of the two cell lines underwent G1/S arrest with ionizing radiation.
The data presented here provide a potential lead into future mechanistic investigations regarding cell cycle control in this system. The mRNA expression data showed significantly higher levels of CCND2 and CDKN1A in SCC-61 and high levels of CDK4 in both SCC-61 and rSCC-61 cells (Fig. 5B). Reed et al. reported that increased CDK4 could titrate the CDKN1A in cells and enable the cells to bypass G1/S arrest when exposed to ionizing radiation.48 Rossig et al. identified AKT as an upstream regulator of CDKN1A, which phosphorylates this protein and inhibits its binding to CDK4.12 Thus, it is feasible that SCC-61 and rSCC-61 cells bypass G1/S arrest by the two distinct mechanisms described by Rossig et al. and Reed et al., but involving the same key components whose expression and activity might be controlled at epigenetic level.
Based on differences in global methylation between radiation-resistant and radiation-sensitive cells and tumors, 5-Aza treatment would be expected to enhance response to radiation. However, the cell viability studies described here using ionizing radiation without or with pretreatment with 5-Aza or AraC did not show increased cell death beyond the expected cytotoxic effects of AraC. These results were consistent with the measurement of the DNA damage marker phospho-γH2AX, which was not increased in cells pretreated with 5-Aza and exposed to radiation compared with cells exposed to radiation alone. Interestingly, treatment of SCC-61 cells with 5-Aza also did not increase cell death beyond the effects of radiation alone. When compared with AraC profiles, it is possible that 5-Aza protected cells from the combined effects of drug cytotoxicity and radiation.
Most studies investigating the effect of 5-Aza treatment on the response to radiation conclude that 5-Aza increases radiation sensitivity of cancer cells,13-18,49 although the opposite has been described as well.50 Our data demonstrate that while radiation resistance phenotype is clearly associated with global hypermethylation, the mechanisms by which DNA methylation drives radiation resistance are complex and involve targeted control of DNA methylation. Cumulatively, the studies reported here add to the literature by bringing into focus the nuanced complexity of relationships between the amplitude and location of DNA methylation within the broad genome context and expression of genes controlling cell growth and response to therapies. While global demethylation may function as radiation sensitizer in some cases, targeted manipulation of CpG methylation would most likely be required to enhance radiation responsiveness.
Materials and Methods
Materials
Antibodies were obtained from the following sources: anti-p-Akt (T308) (Cell Signaling #13038), anti-Akt (Cell Signaling #4691), anti-p-GSK-3α/β (S21/9) (Cell Signaling #9331), anti-PTEN (Cell Signaling #9188), anti-β-actin (Cell Signaling #4970), anti-GAPDH (EMD Millipore ABS16), anti-CCND2 (Santa Cruz Biotechnology sc-181), anti-CDKN1A (Santa Cruz Biotechnology sc-397), and anti-p-γH2AX(S139/Y143) (Cell Signaling #5438). DMEM/F12 (1:1) media (Cat. No. 11330) and Pen/Strep (Cat. No. 15140) were purchased from Gibco (Invitrogen). Fetal Bovine Serum (FBS) was purchased from Atlanta Biologicals (Cat. No. S11150). DNA QIAamp DNA Mini kit (Cat. No. 51304) and RNeasy Plus Mini kit (Cat. No. 74134) were purchased from Qiagen. 5-Aza (Cat. No. A3656), AraC (Cat No. C1768), and SAM (Cat. No. A7007) were purchased from Sigma Aldrich. DMSO was purchased from Fisher Scientific (Cat. No. BP231). MethylFlash Methylated DNA Quantification kit was purchased from Epigentek (Cat. No. P-1034). SuperSignal West Pico Chemiluminescent substrate was purchased from Thermo Scientific (Cat. No. 34080). WesternBright ECL HRP substrate was purchased from Advansta (Cat. No. K-12045-D50). PhosSTOP phosphatase inhibitor cocktail tablets (Cat. No. 04906835001) and cOmplete protease inhibitor cocktail tablets (Cat. No. 04693159001) were purchased from Roche. Protran nitrocellulose transfer membrane (0.45 µm) was purchased from PerkinElmer (Cat. No. NBA085C001EA).
