

Abstract
Deregulation of the phosphatidylinositol 3-kinase (PI3K) pathway is central to many human malignancies while normal cell proliferation requires pathway functionality. Although inhibitors of the PI3K pathway are in clinical trials or approved for therapy, an understanding of the functional activities of pathway members in specific malignancies is needed. In lung cancers, the PI3K pathway is often aberrantly activated by mutation of genes encoding EGFR, KRAS, and PIK3CA proteins. We sought to understand whether class IA PI3K enzymes represent rational therapeutic targets in cells of non-squamous lung cancers by exploring pharmacological and genetic inhibitors of PI3K enzymes in a non-small cell lung cancer (NSCLC) cell line system. We found that class IA PI3K enzymes were expressed in all cell lines tested, but treatment of NSCLC lines with isoform-selective inhibitors (A66, TGX-221, CAL-101 and IC488743) had little effect on cell proliferation or prolonged inhibition of AKT activity. Inhibitory pharmacokinetic and pharmacodynamic responses were observed using these agents at non-isoform selective concentrations and with the pan-class I (ZSTK474) agent. Response to pharmacological inhibition suggested that PI3K isoforms may functionally compensate for one another thus limiting efficacy of single agent treatment. However, combination of ZSTK474 and an EGFR inhibitor (erlotinib) in NSCLC resistant to each single agent reduced cellular proliferation. These studies uncovered unanticipated cellular responses to PI3K isoform inhibition in NSCLC that does not correlate with PI3K mutations, suggesting that patients bearing tumors with wildtype EGFR and KRAS are unlikely to benefit from inhibitors of single isoforms but may respond to pan-isoform inhibition.
Keywords: PI3K, AKT, mTOR, KRAS, EGFR, drug-resistant, non-squamous
Abbreviations
- AKT
protein kinase B
- E545K
amino acid change at position 545 from glutamic acid to lysine
- EGFR
epidermal growth factor receptor
- ERK1
mitogen activated protein kinase 3/p44
- ERK2
mitogen activated protein kinase 1/p42
- FBS
fetal bovine serum, Ser473- serine at position 473
- G188D
amino acid change at position 188 from glycine to aspartic acid
- GI50
drug concentration causing 50% maximum inhibition of cellular proliferation
- Hr
hour
- IC50
drug concentration causing 50% inhibition of activity
- LKB1
liver kinase 1
- MAPK
mitogen activated protein kinase
- mTOR
mammalian target of rapamycin
- NSCLC
non-small cell lung cancer
- PARP
poly ADP ribose polymerase
- PI3K
phosphatidylinositol 3-kinase
- PIP2
phosphatidylinositol 4,5-biphosphate
- PIP3
phosphatidylinositol 3,4,5-triphosphate
- PTEN
phosphatase and tensin homolog
- S6RP
ribosomal subunit S6
- siRNA
small interfering RNA
- μM
micromolar.
Introduction
Lung cancer leads to the greatest morbidity and mortality of all cancer deaths in the United States.1 To combat these diseases, new therapeutic agents and therapeutic strategies are necessary, and these strategies should include patient-specific therapy to minimize toxicity and cost. Targeting therapeutic strategies to actionable mutations found in non-small cell lung carcinomas (NSCLC), such as mutation/amplification of epidermal growth factor receptor2 gene,3 activating point mutations in p110α isoform of PI3K (PIK3CA),4 and in the KRAS gene,5 and deletion of the PTEN gene,6 may help achieve personalized treatments. Importantly, each of these genetic alterations can lead to increased survival signals that converge on the PI3K/AKT/mTOR cascade. The PI3K pathway is a central node for control of both cell growth and cell proliferation in normal epithelial cells, and aberrant pathway activation in lung tumorigenesis dysregulates cell metabolism, proliferation, apoptosis, and angiogenesis.7-9
Because many cancers depend on a hyperactive PI3K pathway, increased attention has been placed on development of pharmacological inhibitors of PI3K enzymes that are often mutated or improperly expressed in tumors.10,11 Specifically, activation of class IA PI3Ks (p110-catalytic and p85-regulatory heterodimers) generates plasma membrane lipid second messenger molecules by phosphorylating the 3′-OH position of phosphatidylinositol 4,5 bisphosphate (PIP2) to produce phosphatidylinositiol-3,4,5-trisphosphate lipid (PIP3). While PIP3 levels are tightly regulated by the cell, net pathway activation is determined by a delicate balance between the opposing activities of PI3K lipid kinases and 3- and 5- lipid phosphatases such as PTEN. Recent evidence suggests that there may be specific cellular contexts and physiological roles for each class IA PI3K isoform: p110α (PIK3CA), p110β (PIK3CB), and p110δ (PIK3CD).12,13 Activated receptor tyrosine kinases, G-protein coupled receptors, RAS and other small GTPases can signal through cell type-specific PI3K isoforms.14-19 Application of these data to drug discovery efforts revealed a role for PIK3CD inhibition for treatment of B-cell malignancies and autoimmune diseases.20,21 Finally, class IA PI3Ks may also function with non-catalytic activity, serving as molecular scaffolds for other signaling proteins.22 Thus, therapeutic targeting of individual PI3K class IA catalytic subunits may have clinical promise by impeding a host of PI3K functions necessary for tumor growth, but the biology underlying PI3K isoform function in specific contexts is essential.
