

Abstract
The IgG1 Fc is a dimeric protein that mediates important antibody effector functions by interacting with Fcγ receptors (FcγRs) and the neonatal Fc receptor (FcRn). Here, we report the discovery of a monomeric IgG1 Fc (mFc) that bound to FcγRI with very high affinity, but not to FcγRIIIa, in contrast to wild-type (dimeric) Fc. The binding of mFc to FcRn was the same as that of dimeric Fc. To test whether the high-affinity binding to FcγRI can be used for targeting of toxins, a fusion protein of mFc with a 38 kDa Pseudomonas exotoxin A fragment (PE38), was generated. This fusion protein killed FcγRI-positive macrophage-like U937 cells but not FcγRI-negative cells, and mFc or PE38 alone had no killing activity. The lack of binding to FcγRIIIa resulted in the absence of Fc-mediated cytotoxicity of a scFv-mFc fusion protein targeting mesothelin. The pharmacokinetics of mFc in mice was very similar to that of dimeric Fc. The mFc's unique FcγRs binding pattern and related functionality, combined with its small size, monovalency and the preservation of FcRn binding which results in relatively long half-life in vivo, suggests that mFc has great potential as a component of therapeutics targeting inflammation mediated by activated macrophages overexpressing FcγRI and related diseases, including cancer.
Keywords: monomeric Fc, Fcγ receptors, FcRn, ADCC, chronic inflammation
Significance Statement
Fc fusions are a well-established class of therapeutics, but their uses have been limited by several factors, such as the relatively large size and unwanted cytotoxic effects. Here, we describe a small monomeric Fc that can be exploited to greatly expand the Fc-related therapeutic applications, owing to its unique receptor binding pattern and related functionality. The mFc 1) binds to FcRn and exhibits a similar in vivo half-life to that of dimeric Fc; 2) lacks binding to FcγRIIIa which results in the absence of Fc-mediated cytotoxicity; 3) possesses high-affinity binding to FcγRI and can be used for FcγRI targeting. The mFc thus has potential as a component of therapeutics targeting inflammation mediated by activated macrophages overexpressing FcγRI.
Introduction
The success of antibody-based therapeutics is to large extent due to the physical and biological properties conferred by the antibody Fc region. The IgG Fc can interact with the neonatal Fc receptor (FcRn), which salvages the antibody from lysosomal degradation and thereby extends its in vivo half-life.1 The Fc can also bind to multiple Fc receptors to induce effector functions, including antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC).2 Consequently, Fc engineering to modulate Fc dependent properties is promising for generating antibody-based therapeutics with improved or novel properties.3-5 One very promising field that could benefit from Fc engineering is the development of therapeutic Fc-fusion proteins.6-8 There are currently six Fc fusion proteins approved by the US Food and Drug Administration for clinical use, and many more are in clinical trials. A major advantage of Fc fusion is the half-life extension to biologically active proteins, which usually display very short half-life and would otherwise be quickly eliminated in vivo. However, the relatively large size of homodimeric Fc can lead to poor tissue penetration, making the fusion proteins ineffective for some applications, e.g., treatment of solid tumors. Moreover, under many circumstances the Fc-induced cytotoxic effects are unwanted, highlighting the importance of Fc engineering to reduce the size of fusion products and to modify effector functions.
Fcγ receptors play a crucial role in mediating Fc effector functions. There are three classes of human Fcγ receptors: FcγRI, FcγRII and FcγRIII. While the critical role of FcγRIIIa in inducing ADCC by natural killer (NK) cells has been well documented, the role of FcγRI, which is only constitutively expressed on macrophages, monocytes, and their progenitors, is far less understood.9 It is notable that FcγRI is strongly and specifically expressed on some inflammation-related cells, such as inflammatory macrophages, and FcγRI has been proposed as a target for the treatment of diseases related to chronic inflammation (e.g., arthritis, multiple sclerosis and cancer).10-14 The role of FcγRI in the promotion of inflammation and anaphylaxis was also recently confirmed in mouse models.15 It is also interesting to note that IgG4, which has been known for some time to be functionally monovalent (i.e., polyclonal IgG4 antibodies do not cross-link two antigens), can bind FcγRI but shows very low affinity to FcγRIIIa and, importantly, differs functionally from the other IgGs in its superior anti-inflammatory activity.16,17 These findings suggest that engineering Fc with selective binding to different Fcγ receptors may lead to unique properties and serve different therapeutic purposes.
In this study, we describe a stable, monomeric IgG1 Fc (mFc) exhibiting high binding affinity to FcγRI, but no measurable binding to FcγRIIIa. The mFc contains only a small subset of mutations at the Fc dimer interface. Although half the size of Fc, its binding to FcRn was not affected, and it exhibited similar serum half-life to that of dimeric Fc in mice. Remarkably, we also found that mFc fusion proteins did not induce Fc-mediated cytotoxicity in vitro, including ADCC and CDC, suggesting that mFc can serve as a safe, non-toxic platform for the delivery of prolonged half-life to therapeutic proteins. Furthermore, a recombinant fusion protein of mFc with a 38 kDa Pseudomonas exotoxin A fragment (PE38) selectively eliminated FcγRI-positive macrophage-like cells in direct cell killing assays in vitro, suggesting that mFc-based fusion proteins could potentially benefit the macrophage-directed therapies. Hence, we have demonstrated that Fc monomers exhibited unique Fcγ receptors binding profile which can be exploited to greatly expand the Fc-related therapeutic applications.
Results
Identification of a monomeric IgG1 Fc
To reduce the size of IgG1 Fc, we have identified three soluble Fc monomers (mFc.1, mFc.23, mFc.67) from a rationally designed Fc mutant library.18 Each Fc monomer contains six to seven point mutations of the wild-type Fc. In the current study, we sought to minimize the number of mutations required to produce a soluble monomer, while leaving FcRn binding activity unaffected, for two reasons: 1) to reduce the possible immunogenicity, and 2) to improve protein stability and thus provide more opportunities for further engineering. Three new monomeric Fc variants were identified in the current study, and their sequences are summarized in Figure 1A. First, the mutations in the previously reported Fc monomers (mFc.1, mFc.23, mFc.67) were reverted back to their wild-type residues, and the resulting variants were expressed separately and assayed for solubility and monomer content (Fig. 1B). Using this approach, we identified an Fc variant (referred to hereafter as “mFc”) that has only four point mutations and is exclusively monomeric as judged by size exclusion chromatography (SEC) (Fig. 1C). A specific combination of residues 351, 366, 368 and 395 is essential to generate pure Fc monomer. The identified mFc was highly expressed in soluble form in E. coli, and 15 mg purified protein could be obtained from 1 L culture under optimal conditions.
Figure 1.