Cell culture and treatment
The development of the SCC-61/rSCC-61 matched model system of radiation resistance in HNSCC was described previously.4 Both cell lines were cultured in the DMEM/F12 (1:1) medium (Gibco Cat. No. 11330) supplemented with 10% FBS (Atlanta Biologicals Cat. No. S11150) and 1% Pen Strep (Gibco Cat. No. 15140) at 37 °C with 5% CO2. Where applicable, radiation treatment was performed using a 444 TBq 12,000 Ci self-shielded 137Cs (Cesium) irradiator. Culture dishes were placed on a styrofoam insert within the chamber of the irradiator, such that the distance from the Cs source results in a homogenous dose distribution over the desired field, with a dose rate of 392 rad/min. From the dose rate, the exposure time required to deliver the desired dose was calculated and input into the irradiator. To manipulate the DNA methylation in SCC-61 and rSCC-61 cells, the cells were incubated with indicated concentrations of 5-Aza (Sigma Aldrich Cat. No. A3656), AraC (Sigma Aldrich Cat. No. C1768), or DMSO (Fisher Scientific Cat. No. BP231) for 4 days, refreshing the medium every other day. For the cell viability assay, cells were treated with 2 µM 5-Aza, 2 µM AraC, or 100 µM SAM (Sigma Aldrich Cat. No. A7007) overnight before irradiation or for 4 days, with the irradiation starting on the second day of treatment. For CCND2 bisulfite pyrosequencing studies, the cells were treated with 1 μM 5-Aza or 1 μM AraC for 4 days, refreshing the medium every other day.
Illumina HumanMethylation450 BeadChip
DNA purified from SCC-61 and rSCC-61 cells using the DNA QIAamp DNA Mini kit (Qiagen Cat. No. 51304) was sent to the core facility at University of Southern California Epigenome Center for HM450 BeadChip (Illumina, Inc.) analysis. DNA extracted from each cell line (1 µg) was modified with bisulfite treatment, amplified, fragmented, and analyzed for CpG methylation using the HM450 BeadChip. The Illumina HM450 methylation assay examines the DNA methylation status of 485,577 CpG sites. Using the R package “FDb.InfiniumMethylation.hg19,” Illumina identifiers were mapped to the hg19 genome build.50 The probes with detection levels above background across all samples (detection P < 0.05) were kept. Methylation levels of each CpG are represented by a β value, computed with (M/(M+U+100)), where M and U represent the methylated and unmethylated signals. CpG sites were considered as hypermethylated and hypomethylated if the methylation level changes (Δβ) between samples were detected greater than 0.2 and less than −0.2, respectively. The annotation of neighborhood regions was obtained by following the guidelines on the UCSC genome browser.51 CpG islands were identified by scoring each dinucleotide (+17 for CG and −1 for others). Segments with highest scores were then analyzed for GC content (>50 %), length (>200 bp), and ratio of observed to expected number of CG dinucleotides (>0.6) according to the formula: Obs/Exp CpG = Number of CpG * N / (Number of C * Number of G) where N = length of sequence.52
Measurement of CpG methylation in the TSS200 promoter region of CCND2 using bisulfite pyrosequencing
Pyrosequencing DNA (400 ng/cell line and treatment) was bisulfite-treated using the Zymo EZ DNA Methylation Lightning kit (Zymo Cat. No. D5030) and PCR primers were designed using the Pyromark Assay Design Software (Qiagen). One μL of bisulfite treated DNA was amplified using the Bioline EPIK™ Amplification kit (BiolineCat. No. BIO-66025) in a BioRad C1000 thermal cycler. The following gene-specific primers were designed to target CpG sites in the TSS200 promoter region of the gene analyzed by the BeadChip: CCND2 (chromosome:NCBI36:12:4251399: 4285377:1) coordinates 4252151 and 4252143. Primers for pyrosequencing were: FWD5′-AGTAGGTTTTTAGGGAGAAAGTTTGG -3′, REV Biot-5′-AAACACCACCACCCCTTCCTTT-3′, SEQ5′- TTTTTAGGGAGAAAGTTTG-3′. Additional CpG sites not analyzed by the BeadChip were assessed in the pyrosequencing assay due to their proximity to the CpG sites of interest. Single stranded products were prepared for pyrosequencing by PyroMark vacuum prep tool (Qiagen). Pyrosequencing reactions were performed using a PyromarkQ24 system (Qiagen) according to the manufacturer's instructions. Data were analyzed using Pyromark Q24 software for percent methylation at the CpG sites interrogated.
