

Abstract
Estrogen receptor (ER) is a major therapeutic target for the treatment of breast cancer, because of the crucial role of estrogen signaling deregulation in the development and progression of breast cancer. In this study, we report the identification of a novel ERα binding compound, cryptotanshinone (CPT), by screening the CADD database. We also show that CPT effectively inhibits estrogen-induced ER transactivation and gene expression of ER target genes. Furthermore, we showed that CPT suppressed breast cancer cell growth mainly in an ERα dependent manner. Finally, we confirmed the potential therapeutic efficiency of CPT using xenograft experiments in vivo. Taken together, our results describe a novel mechanism for the anticancer activity of CPT and provide supporting evidence for its use as a potential therapeutic agent to treat patients with ERα positive breast cancer.
Keywords: breast cancer, cryptotanshinone, estradiol, estrogen receptor
Abbreviations
- ER
estrogen receptor
- CPT
cryptotanshinone
- EREs
estrogen-responsive elements
- SERM
selective estrogen receptor modulator
- CADD
computer-aided drug design
- ROS
reactive oxygen species
Introduction
Breast cancer is the most commonly diagnosed cancer among women specifically in the United States and remains the second leading cause of cancer deaths.1 Estrogen and estrogen receptor (ER) play a pivotal role in the development and progression of breast cancer.2,3 The ERα, a member of the nuclear hormone receptor superfamily and a ligand-regulated transcription factor, is expressed in at least 70% of breast tumors.4-6 The pro-oncogenic effect of ERα is mediated primarily by the binding of estrogen to ERα, which directly activates the transcription of ER target genes by directly binding to the estrogen-responsive elements (EREs) on their promoters. Indirect activation of gene transcription without EREs on their gene promoters also occurs through interaction with other transcription factors that regulate the expression of multiple target genes, which promote cell proliferation or decrease apoptosis.7-11 Because of the importance of estrogen signaling in the carcinogenesis of breast cancer, ERα has become the target of current endocrine therapies for breast cancer.
Salvia miltiorrhiza Bunge (Danshen), a well-known traditional Chinese herb, is commonly used in the clinical treatment of multiple diseases including cardiovascular diseases, diabetes, hepatitis, ischemic stroke, chronic renal failure and menstrual disorders.12,13 Cryptotanshinone (CPT), one of the major representative components isolated from Danshen, has been reported to possess various pharmacological activities, such as anti-inflammatory, anti-angiogenic, anti-oxidative, anti-obese, and anti-diabetic functions.14-17 Recently, CPT has also been reported to have obvious antitumor activity in a variety of cancer cells, including prostate carcinoma, hepatocarcinoma, rhabdomyosarcoma and melanoma cells.18-23 In breast carcinoma cells, CPT has been shown to inhibit MCF7 cell proliferation by suppressing mTOR mediated CyclinD1 expression and Rb phosphorylation that lead to MCF7 cell apoptosis by inducing stress in the endoplasmic reticulum (ER).24,25 However, it is not known whether CPT has any effects on estrogen-stimulated ER signaling and estrogen /ER mediated cancer cell growth.
In this study, we determined the effects of CPT on estrogen signaling and estrogen induced cell growth. We identified that CPT isolated from natural products could be a novel ERα binding compound and show that CPT suppressed E2-induced ER transcriptional activity and the expression of ER target genes through competitive binding of ERα. Consistent with the inhibitory effect of CPT on estrogen signaling, CPT treatment more effectively inhibits the growth of ERα positive breast cancer cells compared ERα negative breast cancer cells. We further confirmed the potential therapeutic efficiency of CPT using xenograft experiments in vivo. Our findings show that CPT is an anti-ERα isolated from a natural source, with potential to be used in the therapeutic treatment of ERα positive breast cancer patients.
Results
Identification of CPT as a novel ERα modulator
To expedite identification of ERα antagonists that may be directly used for the treatment of ER-associated breast cancer, we chose to screen a database of natural products using computer-aided drug design (CADD) methods based on the 3D structure of the ERα protein. The binding pocket of ERα was made of Leu346, Thr347, Ala350, Glu353, Leu387, Met388, Leu391, Arg394, Phe404, Met421, Ile424, Leu525, and His524. Since most of the residues are non-polar, the binding pocket was presumed to be hydrophobic. Among the ∼10,000 natural products screened virtually, CPT ranked among the top 10% of the compounds with the best glide scores for docking to the human ERα protein. Therefore, CPT was selected and subjected to experimental testing. To evaluate the mode of binding between CPT and ERα protein, computer docking was simulated. Since the chemical structure of estradiol (Fig. 1A) is similar to CPT with 3 cyclohexane rings, one cyclopentane ring and 17 carbon atoms, estradiol was chosen for the simulation. As shown in Figure 1B, estradiol can form 2 hydrogen bonds with Glu353 and Arg394 at the entrance of the active sites of ERα. Although CPT has no hydrogen bond with ERα, it was docked into the hydrophobic pocket of this protein, and the binding conformation overlapped well with that of estradiol. Evidently, CPT has interaction energy comparable to that of estradiol with ERα (Fig. 1B). To validate that CPT binds directly to the ERα protein, we examined whether binding of the modulator altered the fluorescence of the protein using the ERα competitive-binding assay. As shown in Figure 1C, CPT significantly quenched ERα fluorescence with the IC50 value of 1.36 nM indicating that CPT binds directly to ERα, and its potential as a promising ERα antagonist for the development of novel anti-breast cancer drug.
Figure 1.
