

Abstract
Expression of recombinant proteins often takes advantage of peptide tags expressed in fusion to allow easy detection and purification of the expressed proteins. However, as the fusion peptides most often are flexible appendages at the N- or C-terminal, proteolytic cleavage may result in removal of the tag sequence. Here, we evaluated the functionality and stability of 14 different combinations of commonly used tags for purification and detection of recombinant antibody fragments. The tag sequences were inserted in fusion with the c-terminal end of a domain antibody based on the HEL4 scaffold in a phagemid vector. This particular antibody fragment was able to refold on the membrane after blotting, allowing us to detect c-terminal tag breakdown by use of protein A in combination with detection of the tags in the specific constructs. The degradation of the c-terminal tags suggested specific sites to be particularly prone to proteolytic cleavage, leaving some of the tag combinations partially or completely degraded. This specific work illustrates the importance of tag design with regard to recombinant antibody expression in E. coli, but also aids the more general understanding of protein expression.
Keywords: antibodies, peptide tags, phage display, protein expression, proteolytic degradation
Abbreviations
- DNA
deoxyribonucleic acid
- TEV protease
tobacco etch virus
- HRP
horseradish peroxidase
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- E. coli
escherichia coli
- Tsp protease
tail-specific protease
- dAb
domain antibody
- scFv
single chain fragment variable
- RCF
relative centrifugal force
- rpm
revolutions per minute
- PCR
polymerase chain reaction
Introduction
Recombinant antibody technology has become an increasingly important tool in biotechnology due to its versatile application. Today, recombinant antibodies can be generated in vitro by applying various display systems that allow for the selection of high affinity binders to virtually any type of antigen.1 One of the most common display formats for recombinant antibodies is phage display using filamentous phage. In phage display, a foreign protein is expressed in fusion with one of the phage capsid proteins by cloning the foreign DNA sequence into the phage genome. This allows for a physical coupling between the phenotype (the fusion protein) and the genotype (the DNA inside the virion encoding the fusion protein); which again allows for high throughput selection of desirable properties of the fusion protein from a library of billions of variants.2 Numerous antibody fragment libraries for phage display have been created, and the creation of new libraries continues as knowledge and technology expands the possibilities.3-10 Improvements concern stability and functionality of the various antibody fragments themselves, regulation of the expression, the fusion partner utilized in the display system and not least the peptide tag sequences applied (Fig. 1).
Figure 1.

The structure of immunoglobulin G in space-filling and cartoon representation with light and heavy chain colored in green and blue, respectively. Lower panel illustrates crystal structures of the antibody fragments FAB (fragment antigen-binding), scFv (single-chain fragment variable) and single domain antibody. A peptide tag of approximately 30 residues is illustrated at the c-terminal of the domain antibody. The immunoglobulin structure used in this figure is based on the RCSB Protein Data Bank entry 1igt.
After applying phage display to select antibody fragments, the function of the soluble antibody (antibodies expressed without the phage fusion protein) must be validated. Expression of soluble antibody usually requires time-consuming subcloning of selected clones into expression vectors. To circumvent this, phagemid libraries have been created with an amber stop codon between the antibody and the phage fusion protein. This facilitates fast and easy production of antibody either as phage antibody fusion (by using an amber suppressor strains) or as soluble antibody (when expressing in a non-suppressor strain).11
To take full advantage of the ability to express soluble antibodies without subcloning, it is essential that the library is designed with appropriate peptide tags on the antibody side of the amber stop codon. The tags used for a library can be pivotal both with regard to purification of soluble antibody and the detection of the recombinant antibody. The human c-Myc tag (myc-tag) is an example of a well-known tag that has been used in a variety of antibody libraries for screening purposes.12,13
When antibodies containing a variable heavy chain of the gene segment family VH3 is used, purification and detection can often be facilitated by the use of staphylococcal protein A.14,15 The use of protein A for purification does, however, entail acid elution, which can cause problems with regard to stability.16 Also the use of protein A for detection and immunostaining can often be problematic because protein A is able to bind a large variety of immunoglobulins.17 There are numerous tags available for purification and detection, but there are significant differences between tags with regard to protein purity, protein yield and cost.2,18 One of the more cost effective tags is the poly-histidine tag (his-tag), which has been used several times in phage displayed libraries. Another possibility could be the use of a biotin accepting sequence like the AviTag, which is a 15 amino acid peptide sequence that can be biotinylated by the biotin ligase. Some of the advantages of this tag are that the biotinylation process can be made to work in vivo during expression, it is very specific, and binding by streptavidin (or avidin) is known to be in the sub-picomolar range. The strength of the streptavidin biotin binding makes it very attractive for immobilization onto solid supports. In combination with biotinylation of the recombinant protein, it is often necessary to cleave off the AviTag, e.g., during purification. By inserting the recognition sequence cleaved by a specific protease, release becomes possible. One such protease is the tobacco etch virus (TEV) protease.19,20
Because it can be difficult to predict how tags behave in a given setting, the tag combinations need to be tested for functionality and possible proteolysis during expression in the relevant host strain.21 It is well known that E. coli contains several proteases that can degrade recombinant protein expressed in the bacteria.22,23 Tsp is a periplasmic serine protease that degrades protein from the C-terminal end based on the recognition of the C-terminal residues. A range of amino acids are substrates for Tsp when placed at the C-terminal, but there is a preference for substrates with non-polar carboxyl termini and preferably at least 2 alanines out of the 3 C-terminal positions. The proteolytic activity of Tsp works as an endopeptidase that cleaves preferentially 8 amino acids before the C-terminal. Tsp is promiscuous with regard to cleavage site, but alanine, serine and valine are often found on either side of the digested peptide bond. The site of cleavage for Tsp often results in a new recognizable carboxy terminus, which leads to consecutive degradation.24
To evaluate the functionality and the level of degradation of tags, western blotting is the method of choice. It is generally accepted that linear epitopes are detected in protein gel blot, although several studies have confirmed that this is not always the case.25,26 Some conformational epitopes can be detected in western blots if reducing agents are omitted in the sample preparation.25,27 This indicates that the intramolecular disulphide bonds (for some antigens) can serve to maintain essential epitope integrity and thereby enable detection by conformational binders. On the contrary, it has been reported that detection of linear epitopes can be obstructed by incomplete denaturation or high renaturation propensity of antigens during protein gel blotting, which makes it difficult to detect internal linear epitopes.28
Here, we demonstrate that a domain antibody based on the Hel4 scaffold can be detected by protein A in western blots due to its ability to refold following denaturation. This antibody was used to test 14 different c-terminal fused tag combinations for functionality, degradation and influence on the functionality of the antibody. Protein A was used for the purification of expressed antibody constructs, allowing us to assay the influence of the various tags on the output of functional protein and level the breakdown of C-terminal tags. We further demonstrate that Tsp is involved in the proteolytic degradation of the C-terminal tags and that the amino acid composition is relevant for the level of degradation.