We began this study with a hypothesis that PI3K isoform-selective inhibitors may have clinical value for personalized treatment of lung cancers due to activation of PIK3CA in some lung cancers. Herein, we describe the treatment of genetically diverse NSCLC cell lines with inhibitors of class IA PI3K enzymes on pathway activation, proliferation, and viability. Importantly, our results demonstrate the apparent compensatory activity of PI3K isoforms, independent of the genetic background of the cells, that may derail clinical use of single-agent PI3K inhibitors in lung cancers in favor of pan-PI3K inhibitors in combination with other agents.
Results
Neither PI3K isoform expression levels nor mutation status alone dictates response to PI3K inhibitors
The experiments were initiated by determining gene and protein expression levels for class IA PI3K enzymes in a panel of NSCLC cell lines (Fig. 1A). Mutation status for important driver mutations (EGFR, KRAS, PIK3CA, LKB1, TP53, and PTEN) is also indicated for the cell lines utilized as reported by the COSMIC database and others (cancer.sanger.ac.uk).23,24 Gene expression levels of PI3K isoforms were measured in NSCLC cells using Affymetrix DNA microarray technology. Normalized Affymetrix signal intensity values revealed that the p110β isoform is the most highly expressed of the 3 isoforms assayed while the p110δ isoform was poorly expressed. PTEN demonstrated varying levels of expression among cell lines.25 Even though gene expression levels vary among PI3K isoforms, levels of protein expression for PI3K isoforms change only modestly (Fig. 1B). PTEN expression is the most variable among pathway members assessed. All cell lines show activated AKT (S473) and varying levels of activated ERK1/2 activity under normal growth conditions.
Figure 1.

Expression of class IA PI3K p110 isoforms and PTEN among NSCLC cell lines. (A) Relative mRNA expression of p110 isoforms and PTEN were measured by Affymetrix microarray analysis. NSCLC cell line mutation status and histopathology are indicated below. (B) PI3K p110/p85 isoform and pathway effector protein expression in NSCLC cell lines was determined by immunoblotting analysis.
To assess the contribution of each Class IA PI3K enzyme in lung cancer cells to proliferation and AKT phosphorylation, we chose pharmacological inhibitors of PI3K isoform activity. Inhibitors of 3 chemotypes, previously shown to possess isoform-selective activity were selected: A66 (aminothiazole) 26; TGX-221 (morpholinochromone) 27; CAL-101 (quinazolinone purine),28 and IC488743 29 (Table 1). Briefly, A66 preferentially inhibits p110α; TGX-221 preferentially inhibits p110β isoform; CAL-101 and IC488743 are potent p110δ inhibitors, and the latter also had significant inhibitory activity against p110β. We used the pan-PI3K inhibitor, ZSTK474, which has low nanomolar IC50 values for all 3 class IA isoforms as a control.30 Although the IC50 values for these inhibitors have been independently reported, we independently quantified the kinase activity of A66, TGX-221, CAL-101 and IC488743 using an ELISA assay.26-29 The resulting IC50 values are consistent with those previously reported (Fig. S1).
Table 1.