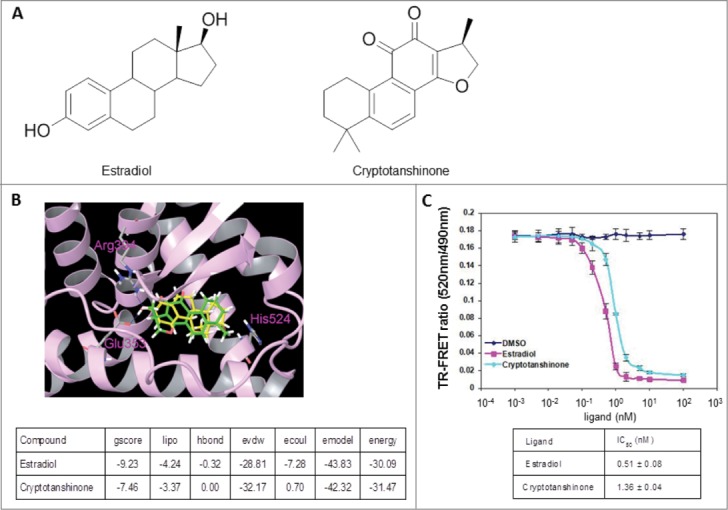
CPT is an ERα modulator. (A) Chemical structure of estradiol and CPT. (B) Molecular model of estradiol and CPT docking with ERα. The protein structure is shown in plum color (ribbon), CPT and estradiol are shown in green and yellow carbon scheme, respectively. Hydrogen bonds are shown in blue dashed lines. (C) Direct binding of CPT to ERα in the ERα competitive-binding assay. Assay composition consisted of serial dilutions of the indicated compounds in 1% final DMSO concentration, 2 nM ERα-LBD, 3 nM Fluormone™ ES2 Green, and 2 nM Tb anti-GST antibody. Results are shown with the corresponding IC50 values. Experiments were repeated 3 times. Data shown are the mean ± SD.
CPT inhibited ERα-mediated transcriptional activity but not ERα protein levels
Since the previous results demonstrated that CPT could bind directly to the ERα protein, we next tested whether CPT could modulate ERα function. For this, we first investigated the ability of CPT to regulate the ER-mediated transactivation activity. To test this possibility, ERα positive breast cancer cells (ZR-75-1 and MCF7) were transiently transfected with an ERE reporter construct. In both ZR-75-1 and MCF7 cells, E2 stimulated an ERE-driven luciferase reporter activity, and CPT effectively suppressed the E2-induced transcriptional activity in a concentrations dependent manner (Fig. 2A). To further illustrate the ability of CPT to regulate ER-mediated transactivation activity, we explored the effect of CPT on mRNA as well as protein levels of ER target genes in both ZR-75-1 and MCF7 cells. Our data showed that the ER target gene (pS2, and Cat D) mRNA levels were induced by 10 nM E2, and addition of 2 μM CPT significantly inhibited E2-induced pS2, and Cat D mRNA levels in both ZR-75-1 and MCF7 cells (Fig. 2B). In addition, CPT also inhibited E2-induced pS2, and Cat D protein levels in both ZR-75-1 and MCF7 cells in a concentration dependent manner (Fig. 2C). However, we found that CPT has little effect on ERα protein levels in both ZR-75-1 and MCF7 cells (Fig. 2C), indicating that CPT may inhibit ER transcriptional activity through competitive binding of ERα, rather than by down-regulating ERα expression levels.
Figure 2.
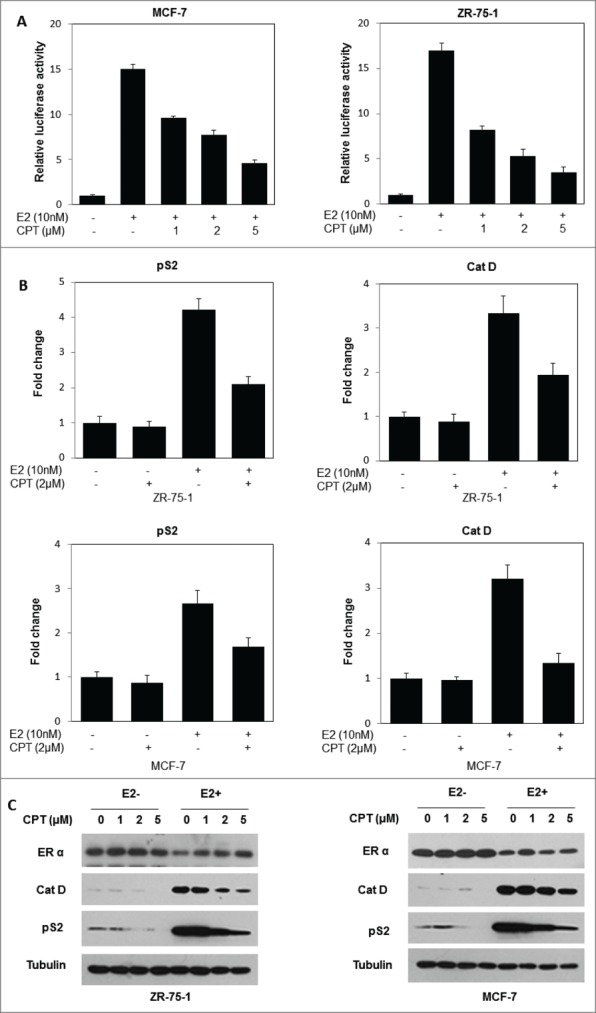
CPT inhibits ER-mediated transcriptional activity but not ERα protein levels. (A) CPT inhibits E2-induced ERE reporter gene activity. ZR-75-1 and MCF7 cells were transiently transfected with an ERE reporter construct or a control Renilla luciferase construct. After 24 h of transfection, cells were treated with various concentrations of CPT with 10 nM E2 or left untreated for 24 h. Results are shown as mean ± SD of 3 independent experiments. (B) ZR-75-1 and MCF7 cells were treated with or without 10 nM E2, 2 μM CPT or a combination of both for 24 h and then the mRNA expression of ER target genes (pS2, and Cat D) was analyzed using real-time RT-PCR. Data shown are mean ± SD of 3 independent experiments with 3 replicates in each experiment. (C) ZR-75-1 and MCF7 cells were treated with various concentrations of CPT with or without 10 nM E2. After treatment for 48 h, the cells were harvested and were analyzed for protein expressions of ER target genes (pS2, and Cat D) using Western blot. Experiments were repeated 3 times and similar results were obtained in all. Therefore, representative result from one experiment is shown.