HumanHT-12 v4 expression beadChip
Total RNA was extracted and purified from SCC-61 and rSCC-61 cells using the RNeasy Plus Mini kit (Qiagen Cat. No. 74134). The quality of extracted total RNA was assessed by the 260/280 absorbance ratios and by RIN (RNA integrity number) measured using the Agilent BioAnalyzer System. The analysis was based on 3 biological replicates for each cell line. The 260/280 absorbance ratios varied between 2 and 2.1 and the RIN values between 9.4 and 10. All samples were assayed with the HumanHT-12 v4 Expression BeadChip (Illumina, Inc.) and read on an iScan array reader (Illumina, Inc.). This microarray assays over 47,000 probes spanning approximately 30,000 genes. Sample intensities were determined and preliminary quality control analyses were performed with the GenomeStudio software (Illumina, Inc.). Probes with detection P-values less than 0.05 were kept for future analysis and annotated with human genome version hg19 of the human genome. Gene expression comparisons between samples were calculated with Limma module of Bioconductor, using linear models and Bayes methods to assess differential expression. Genes were considered to be significantly expressed if the associated P-values were less than 0.05 and log2-based expression change was greater than 1.5 or less than −1.5. The P-values for gene expression changes were corrected for multiple tests using the Benjamini and Hochberg method.53
5-methylcytosine (5mC) measurement
DNA purified from cells was subjected to DNA methylation analysis using MethylFlash Methylated DNA Quantification Kit (Epigentek Cat. No. P-1034), following the manufacturer's instructions.
Western blot analysis
Cells were lysed with RIPA buffer (50 mM Tris-HCl, pH 7.4; 1% NP40; 150 mM NaCl; 0.5% sodium deoxycholate; 0.1% SDS) supplemented with protease and phosphatase inhibitors (Roche Cat. No. 04693159001 and 04906835001). The lysates were incubated on ice for 1 h followed by centrifugation at 10,000 g for 10 min. Equal amounts of total protein were subjected to SDS-PAGE and then transferred to a Protran nitrocellulose transfer membrane (0.45 µm, PerkinElmer Cat. No. NBA085C001EA). Blots were blocked in 5% non-fat milk or BSA and probed with the appropriate antibodies. Immunoreactive bands were visualized using SuperSignal West Pico (Thermo Scientific Cat. No. 34080) or WesternBright ECL (Advansta Cat. No. K-12045-D50). Band intensity was quantified using ImageJ densitometric analysis. Statistical analysis (Student's t-test, one-tailed, paired) was based on 3 biological replicates using Excel 2010. Asterisks indicate statistically significant changes [α = 0.05, P-values of 0.01–0.05 (*), 0.001–0.01 (**), or <0.001 (***)].
Correlation of gene expression with DNA methylation
Genes with significant changes in expression (P-value < 0.05) were categorized into 4 groups based on their changes in gene expression and methylation levels (Δβ): 1) hypermethylated and upregulated genes (Δβ > 0.2 and log2 fold change > 1.5), 2) hypermethylated and downregulated genes (Δβ > 0.2 and log2 fold change < −1.5), 3) hypomethylated and upregulated genes (Δβ < −0.2 and log2 fold change > 1.5) and hypomethylated and downregulated genes (Δβ < −0.2 and log2 fold change < −1.5).