PI3K inhibitor IC50 selectivity profile- Summary of previously published biochemical selectivity for PI3K class I isoform inhibitors used in this study.
| IC50(µM) | p110 | p110 | p110 | p110 | mTOR |
|---|---|---|---|---|---|
| Pan-PI3K inhibitor | |||||
| ZSTK474 | 0.017 | 0.053 | 0.046 | 0.049 | >10 |
| Isoform-selective PI3K inhibitors | |||||
| A66 | 0.032 | >12.5 | >1.25 | 3.48 | >50 |
| TGX-221 | 5 | 0.005 | 0.100 | >35 | >10 |
| CAL-101 | 0.82 | 0.56 | 0.002 | 0.089 | >1 |
| IC488743 | >20 | 1.2 | 0.005 | — | — |
To determine whether isoform-selective PI3K inhibitors affect NSCLC cell proliferation and survival, Alamar Blue and Trypan Blue exclusion assays were used. We tested cell lines in asynchronous and synchronized growth conditions to determine if cell cycle position affected activity of the agents. First, asynchronously growing cells were treated for 72hr with the pan-PI3K inhibitor, ZSTK474, as a control, or each of 4 isoform-selective inhibitors to determine the half-maximal concentration for growth inhibition (GI50) using the Alamar Blue assay (Fig. 2, Table 2). In cells synchronized by serum starvation, treatment with PI3K inhibitors was carried out for 72 hr and assayed by Trypan Blue exclusion (Table S1). There was significant agreement between inhibition of asynchronous and synchronous cells as determined by match-ranking the inhibitors based on the calculated GI50(r = 0.651, p = 0.001).
Figure 2.
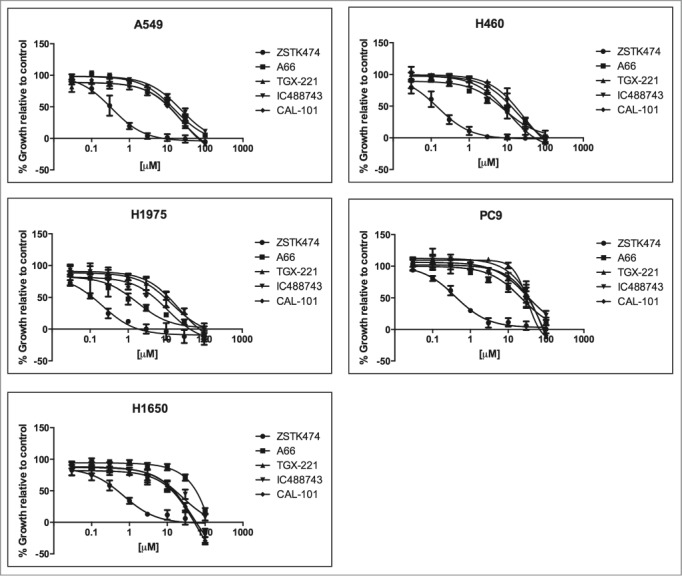
Growth inhibition activity PI3K inhibitors against a panel of 5 NSCLC cell lines. A549, H460, H1975, PC9, and H1650 were treated with increasing concentrations of PI3K inhibitors; ZSTK474, A66, TGX-221, IC488743, and CAL-101 for 72hr (0.03 – 100μM). Cell proliferation was determined by Alamar Blue viability assay. Each experiment was performed with triplicate cultures, for 3 independent experiments (n=3). Error bars represent standard deviation (SD). Experimental results were normalized to 24 hr plated cells and divided by untreated control to determine (% growth relative to control). Non-linear curve fitting and GI50 values were generated using Graphpad Prism.
Table 2.
GI50 values of PI3K inhibitors for NSCLC cell line. Each cell lines was treated with increasing concentrations of indicated PI3K inhibitors for 72 hr. Cell proliferations was determined by Alamar Blue viability assay. Non-linear curve fitting and GI50 values were generated using Graphpad Prism.
| GI50(µM) | A549 | H460 | H1975 | PC9 | H1650 |
|---|---|---|---|---|---|
| Pan-PI3K inhibitor | |||||
| ZSTK474 | 0.33 | 0.15 | 0.21 | 0.40 | 0.69 |
| Isoform-selective PI3K inhibitors | |||||
| A66 | 15.64 | 8.10 | 1.74 | 23.31 | >200 |
| TGX-221 | 24.19 | 26.78 | 21.88 | 120.5 | 50.14 |
| CAL-101 | 22.09 | 10.66 | 10.85 | 131.6 | 62.10 |
| IC488743 | 22.80 | 17.45 | 14.28 | 32.15 | 24.45 |
In summary, pan-PI3K isoform inhibition with ZSTK474 was effective at blocking proliferation in all NSCLC cell lines achieving GI50 values at nanomolar concentrations. Treatment with isoform-selective inhibitors for p110α, p110β, and p110δ enzymes was largely ineffective at impairing proliferation at concentrations conferring isoform selectivity. Specifically, H460 and H1975 cells have the greatest sensitivity to the PIK3CA inhibitor, A66 (GI508.1 µM and 1.7µM) and demonstrated the greatest overall responsiveness to PI3K inhibition of proliferation compared to other cell lines tested. Conversely, PC9 and H1650 cell lines were found to be the least sensitive to all PI3K inhibitors tested. In particular, PC9 cells are most resistant to TGX-221 and CAL-101 (GI50>100µM), and H1650 cells are most resistant to A66 (GI50 >200µM). Finally, cell cycle position does not appear to alter sensitivity in any cell line.