Results
A total of 14 different combinations of the myc-tag, his-tag, AviTag and TEV protease recognition site were constructed (Table 1). The different constructs were tested for functionality in display from phage and production of free soluble antibody. A single domain antibody clone (8H) previously selected from a library based on the stable HEL4 VH3 scaffold, was used as model antibody.29 Phage particles were produced in TG1 using the KM13 helper phage for packaging. The results show that the tag sequence does not have any major influence on the display of the antibody from phage (Fig. 2). For constructs containing a TEV protease recognition site, digestions of packaged phage particles were made to determine whether TEV site was accessible for digestion when located between the antibody and the phage protein III. The samples treated with TEV protease had the antibody portion of the antibody-pIII fusion protein cleaved off. Thus, the TEV recognition site is readily accessible in all the TEV site-containing constructs (Fig. 3). The efficiency of in vivo biotinylation was determined by protein gel blot using streptavidin-HRP for detection. The results show that in vivo biotinylation is not occurring on antibodies purified from the supernatant, while antibody trapped in the pellet was in vivo biotinylated (Fig. 4A). Detection of the antibody with anti-his antibody, showed that the full sized tag was present on both the antibody from pellet and the supernatant (Fig. 4B). To get an estimate of the degradation level of the antibodies not fused to phage protein III, antibody from all the different constructs were expressed in HB2151 from the pHEN1 vector. The antibodies were purified from the supernatant by protein A affinity chromatography. Coomassie staining of the purified antibodies showed that there were several bands below the expected size of the antibodies (Fig. 5A). To determine the identity of the lower bands, western blots were made using protein A-HRP to detect for the presence of antibody. The results show that antibody was present in the lower bands indicating degradation of the protein was occurring (Fig. 5B). To determine whether degradation were occurring in the C-terminal tag region the presence of His-tag was examined using protein gel blot on all the constructs containing his-tag. The absence of some bands indicated that the tag region was being degraded (Fig. 6). To determine if the breakdown occurred in the supernatant and whether increased degradation occurred as expression time increased, expressions from TG1 was performed with construct A5:HA. The A5:HA TG1 cultures were incubated for either 3.5 hours, 10.5 hours or 22.5 hours after IPTG induction, and then antibody was purified by protein A affinity chromatography. As the ratio between the bands corresponding to full size and degraded antibody appears to be independent of expression time, the degradation is likely to occur in the cytoplasm or the periplasmic compartment rather than in the medium (Fig. 7).
Table 1.
Schematic overview of peptide tag construct investigated for stability. The NotI restriction site denotes the c-terminal end of the model antibody. The different peptide tags and linker sequences are given in single letter code. Sizes given in the table are including the model domain antibody
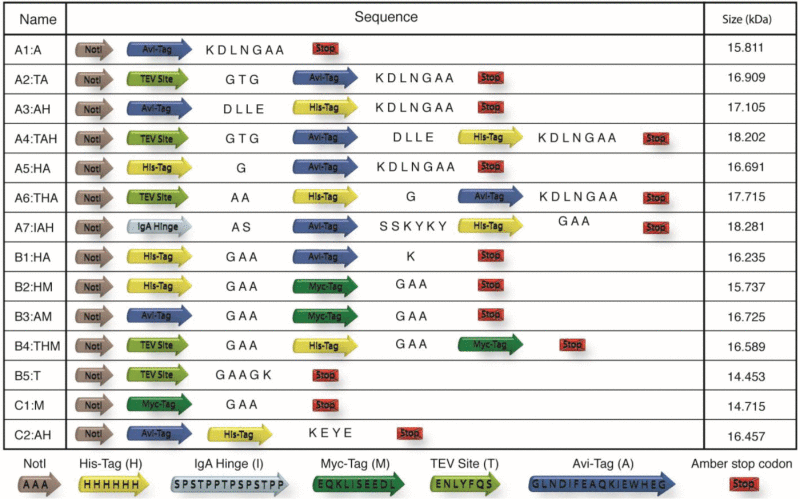 |
Figure 2.