Selection of HNSCC patients from the TCGA database
To illustrate the consistency between our observations based on cell lines and patient samples, we selected patient data from TCGA based on the available clinical information and following criteria: HPV-, Perspective collection: yes, Primary tumors: yes. We also removed the patients that had a conflict between Tumor status and Treatment outcome first course (e.g., With Tumor/Complete Response) and patients who did not have both DNA methylation and gene expression data collected. Filtering by these criteria yielded data from 4 patients with radiation-resistant tumors and 23 patients with radiation-responsive tumors (Supplemental File 4), which were used for the analysis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Dr. Daniel J Weisenberger from USC Epigenome Center, and Drs. Siqun Zheng, Tim Howard and Greg Hawkins from the Center for Genomics & Personalized Medicine Research at Wake Forest School of Medicine, for providing core support for DNA methylation and mRNA expression studies.
Funding
Research reported in this paper was supported by the National Cancer Institute of the National Institutes of Health under award number R01 CA136810 to CMF and R01LM010185 to XZ. The authors acknowledge support from Wake Forest School of Medicine (development funds to CMF), the Wake Forest University Structural and Computational Biophysics training program (T32 GM095440, predoctoral fellowship to JM), and Comprehensive Cancer Center of Wake Forest University NCI CCSG P30CA012197 grant.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9-29; PMID:24399786; http://dx.doi.org/ 10.3322/caac.21208 [DOI] [PubMed] [Google Scholar]
- 2.Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005; 104:1129-37; PMID:16080176; http://dx.doi.org/ 10.1002/cncr.21324 [DOI] [PubMed] [Google Scholar]
- 3.Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer 2011; 11:239-53; PMID:21430696; http://dx.doi.org/ 10.1038/nrc3007 [DOI] [PubMed] [Google Scholar]
- 4.Bansal N, Mims J, Kuremsky JG, Olex AL, Zhao W, Yin L, Wani R, Qian J, Center B, Marrs GS, et al.. Broad phenotypic changes associated with gain of radiation resistance in head and neck squamous cell cancer. Antioxid Redox Signal 2014; 21:221-36; PMID:24597745; http://dx.doi.org/ 10.1089/ars.2013.5690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358:1148-59; PMID:18337604; http://dx.doi.org/ 10.1056/NEJMra072067 [DOI] [PubMed] [Google Scholar]
- 6.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415-28; PMID:12042769; http://dx.doi.org/ 10.1038/nrg962 [DOI] [PubMed] [Google Scholar]
- 7.Singal R, Ginder GD. DNA methylation. Blood 1999; 93:4059-70; PMID:10361102 [PubMed] [Google Scholar]
- 8.Das PM, Singal R. DNA methylation and cancer. J Clin Oncol 2004; 22:4632-42; PMID:15542813; http://dx.doi.org/ 10.1200/JCO.2004.07.151 [DOI] [PubMed] [Google Scholar]
- 9.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al.. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288-95; PMID:21839163; http://dx.doi.org/ 10.1016/j.ygeno.2011.07.007 [DOI] [PubMed] [Google Scholar]
- 10.Takano Y, Kato Y, van Diest PJ, Masuda M, Mitomi H, Okayasu I. Cyclin D2 overexpression and lack of p27 correlate positively and cyclin E inversely with a poor prognosis in gastric cancer cases. Amer J Pathol 2000; 156:585-94; http://dx.doi.org/ 10.1016/S0002-9440(10)64763-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li R, Waga S, Hannon GJ, Beach D, Stillman B. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature 1994; 371:534-7; PMID:7935768; http://dx.doi.org/ 10.1038/371534a0 [DOI] [PubMed] [Google Scholar]