Inhibition of PI3K/AKT blocks survival signals necessary for proliferation
To assess whether the lack of proliferative response of the cell lines to isoform-selective inhibitors of the PI3K enzymes was due to lack of target inhibition, we tested AKT activation (Ser473) over a range of inhibitor concentrations by western analysis after 3 hr of treatment. The ratio of pAKT/tAKT was determined by densitometric evaluation of the immunoblots and plotted for each cell line and isoform selective inhibitor concentrations (Fig. 3).
Figure 3.

Pharmacodynamic responses to PI3K isoform selective inhibition. The relative levels of phosphorylated AKT in NSCLC cell lines (A549, H460, H1975, PC9, H1650) treated with increasing concentrations of PI3K inhibitors (A) A66 (B) TGX-221 (C) IC488743 and (D) CAL-101. Cells were synchronized by serum-starved for 24hr prior to release in RPMI 1640 media containing 1% serum with or without drug for 3 hr. Cell lysates were collected for immunoblotting analysis with pAKT (S473) and tAKT antibodies. α-tubulin serves as the normalization control. ImageJ software was used for densitometric analysis of pAKT and tAKT levels. Three independent experiments were quantified, pAKT (S473) was normalized to tAKT , and data were averaged to reflect relative changes in pAKT (S473) activation. Error bars represent mean ± SEM. PIK3CA mutant cell lines are indicated.
Consistent with the proliferation assays, H1975 and H460 cell lines demonstrate the greatest reduction of activated AKT at nanomolar concentrations of A66 while H1650 demonstrated only minor reduction in pAKT, even at high concentrations (Fig. 3A). Treatment with the p110β inhibitor, TGX-221, had little effect on AKT phosphorylation in all cell lines except for H1650 cells (Fig. 3B). H1650 cells demonstrated nearly 75% reduction in phosphorylated AKT at nanomolar levels of TGX-221. Treatment with the p110δ inhibitors (CAL-101 and IC488743) reduced AKT phosphorylation more than 50% at the highest concentrations used for most cell lines (Figs. 3C and 3D).
To determine the duration of inhibition of effector molecule (pAKT and pS6RP) phosphorylation, a treatment time course was performed using 1μM of each inhibitor on 3 representative NSCLC lines (A549, PC9, H1650) (Fig. 4, top). At this concentration (1μM), each compound is predicted to be on-target, having minimal impact on other PI3K isoforms. The duration of activity was consistent with both the proliferation and concentration-dependent treatment assays. Specifically, the effect of each inhibitor on A549 cells was sustained over time, as measured by reduction in phosphorylation of AKT and S6. Except for A66, PC9 cells displayed minimal response to each agent as demonstrated by modest reduction of pAKT. However, phosphorylated S6 signal was reduced over time with each inhibitor relative to untreated PC9 cells. H1650 cells also demonstrated sustained responses to each agent as measured by absence of S6 phosphorylation. However, in H1650 cells, A66 was ineffective in reducing pAKT levels, relative to the untreated control.
Figure 4.

Isoform-selective PI3K inhibitor treatment demonstrates time-dependent inhibition of PI3K signaling. (Top) A549, PC9 and H1650 cells were treated with 1μM isoform selective inhibitors in RPMI 1640 containing 1% serum and collected at indicated times after drug addition. Untreated controls represent serum-starved cells collected at times post-serum addition. (Bottom) Cell lines were treated with 1μM ZSTK474 as in the top panel. Samples were collected for immunoblotting with indicated antibodies as described above. α-tubulin serves as the normalization control.