Western blot analysis with anti-pIII antibody were made in order to compare the display levels of the different constructs with another single domain library (Garvan) and a scFv library (Tomlinson). The KM13 helper phage was also included as a reference for the pIII size. Intensities of the lower bands compared to the upper bands give the ratio of displayed pIII-proteins and pIII-fusion proteins. Similar display ratios are observed for all constructs including the Garvan library, whereas, the Tomlinson library had lower display. The phage concentrations were normalized so the same amount of phage were loaded in each lane.
Figure 3.

Western blot analysis with anti-pIII antibody confirms the accessibility of the TEV site located within the tag sequence on the pIII-fusion protein. Phages produced with tag construct A2:TA, A4:TAH and A6:THA gives bands corresponding to the size of pIII-fusion protein. After incubation with TEV protease the fusion proteins are cleaved off and only pIII without fusion protein is present.
Figure 4.

Western blot of in vivo biotinylated antibody detected with HRP conjugated streptavidin (A) and western blot detected with HRP conjugated anti-his antibody (B). The biotin accepting sites of constructs A7:IAH, B3:AM and C2:AH are not biotinylated in vivo when the antibody is directed to the periplasm. However, the pellet fraction does contain in vivo biotinylated antibody. Purified antibody from the A7:IAH construct detected with anti-his antibody (B) revealed that the terminal his-tag was intact on both antibody from the pellet and the supernatant. Thus, establishing that the AviTag is present, as it is flanked by the his-tag. The A7:IAH supernatant samples were purified from the same expression batch and loaded in same concentration.
Figure 5.

Degradation of the c-terminal tag sequence analyzed by SDS-PAGE (A) and protein gel blot detected with protein-A HRP (B). In each lane of the SDS-PAGE and the western blot 4 μg and 1 μg of HB2151 expressed and purified antibody was loaded, respectively. All the constructs showed to be prone to degradation at specific positions leaving distinct breakdown patterns in both the SDS-PAGE and protein gel blot analysis.
Figure 6.

Western blot analysis of all the His-tag containing constructs with anti-His antibody, showed different amount of degradation. Equal amounts of protein were loaded in the lanes and the western blots were developed using the same exposure times.
Figure 7.

Analysis of proteolytic breakdown of c-terminal tag sequence and expression time by Coomassie blue stained SDS-PAGE. Construct A5:HA expressed for 3.5, 10.5 and 22.5 hours in TG1 were purified and equal amounts were loaded in each lane. Upper band corresponds to full length antibody plus tag sequence. Lower band corresponds to antibody without tag sequence and the intermediate bands correspond to partially degraded protein.
A large number of different proteases are expressed by the bacteria and substantial efforts have gone into creation of bacterial strains in which proteases are knocked out or inactivated by mutation. By searching the literature and applying online sequence analysis tools such as the MEROPS peptidase database (http://merops.sanger.ac.uk/) the amino acid sequence of the tags utilized in this study were analyzed. We identified one potential protease that could be responsible for the degradation, namely the Prc protease also known as Tsp protease. The A5:HA construct was transfected into the E. coli strains KS1000 in which the Prc protease was inactivated by mutation To compare the tag degradation from KS1000 the same construct was expressed in TG1, CAG597 and ER2738. Antibody was purified from supernatant using protein A affinity chromatography (Fig. 8).
Figure 8.

Coomassie blue stained SDS-PAGE with construct A5:HA expressed in 4 different E. coli strains. The antibodies were purified and normalized. KS1000 is defective in the tail-specific protease (Tsp), CAG597 is defective in stress-induced proteases at high temperature and ER2738 is a commonly used strain in phage antibody display (provided with the Ph.D. Phage Display Kit). KS1000 showed to have the least amount of degradation compared to the other strains.
The robustness of detecting domain antibody in western blots using HRP conjugated protein A, was further verified using 4 of the constructs, either with or without reducing agent in the sample buffer. The protein gel blots were blocked for 2 hours before incubating with protein A-HRP. The membrane was then stripped and blocked again, followed by incubation with Anti-c-Myc antibody to verify that the protein was indeed present (data not shown).
Discussion
We designed 14 different library tag combinations that we tested for display on phage particles (Table 1). All tag constructs gave similar display levels when compared to existing scFv and dAb libraries in western blots (Fig. 2). The constructs containing TEV sites were investigated to determine whether these were functional (Fig. 3). The TEV sites were accessible by protease in all the constructs.