CPT inhibits estrogen-stimulated cell growth and survival in ERα-positive breast cancer cells
Because of the critical role of ER signaling in breast cancer cell proliferation and survival, we next investigated the effect of CPT on cell proliferation and survival of ERα positive breast cancer cells (ZR-75-1 and MCF7) in the presence or absence of E2. As shown in Figure 3A, cell growth was assessed by CCK8 assay. In the presence of 10 nM E2, treatment with CPT for 48 h inhibited cell growth in a dose-dependent manner. However, in the absence of E2, the inhibitory effect of different CPT concentrations on cell growth was not significantly antagonized in both cells. We then examined the effects of CPT on cell survival using Colony-Forming Unit assay, and obtained results similar to cell proliferation (Fig. 3B). These findings suggest that CPT could inhibit estrogen-stimulated cell growth and survival in ERα-positive breast cancer cells.
Figure 3.
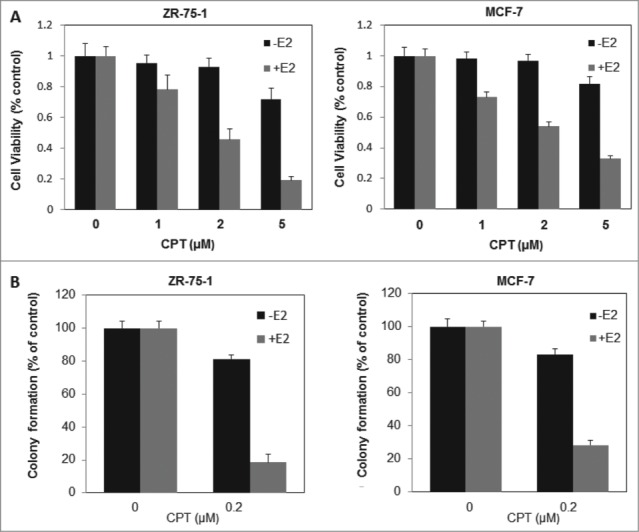
CPT inhibits estrogen-stimulated cell growth and survival in ERα-positive breast cancer cells. (A) ZR-75-1 and MCF7 cells were grown in phenol red-free DMEM supplemented with 10% charcoal/dextran without FBS for 24 h and then treated with various concentrations of CPT with or without 10 nM E2. After treatment for 48 h, cell growth was analyzed by CCK8 assay. Bars (mean ± SD) indicate the average of 3 independent experiments. (B) ZR-75-1 and MCF7 cells were plated in phenol red-free DMEM supplemented with 10% charcoal/dextran without FBS. When attached, the cells were treated with or without 10 nM E2, 0.2 μM CPT or a combination of both. Colonies were enumerated 10 days (ZR-75-1 cells) or 16 days (MCF7 cells) later and normalized against the number of colonies derived from control cells without CPT treatment. Data are shown as mean ± SD. Representative results from 2 independent experiments are shown.
CPT inhibited breast cancer cell growth in an ERα dependent manner
Previous results showed that CPT effectively inhibited the cell growth and survival of ERα-positive human breast cancer cells in the presence of E2, compared to the absence of E2, which indicates that CPT may inhibit cell growth and survival through its antiestrogen activity and that ERα is required for its inhibitory effect on cell growth. To further test this hypothesis, the half maximal inhibitory concentration (IC50) of CPT on ERα positive breast cancer cells (ZR-75-1 and MCF7) and ERα negative breast cancer cells (MDA-MB-231 and MDA-MB-435) was determined by half-inhibition of cell growth 48 h after CPT treatments with or without 10 nM E2. In the presence of E2, the IC50 concentrations of CPT in ZR-75-1, MCF7, MDA-MB-231 and MDA-MB-435 were 1.7 ± 0.1 μM, 1.1 ± 0.1 μM, 21.9 ± 1.5 μM, and 17.9 ± 1.2 μM, respectively (Fig. 4A). In the absence of E2, the IC50 concentrations of CPT in ZR-75-1, MCF7, MDA-MB-231 and MDA-MB-435 were 15.8 ± 0.8 μM, 12.2 ± 0.6 μM, 18.8 ± 1.1 μM, and 19.3 ± 1.6 μM, respectively (Fig. 4A). These results indicate that CPT more effectively inhibited the cell growth of ERα-positive human breast cancer cells compared to ERα negative breast cancer cells, confirming the hypothesis that ER is required for the inhibitory effect of CPT on cell growth. To further validate this hypothesis, we suppressed the expression of ERα in both ZR-75-1 and MCF7 cells and investigated whether the depletion of ERα alters the inhibitory effect of CPT on cell growth in the presence of E2. Figure 4B and C shows that ERα knockdown suppressed the E2-induced transcriptional activity. As shown in Figure 4D, ERα knockdown suppressed the inhibitory effect of CPT on cell growth, confirming that CPT inhibits breast cancer cell growth by inhibiting ERα.
Figure 4.