Engineering monomeric and stable IgG1 Fc. (A) Amino acid sequence alignment of human IgG1 Fc, mFc, smFc and ssmFc. The differences of mFc, smFc and ssmFc to the wild-type IgG1 Fc are colored in red, gold and brown, respectively. (B) The Fc monomers mFc.1, mFc.23 and mFc.67, previously identified from a large rationally designed Fc mutant library, were mutated back to wild-type amino acids as indicated. The insoluble, oligomeric and pure monomeric mutants are shown in white gray, gray and red, respectively. The identified monomeric Fc with fewest mutation numbers (the Fc L351S/T366R/L368H/P395K mutant) is named as “mFc.” (C) Size exclusion chromatography of Fc, mFc, smFc and ssmFc. Fc is dimeric with an elution volume of 9.8 mL while the other three proteins are exclusively monomeric with elution volumes from 11.1 to 11.4 mL. (D) Plots of the change in fraction folded (calculated from CD molar ellipticity at 216 nm) for mFc, smFc and ssmFc.
Stability of mFc
We previously found that Fc monomers have reduced stabilities compared with the wild-type Fc, probably due to lack of the stabilization effect of the dimerization.18 Additionally, we speculated that the introduced mutations could also contribute to the destabilization. To assess protein stability, changes in the ellipticity of circular dichroism (CD) spectra at 216 nm at increasing temperature were monitored (Fig. 1D). Indeed, the midpoint transition (melting) temperature (Tm) of mFc was found to be 54.3 ± 0.1°C, which is 9°C higher than the Fc monomers that have seven point mutations (mFc.1, 45.0 ± 0.6°C; mFc.23, 45.2 ± 0.6°C) and 3 °C higher than the Fc monomer that have six point mutations to Fc (mFc.67, 51.0 ± 0.5°C). We also explored other strategies to further stabilize mFc (Fig. S1), and found that the introduction of additional disulfide bonds effectively improved its stability. By mutating two residues to cysteines in the CH2 domain of mFc, the resulting variant, smFc, was found to have a Tm of 59.8 ± 0.6°C, 6°C higher than that of mFc. Another remarkable increase in Tm was achieved by engineering an additional disulfide bond in the CH3 domain. The resulting mutant was named ssmFc, and it had a Tm of 71.7 ± 0.2°C, which is only slightly lower than that of the wild-type Fc (75.1 ± 0.5°C). The mutants were confirmed as monomeric by SEC (Fig. 1C). Thus, these results indicate that a monomeric IgG1 Fc could be developed through use of only a small subset of mutations, and the destabilization effect due to disruption of Fc dimerization could be compensated by antibody engineering techniques.
Pharmacokinetics of mFc in mice
To determine whether the pharmacokinetics of mFc are not affected by the mutations and its smaller size, we intravenously administered mFc and dimeric Fc side-by-side in two groups of mice (4 mice in each group). The mean plasma concentrations vs. time profiles following administration are shown in Figure 2. Importantly, mFc exhibited a similar pharmacokinetic profile compared with dimeric Fc in mice, and its elimination phase half-life was 23.3 h, which is only slightly lower that of the wild-type Fc (28.8 h) (Table 1). To determine whether the similarity of the half-lives is due to unaltered binding to the FcRn, we measured the affinity of mFc to recombinant human FcRn by surface plasmon resonance (SPR) analysis (Fig. S2). At pH 6.0, the calculated binding affinity (KD) was 38 nM, which is comparable to that of the wild-type Fc (111 nM) and the three previously identified Fc monomers (59 to 204 nM). No binding was observed at pH 7.4, indicating that mFc retained the pH-dependent FcRn binding capability of the wild-type Fc.
Figure 2.
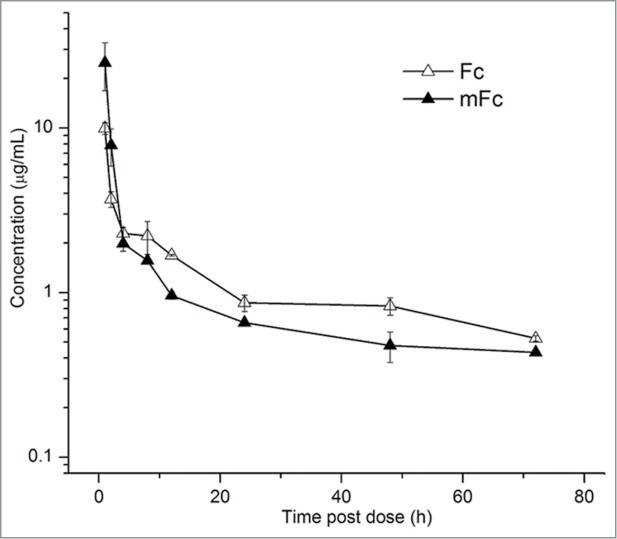
Plasma concentration-time profiles for mFc and the wild-type Fc after intravenous administration to mice. The experimental points are connected by solid linear lines and are represented as mean ± standard deviation (n = 4).
Table 1.
Pharmacokinetics of Fc and mFc in B6 mice intravenously administered 5 mg/kg doses
| Size (kDa) | alpha-phase (h) | beta-phase (h) | |
|---|---|---|---|
| Fc | 55 | 0.64 | 28.8 |
| mFc | 27 | 0.56 | 23.3 |
mFc binds selectively to FcγRI, but not to FcγRIIIa
The distinct effector function patterns of the four human IgG subclasses (IgG1, IgG2, IgG3 and IgG4) are due to their differential binding affinities to different Fcγ receptors.2,19 Interestingly, the monomeric behavior of mFc is reminiscent of IgG4, which is unique among the IgG subclasses for its ability to form monovalent half molecules in vivo due to the relatively weak interaction between homodimeric heavy chains.16 IgG4 has reduced FcγRI and much reduced FcγRIIIa binding properties compared with IgG1, leading to its lack of effector functions. Thus, we sought to determine whether a similar FcγR-binding profile could be observed in mFc.
We first evaluated the FcγRI binding capacity of mFc by SPR. It is well known that the binding of IgG Fc to Fcγ receptors is dependent on its glycosylation at asparagine residue 297 (N297).2 Therefore, mFc was also produced in HEK-293F cells using a mammalian expression vector. Moreover, we generated a scFv-mFc fusion protein by fusing an anti-mesothelin human scFv antibody (m912) to mFc. The m912 IgG has been shown to bind specifically to cancer cells that express the tumor differentiation antigen mesothelin, and kill them by ADCC.20 Strikingly, we found that mFc and scFv-mFc, either produced in bacterial or mammalian expression systems, bound with high affinity to FcγRI (Fig. 3 and Fig. S3). The mammalian-expressed wild-type Fc and IgG1 exhibited similar binding affinity to FcγRI with KD of 67 nM and 75 nM, respectively, which is consistent with previous findings that FcγRI is a high affinity receptor for IgG with KD ∼tens of nM.21 Notably, both bacterial and mammalian-expressed scFv-mFc displayed high affinity to FcγRI with KD of 9.8 and 7.8 nM, respectively, which are ∼8-fold improved over Fc or IgG1 (Fig. 3 and Fig. S3). The mFc also displayed nanomolar affinity for FcγRI although KD could not be accurately estimated due to the low signal-to-noise ratio (Fig. S3). These results indicate that mFc and mFc fusion proteins can bind strongly to FcγRI and this interaction is independent of its N-glycosylation.