The A7:IAH, B3:AM and C2:AH constructs were tested for their biotinylation capacity. Construct A7:IAH was designed to match a previously published tag that had been used for in vivo biotinylation.30,31 The streptavidin-HRP protein gel blot shows that in vivo biotinylation of the Avitag does happen in the antibody located in the pellet. No in vivo biotinylation could be detected in antibody purified from the supernatant (Fig. 4A). The Avitag was, however, present on the antibody purified from the supernatant, as, following the purification, the c-terminal his-tag, flanking the Avitag, could be detected (Fig. 4B). In vitro biotinylation of the samples revealed that the AviTag on antibody from the supernatant was functional (data not shown). These results show that in vivo biotinylation is very inefficient when protein is directed to the periplasmic compartment by a leader peptide. This is likely due to the cytoplasmic localization of the biotin ligase, which as a consequence is only able to biotinylate proteins while these are in the cytoplasm.32 We suspect that the dAb are less effectively biotinylated compared to larger fragments due to their smaller size, and thereby faster translocation to the periplasm. The smaller size of the dAb implicates that a C-terminal AviTag stays a shorter time in the cytoplasm than the corresponding C-terminal AviTag of a larger fragment. As reported in both Thie et al. and Cloutier et al., in vivo biotinylation should be feasible using the A7:IAH tag construct on scFv.30,31
To test the functionality and stability of the tags on soluble antibodies, expression was induced from the phagemid in a non-suppressor E. coli strain. Incubating the induced cultures for more than 12 hours led to a considerable buildup of antibody in the supernatant where less bacterial protein contaminants are present.33,34 Protein A purification of expressed protein showed that most of the tag constructs suffered from considerable breakdown because clearly visible bands appeared in the Coomassie staining below the expected full-size antibody (Fig. 5A). Reducing or increasing expression times did not seem to have an influence on the level of degradation (Fig. 7). The A5:HA construct was purified from the medium by binding to protein A sepharose. Hence, the protein analyzed in Fig. 7 had been subjected to equal amount of proteolytic degradation in the cytoplasm and the periplasmic compartment before it leaks out into the medium. This indicates that the breakdown did not occur in the medium. This is not surprising, as the concentration of endogenous proteases in the periplasm is high compared to the medium.
The Hel4 domain antibody is known to fold very efficiently. We therefore tried to determine if our dAb antibody could be detected by protein A after western blotting. The results showed that the dAb was clearly detected by protein A, thus showing that the antibody was refolding after blotting onto the membrane (Fig. 5B). This resembles the method of far-western, except no denaturation/renaturation step is needed.35 The purified antibody could be detected using protein A-HRP whether or not reducing agent had been added to the sample.. When reducing agent was omitted the antibody could clearly be detected after just 4 hours incubation ( Fig. S1). When reducing agent was included, development overnight was needed to obtain weak signals indicating that refolding of the domain antibody was significantly promoted by the internal disulfide bond(data not shown).
The D domain of Protein A is binding to a non-linear epitope on the variable region of the heavy chain, which means that the antibody must be folded in order for protein A to recognize it.36 The above experiments show that antibody fragments are capable of refolding or partially refolding on the membrane after protein gel blotting. This is in line with what has previously been reported about the folding characteristics of the Hel4 antibody and VH3 gene family containing scFv.37,38
These results showed that the breakdown was the result of terminal degradation. It was further indicated that the tags were cleaved at specific sites, as the Coomassie staining and the protein A western blot gave rise to well-defined bands. The purified protein was subsequently subjected to protein gel blotting and detected with anti-his tag antibody. This confirmed that the degradation was happening at the C-terminal tag region (Fig. 6). When comparing the results from anti his-tag western with the results from the protein A western, it appears that some tags give less signal than could be expected from the amount of protein present. Especially the detection of A4:TAH with anti-his antibody (Fig. 6) showed to be problematic. Only a weak band could be observed after prolonged exposure time (data not shown). This could be due to degradation within the his-tag, but it is more likely a consequence of the placement of the his-tag and the resulting accessibility of the tag to the detection antibody. As increased expression time did not seem to increase degradation, we suspect the degradation to happen in the cytoplasm or in the periplasm before the protein leaks out into the supernatant (Fig. 7), Based on the observed degradation pattern giving rise to distinct bands, the degradation was furthermore expected to be caused by proteolytic degradation. There did not seem to be notable differences between the constructs when displayed on pIII, indicating that the C-terminal composition of the protein had an influence on the degradation. The tail-specific protease Tsp (also known as Prc) seemed to be a good candidate with regard to this assumption.24 To test if Tsp was implicated in the degradation of the tag constructs, we used the KS1000 strain, which has a deletion of the Tsp gene.39 Protein expressions of the A5:HA tag construct from different strains were compared (Fig. 8). TG1 and ER2738 showed to have equal ratio between antibody with full size tag sequence and completely degraded tag. These strains are commonly used for M13 phage display library creation and screening due to their high transformation efficiency. Because these 2 strains are not protease-deficient, the heavy degradation of the tag sequence observed in Fig. 8 was expected. On the contrary, CAG597 is defective in stress-induced proteases at high temperatures. Although CAG597 showed lower levels of tag degradation compared to TG1 and ER2738, the highest improvement in the yield of antibody with full-sized tag, was observed when the expression was made in the KS1000. The degradation was, however, only reduced and not completely eliminated, which is not surprising as a wide range of other proteases both cytoplasmic and periplasmic exists in E. coli.21c There are several other proteases that recognizes their substrates based on the composition of the carboxy terminus, and the location of especially non-polar residues has been linked to the susceptibility of proteolysis of proteins in E. coli 40c