- 12.Rossig L, Jadidi AS, Urbich C, Badorff C, Zeiher AM, Dimmeler S. Akt-dependent phosphorylation of p21(Cip1) regulates PCNA binding and proliferation of endothelial cells. Mol Cell Biol 2001; 21:5644-57; PMID:11463845; http://dx.doi.org/ 10.1128/MCB.21.16.5644-5657.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brieger J, Mann SA, Pongsapich W, Koutsimpelas D, Fruth K, Mann WJ. Pharmacological genome demethylation increases radiosensitivity of head and neck squamous carcinoma cells. Int J Mol Med 2012; 29:505-9; PMID:22109647 [DOI] [PubMed] [Google Scholar]
- 14.Hofstetter B, Niemierko A, Forrer C, Benhattar J, Albertini V, Pruschy M, Bosman FT, Catapano CV, Ciernik IF. Impact of genomic methylation on radiation sensitivity of colorectal carcinoma. Int J Radiat Oncol Biol Phys 2010; 76:1512-9; PMID:20338477; http://dx.doi.org/ 10.1016/j.ijrobp.2009.10.037 [DOI] [PubMed] [Google Scholar]
- 15.Jiang W, Li YQ, Liu N, Sun Y, He QM, Jiang N, Xu YF, Chen L, Ma J. Five-Azacytidine enhances the radiosensitivity of CNE2 and SUNE1 cells in vitro and in vivo possibly by altering DNA methylation. PLoS One 2014; 9:e93273; PMID:24691157; http://dx.doi.org/ 10.1371/journal.pone.0093273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Geng P, Jiang W, Wang Y, Yao J, Lin X, Liu J, Huang L, Su B, Chen H. Enhancement of radiosensitivity by 5-Aza-CdR through activation of G2/M checkpoint response and apoptosis in osteosarcoma cells. Tumour Biol 2014; 35:4831-9; PMID:24474250; http://dx.doi.org/ 10.1007/s13277-014-1634-5 [DOI] [PubMed] [Google Scholar]
- 17.Qiu H, Yashiro M, Shinto O, Matsuzaki T, Hirakawa K. DNA methyltransferase inhibitor 5-aza-CdR enhances the radiosensitivity of gastric cancer cells. Cancer Sci 2009; 100:181-8; PMID:19037991; http://dx.doi.org/ 10.1111/j.1349-7006.2008.01004.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Zhang Y, Li R, Chen Y, Pan X, Li G, Dai F, Yang J. Five-aza-2'-Deoxycytidine enhances the radiosensitivity of breast cancer cells. Cancer Biother Radiopharm 2013; 28:34-44; PMID:22917213; http://dx.doi.org/ 10.1089/cbr.2012.1170 [DOI] [PubMed] [Google Scholar]
- 19.Fuso A, Nicolia V, Pasqualato A, Fiorenza MT, Cavallaro RA, Scarpa S. Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol Aging 2011; 32:187-99; PMID:19329227; http://dx.doi.org/ 10.1016/j.neurobiolaging.2009.02.013 [DOI] [PubMed] [Google Scholar]
- 20.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol 2010; 28:1057-68; PMID:20944598; http://dx.doi.org/ 10.1038/nbt.1685 [DOI] [PubMed] [Google Scholar]
- 21.Chaudhry MA, Omaruddin RA. Differential DNA methylation alterations in radiation-sensitive and -resistant cells. DNA Cell Biol 2012; 31:908-16; PMID:22185261; http://dx.doi.org/ 10.1089/dna.2011.1509 [DOI] [PubMed] [Google Scholar]
- 22.Antwih DA, Gabbara KM, Lancaster WD, Ruden DM, Zielske SP. Radiation-induced epigenetic DNA methylation modification of radiation-response pathways. Epigenetics 2013; 8:839-48; PMID:23880508; http://dx.doi.org/ 10.4161/epi.25498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pogribny I, Raiche J, Slovack M, Kovalchuk O. Dose-dependence, sex- and tissue-specificity, and persistence of radiation-induced genomic DNA methylation changes. Biochem Biophys Res Commun 2004; 320:1253-61; PMID:15249225; http://dx.doi.org/ 10.1016/j.bbrc.2004.06.081 [DOI] [PubMed] [Google Scholar]
- 24.Kuhmann C, Weichenhan D, Rehli M, Plass C, Schmezer P, Popanda O. DNA methylation changes in cells regrowing after fractioned ionizing radiation. Radiother Oncol 2011; 101:116-21; PMID:21704414; http://dx.doi.org/ 10.1016/j.radonc.2011.05.048 [DOI] [PubMed] [Google Scholar]