Figure 3.
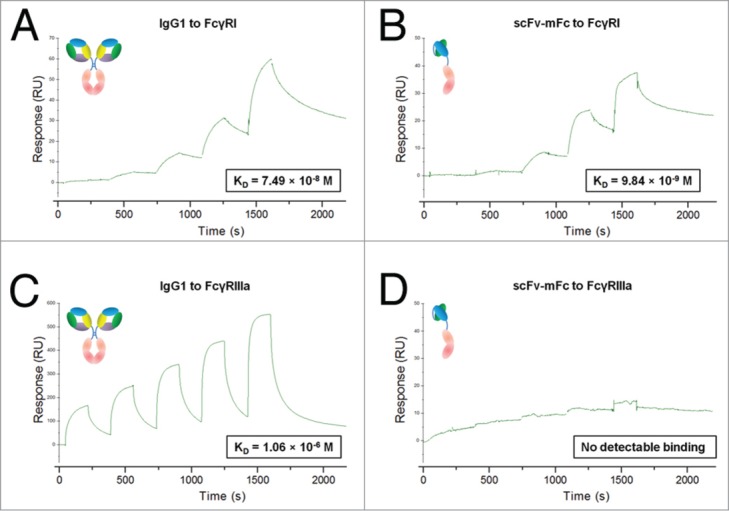
The selective Fcγ receptors binding pattern of scFv-mFc measured by BIAcore. Gradient concentrations of IgG1 or scFv-mFc were injected over the CM5 sensor chip immobilized with FcγRI or FcγRIIIa. The kinetic profiles are shown. IgG1 binds to FcγRI (A) and FcγRIIIa (B) with affinities of 75 nM and 1.1 μM, respectively. scFv-mFc binds to FcγRI (C) with a affinity of 9.8 nM and has no detectable binding to FcγRIIIa (D).
We next examined the binding of mFc to FcγRIIIa, which is critical for NK cell-mediated ADCC effector function (Fig. 3C, D). The binding affinity of IgG1 to FcγRIIIa is 1.1 μM as determined by SPR, which is consistent with previous reports (0.1 to 10 μM).22 In contrast, mFc and scFv-mFc did not show any detectable binding to FcγRIIIa. This selective binding pattern was also confirmed by cells expressing Fcγ receptors, and there was also no association between mFc and FcγRIIa or FcγRIIb-expressing cells (Fig. S4). Thus, it is clear that unlike IgG4, which shows weak binding to both FcγRI and FcγRIIIa, mFc binds selectively to FcγRI with high affinity, but does not bind to FcγRIIIa.
mFc does not induce Fc-mediated cytotoxicity in vitro
Having shown that mFc is not involved in the interaction with FcγRIIIa, we next sought to determine whether this behavior correlates with the absence of ADCC when mFc is used as a fusion partner. For this purpose, the killing effect of the anti-mesothelin m912 scFv-mFc fusion protein on H9 cells, which is a transfected A431 human epithelial carcinoma cell line that stably expresses human mesothelin,23 was examined. As shown in Figure 4A, scFv-mFc and the m912 IgG1 bound highly specifically to H9 cells, but not to mesothelin-negative (A431) cells.
Figure 4.
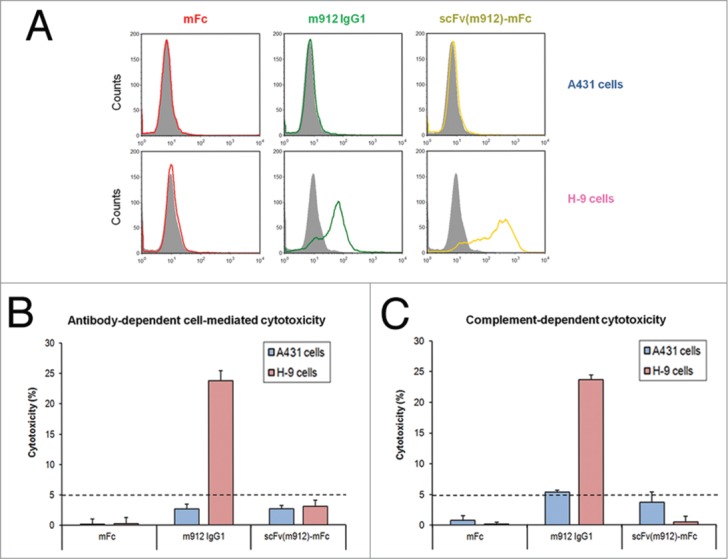
Summary of ADCC and CDC activity in H9 and A431 cells. (A) Mesothelin-negative A431 cells (left panel) and mesothelin-positive H9 cells (right panel) were incubated with 0.3 μM negative scFv (gray), mFc (red), m912 IgG1 (green) and scFv(m912)-mFc (gold). The cells were washed and further incubated with FITC-conjugated goat F(ab’)2 anti-human IgG, then washed and analyzed by flow cytometry. (B) IgG1 m912 induced ADCC in mesothelin-positive H9 cells but not in mesothelin-negative A431 cells. The scFv(m912)-mFc fusion protein did not induce detectable cytotoxicity. (C) IgG1 m912 induced CDC in mesothelin-positive H9 cells but not in mesothelin-negative A431 cells. The scFv(m912)-mFc fusion protein did not induce detectable cytotoxicity. The detection limit of the assays is indicated (dotted line) and represents 5% of lysis. Error bars show s.d. from an average of three separate experiments.
The ADCC activity of m912 scFv-mFc was first assessed using peripheral blood mononucleated cells (PBMCs) as effector cells at an effector-to-target ratio of 50:1. As shown in Figure 4B, m912 IgG1 efficiently and specifically lysed mesothelin-positive (H9) cells at the same level as reported previously, but not mesothelin-negative (A431) cells. In contrast and as expected, scFv-mFc exhibited no ADCC activity at any concentration, although it had potent binding to H9 cells. Moreover, we also performed ADCC assays using macrophage-like U937 cells as effector cells. This is to rule out the possibility that unwanted cytotoxicity might be elicited by mFc under certain circumstances where FcγRI-positive cells occupy the major cell population. Similarly, no cell killing was observed (data not shown), suggesting that only the FcγRI binding is not sufficient to elicit ADCC.
CDC is another Fc-mediated effector function triggered by binding of C1q to the Fc domain of antibody or Fc fusion protein bound on the target cells. The binding sites on Fc for C1q and Fcγ receptors are overlapping. To assess the CDC activity of scFv-mFc, the H9 and A431 cell lines were used as target cells and healthy human serum as the source of complement (Fig. 4C). The m912 IgG1 showed significant cytotoxic activity against mesothelin-positive H9 cells, and showed no activity against A431 cells. In contrast, no substantial cell lysis was observed when scFv-mFc was administered to mesothelin positive or negative target cells. Collectively, these results suggest that mFc may serve as platform for development of engineered mFc-based antibodies and fusion proteins without introducing unwanted cytotoxicity.