Comparing the differences in degradation of the tag constructs, the level of degradation is less pronounced for C2:AH compared to the very similar A3:AH construct. This is likely due to the differences in C-terminal residues and the residues between the tags. Both A5:HA and B1:HA give rice to 2 distinct bands in the anti-his detected western blot (Fig. 6). Because AviTag is the terminal tag of the protein, the upper bands corresponds to full size protein and the lower bands corresponds to protein with partially or completely degraded AviTag. However, B1:HA appears to have fewer bands from degradation than A5:HA, which indicates that there is an effect of changing the C-terminal residue and the amino acids between the tags. B2:HM is less degraded than B3:AM, indicating that the AviTag might be problematic due to degradation. This is further underscored when comparing the degradation with C1:M to A1:A and B4:THM to A6:THA (Fig. 5). Due to the small size of the TEV recognition site, it is difficult to conclude whether there is degradation of B5:T or not, there is, however, a tendency for more degradation when the TEV recognition site is included in combination with other tags. The his-tag and the myc-tag do not appear to visibly increase the level of degradation, as can be seen from the B2:HM construct. Interestingly, A7:IAH was the only construct that did not give rise to a band corresponding to antibody without tag sequence at approximately 13 kDA (Fig. 5). Instead the lowest band observed corresponds to antibody with IgA hinge, indicating that this rigid proline-rich region is a pore substrate for proteases. The protease resistance of proline-rich regions has been observed previously and could be the consequence of a fixed backbone confirmation preventing degradation by more promiscuous proteases.41
For proteases with endopeptidase activity, like Tsp, the sequence within the tag region is important for the digestion efficiency. There is most likely also an effect of the distance from the C-terminal to the first accessible cleavage site in the tag sequence. As tag sequences in general are fairly small peptide motifs recognized by antibodies without any particular structure, they will inherently also be accessible to proteases. There does, however, seem to be more degradation when AviTag and the TEV site are included and less so when his-tag and myc-tag are included without the other 2 sequences. The length of the tag region seems to influence the amount of degradation, with longer tags giving more degradation (Fig. 5). The construct C2:AH was designed to reduce the degradation by removing superfluous amino acids between the tags and thereby make the tag sequence smaller. Furthermore, C-terminal amino acids were changed to charged bulky amino acids because this should generally reduce the recognition by C-terminal specific proteases like Tsp.21,42 Based on the data presented here, we conclude that there is an effect from the carboxy terminal residues on the level of degradation. More work is warranted to further elucidate the exact mechanisms behind this specific degradation.
Materials and Methods
Bacterial strains
TG1: E. coli K12 strain Δ(lac-proAB) supE thi hsdD5/F’ traD36 proA+BlacIq lacZΔM15 (Source Bioscience). HB2151: E. coli K12 strain ara Δ(lac-proAB) thi/F’ proA+BlacIq lacZΔM15 (Source Bioscience). CAG597: E. coli K12 strain )F− lacZ(am) pho(am) tyrT[supC(ts)] trp(am) rpsL(StrR) rpoH(am)165 zhg::Tn10 mal(am)) (New England Biolabs). ER2738: E. coli K12 strain (F´proA+B+ lacIqΔ(lacZ)M15 zzf::Tn10(TetR)/ fhuA2 glnV Δ(lac-proAB) thi-1 Δ(hsdS-mcrB)5) (New England Biolabs). KS1000: E. coli K12 strain (F' lacIq lac+ pro+/ ara Δ(lac-pro) Δ(tsp)= Δ(prc)::KanR eda51::Tn10(TetR) gyrA(NalR) rpoB thi-1 argE(am)) (New England Biolabs).
Expression and purification of recombinant protein
Expression of soluble antibody was done by transformation of HB2151 electrocompetent cells with plasmid DNA from the 14 constructs. Protein expression was induced by addition of IPTG at OD600 = 0.7–0.9 and cultures were incubated for 18 hours at 30⁰C in shaking incubator (200 rpm). When performing in vivo biotinylation arabinose and biotin were added at the time of induction. Prior to protein purification from the bacterial expression medium, the cultures were centrifuged at 4000 RCF for 30 minutes with subsequent sterile filtration of the supernatant (0.45 μm syringe filters). Antibodies were purified by means of HiTrap protein A HP columns (GE Healthcare, Cat.No.17–0402–01) according to manufacturer's protocol. Protein concentrations were measured using a Nanodrop 1000 spectrophotometer (Thermo Fisher Scientific).
SDS-PAGE, Western blotting and reagents
SDS-PAGE were run on precast 12% Bis-Tris SDS polyacrylamide gels (Bio-Rad, Cat.No.345–0124). Gels were run in XT MES buffer using Precision plus dual color (Bio-Rad) for determining protein sizes.
Western blots were blocked in 2% MPBS and incubated with the appropriate detection agent diluted in 2% MPBS. Western blots were then washed 3 times in PBS and protein was visualized using ECL Prime reagent (GE Healthcare, Cat.No.RPN2232). When blots were developed using multiple detection agents, the blots were treated with Restore protein gel blot stripping buffer (Thermo Fisher scientific, Cat. No. 21059) to remove the previously used detection agent. For detection of tagged dAb, the following were used: Horseradish peroxidase (HRP)-conjugated mouse anti c-Myc antibody clone 9E10 (Aarhus University core facility), HRP conjugated mouse anti-his antibody (Sigma-Aldrich, Cat.No.A7058–1VL), and HRP-conjugated protein A (Life technologies, Cat No. Ten–1023).