CPT inhibited breast cancer cell growth in an ER dependent manner. (A) IC50 of CPT in different breast cancer cells. We determined the IC50 values of CPT in ZR-75-1, MCF7, MDA-MB-231 and MDA-MB-435 cells. All cells were seeded on 96 well plates in phenol red-free DMEM supplemented with 10% charcoal/dextran without FBS for 24 h and then treated with a range of CPT concentrations alone or with 10 nM E2 for another 48 h. IC50 values were determined by CCK8 assay. Data represent mean ± SD of 3 independent experiments with 3 replicates in each experiment. (B and C) ERα knockdown suppressed the E2-induced transcriptional activity. Control and ERα-depleted ZR-75-1 and MCF7 cells were transiently transfected with an ERE reporter construct or a control Renilla luciferase construct. After 24 h of transfection, cells were treated with various concentrations of CPT with 10 nM E2 or left untreated for 24 h. Results are shown as mean ± SD of 3 independent experiments. (D) The inhibitory effect of CPT on cell growth was suppressed by silencing ERα. Control and ERα-depleted ZR-75-1 and MCF7 cells were grown in phenol red-free DMEM supplemented with 10% charcoal/dextran without FBS for 24 h and then treated with various concentrations of CPT in the presence of 10 nM E2. Two days later, cell growth was analyzed by CCK8 assay. Data are shown as mean ± SD of 3 independent experiments.
CPT can effectively inhibit breast cancer growth in vivo
We next determined the therapeutic efficiency of CPT using xenograft experiments in vivo. To test the CPT anti-cancer effect in vivo, the ZR-75-1 xenograft model was established. When tumors reached an approximate volume of 100 mm3, mice were randomly allocated to 2 groups and treated with control oil or 15 mg/kg CPT every 2 days by intra-peritoneal injection. After 3 weeks of treatment, the mice were sacrificed and tumors were collected. The results show that CPT treatments effectively inhibited tumor growth in BALB/c nude mice (Fig. 5A–C), but has little effect on mouse body weight (Fig. 5D). Moreover, we found that the ER target gene protein expression (pS2, and Cat D) was significantly suppressed in CPT treated group without affecting the ERα protein level (Fig. 5E), indicating that CPT can effectively inhibit ER signaling in an in vivo xenograft model.
Figure 5.
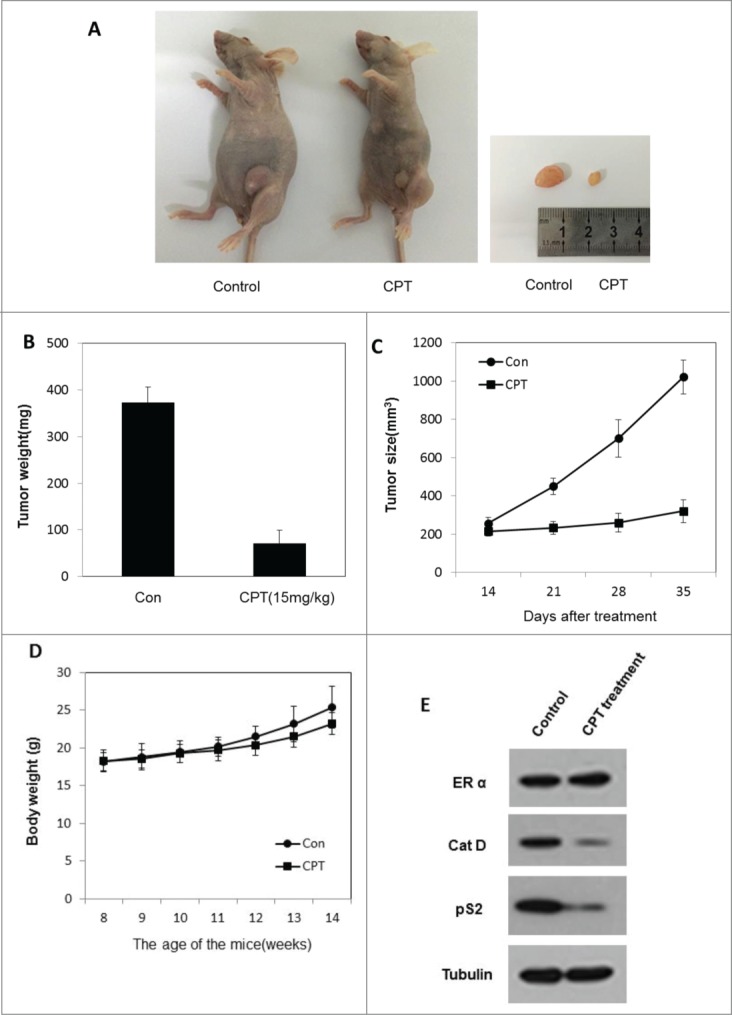
CPT effectively inhibited breast cancer growth in vivo. To test the therapeutic effect of CPT, 1 × 107 ZR-75-1 cells were injected into the abdominal mammary fat pad of 6-weeks old female BALB/c nude mice after implantation of estrogen pellets for 2 days. After 2–3 weeks, when tumor sizes were approximately 100 mm3, mice were randomly allocated to 2 experimental groups and intraperinoteally injected with control oil or 15 mg/kg CPT every 2 days for 3 weeks. The mice were then sacrificed and the tumors were collected for data analysis. (A and B) CPT inhibited tumor growth in the in vivo ZR-75-1 xenograft model. (C) CPT treatment inhibited ZR-75-1 tumor weights. Data are shown as mean ± SD (n = 6 for each group, P < 0.05). (D) CPT treatments had little effect on mouse body weights. Data are shown as mean ± SD (n = 6 for each group, P < 0.05). (E) CPT treatment inhibited the protein expression of ER target genes, pS2, and Cat D, in the in vivo xenografted ZR-75-1 tumors.
Discussion
Down-regulated estrogen signaling is crucial for the development and progression of breast cancer. Estrogen mediates its biological effects primarily by binding to ER, that results in conformational changes in ER protein structure, followed by ER dimerization, and binding to estrogen response elements (ERE) on the promoters of estrogen-responsive genes. Increased transcription of ER target genes promotes cell proliferation and antagonizes cell apoptosis,9,11,26 which contribute to tumorigenesis.