The mFc-toxin fusion protein specifically kills cells expressing FcγRI in vitro
FcγRI is unique among Fcγ receptors not only because its ability to bind and internalize monomeric IgG (as opposed to IgG-containing immune complexes), but also because it is constitutively expressed solely on macrophages, monocytes, and their progenitors.10 Consequently, FcγRI has been the focus of considerable interest as a therapeutic target in chronic inflammatory diseases, where inflammatory macrophages, characterized by strongly enhanced FcγRI expression, play a significant role throughout all phases of disease progression.9,10,13-15,24 Indeed, elimination of macrophages using anti-FcγRI scFv immunotoxins has been shown to effectively resolve inflammation in vitro and in animal models.11,12,25,26 Given the finding that mFc can selectively and potently bind to FcγRI, we therefore sought to determine whether mFc-based fusion proteins could be capable of FcγRI-mediated targeting of macrophages, offering additional therapeutic benefits in treatment of chronic inflammation-related diseases.
For this proof-of-concept study, we generated an mFc-based toxin fusion protein by attaching PE38 to the C-terminus of mFc. PE38 has been widely used to construct immunotoxins in which PE38 is attached to specific antibody in scFv or Fab format, and several such immunotoxins are being evaluated in clinical trials.27,28 The immunotoxin is internalized by the targeting of the antibody to its cell-surface antigen, then proteolytic processing releases PE38 into cytosol and cell death is induced. We hypothesized that mFc, with its demonstrated high affinity binding to FcγRI, would also function as an FcγRI-targeting unit and deliver the fused PE38 to FcγRI-expressing cells. Thus, we evaluated the cell killing effect of mFc-PE38 both on FcγRI-negative HEK-293F cells and FcγRI-positive U937 cells. The human U937 macrophage-like cell line is a well-characterized model for investigating macrophage biology; its high-level of FcγRI expression was confirmed by flow cytometry (Fig. S5). The cells were treated for 48 h with different concentration of mFc, PE38 and the mFc-PE38 fusion protein, and cell viability was evaluated by cell proliferation assay (Fig. 5). Our results confirmed that mFc had no killing effect on either cell line at any concentration, and only moderate non-specific killing activity was observed for PE38 alone at the highest concentration tested. In contrast, it is notable that the mFc-PE38 fusion protein was efficient in killing FcγRI-positive U937 cells with an IC50 of 97 nM, while it had no effect on FcγRI-negative cells. Taken together, these results indicate that the mFc-toxin is selectively toxic to FcγRI-expressing cells, and that mFc is able to confer the macrophage-targeting capability (mediated by FcγRI binding) to mFc-based fusion proteins.
Figure 5.
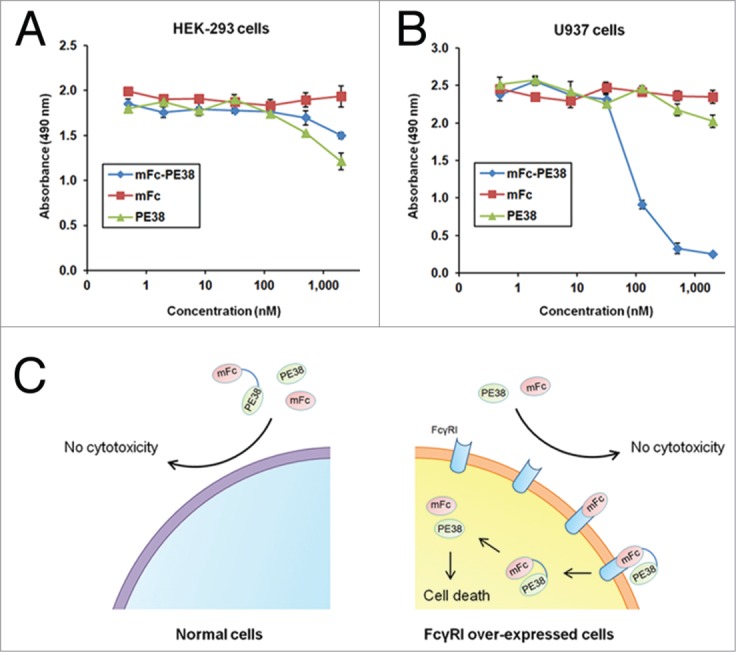
FcγRI specific cell killing. The HEK-293F cells (A) and the FcγRI-positive macrophage-like U937 cells (B) were cultured with gradient concentrations of PE38 (green), mFc-PE38 (blue) and mFc (red) at 37°C for 48 h. Cell viability assays were then performed to evaluate cell survival in response to the proteins. The mFc-PE38 fusion protein has potent cell killing to U937 cells but no killing effect to HEK-293F cells. (C) Schematic illustration of FcγRI specific cell killing mechanism. PE38 and mFc-PE38 cannot enter normal cells (left panel). The mFc-PE38 fusion protein binds to FcγRI, enters the FcγRI overexpressed cells by endocytosis, and is processed to release PE38 and causes cell death (right panel).
Discussion
The mFc presented here provides at least three advantageous features compared with that of the IgG1 Fc fragment. First, the small size of resulting fusion proteins would enable them to better penetrate tissues. This is particularly essential for use in cancer therapy, since the clinical efficacy of anti-cancer drugs is largely limited by their poor penetration into tumor tissues. Second, mFc does not induce any cytotoxicity, and thus represents a robust platform for generating mFc-based fusion therapeutics for safer clinical applications. For most therapeutic proteins, direct cytotoxicity is not involved in their mechanism of action and in many cases should be avoided.29,30 For example, many protein therapeutics function through enzymatic or regulatory activity, and many drugs bind to cells to block a surface antigen or stimulate a signaling pathway instead of depleting the target cells. Currently the IgG2 and IgG4 Fc fragments, which have reduced effector functions compared with the IgG1 Fc, are being pursued to generate fusion proteins, in order to reduce unwanted Fc-mediated cytotoxicity.7 Some of such Fc-fusion proteins have been suggested to possess superior properties. In these cases, mFc might be more suitable for fusion protein construction than IgG2 and IgG4 Fc because of its minimal Fc-mediated cytotoxicity, as well as other advantages demonstrated here. Finally, the unique FcγR-binding profile of mFc might offer additional benefits to the fusion proteins, leading to improved therapeutic efficacy in some clinical applications, e.g., the treatment of chronic inflammation-related diseases.