Construction of tags
To construct the tags, a combinations of the following primers were used in PCR A1-A5; Forward A1, A2, A5 and Reverse A1, A3 (Table S1). The PCRs was run using either of the plasmids pMCSG15 or pMCSG16 as template.20 The PCR products were double-digested with NotI and BglII and gel purified on a 1% agarose gel followed by electroelution in Spectra/Por 12–14,000 kDa dialysis tube (Spectrum laboratories, Cat. No. 132700), phenol extraction and alcohol precipitation of the DNA, all procedures according to standard protocol.43 An antibody clone called 8H previously selected in the lab from a domain antibody library kindly supplied by Dr. Daniel Christ at the Garvan Institute (Sydney, Australia) was used for testing the tags. A BglII site were introduced in the pHEN111 vector by site directed mutagenesis PCR just after the myc-tag by using the Forward BglII and Reverse BglII (Table S1). The modified vector was double-digested with NotI and BglII, gel purified and electroeluted. The constructs A1 to A5 were created by ligating the PCR tag combination products into the prepared vector. The A6 construct was made in the same way using the A5 vector as template and the primers; Forward A6 along with Reverse A1 (Table S1). The A7 construct was synthesized at Mr. Gene (Mr. Gene, Regensburg, Germany) and placed in the vector. The tag of A7 was replicated from a previously published paper showing successful in vivo biotinylation.30 All tag sequences of the constructs, except A7 were verified by sequencing using the LMB3 primer (Table S1). To make the B1-B5 and the C2 constructs, forward and reverse oligonucleotides for each tag were ordered with an overlapping sequence (Table S1). The overlapping sequences were designed to have a melting temperature of ca. 60⁰C. The 2 matching oligonucleotides for each of the 5 reactions were paired 1:1 and mixed together in a PCR reaction mix with Taq polymerase. Second strand synthesis was carried out in a thermocycler. In the PCR, each oligonucleotide works both as a template and a primer. In the PCR program, the annealing temperature was set to 54⁰C. The 5 PCR products were double-digested with NotI and BlpI restriction enzymes and gel purified as previously described. The C1 tag was identical to the original pHEN1 tag (Table S1).
In vitro enzymatic biotinylation
In vitro biotinylation of antibodies was carried out using commercial BirA biotin ligase enzyme with corresponding buffers (Avidity, Cat.No.EC 6.3.4.15), according to product protocol, unless otherwise specified.
TEV digestion
TEV digestion was carried out using a 1:50 molar ratio between phage particles and TEV enzyme. The TEV enzyme was a kind gift from Dr. Gregers Rom Andersen, Department of Molecular biology and Genetics at Aarhus University.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was partly supported by grants from the The Danish Council for Independent Research | Technology and Production Sciences (Grant no. 0602–02377B) to PK, The Lundbeck Foundation (Grant no. R126–2012–12143) to PK and Sino-Danish Center to SL.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Bradbury AR, Sidhu S, Dubel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol 2011; 29:245-54; PMID:21390033; http://dx.doi.org/ 10.1038/nbt.1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bradbury AR, Marks JD. Antibodies from phage antibody libraries. J Immunol Methods 2004; 290:29-49; PMID:15261570; http://dx.doi.org/ 10.1016/j.jim.2004.04.007 [DOI] [PubMed] [Google Scholar]
- 3.Pansri P, Jaruseranee N, Rangnoi K, Kristensen P, Yamabhai M. A compact phage display human scFv library for selection of antibodies to a wide variety of antigens. BMC Biotechnol 2009; 9:6; PMID:19175944; http://dx.doi.org/ 10.1186/1472-6750-9-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silacci M, Brack S, Schirru G, Marlind J, Ettorre A, Merlo A, Viti F, Neri D. Design, construction, and characterization of a large synthetic human antibody phage display library. Proteomics 2005; 5:2340-50; PMID:15880779; http://dx.doi.org/ 10.1002/pmic.200401273 [DOI] [PubMed] [Google Scholar]
- 5.Rothe C, Urlinger S, Lohning C, Prassler J, Stark Y, Jager U, Hubner B, Bardroff M, Pradel I, Boss M, et al. The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. J Mol Biol 2008; 376:1182-200; PMID:18191144; http://dx.doi.org/ 10.1016/j.jmb.2007.12.018 [DOI] [PubMed] [Google Scholar]
- 6.Christ D, Famm K, Winter G. Repertoires of aggregation-resistant human antibody domains. Protein Eng Des Sel 2007; 20:413-6; PMID:17720749; http://dx.doi.org/ 10.1093/protein/gzm037 [DOI] [PubMed] [Google Scholar]
- 7.Brockmann EC, Akter S, Savukoski T, Huovinen T, Lehmusvuori A, Leivo J, Saavalainen O, Azhayev A, Lovgren T, Hellman J, et al. Synthetic single-framework antibody library integrated with rapid affinity maturation by VL shuffling. Protein Eng Des Sel 2011; 24:691-700; PMID:21680620; http://dx.doi.org/ 10.1093/protein/gzr023 [DOI] [PubMed] [Google Scholar]