In this study, using CADD database screening of natural products, we identified CPT as a novel ERα binding compound that is structurally similar to estradiol. Molecular modeling predicted that CPT could target the hydrophobic pocket of ERα and has interaction energy comparable to estradiol with ERα. We further confirmed that CPT directly binds to ERα through fluorescence quenching experiments. To test the effect of CPT on ER transcriptional activity, we determined the relative luciferase activity within ERα positive breast cancer cells (ZR-75-1 and MCF7) transiently transfected with an ERE reporter construct. The results showed that CPT suppressed the ERE-driven luciferase reporter activity induced by E2. To further confirm the ability of CPT to regulate ER transcriptional activity, expression of ER target genes was assayed in ERα positive breast cancer cells treated with CPT and E2. Our data showed that the ER target gene (pS2 and Cat D) mRNA and protein levels were induced by E2, and CPT significantly suppressed the E2-induced ER target gene expression in ZR-75-1 and MCF7 cells. Further results showed that CPT had little effect on the protein levels of ERα, indicating that CPT may inhibit ER transcriptional activity through competitive binding of ERα, rather than by down-regulating ERα expression levels. We then investigated the effects of CPT on the cell proliferation of ERα positive breast cancer cells (ZR-75-1 and MCF7) in the presence or absence of E2. The results showed that CPT inhibits cell growth in a dose-dependent manner in the presence of E2, but had moderate inhibitory effect on cell growth in the absence of E2, indicating that CPT affected breast cancer cell growth through ER dependent signaling. To confirm the inhibition of CPT to breast cancer cell growth in a ER signaling dependent manner, we compared IC50 values of CPT in ERα positive (ZR-75-1 and MCF7) and ERα negative (MDA-MB-231 and MDA-MB-435) breast cancer cells in the presence or absence of E2. We found that that CPT more effectively inhibited the growth of ERα positive breast cancer cells. The result that depletion of ERα antagonized the inhibitory effect of CPT to ERα positive breast cancer cell growth further validated that CPT inhibits breast cancer cell growth by inhibiting ERα. Recently, CPT was reported to inhibit the androgen receptor activity and suppress prostate cancer growth in androgen-dependent manner, but the reason for that is not due to the direct bind to androgen receptor and is attributed to its functional inhibition of LSD1-mediated demethylation of H3K9.19,20 Thanshinone I has been reported to induce apoptosis of breast cancer cells,27 so it is possible that Thanshinone I also could inhibit ER transcriptional activity due to the similarity of the chemical structures of Thanshinone I and CPT. However, we found that anshinones I less effectively inhibited E2-induced transcriptional activity and expression of the ER target gene (pS2, and Cat D) in the presence of E2, compared with CPT, which is consistent with the same inhibitory effect of anshinones I on ER-positive as well as ER-negative cancer cells. Taken together, we think that the effect of CPT on the binding to ERa and inhibition of ER signaling is specific to a certain extent.
Recently, CPT has been reported to exert potential anticancer activity in multiple cancer cells through different mechanisms. CPT has been found to suppress DU145 cell growth by inhibiting STAT3 signaling, mTOR-mediated cyclin D1 expression and Rb phosphorylation.18,24 In addition, CPT has been found to inhibit HepG2 cell proliferation by arresting cell cycle at the G1 phase in an AMPK-dependent manner.28 It has been found that CPT leads to caspase-independent cell death in Rh30 and DU145 cells by activating p38/JNK signaling and inhibiting Erk1/2 signaling in a reactive oxygen species (ROS) dependent manner.22 CPT has been found to induce MCF7 cell apoptosis in a similar manner. Additionally, recent studies demonstrated that CPT induces endoplasmic reticulum (ER) stress-mediated apoptosis in MCF7 cell via ROS generation.25 However, the effects of CPT on estrogen signaling and estrogen induced cell growth remain unclear, although estrogen signaling has been recognized as a crucial carcinogenic factor in breast cancer. Our study demonstrated that CPT inhibits ER transcriptional activity and affects breast cancer cell growth in an ER dependent manner, indicating that CPT has the potential to be developed as an anti-ERα drug for the treatment of ERα positive breast cancer patients. Further, higher concentrations of CPT (more than 5 μM) was found to inhibit the cell proliferation of ERα positive breast cancer cells (ZR-75-1 and MCF7) in the absence of E2 and ERα negative breast cancer cells (MDA-MB-231 and MDA-MB-435) in the presence or absence of E2. Based on these results, we presume that CPT could control breast cancer cell growth by regulating other signaling pathways reported previously, such as mTOR, p38/JNK, Erk1/2 signaling or induction of endoplasmic reticulum (ER) stress, rather than estrogen signaling. Therefore, it is also possible that CPT has the potential to treat ERα negative breast cancer patients, such as those resistant to tamoxifen.
In summary, our data reveal that CPT is a novel potential estrogen signaling inhibitor, which could suppress E2-induced ER transcriptional activity through competitive binding and E2-stimulated cell growth and survival in ERα-positive breast cancer cells. These results describe a novel mechanism of anticancer activity for CPT and expand our understanding of the signaling pathways regulated by CPT. More importantly the data presented in this study suggest the possibility for the use of CPT as a novel potential antiestrogen drug to treat patients with ERα positive breast cancer.
Materials and Methods
Antibodies and chemicals
Antibodies from Santa Cruz (Santa Cruz, CA, USA) included ER-α and tubulin. Anti-pS2 was Abcam. Anti-Cathepsin D antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). Cryptotanshinone was obtained from Xi’an HaoXuan Bio-Tech Co., Ltd (Xi’ an, China), and dissolved in DMSO to prepare stock solutions (20 mM) for subsequent experiments. 17 b-estradiol (E2) was purchased from Sigma (St. Louis, MO, USA). All test compounds possess a purity of ≥98% by HPLC according to the manufacturer.