Antibodies have evolved to achieve fine-tuning of various immunological functions by differential binding to specific Fcγ receptors. Unlike FcRn, which binds to the CH2-CH3 domain interface of the IgG1 Fc, Fcγ receptors interact with the upper CH2 and lower hinge region of the Fc domain. Appropriate glycosylation at the asparagine 297 residue in the CH2 domain of Fc is essential for its interaction with Fcγ receptors.2 Interestingly, the N297 is not directly involved in the interaction, but its glycosylation may stabilize a favorable lower hinge “open” conformation for the receptor binding.31 We have shown in this study that mFc and its fusion proteins, either from bacterial or mammalian sources, can bind to FcγRI with very high affinity. We assume that mFc could also adopt an “open” conformation, which is similar to that induced by N-glycosylation in Fc, driven by the substitutions in its CH3 domain. The FcRn binding of mFc is unaffected, suggesting that the conformational change is confined to the flexible lower hinge region. It is also interesting to note that all the substituted residues in mFc are quite distant from the upper CH2 region. This kind of phenomenon was also observed in another study,32 which identified aglycosylated Fc variants that possessed FcγRI binding with several tens of nanomolar affinities, as well as weakened FcγRIIIa binding compared with mammalian Fc but higher than aglycosylated wild-type Fc (which has no binding to the receptors). FcγRIIIa binding affinity was restored to a value similar to that of mammalian Fc by expressing the Fc variants in mammalian cells. This is distinct from mFc, the glycosylated form of which did not show any detectable binding to FcγRIIIa. It is probable that more extensive perturbations occurred at the N-terminus of mFc compared with those Fc variants, leading to a conformation more favorable to FcγRI binding while entirely free from FcγRIIIa interaction. Taken together, these findings suggest that a local conformational change in the upper CH2 domain of Fc could be triggered by changes in its CH3 domain, and that conformational changes in IgG Fc might be a strategy employed by antibodies to modulate their binding affinities and selectivities to various Fc receptors.
On the other hand, the surprisingly high selectivity of mFc binding to Fcγ receptors implies that FcγRI binds to IgG1 in a different manner from that of the FcγRIIIa to IgG1. This is contrary to the previously held view that FcγRI and its low affinity counterparts use a similar mode to interact with IgG Fc.31,33,34 FcγRIII has been shown to bind asymmetrically to two Fc chains, resulting in a 1:1 receptor:Fc stoichiometry. In contrast, we report here the first evidence, to the best of our knowledge, that only one Fc monomer is sufficient to interact with FcγRI with very high affinity. This does not necessarily mean that FcγRI interacts with only one of the two Fc chains when binding to antibodies, but at least compared with FcγRIII, it has more extensive contacts with one of the Fc chains, making the interaction more stable and not absolutely dependent on the dimeric form of Fc. Figure 6 depicts the FcγR binding site, FcRn binding site as well as the Fc dimerization interface in IgG1 Fc.
Figure 6.
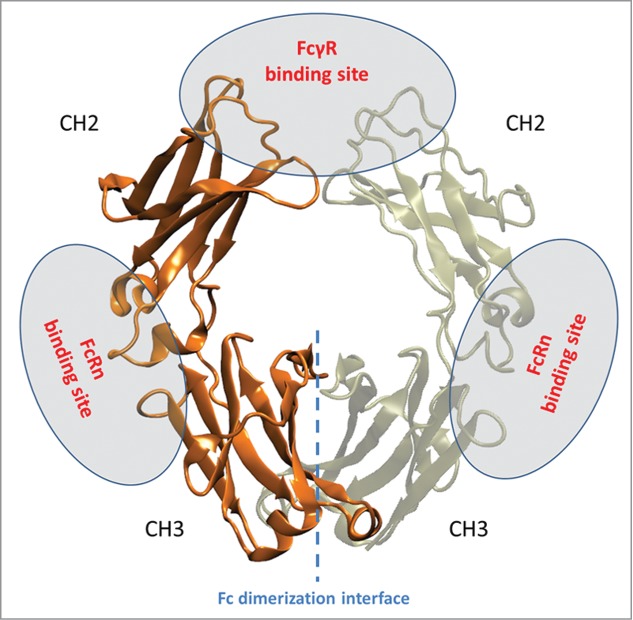
Structure of IgG1 Fc with the two chains colored orange and tan. FcγRs bind at the upper CH2 and lower hinge region of the Fc domain. FcRn binds at the junction between CH2 and CH3 domains. The mFc contains four mutations at the Fc dimerization interface, which is indicated by a blue dotted line.
Importantly, the ability of the mFc fusion proteins to target FcγRI, as demonstrated by our proof-of-concept studies, opens exciting therapeutic opportunities for the treatment of many diseases where macrophage inflammatory responses play essential roles. The presence of inflammatory macrophages, which contribute to disease progression by producing a variety of pro-inflammatory mediators, is a prominent feature of many chronic inflammatory and autoimmune diseases.10,24 Moreover, accumulated evidences support that, in most cancer types, macrophages promote tumor initiation, progression and metastasis, which is counter to previously-held views that macrophages could be part of anti-tumor response.35 Therefore, FcγRI, the overexpression of which is a hallmark of inflammatory macrophages, represents a suitable target for therapy of these diseases. Indeed, a number of anti-FcγRI immunotoxins, composed of anti-FcγRI antibodies attached to various toxins, have recently been developed, and their efficacy has been demonstrated in vitro and in mouse models of different diseases, such as chronic cutaneous inflammation, rheumatoid arthritis, acute myeloid leukemia.11,12,25,26,36,37 In this regard, the use of mFc to deliver therapeutic molecules could be particularly valuable because it provides extended circulating half-life and improved biodistribution by interacting with FcRn,38,39 and it offers additional targeting to FcγRI-overexpressed cells, which in certain cases could lead to greater therapeutic benefit. For instance, one immediate application is to fuse mFc with anti-FcγRI immunotoxins. Considering that both mFc and the anti-FcγRI antibody can bind the receptor, it is reasonable to expect that such a bispecific fusion protein would target macrophages more effectively through avidity effects.
Moreover, mFc could also be fused to a Fab fragment or scFv to generate a novel type of “half-antibody.” IgG4 antibodies are functionally monovalent, and are preferred for immunotherapy in which effector functions are not desired. However, the therapeutic IgG4 antibodies were recently found to be able to engage with plasma IgG4 in humans by so called “Fab-arm exchange.”17 This could have contributed to some of the adverse events of IgG4 antibody therapeutics, e.g., a humanized IgG4, TGN1412, caused severe adverse effects in healthy individuals. It is conceivable that this issue could be avoided by using mFc-based half-antibodies. A half-antibody is also desirable when monovalent binding is necessary to avoid unwanted activation of the target. It is interesting to note that IgG4 possesses superior anti-inflammation activity, probably by interfering with the immune complexes formation by other antibody isotypes.16 The same mechanism may be shared by mFc-based half-antibodies. This, combined with the capability of mFc to target FcγRI-positive macrophages, makes the mFc-based fusion proteins promising candidates for the treatment of inflammation-associated diseases.
Overall, our study shows that Fc monomers, engineered with a unique Fcγ receptors binding profile, can be exploited to greatly expand the therapeutic applications of current Fc fusion technology. The absence of cytotoxicity demonstrated here should contribute to exploration of a safe platform for the delivery of prolonged half-life to therapeutic proteins. The FcγRI targeting capability of the mFc-based fusion proteins could potentially benefit the macrophage-directed therapies. Additional benefits of mFc fusion include the enhancement of solubility and stability, the easy and cost-effective purification through protein A/G. We anticipate that further engineering to modulate mFc dependent properties will increase the power of this technique, and that it will find wide application in biopharmaceutical research and development.