- 8.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, et al. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J Mol Biol 2007; 373:924-40; PMID:17825836; http://dx.doi.org/ 10.1016/j.jmb.2007.08.005 [DOI] [PubMed] [Google Scholar]
- 9.Mandrup OA, Friis NA, Lykkemark S, Just J, Kristensen P. A novel heavy domain antibody library with functionally optimized complementarity determining regions. PLoS One 2013; Accepted; PMID:24116173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prassler J, Thiel S, Pracht C, Polzer A, Peters S, Bauer M, Norenberg S, Stark Y, Kolln J, Popp A, et al. HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J Mol Biol 2011; 413:261-78; PMID:21856311; http://dx.doi.org/ 10.1016/j.jmb.2011.08.012 [DOI] [PubMed] [Google Scholar]
- 11.Hoogenboom HR, Griffiths AD, Johnson KS, Chiswell DJ, Hudson P, Winter G. Multi-subunit proteins on the surface of filamentous phage: methodologies for displaying antibody (Fab) heavy and light chains. Nucleic Acids Res 1991; 19:4133-7; PMID:1908075; http://dx.doi.org/ 10.1093/nar/19.15.4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffiths AD, Williams SC, Hartley O, Tomlinson IM, Waterhouse P, Crosby WL, Kontermann RE, Jones PT, Low NM, Allison TJ, et al. Isolation of high affinity human antibodies directly from large synthetic repertoires. EMBO J 1994; 13:3245-60; PMID:8045255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Wildt RM, Mundy CR, Gorick BD, Tomlinson IM. Antibody arrays for high-throughput screening of antibody-antigen interactions. Nat Biotechnol 2000; 18:989-94; PMID:10973222; http://dx.doi.org/ 10.1038/79494 [DOI] [PubMed] [Google Scholar]
- 14.Potter KN, Li Y, Pascual V, Capra JD. Staphylococcal protein A binding to VH3 encoded immunoglobulins. Int Rev Immunol 1997; 14:291-308; PMID:9186782; http://dx.doi.org/ 10.3109/08830189709116521 [DOI] [PubMed] [Google Scholar]
- 15.Meininger DP, Rance M, Starovasnik MA, Fairbrother WJ, Skelton NJ. Characterization of the binding interface between the E-domain of Staphylococcal protein A and an antibody Fv-fragment. Biochemistry 2000; 39:26-36; PMID:10625476; http://dx.doi.org/ 10.1021/bi9920174 [DOI] [PubMed] [Google Scholar]
- 16.Conley GP, Viswanathan M, Hou Y, Rank DL, Lindberg AP, Cramer SM, Ladner RC, Nixon AE, Chen J. Evaluation of protein engineering and process optimization approaches to enhance antibody drug manufacturability. Biotechnol Bioeng 2011; 108:2634-44; PMID:21618474; http://dx.doi.org/ 10.1002/bit.23220 [DOI] [PubMed] [Google Scholar]
- 17.Langone JJ. Applications of immobilized protein A in immunochemical techniques. J Immunol Methods 1982; 55:277-96; PMID:6762396; http://dx.doi.org/ 10.1016/0022-1759(82)90088-6 [DOI] [PubMed] [Google Scholar]
- 18.Lichty JJ, Malecki JL, Agnew HD, Michelson-Horowitz DJ, Tan S. Comparison of affinity tags for protein purification. Protein Expr Purif 2005; 41:98-105; PMID:15802226; http://dx.doi.org/ 10.1016/j.pep.2005.01.019 [DOI] [PubMed] [Google Scholar]
- 19.Schatz PJ. Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: a 13 residue consensus peptide specifies biotinylation in Escherichia coli. Biotechnology (N Y) 1993; 11:1138-43; PMID:7764094; http://dx.doi.org/ 10.1038/nbt1093-1138 [DOI] [PubMed] [Google Scholar]
- 20.Scholle MD, Collart FR, Kay BK. In vivo biotinylated proteins as targets for phage-display selection experiments. Protein Expr Purif 2004; 37:243-52; PMID:15294305; http://dx.doi.org/ 10.1016/j.pep.2004.05.012 [DOI] [PubMed] [Google Scholar]
- 21.Rozkov A, Enfors SO. Analysis and control of proteolysis of recombinant proteins in Escherichia coli. Adv Biochem Eng Biotechnol 2004; 89:163-95; PMID:15217159 [DOI] [PubMed] [Google Scholar]
- 22.Chen C, Snedecor B, Nishihara JC, Joly JC, McFarland N, Andersen DC, Battersby JE, Champion KM. High-level accumulation of a recombinant antibody fragment in the periplasm of Escherichia coli requires a triple-mutant (degP prc spr) host strain. Biotechnol Bioeng 2004; 85:463-74; PMID:14760686; http://dx.doi.org/ 10.1002/bit.20014 [DOI] [PubMed] [Google Scholar]
- 23.Hwang BY, Varadarajan N, Li H, Rodriguez S, Iverson BL, Georgiou G. Substrate specificity of the Escherichia coli outer membrane protease OmpP. J Bacteriol 2007; 189:522-30; PMID:17085556; http://dx.doi.org/ 10.1128/JB.01493-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keiler KC, Sauer RT. 13 Tsp and related tail-specific proteases. Enzymes 2002:373-86. [Google Scholar]