Computational modeling of the CPT docking pose
Briefly, the 3-dimensional (3D) crystal structure of ERα was downloaded from Protein Data Bank database (http://rcsb.org) using the ID code 1ERR. Chain A was used for molecular docking, hydrogen atoms were added using the Protein Preparation Wizard in the Schrödinger program, and the force field used was OPLS_2005. The center of mass (COM) of Glu353, Arg394, and His524 was defined as the center of the binding pockets with a box size of 10 Å radius to produce grid parameters. Van der Waals radius correction factor was 1.0, and partial charge cutoff was 0.25. The structures of CPT and estradiol were built in Maestro and processed by Ligprep module. Docking calculation was performed by Glide module in the Schrödinger program with default parameters.
In vitro estrogen receptor (ER) competitive-binding assay
This assay was performed as described in the protocol of the LanthaScreen® TR-FRET Estrogen Receptor α Competitive Binding Assay Kit (Carlsbad, CA, USA). Briefly, 5 μL of Fluormone™ ES2 Green tracer (3 nM) was added to 10 μL of each of the compound serial dilutions (1% final DMSO concentration) or solvent controls in Nuclear Receptor Buffer K (containing 5 mM DTT) followed by addition of a mixture of 5 μL ERα -LBD (2 nM) and terbium anti-GST antibody (2 nM) in a black 384-well plate. After an incubation period at room temperature for 2 h, the excitation wavelength was 340 nm and fluorescence was monitored from 495 to 520 nm using a fluorescence plate reader and the TR-FRET ratio of 520:495 emissions was calculated and can be used to determine the IC50 from a dose response curve by plotting the emission vs. the log [ligand].
Cell culture
ZR-75-1, MCF7, MDA-MB-231 and MDA-MB-435 breast cancer cells were routinely cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Hyclone, Logan, UT, USA). For hormone treatment experiments, cells were maintained in phenol red–free DMEM containing 10% charcoal/dextran stripped FBS (Hyclone). Cells were cultured in an incubator at 37°C and 5% CO2.
To decrease the expression of ER, ZR-75-1 cells were infected with appropriate amounts of lentiviral particles carrying ER shRNA or control shRNA (lentivirus obtained from GeneChem Co., Shanghai, China) with 8 ug/ml polybrene. Twelve hours later, virus-containing medium was refreshed with fresh medium. Infected cells were selected for stable cell lines in 1 ug/ml puromycin 48 h later.
Luciferase reporter assays
Cells were seeded in triplicate in 96-well culture plates with 10% charcoal stripped (CS)-FBS in phenol red-free DMEM medium for 24 h and transiently transfected with pGL3-(ERE)3-Luc and the pRL-TK-Luc plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. After 24 h post-transfection, the cells were treated with serial concentrations of CPT with 10 nM E2 or left untreated for 24 h. Then, cell lysates were prepared and assayed for luciferase activity using the dual luciferase assay system (Promega, Madison, WI, USA) according to the manufacturer's instructions.
mRNA isolation and quantitative real-time PCR
Total RNA was isolated from breast cancer cells using TRIzol reagent (Invitrogen, Carlsbad, CA,USA) and reverse transcription was performed using SuperScript II Reverse Transcriptase (Invitrogen) following the manufacturer's instructions. Quantitative real-time PCR analyses were performed using the SYBR Green PCR Master Mix kit (Perkin Elmer, Applied Biosystems, Wellesley, MA, USA) according to the manufacturer's instructions. The pS2 primer pairs were 5′-CATCGACGTCCCTCCAGAAGAG-3′ and 5′-CTCTGGGACTAATCACCGTGCTG-3′. The Cat-D primer pairs were 5′-CGCGATCACACTGAAGCTGG-3′ and 5′-ACAGTGTAGTAGCGGCCGATG-3′. The β-actin control primers used were 5′-ATCACCATTGGCAATGAGCG-3′ and 5′-TTGAAGGTAGTTTCGTGGAT-3′.
Western blot
Western blot were performed as described previously.29 Briefly, the whole cell lysates were prepared and the concentration of protein was measured using the BCA protein reagent (Pierce Chemical, Rockford, IL, USA). Approximately 50 ug of protein were resolved by 10% SDS-PAGE gel and then transferred to nitrocellulose membranes. After being blocked with 5% nonfat milk for 2 h at room temperature, the membranes were incubated with primary antibodies overnight at 4°C, followed by incubation with secondary antibodies for 2 h at room temperature. Protein expressions were visualized using enhanced chemiluminescence (ECL) detection reagent.
Cell growth assay
Cells were seeded at a density of 1500 cells per well in triplicate in 96-well culture plates, and cultured in phenol red-free DMEM supplemented with 10% charcoal/dextran stripped FBS. Once attached, the cells were treated with serial concentrations of CPT with or without 10 nM E2. After treatment for 48 h, cell growth was assessed using the Cell Counting Kit 8 (Dojindo Laboratories, Tokyo, Japan) according to the manufacturer's instructions. Data are presented as means ± s.d.
Colony-Forming Unit Assay
ZR-75-1 cells (500 cells per well) or MCF7 cells (2500 cells per well) were in triplicate in 6-well culture plates and cultured in phenol red-free DMEM supplemented with 10% charcoal/dextran stripped FBS. Once attached, the cells were treated with or without 10 nM E2, 0.2 uM CPT or a combination. After 10 days (ZR-75-1 cells) or 16 days (MCF7 cells) of culture at 37°C and 5% CO2, colony forming units (CFUs) were assayed.