Methods
Cell culture
A431 cells (ATCC) were maintained in RPMI medium supplemented with 10% fetal bovine serum. H9 cells (a gift from Dr. Mitchell Ho, National Cancer Institute) were stable clone cells established from A431 cells that have been transfected with a vector carrying full-length mesothelin cDNA. H9 cells were maintained in RPMI supplemented with 10% fetal bovine serum and 0.75 mg/mL G418. The human embryonic kidney (HEK) 293F cells (HEK-293F, Invitrogen) were cultured in the 293 Freestyle serum-free medium (Invitrogen). The HEK-293T cells (Invitrogen) were cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum. The human macrophage-like U937 cells (clone P) (a gift from Dr. Anu Puri, National Cancer Institute) were maintained in RPMI supplemented with 10% fetal bovine serum and 30 ng/mL phorbol 12-myristate 13-acetate (PMA). To generate the Fcγ receptor expression cells, the HEK-293T cells were transiently transfected with FcγRI, FcγRIIa, FcγRIIb, and FcγRIIIa vectors (Genscript, Piscataway, NJ) using PolyFect (Qiagen) transfection reagent according to the manufacturer's protocol.
Plasmid construction
The construction of mFc.1, mFc.23, mFc.67 and m912 IgG1 expression vectors were described previously.18,20 The monomeric Fc reverse mutants, smFc (mFc L242C/K334C) and ssmFc (mFc L242C/K334C/P343C/A431C) variants were generated using site-directed mutagenesis (QuikChange Site-Directed Mutagenesis Kit, Stratagene). For construction of scFv(m912)-mFc, the m912 scFv and mFc gene was amplified by PCR separately and joined together with a (G4S)3 linker inserted between m912 and mFc by overlap-extension PCR, and cloned into pComb3x (a gift from Dr. Dennis Burton, The Scripps Research Institute). For mammalian expression, mFc, Fc and scFv-mFc gene were amplified and cloned into pSecTag2A (Invitrogen). For construction of mFc-PE38, the synthesized PE38 gene (Genscript, Piscataway, NJ) was amplified and joined to the C-terminal of mFc with a (G4S)3 linker inserted between mFc and PE38, and cloned into pComb3x. All restriction enzymes were purchase from NEB (Beverly, MA). Oligonucleotides were purchased from Invitrogen and all constructs were verified by DNA sequencing.
Protein expression and purification
The bacterial-expressed monomeric Fc reverse mutants, mFc, smFc, ssmFc, scFv(m912)-mFc and mFc-PE38 were expressed in E. coli HB2151 by using a procedure similar to that described previously.18 The mammalian-expressed Fc, mFc, scFv(m912)-mFc and IgG1 were expressed by transient transfection of HEK-293F cells with expression vectors. Protein purity was monitored by SDS-PAGE, and protein concentration was measured spectrophotometrically (NanoVue, GE Healthcare).
Size exclusion chromatography
Purified mFc, smFc and ssmFc proteins were loaded onto a Superdex 75 10/300 GL column running on an FPLC AKTA BASIC pH/C system (GE Healthcare). PBS was used as the running buffer at the flow rate 0.5 mL/min, and eluted proteins were monitored at 280 nm. The molecular mass standards used were ribonuclease A (13.7 kDa), chymotrypsinogen A (25 kDa), ovalbumin (44 kDa), bovine serum albumin (67 kDa) and aldolase (158 kDa).
Circular dichroism (CD)
The CD spectra were collected with an AVIV Model 202 spectropolarimeter (Aviv Biomedical). Purified Fc and monomeric Fc proteins were dissolved in PBS, pH 7.4 at the final concentration of 0.25 mg/mL. CD signals at 216 nm were collected (0.1 cm path length). The instrument was programmed to acquire spectra at 1 °C intervals over the range 25–90°C.
Surface Plasmon Resonance binding experiments
SPR measurements were performed using a BIAcore X100 instrument (GE Healthcare). For Fcγ receptors binding test, the FcγRI or FcγRIIIa protein (R&D Systems) diluted in 10 mM sodium acetate buffer (pH 5.5) was immobilized on a CM5 biosensor chip using a primary amine coupling method. The running buffer was allowed to flow through the cells at a rate of 30 μL/min. The analytes consisted of serial dilution of proteins between 500 nM to 0.8 nM for FcγRI tests and 3 μM to 0.2 μM for FcγRIIIa tests. For FcRn binding test, the purified human soluble single-chain FcRn was immobilized on a CM5 chip. The proteins were diluted in PBS plus 0.005% Tween 20 at pH 7.4 first for testing binding at pH 7.4, while the same running buffer was adjusted to pH 6.0 with HCl for testing binding at pH 6.0. The analytes consisted of serial dilution of proteins between 1 μM and 62.5 nM. The chip was regenerated with pH 8.0 buffer (100 mM Tris, 50 mM NaCl, pH 8.0) after 10 min of dissociation.
Flow cytometry
To measure the interactions of proteins with mesothelin, aliquots of A431 and H9 cells were incubated with 0.3 μM proteins in 250 μL of RPMI supplemented with 10% fetal bovine serum for 1 h on ice. Unbound antibodies were washed away with medium. The secondary antibody FITC-conjugated goat F(ab’)2 anti-human IgG (Fc-specific) antibody or (Sigma-Aldrich) was incubated with cells for 30 min. Cells were washed and resuspended in PBS plus 0.5% bovine serum albumin (BSA) for flow cytometry on FACSCalibur (Becton Dickinson). To measure the interactions of mFc and IgG1 with Fcγ receptor expressing cells, the HEK-293T cells transfected with FcγRI, FcγRIIa, FcγRIIb and FcγRIIIa were incubate with 1 μM proteins in 200 μL PBS containing 0.1% BSA, and after the wash, the bindings were detected by FITC-conjugated goat F(ab’)2 anti-human IgG (Fc-specific) antibody. To measure the expression of FcγRI, HEK-293F and PMA-stimulated U937 cells in 200 μL PBSA were mixed with FITC-conjugated mouse anti-human FcγRI antibody (Invitrogen) and incubated for 30 min on ice.
Antibody-dependent cell-mediated cytotoxicity assay
The ADCC assay was performed as previously described.20 Briefly, mesothelin-negative A431 or mesothelin-positive H9 cells were incubated with 50 nM m912 IgG1, scFv(m912)-mFc or mFc for 30 min, followed by the addition of the target cells to wells with the effector cells (PBMC cells or U937 cells) attached, at an effector to target cell ratio of 50:1. After 24 h incubation, the lysis of the target cells was measured using CytoTox-ONE Homogeneous Membrane Integrity Assay (Promega) according to the manufacturer's protocol.