Because of the poor response of A549, PC9 and H1650 cells to the isoform-selective inhibitors, we also assessed the pharmacodynamic response to ZSTK474 on all 5 cell lines originally tested for proliferation defects in Figure 2. In Figure 4, bottom, we found that all 5 cell lines responded to treatment of 1μM ZST474 with diminished pAKT473 and pS6 that was sustained for the entire timecourse, suggesting that inhibition of each of the PI3K isoforms is necessary for a robust and sustained response. We recognize that this concentration of drug is well above the GI50 for each of these cell lines. Thus, the accumulated data suggest that loss of a single PI3K class I isoform might be compensated by remaining isoforms, or alternative pathways, as measured by maintenance of activated AKT. To test the hypothesis of isoform compensation, we undertook an experiment to couple gene silencing with a pharmacological inhibitor of PIK3CB/D.
siRNA silencing of p110β does not phenocopy effects observed with PIK3CB and D inhibition
The previous experiments provided evidence that inhibition of proliferation and loss of AKT and S6 signaling were only apparent at concentrations of each inhibitor that exceeded selectivity for that compound. Only the pan-inhibitor, ZSTK474, was effective at inhibiting proliferation at sub-micromolar concentrations, suggesting that in most cells complete PI3K activity was necessary for growth. Further, the cell lines with activating PIK3CA mutations (H460 and H1975) were the most sensitive to A66, as previously described.31 We then postulated that PI3K isoforms may compensate for one another. To test this hypothesis, we chose cells least sensitive to PIK3CA inhibition and most sensitive to PIK3CB and PIK3CD inhibitors (A549, PC9, H1650). Expression of p110β was reduced using short-interfering RNA (siRNA) then cells were treated with either CAL-101 or IC488743 for 3hr at 1μM concentration then effector molecule expression was tested. IC488743 is expected to have off-target activity relative to CAL-101 at this concentration (Table 1).
Silencing of p110β isoform reduced expression in A549 by 80%, in PC9 by 55%, and in H1650 by 61%, as measured by densitometry (Fig. 5A). Partial ablation of the p110β isoform alone did not significantly reduce phosphorylation of AKT at S473 compared with IC488743 or CAL-101 treatment alone in all cells, suggesting that p110α and p110δ isoforms, or residual p110β, remain active. Importantly, the observed decrease in pAKT after p110β ablation with CAL-101 treatment was not superior to treatment with CAL-101 alone. However, treatment of p110β-depleted cells with IC488743 further reduced pAKT levels compared with the either p110β-depletion or IC488743 treatment, suggesting that PIK3CA may responsible for the remaining activity (Fig. 5B).
Figure 5.

siRNA-mediated ablation of p110β does not phenocopy pharmacological IC488743 inhibition in NSCLC. (A) A549, PC9, and H1650 cells were transiently-transfected with siRNA specific for p110β for 24hr, then treated with CAL-101 or IC488743 (1µM) for 3 hours. Cell lysates were prepared for immunoblotting with indicated antibodies. (B) Densitometric analysis of phosphor-AKT (Ser 473) levels in treated cells. Signals normalized to tAKT.
Interestingly, pERK1/2 signaling was reduced in H1650 cells that were treated with genetic and pharmacological inhibitors but not the other lines (Fig. 5A). Densitometric analysis of pERK1/2 showed reduction of signal by nearly 50%, suggesting that one or both isoforms play a role in RAS/MAPK signaling in H1650 cells perhaps independently of AKT phosphorylation. These data suggest that activity of both the AKT and RAS/MAPK signaling cascades are controlled by multiple PI3K isoforms.
Treatment with IC488743 or ZSTK474 promotes transient cell cycle arrest but not apoptosis NSCLC cells
Inhibition of p110β and p110δ isoforms by genetic and/or pharmacological treatment reduced cell proliferation and inhibited PI3K signaling in A549, PC9, and H1650 NSCLC lines better than isoform-specific inhibitors alone but not as well as the pan-inhibitor. We sought to understand whether reduced proliferation associated with increased concentration of the isoform-selective inhibitors was due to cell cycle arrest or induction of apoptosis, given the potential clinical consequences on normal and tumor cells.
When we initiated these studies, we operated under that hypothesis that isoform-selective inhibition of PI3K species may have a more favorable clinical path than pan-inhibition if inhibition resulted in tumor stasis or death. However, given that the data generated thus far suggests that inhibition of multiple PI3K isoforms in NSCLC does not appear to induce cell death, we chose to assay the cell cycle profile of cells treated with the IC488743 compared to the pan-isoform inhibitor ZSTK474 at the individual GI75 concentrations for the cell lines to determine cell fate. The GI75 was selected because this represents the concentration where loss of proliferation was observed with IC488743. Importantly, when evaluating differences in sensitivity between the 2 inhibitors, ZSTK474 was >50-fold more potent than IC488743 (1.3 µM vs 73.3 µM) among the cell lines. After 24 hr of treatment, treatment with either compound led to significant cellular accumulation in G1 phase for each cell line with concomitant loss in the S phase compartment (Fig. 6A). At 48 hr, we observed a sustained arrest in G1 in A549 and H1650, however PC9 cells appear to exit G1 and re-enter the cell cycle. Additionally, at both 24 and 48 hr, we detected no change in the % sub-G1 population suggesting apoptosis was not being induced by inhibitor treatments for the cell lines tested (Fig. 6B). We then treated each of the original cell lines at the GI75 for 72h and found insignificant changes in cell cycle position relative to control cells which remained in 1%-serum containing media for the length of the experiment (Fig. S3A).