IC50 assay
To determine the IC50 value of CPT, 4 breast cancer cells, ZR75-1, MCF7, MDA-MB-231 and MDA-MB-435 were plated at a density of 5000 cells per well in 96-well culture plates. The cells were grown in phenol red-free DMEM supplemented with 10% charcoal/dextran without FBS. When attached, the cells were treated with various concentrations of CPT alone or with 10 nM E2 for 48 h. Then, cell viability was determined using the Cell Counting Kit 8 assay. IC50 value was analyzed using the program CompuSyn (Developer). All experiments were performed in triplicates.
Xenograft animal model
About 6-weeks old female BALB/c nude mice (Vital River Laboratories, Beijing, China) were implanted with estrogen pellets (E2, 0.36 mg/pellet, 60-day release) (Innovative Research of America, Sarasota, FL, USA). Two days after implantation, mice were injected 1 × 107 ZR-75-1 cells into the abdominal mammary fat pad. When the tumors reached an approximate size of 100 mm3, which took about 2–3 weeks, the mice were randomly allocated to 2 experimental groups (n = 6); while one group was treated with control oil the other group was treated with 15 mg/kg CPT by intra-peritoneal (i.p.) injection every 2 days for 3 weeks. The body weights of mice and tumor growth were monitored every week. After 3 weeks of treatment, all mice were sacrificed and the tumors were weighed. To detect CPT effects on ERα and the protein levels of its target genes, proteins were extracted from the tumor samples stored in liquid nitrogen. Western blot analyses were performed as described in Section 2.6.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Dr. Qinong Ye for providing pGL3-(ERE)3-Luc plasmid.
Funding
This work was supported by National Science and Technology Major Project of the Ministry of Science and Technology of China (2012ZX10003008002, to SH Li), National Natural Science Foundation of China (81372770, to JG Zhou).
References
- 1. Ma J, Jemal A. Breast Cancer Statistics Breast Cancer Metastasis and Drug Resistance. New York: Springer; 2013; 1-18 [Google Scholar]
- 2. Platet N, Cathiard AM, Gleizes M, Garcia M. Estrogens and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion. Crit Rev Oncol Hematol 2004; 51:55-67; PMID:15207254; http://dx.doi.org/ 10.1016/j.critrevonc.2004.02.001 [DOI] [PubMed] [Google Scholar]
- 3. Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. New Engl J Med 2006; 354:270-82; http://dx.doi.org/ 10.1056/NEJMra050776 [DOI] [PubMed] [Google Scholar]
- 4. Osborne CK, Schiff R, Fuqua SA, Shou J. Estrogen receptor: current understanding of its activation and modulation. Clin Cancer Res 2001; 7:4338s-42s; PMID:11916222 [PubMed] [Google Scholar]
- 5. Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res 2001; 29:2905-19; PMID:11452016; http://dx.doi.org/ 10.1093/nar/29.14.2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nadji M, Gomez-Fernandez C, Ganjei-Azar P, Morales AR. Immunohistochemistry of estrogen and progesterone receptors reconsidered: experience with 5,993 breast cancers. Am J Clin Pathol 2005; 123:21-7; PMID:15762276; http://dx.doi.org/ 10.1309/4WV79-N2GHJ3X1841 [DOI] [PubMed] [Google Scholar]
- 7. Shang Y. Molecular mechanisms of oestrogen and SERMs in endometrial carcinogenesis. Nat Rev Cancer 2006; 6:360-8; PMID:16633364; http://dx.doi.org/ 10.1038/nrc1879 [DOI] [PubMed] [Google Scholar]
- 8. Liang J, Shang Y. Estrogen and cancer. Annu Rev Physiol 2013; 75:225-40; PMID:23043248; http://dx.doi.org/ 10.1146/annurev-physiol-030212-183708 [DOI] [PubMed] [Google Scholar]
- 9. Caldon CE, Sutherland RL, Musgrove E. Cell cycle proteins in epithelial cell differentiation: implications for breast cancer. Cell Cycle 2010; 9:1918-28; PMID:20473028; http://dx.doi.org/ 10.4161/cc.9.10.11474 [DOI] [PubMed] [Google Scholar]
- 10. Butt AJ, McNeil CM, Musgrove EA, Sutherland RL. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: the potential roles of c-Myc, cyclin D1 and cyclin E. Endocr-Relat Cancer 2005; 12 (Suppl 1):S47-59; PMID:16113099 [DOI] [PubMed] [Google Scholar]
- 11. Gompel A, Somai S, Chaouat M, Kazem A, Kloosterboer HJ, Beusman I, Forgez P, Mimoun M, Rostène W. Hormonal regulation of apoptosis in breast cells and tissues. Steroids 2000; 65:593-8; PMID:11108864; http://dx.doi.org/ 10.1016/S0039-128X(00)00172-0 [DOI] [PubMed] [Google Scholar]
- 12. Chan K, Chui SH, Wong DY, Ha WY, Chan CL, Wong RN. Protective effects of Danshensu from the aqueous extract of Salvia miltiorrhiza (Danshen) against homocysteine-induced endothelial dysfunction. Life Sci 2004; 75:3157-71; PMID:15488895; http://dx.doi.org/ 10.1016/j.lfs.2004.06.010 [DOI] [PubMed] [Google Scholar]
- 13. Wang X, Morris-Natschke SL, Lee KH. New developments in the chemistry and biology of the bioactive constituents of Tanshen. Med Res Rev 2007; 27:133-48; PMID:16888751; http://dx.doi.org/ 10.1002/med.20077 [DOI] [PubMed] [Google Scholar]