Complement-dependent cytotoxicity assay
To perform the CDC assay, A431 and H9 cells were washed in serum-free RPMI and density adjusted to 1 million/mL in serum-free RPMI. Fifty μL of cell suspension was incubated with 50 μL of antibody dilution in RPMI. Fresh human plasma was diluted in PBS to 1:4 and clarified with centrifugation. Fifty μL of diluted plasma was added to each cell/antibody mixture and incubated in 96-well plates (Costar, Corning) in 37°C to facilitate complement-mediated cell lysis. After 3 h of incubation, 100 μL of the supernatant was transferred to a white plate and 100 μL substrate from CytoTox-ONE Homogeneous Membrane Integrity Assay Kit was added. The plate was incubated at room temperature for 10 min, and fluorescent signals were read in a fluorometer (SpectraMax, Molecular Devices, Sunnyvale, CA) with excitation wavelength at 530 nm and emission wavelength at 590 nm. Each antibody concentration was tested with 6 well-duplicates. The negative control was 50 μL of RPMI in the place of antibody dilution. The positive control was target cells lysed with 1% Triton X-100 in 150 μL final volume. The CDC of target cells was expressed as the percent of the experimental sample to the positive control. Plasma used for the study was collected under the NIC-Frederick Research Donor Program from healthy donors.
Cell proliferation assay
The HEK-293F cells or the FcγRI-positive macrophage-like U937 cells were plated at 5,000 cells/50 μL in round-bottom 96-well cell culture plates (Costar, Corning). Then, equal volume of culturing medium containing PE38, mFc-PE38 and mFc at various concentrations was added to each well. The plates were incubated at 37°C for 48 h. The cell viability was evaluated with the CellTiter 96 AQueous One Solution Cell Proliferation Assay Kit (Promega) according to the manufacturer's protocol.
Pharmacokinetic study
Studies were performed at NCI-Frederick and approved by the Institutional Animal Car and Use Committee (IACUC). Female 4–6 wk C57BL/6 mice (4/group) were given intravenous injections via the tail vein with the tested proteins (IgG1 Fc or mFc, 100 μg/mouse). Blood (20 μl) was collected at pre-dose and at various time-points. Blood was pooled for each group, processed to serum and analyzed by ELISA. The human IgG1 CH2 specific mouse monoclonal antibody (AdD serotec, Oxford, UK) was coated on ELISA wells (Corning, Corning, NY) at 50-ng per well and blocked with protein-free blocking buffer (Pierce, Rockford, IL). After washing, the diluted serum/PBS samples at different time points were added and incubated at 37°C. The purified Fc or mFc proteins serially diluted in pre-dose serum/PBS were used to make standard curves. Bound proteins were detected by HRP-conjugated anti-FLAG tag antibody (Sigma-Aldrich). The assay was developed at 37°C with ABST substrate (Roche, Indianapolis, IN) and monitored at 405 nm. All PK analyses were performed using ELISA concentration/timepoint data using the PK Solutions 2.0, noncompartmental PK data analysis software from Summit Research Services.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Competing Financial Interests
The authors declare no competing financial interests.
Acknowledgments
We thank Dr. Sergey G. Tarasov and Ms. Marzena A. Dyba of the Biophysics Resource in the Structural Biophysics Laboratory, NCI-Frederick for technical support.
Funding
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and by Federal funds from the NIH, National Cancer Institute, under Contract No. N01-CO-12400.
Author Contributions
TY and DSD designed research; TY, YF, YW and WC performed experiments; and TY and DSD analyzed the data and wrote the paper.
Supplemental Materials
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715-25; PMID:17703228; http://dx.doi.org/ 10.1038/nri2155 [DOI] [PubMed] [Google Scholar]
- 2. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 2008; 8:34-47; PMID:18064051; http://dx.doi.org/ 10.1038/nri2206 [DOI] [PubMed] [Google Scholar]
- 3. Strohl WR. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr Opin Biotechnol 2009; 20:685-91; PMID:19896358; http://dx.doi.org/ 10.1016/j.copbio.2009.10.011 [DOI] [PubMed] [Google Scholar]
- 4. Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol 2010; 28:157-9; PMID:20081867; http://dx.doi.org/ 10.1038/nbt.1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A 2006; 103:4005-10; PMID:16537476; http://dx.doi.org/ 10.1073/pnas.0508123103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rath T, Baker K, Dumont JA, Peters RT, Jiang H, Qiao SW, Lencer WI, Pierce GF, Blumberg RS. Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Crit Rev Biotechnol 2013;In press; PMID:24156398; http://dx.doi.org/ 10.3109/07388551.2013.834293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Czajkowsky DM, Hu J, Shao Z, Pleass RJ. Fc-fusion proteins: new developments and future perspectives. EMBO Mol Med 2012; 4:1015-28; PMID:22837174; http://dx.doi.org/ 10.1002/emmm.201201379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beck A, Reichert JM. Therapeutic Fc-fusion proteins and peptides as successful alternatives to antibodies. MAbs 2011; 3:415-6; PMID:21785279; http://dx.doi.org/ 10.4161/mabs.3.5.17334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van der Poel CE, Spaapen RM, van de Winkel JG, Leusen JH. Functional characteristics of the high affinity IgG receptor, FcγRI. J Immunol 2011; 186:2699-704; PMID:21325219; http://dx.doi.org/ 10.4049/jimmunol.1003526 [DOI] [PubMed] [Google Scholar]
- 10. Thepen T, Huhn M, Melmer G, Tur MK, Barth S. Fcgamma receptor 1 (CD64), a target beyond cancer. Curr Pharm Des 2009; 15:2712-8; PMID:19689341; http://dx.doi.org/ 10.2174/138161209788923967 [DOI] [PubMed] [Google Scholar]
- 11. Thepen T, van Vuuren AJ, Kiekens RC, Damen CA, Vooijs WC, van De Winkel JG. Resolution of cutaneous inflammation after local elimination of macrophages. Nat Biotechnol 2000; 18:48-51; PMID:10625390; http://dx.doi.org/ 10.1038/71908 [DOI] [PubMed] [Google Scholar]
- 12. Tur MK, Huhn M, Thepen T, Stöcker M, Krohn R, Vogel S, Jost E, Osieka R, van de Winkel JG, Fischer R, et al. Recombinant CD64-specific single chain immunotoxin exhibits specific cytotoxicity against acute myeloid leukemia cells. Cancer Res 2003; 63:8414-9; PMID:14679004 [PubMed] [Google Scholar]
- 13. Nuutila J. The novel applications of the quantitative analysis of neutrophil cell surface FcgammaRI (CD64) to the diagnosis of infectious and inflammatory diseases. Curr Opin Infect Dis 2010; 23:268-74; PMID:20407370; http://dx.doi.org/ 10.1097/QCO.0b013e32833939b0 [DOI] [PubMed] [Google Scholar]