- 25.Ozyoruk F, Cheevers WP, Hullinger GA, McGuire TC, Hutton M, Knowles DP. Monoclonal antibodies to conformational epitopes of the surface glycoprotein of caprine arthritis-encephalitis virus: potential application to competitive-inhibition enzyme-linked immunosorbent assay for detecting antibodies in goat sera. Clin Diagn Lab Immunol 2001; 8:44-51; PMID:11139194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou YH, Chen Z, Purcell RH, Emerson SU. Positive reactions on Western blots do not necessarily indicate the epitopes on antigens are continuous. Immunol Cell Biol 2007; 85:73-8; PMID:17130902; http://dx.doi.org/ 10.1038/sj.icb.7100004 [DOI] [PubMed] [Google Scholar]
- 27.Clark S, Eckardt G, Siddle K, Harrison LC. Changes in insulin-receptor structure associated with trypsin-induced activation of the receptor tyrosine kinase. Biochem J 1991; 276(Pt 1):27-33; PMID:1645531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robishaw JD, Balcueva EA. A high temperature transfer procedure for detection of G protein gamma subunits by immunoblotting. Anal Biochem 1993; 208:283-7; PMID:7680843; http://dx.doi.org/ 10.1006/abio.1993.1047 [DOI] [PubMed] [Google Scholar]
- 29.Dudgeon K, Famm K, Christ D. Sequence determinants of protein aggregation in human VH domains. Protein Eng Des Sel 2009; 22:217-20; PMID:18957405; http://dx.doi.org/ 10.1093/protein/gzn059 [DOI] [PubMed] [Google Scholar]
- 30.Cloutier SM, Couty S, Terskikh A, Marguerat L, Crivelli V, Pugnieres M, Mani JC, Leisinger HJ, Mach JP, Deperthes D. Streptabody, a high avidity molecule made by tetramerization of in vivo biotinylated, phage display-selected scFv fragments on streptavidin. Mol Immunol 2000; 37:1067-77; PMID:11399324; http://dx.doi.org/ 10.1016/S0161-5890(01)00023-2 [DOI] [PubMed] [Google Scholar]
- 31.Thie H, Binius S, Schirrmann T, Hust M, Dubel S. Multimerization domains for antibody phage display and antibody production. N Biotechnol 2009; 26:314-21; PMID:19631299; http://dx.doi.org/ 10.1016/j.nbt.2009.07.005 [DOI] [PubMed] [Google Scholar]
- 32.Jander G, Cronan JE Jr., Beckwith J. Biotinylation in vivo as a sensitive indicator of protein secretion and membrane protein insertion. J Bacteriol 1996; 178:3049-58; PMID:8655479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lowe D, Dudgeon K, Rouet R, Schofield P, Jermutus L, Christ D. Aggregation, stability, and formulation of human antibody therapeutics. Adv Protein Chem Struct Biol 2011; 84:41-61; PMID:21846562; http://dx.doi.org/ 10.1016/B978-0-12-386483-3.00004-5 [DOI] [PubMed] [Google Scholar]
- 34.Rouet R, Lowe D, Dudgeon K, Roome B, Schofield P, Langley D, Andrews J, Whitfeld P, Jermutus L, Christ D. Expression of high-affinity human antibody fragments in bacteria. Nat Protoc 2012; 7:364-73; PMID:22301775; http://dx.doi.org/ 10.1038/nprot.2011.448 [DOI] [PubMed] [Google Scholar]
- 35.Wu Y, Li Q, Chen XZ. Detecting protein-protein interactions by Far western blotting. Nat Protoc 2007; 2:3278-84; PMID:18079728; http://dx.doi.org/ 10.1038/nprot.2007.459 [DOI] [PubMed] [Google Scholar]
- 36.Graille M, Stura EA, Corper AL, Sutton BJ, Taussig MJ, Charbonnier JB, Silverman GJ. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc Natl Acad Sci U S A 2000; 97:5399-404; PMID:10805799; http://dx.doi.org/ 10.1073/pnas.97.10.5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jespers L, Schon O, James LC, Veprintsev D, Winter G. Crystal structure of HEL4, a soluble, refoldable human V(H) single domain with a germ-line scaffold. J Mol Biol 2004; 337:893-903; PMID:15033359; http://dx.doi.org/ 10.1016/j.jmb.2004.02.013 [DOI] [PubMed] [Google Scholar]
- 38.Akerstrom B, Nilson BH, Hoogenboom HR, Bjorck L. On the interaction between single chain Fv antibodies and bacterial immunoglobulin-binding proteins. J Immunol Methods 1994; 177:151-63; PMID:7822821; http://dx.doi.org/ 10.1016/0022-1759(94)90152-X [DOI] [PubMed] [Google Scholar]
- 39.Silber KR, Sauer RT. Deletion of the prc (tsp) gene provides evidence for additional tail-specific proteolytic activity in Escherichia coli K-12. Mol Gen Genet 1994; 242:237-40; PMID:8159175; http://dx.doi.org/ 10.1007/BF00391018 [DOI] [PubMed] [Google Scholar]
- 40.Parsell DA, Silber KR, Sauer RT. Carboxy-terminal determinants of intracellular protein degradation. Genes Dev 1990; 4:277-86; PMID:2186965; http://dx.doi.org/ 10.1101/gad.4.2.277 [DOI] [PubMed] [Google Scholar]
- 41.Shinnar AE, Butler KL, Park HJ. Cathelicidin family of antimicrobial peptides: proteolytic processing and protease resistance. Bioorganic Chem 2003; 31:425-36; PMID:14613764; http://dx.doi.org/ 10.1016/S0045-2068(03)00080-4 [DOI] [PubMed] [Google Scholar]
- 42.Keiler KC, Sauer RT. Sequence determinants of C-terminal substrate recognition by the Tsp protease. J Biol Chem 1996; 271:2589-93; PMID:8576225; http://dx.doi.org/ 10.1074/jbc.271.5.2589 [DOI] [PubMed] [Google Scholar]
- 43.Moore D, Dowhan D. Purification and concentration of DNA from aqueous solutions. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current protocols in molecular biology. New York: John Wiley and Sons; 2002. Chapter 2, Unit 2 1A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.