- 14. Kang BY, Chung SW, Kim SH, Ryu SY, Kim TS. Inhibition of interleukin-12 and interferon-gamma production in immune cells by tanshinones from Salvia miltiorrhiza. Immunopharmacology 2000; 49:355-61; PMID:10996033; http://dx.doi.org/ 10.1016/S0162-3109(00)00256-3 [DOI] [PubMed] [Google Scholar]
- 15. Hur JM, Shim JS, Jung HJ, Kwon HJ. Cryptotanshinone but not tanshinone IIA inhibits angiogenesisin vitro. Exp Mol Med 2005; 37:133-7; PMID:15886527; http://dx.doi.org/ 10.1038/emm.2005.18 [DOI] [PubMed] [Google Scholar]
- 16. Park EJ, Zhao YZ, Kim YC, Sohn DH. Preventive effects of a purified extract isolated from Salvia miltiorrhiza enriched with tanshinone I, tanshinone IIA and cryptotanshinone on hepatocyte injury in vitro and in vivo. Food Chem Toxicol 2009; 47:2742-8; PMID:19695300; http://dx.doi.org/ 10.1016/j.fct.2009.08.007 [DOI] [PubMed] [Google Scholar]
- 17. Kim EJ, Jung SN, Son KH, Kim SR, Ha TY, Park MG, Jo IG, Park JG, Choe W, Kim SS, et al. Antidiabetes and antiobesity effect of cryptotanshinone via activation of AMP-activated protein kinase. Mol Pharmacol 2007; 72:62-72; PMID:17429005; http://dx.doi.org/ 10.1124/mol.107.034447 [DOI] [PubMed] [Google Scholar]
- 18. Shin DS, Kim HN, Shin KD, Yoon YJ, Kim SJ, Han DC, Kwon BM. Cryptotanshinone inhibits constitutive signal transducer and activator of transcription 3 function through blocking the dimerization in DU145 prostate cancer cells. Cancer Res 2009; 69:193-202; PMID:19118003; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2575 [DOI] [PubMed] [Google Scholar]
- 19. Wu CY, Hsieh CY, Huang KE, Chang C, Kang HY. Cryptotanshinone down-regulates androgen receptor signaling by modulating lysine-specific demethylase 1 function. Int J Cancer 2012; 131:1423-34; PMID:22052438; http://dx.doi.org/ 10.1002/ijc.27343 [DOI] [PubMed] [Google Scholar]
- 20. Xu D, Lin TH, Li S, Da J, Wen XQ, Ding J, Chang C, Yeh S. Cryptotanshinone suppresses androgen receptor-mediated growth in androgen dependent and castration resistant prostate cancer cells. Cancer Lett 2012; 316:11-22; PMID:22154085; http://dx.doi.org/ 10.1016/j.canlet.2011.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee WY, Liu KW, Yeung JH. Reactive oxygen species-mediated kinase activation by dihydrotanshinone in tanshinones-induced apoptosis in HepG2 cells. Cancer Lett 2009; 285:46-57; PMID:19467570; http://dx.doi.org/ 10.1016/j.canlet.2009.04.040 [DOI] [PubMed] [Google Scholar]
- 22. Chen W, Liu L, Luo Y, Odaka Y, Awate S, Zhou H, Shen T, Zheng S, Lu Y, Huang S. Cryptotanshinone activates p38JNK and inhibits Erk12 leading to caspase-independent cell death in tumor cells. Cancer Prev Res 2012; 5:778-87; http://dx.doi.org/ 10.1158/1940-6207.CAPR-11-0551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen L, Zheng SZ, Sun ZG, Wang AY, Huang CH, Punchard NA, Huang SL, Gao X, Lu Y. Cryptotanshinone has diverse effects on cell cycle events in melanoma cell lines with different metastatic capacity. Cancer Chemoth Pharm 2011; 68:17-27; http://dx.doi.org/ 10.1007/s00280-010-1440-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen W, Luo Y, Liu L, Zhou H, Xu B, Han X, Shen T, Liu Z, Lu Y, Huang S. Cryptotanshinone inhibits cancer cell proliferation by suppressing Mammalian target of rapamycin-mediated cyclin D1 expression and Rb phosphorylation. Cancer Prev Res 2010; 3:1015-25; http://dx.doi.org/ 10.1158/1940-6207.CAPR-10-0020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park IJ, Kim MJ, Park OJ, Choe W, Kang I, Kim SS, Ha J. Cryptotanshinone induces ER stress-mediated apoptosis in HepG2 and MCF7 cells. Apoptosis 2012; 17:248-57; PMID:22113823; http://dx.doi.org/ 10.1007/s10495-011-0680-3 [DOI] [PubMed] [Google Scholar]
- 26. Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 2011; 11:558-72; PMID:21734724; http://dx.doi.org/ 10.1038/nrc3090 [DOI] [PubMed] [Google Scholar]
- 27. Nizamutdinova IT, Lee GW, Son KH, Jeon SJ, Kang SS, Kim YS, Lee JH, Seo HG, Chang KC, Kim HJ. Tanshinone I effectively induces apoptosis in estrogen receptor-positive (MCF-7) and estrogen receptor-negative (MDA-MB-231) breast cancer cells. Int J Oncol 2008; 33(3):485-91; PMID:18695877 [PubMed] [Google Scholar]
- 28. Park IJ, Yang WK, Nam SH, Hong J, Yang KR, Kim J, Kim SS, Choe W, Kang I, Ha J. Cryptotanshinone induces G1 cell cycle arrest and autophagic cell death by activating the AMP-activated protein kinase signal pathway in HepG2 hepatoma. Apoptosis 2014; 19:615-18; PMID:24173372; http://dx.doi.org/ 10.1007/s10495-013-0929-0 [DOI] [PubMed] [Google Scholar]
- 29. Wang H, Wu R, Yu L, Wu F, Li S, Zhao Y, Li H, Luo G, Wang J, Zhou J. SGEF is overexpressed in prostate cancer and contributes to prostate cancer progression. Oncol Rep 2012; 28:1468-74; PMID:22824926 [DOI] [PubMed] [Google Scholar]