- 25.Loree J, Koturbash I, Kutanzi K, Baker M, Pogribny I, Kovalchuk O. Radiation-induced molecular changes in rat mammary tissue: possible implications for radiation-induced carcinogenesis. Int J Radiat Biol 2006; 82:805-15; PMID:17148264; http://dx.doi.org/ 10.1080/09553000600960027 [DOI] [PubMed] [Google Scholar]
- 26.Kim EH, Park AK, Dong SM, Ahn JH, Park WY. Global analysis of CpG methylation reveals epigenetic control of the radiosensitivity in lung cancer cell lines. Oncogene 2010; 29:4725-31; PMID:20531302; http://dx.doi.org/ 10.1038/onc.2010.223 [DOI] [PubMed] [Google Scholar]
- 27.Wang KY, Chen CC, Shen CK. Active DNA demethylation of the vertebrate genomes by DNA methyltransferases: deaminase, dehydroxymethylase or demethylase? Epigenomics 2014; 6:353-63; PMID:25111488; http://dx.doi.org/ 10.2217/epi.14.21 [DOI] [PubMed] [Google Scholar]
- 28.Chen CC, Wang KY, Shen CK. The mammalian de novo DNA methyltransferases DNMT3A and DNMT3B are also DNA 5-hydroxymethylcytosine dehydroxymethylases. J Biol Chem 2012; 287:33116-21; PMID:22898819; http://dx.doi.org/ 10.1074/jbc.C112.406975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turk PW, Laayoun A, Smith SS, Weitzman SA. DNA adduct 8-hydroxyl-2'-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis 1995; 16:1253-5; PMID:7767994; http://dx.doi.org/ 10.1093/carcin/16.5.1253 [DOI] [PubMed] [Google Scholar]
- 30.Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, et al.. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet 2009; 41:1350-3; PMID:19881528; http://dx.doi.org/ 10.1038/ng.471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al.. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178-86; PMID:19151715; http://dx.doi.org/ 10.1038/ng.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13:484-92; PMID:22641018; http://dx.doi.org/ 10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- 33.Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys 2004; 59:928-42; PMID:15234026; http://dx.doi.org/ 10.1016/j.ijrobp.2004.03.005 [DOI] [PubMed] [Google Scholar]
- 34.Edwards PA, Kennedy MA, Mak PA. LXRs; oxysterol-activated nuclear receptors that regulate genes controlling lipid homeostasis. Vascul Pharmacol 2002; 38:249-56; PMID:12449021; http://dx.doi.org/ 10.1016/S1537-1891(02)00175-1 [DOI] [PubMed] [Google Scholar]
- 35.Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer 2008; 123:8-13; PMID:18425818; http://dx.doi.org/ 10.1002/ijc.23607 [DOI] [PubMed] [Google Scholar]
- 36.Christman JK. Five-Azacytidine and 5-aza-2'-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 2002; 21:5483-95; PMID:12154409; http://dx.doi.org/ 10.1038/sj.onc.1205699 [DOI] [PubMed] [Google Scholar]
- 37.Yuan BZ, Jefferson AM, Popescu NC, Reynolds SH. Aberrant gene expression in human non small cell lung carcinoma cells exposed to demethylating agent 5-aza-2'-deoxycytidine. Neoplasia 2004; 6:412-9; PMID:15256063; http://dx.doi.org/ 10.1593/neo.03490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Menendez L, Walker D, Matyunina LV, Dickerson EB, Bowen NJ, Polavarapu N, Benigno BB, McDonald JF. Identification of candidate methylation-responsive genes in ovarian cancer. Mol Cancer 2007; 6:10; PMID:17254359; http://dx.doi.org/ 10.1186/1476-4598-6-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ando T, Nishimura M, Oka Y. Decitabine (5-Aza-2'-deoxycytidine) decreased DNA methylation and expression of MDR-1 gene in K562/ADM cells. Leukemia 2000; 14:1915-20; PMID:11069027; http://dx.doi.org/ 10.1038/sj.leu.2401914 [DOI] [PubMed] [Google Scholar]