- 14. Hulse KE, Woodfolk JA. Targeting allergen to Fc gammaRI: a strategy to treat allergic disease? Curr Opin Allergy Clin Immunol 2008; 8:547-52; PMID:18978470; http://dx.doi.org/ 10.1097/ACI.0b013e32831665d2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mancardi DA, Albanesi M, Jönsson F, Iannascoli B, Van Rooijen N, Kang X, England P, Daëron M, Bruhns P. The high-affinity human IgG receptor FcγRI (CD64) promotes IgG-mediated inflammation, anaphylaxis, and antitumor immunotherapy. Blood 2013; 121:1563-73; PMID:23293080; http://dx.doi.org/ 10.1182/blood-2012-07-442541 [DOI] [PubMed] [Google Scholar]
- 16. van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martínez-Martínez P, Vermeulen E, den Bleker TH, Wiegman L, Vink T, Aarden LA, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 2007; 317:1554-7; PMID:17872445; http://dx.doi.org/ 10.1126/science.1144603 [DOI] [PubMed] [Google Scholar]
- 17. Labrijn AF, Buijsse AO, van den Bremer ET, Verwilligen AY, Bleeker WK, Thorpe SJ, Killestein J, Polman CH, Aalberse RC, Schuurman J, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol 2009; 27:767-71; PMID:19620983; http://dx.doi.org/ 10.1038/nbt.1553 [DOI] [PubMed] [Google Scholar]
- 18. Ying T, Chen W, Gong R, Feng Y, Dimitrov DS. Soluble monomeric IgG1 Fc. J Biol Chem 2012; 287:19399-408; PMID:22518843; http://dx.doi.org/ 10.1074/jbc.M112.368647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daëron M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009; 113:3716-25; PMID:19018092; http://dx.doi.org/ 10.1182/blood-2008-09-179754 [DOI] [PubMed] [Google Scholar]
- 20. Feng Y, Xiao X, Zhu Z, Streaker E, Ho M, Pastan I, Dimitrov DS. A novel human monoclonal antibody that binds with high affinity to mesothelin-expressing cells and kills them by antibody-dependent cell-mediated cytotoxicity. Mol Cancer Ther 2009; 8:1113-8; PMID:19417159; http://dx.doi.org/ 10.1158/1535-7163.MCT-08-0945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miller KL, Duchemin AM, Anderson CL. A novel role for the Fc receptor gamma subunit: enhancement of Fc gamma R ligand affinity. J Exp Med 1996; 183:2227-33; PMID:8642332; http://dx.doi.org/ 10.1084/jem.183.5.2227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li P, Jiang N, Nagarajan S, Wohlhueter R, Selvaraj P, Zhu C. Affinity and kinetic analysis of Fcgamma receptor IIIa (CD16a) binding to IgG ligands. J Biol Chem 2007; 282:6210-21; PMID:17202140; http://dx.doi.org/ 10.1074/jbc.M609064200 [DOI] [PubMed] [Google Scholar]
- 23. Ho M, Hassan R, Zhang J, Wang QC, Onda M, Bera T, Pastan I. Humoral immune response to mesothelin in mesothelioma and ovarian cancer patients. Clin Cancer Res 2005; 11:3814-20; PMID:15897581; http://dx.doi.org/ 10.1158/1078-0432.CCR-04-2304 [DOI] [PubMed] [Google Scholar]
- 24. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature 2013; 496:445-55; PMID:23619691; http://dx.doi.org/ 10.1038/nature12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stahnke B, Thepen T, Stöcker M, Rosinke R, Jost E, Fischer R, Tur MK, Barth S. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol Cancer Ther 2008; 7:2924-32; PMID:18790773; http://dx.doi.org/ 10.1158/1535-7163.MCT-08-0554 [DOI] [PubMed] [Google Scholar]
- 26. Tur MK, Huhn M, Jost E, Thepen T, Brümmendorf TH, Barth S. In vivo efficacy of the recombinant anti-CD64 immunotoxin H22(scFv)-ETA’ in a human acute myeloid leukemia xenograft tumor model. Int J Cancer 2011; 129:1277-82; PMID:21077160; http://dx.doi.org/ 10.1002/ijc.25766 [DOI] [PubMed] [Google Scholar]
- 27. Kreitman RJ, Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res 2011; 17:6398-405; PMID:22003067; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wayne AS, Kreitman RJ, Findley HW, Lew G, Delbrook C, Steinberg SM, Stetler-Stevenson M, Fitzgerald DJ, Pastan I. Anti-CD22 immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-positive hematologic malignancies of childhood: preclinical studies and phase I clinical trial. Clin Cancer Res 2010; 16:1894-903; PMID:20215554; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dimitrov DS. Therapeutic proteins. Methods Mol Biol 2012; 899:1-26; PMID:22735943; http://dx.doi.org/ 10.1007/978-1-61779-921-1_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov 2008; 7:21-39; PMID:18097458; http://dx.doi.org/ 10.1038/nrd2399 [DOI] [PubMed] [Google Scholar]
- 31. Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature 2000; 406:267-73; PMID:10917521; http://dx.doi.org/ 10.1038/35018508 [DOI] [PubMed] [Google Scholar]
- 32. Jung ST, Reddy ST, Kang TH, Borrok MJ, Sandlie I, Tucker PW, Georgiou G. Aglycosylated IgG variants expressed in bacteria that selectively bind FcgammaRI potentiate tumor cell killing by monocyte-dendritic cells. Proc Natl Acad Sci U S A 2010; 107:604-9; PMID:20080725; http://dx.doi.org/ 10.1073/pnas.0908590107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Radaev S, Motyka S, Fridman WH, Sautes-Fridman C, Sun PD. The structure of a human type III Fcgamma receptor in complex with Fc. J Biol Chem 2001; 276:16469-77; PMID:11297532; http://dx.doi.org/ 10.1074/jbc.M100350200 [DOI] [PubMed] [Google Scholar]
- 34. Lu J, Ellsworth JL, Hamacher N, Oak SW, Sun PD. Crystal structure of Fcγ receptor I and its implication in high affinity γ-immunoglobulin binding. J Biol Chem 2011; 286:40608-13; PMID:21965667; http://dx.doi.org/ 10.1074/jbc.M111.257550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 2010; 141:39-51; PMID:20371344; http://dx.doi.org/ 10.1016/j.cell.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Vuuren AJ, van Roon JA, Walraven V, Stuij I, Harmsen MC, McLaughlin PM, van de Winkel JG, Thepen T. CD64-directed immunotoxin inhibits arthritis in a novel CD64 transgenic rat model. J Immunol 2006; 176:5833-8; PMID:16670289; http://dx.doi.org/ 10.4049/jimmunol.176.10.5833 [DOI] [PubMed] [Google Scholar]
- 37. van Roon JA, van Vuuren AJ, Wijngaarden S, Jacobs KM, Bijlsma JW, Lafeber FP, Thepen T, van de Winkel JG. Selective elimination of synovial inflammatory macrophages in rheumatoid arthritis by an Fcgamma receptor I-directed immunotoxin. Arthritis Rheum 2003; 48:1229-38; PMID:12746896; http://dx.doi.org/ 10.1002/art.10940 [DOI] [PubMed] [Google Scholar]
- 38. Ying T, Ju TW, Wang Y, Prabakaran P, Dimitrov DS. Interactions of IgG1 CH2 and CH3 Domains with FcRn. Front Immunol 2014; 5:146; PMID:24765095; http://dx.doi.org/ 10.3389/fimmu.2014.00146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ying T, Gong R, Ju TW, Prabakaran P, Dimitrov DS. Engineered Fc based antibody domains and fragments as novel scaffolds. [epub ahead of print]. Biochim Biophys Acta 2014; PMID:24792384; http://dx.doi.org/ 10.1016/j.bbapap.2014.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.