- 40.Kitagawa Y, Kyo S, Takakura M, Kanaya T, Koshida K, Namiki M, Inoue M. Demethylating reagent 5-azacytidine inhibits telomerase activity in human prostate cancer cells through transcriptional repression of hTERT. Clin Cancer Res 2000; 6:2868-75; PMID:10914736 [PubMed] [Google Scholar]
- 41.Wong J, Sia YY, Misso NL, Aggarwal S, Ng A, Bhoola KD. Effects of the demethylating agent, 5-azacytidine, on expression of the kallikrein-kinin genes in carcinoma cells of the lung and pleura. Patholog Res Int 2011; 2011:167046; PMID:21904690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang K, Jin W, Knuefermann C, Schmidt M, Mills GB, Ang KK, Milas L, Fan Z. Targeting the phosphatidylinositol 3-kinase/Akt pathway for enhancing breast cancer cells to radiotherapy. Mol Cancer Ther 2003; 2:353-60; PMID:12700279 [PubMed] [Google Scholar]
- 43.Gupta AK, Cerniglia GJ, Mick R, Ahmed MS, Bakanauskas VJ, Muschel RJ, McKenna WG. Radiation sensitization of human cancer cells in vivo by inhibiting the activity of PI3K using LY294002. Int J Rad Oncol Biol Phys 2003; 56:846-53; http://dx.doi.org/ 10.1016/S0360-3016(03)00214-1 [DOI] [PubMed] [Google Scholar]
- 44.Gupta AK, McKenna WG, Weber CN, Feldman MD, Goldsmith JD, Mick R, Machtay M, Rosenthal DI, Bakanauskas VJ, Cerniglia GJ, et al.. Local recurrence in head and neck cancer: relationship to radiation resistance and signal transduction. Clin Cancer Res 2002; 8:885-92; PMID:11895923 [PubMed] [Google Scholar]
- 45.Bussink J, van der Kogel AJ, Kaanders JH. Activation of the PI3-K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer. Lancet Oncol 2008; 9:288-96; PMID:18308254; http://dx.doi.org/ 10.1016/S1470-2045(08)70073-1 [DOI] [PubMed] [Google Scholar]
- 46.Huang KH, Huang SF, Chen IH, Liao CT, Wang HM, Hsieh LL. Methylation of RASSF1A, RASSF2A, and HIN-1 is associated with poor outcome after radiotherapy, but not surgery, in oral squamous cell carcinoma. Clin Cancer Res 2009; 15:4174-80; PMID:19509163; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-2929 [DOI] [PubMed] [Google Scholar]
- 47.Mims J, Bansal N, Bharadwaj MS, Chen X, Molina AJ, Tsang AW, Furdui CM. Energy metabolism in a matched model of radiation resistance for head and neck squamous cell cancer. Radiat Res 2015; 183:291-304; PMID:25738895; http://dx.doi.org/ 10.1667/RR13828.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reed MF, Liu VF, Ladha MH, Ando K, Griffin JD, Weaver DT, Ewen ME. Enforced CDK4 expression in a hematopoietic cell line confers resistance to the G1 arrest induced by ionizing radiation. Oncogene 1998; 17:2961-71; PMID:9881698; http://dx.doi.org/ 10.1038/sj.onc.1202450 [DOI] [PubMed] [Google Scholar]
- 49.Kim HJ, Kim JH, Chie EK, Young PD, Kim IA, Kim IH. DNMT (DNA methyltransferase) inhibitors radiosensitize human cancer cells by suppressing DNA repair activity. Radiat Oncol 2012; 7:39; PMID:22429326; http://dx.doi.org/ 10.1186/1748-717X-7-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roy K, Wang L, Makrigiorgos GM, Price BD. Methylation of the ATM promoter in glioma cells alters ionizing radiation sensitivity. Biochem Biophys Res Commun 2006; 344:821-6; PMID:16631604; http://dx.doi.org/ 10.1016/j.bbrc.2006.03.222 [DOI] [PubMed] [Google Scholar]
- 51.Karolchik D, Barber GP, Casper J, Clawson H, Cline MS, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, et al.. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res 2014; 42:D764-70; PMID:24270787; http://dx.doi.org/ 10.1093/nar/gkt1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol 1987; 196:261-82; PMID:3656447; http://dx.doi.org/ 10.1016/0022-2836(87)90689-9 [DOI] [PubMed] [Google Scholar]
- 53.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J Roy Stat Soc B Met 1995; 57:289-300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.