Figure 6.
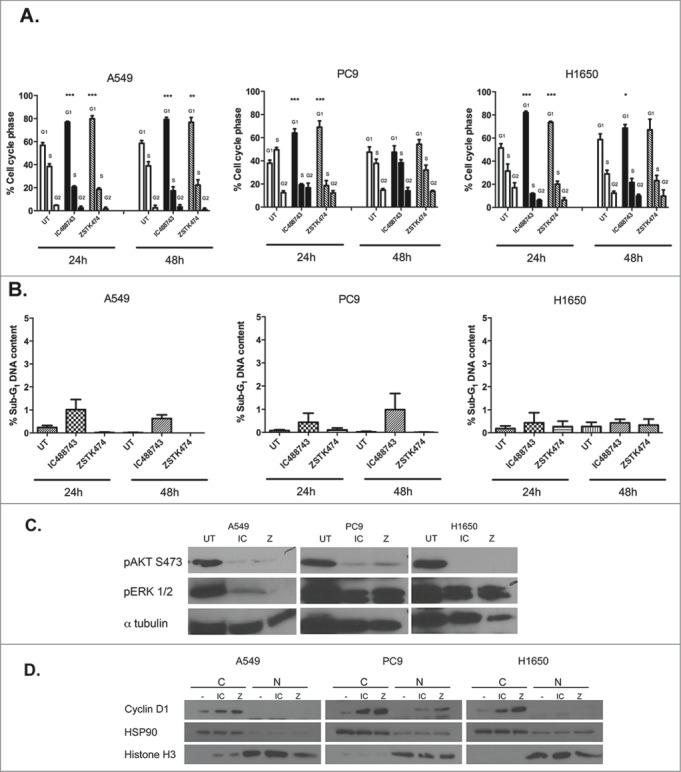
IC488743 and ZSTK474 treatment alters G1 cell cycle progression without inducing apoptosis. (A) Control, IC488743 and ZSTK474 treatment at GI75 concentrations for A549 (IC 65µM and Z 1µM), PC9 (IC 100µM, Z 1.3µM), and H1650 (IC 55µM, Z 1.6µM) cells were assessed for relative cell cycle distributions by propidium iodide (PI) staining of DNA following 24hr and 48hr of treatment. Mean percentages of cells in G0/G1, S, and G2/M are indicated. Error bars represent standard deviation, (n=3) and Student t test showed significant difference (*p <0.05). (B) Control, IC488743, and ZSTK474 treated A549, PC9 and H1650 cells were analyzed for % sub-G1 population indicating apoptosis. Mean percentages of sub-G1 cells are displayed. Error bars represent standard deviation, (n=3). (C) Markers for pathway activation and apoptosis assayed in A549, PC9, and H1650 cells treated at GI75 concentrations of IC488743 (IC) or ZSTK474 (Z) for 24 and 48hr then harvested for western blot analysis. (D) Cells treated for 48hr with GI75 concentrations of IC488743 or ZSTK474 were subjected nuclear and cytosolic fractionation then western blot analysis to determine the sub cellular localization of G1 to S-phase regulatory protein cyclin D1.
We also assessed pathway activity after 48 hr of treatment with IC488743 and ZSTK474. As expected, both drugs inhibited phosphorylation of AKT S473 and phosphorylation of ERK1/2 (Fig. 6C). We evaluated cleaved PARP as an apoptotic signaling molecule. We observe no change in cleaved PARP levels with treatment in any cell line consistent with no significant change in sub-G1 fraction after treatment (Fig. 6B and Fig. S3B).
To further characterize treatment effects on cell cycle progression, treated cells were fractionated into sub-cellular compartments and we evaluated nuclear and cytosolic levels of cyclin D1 after 48 hr treatments (Fig. 6D). Cyclin D1 should be extruded from the nucleus of cycling cells. After 48 hr, cyclin D1 accumulated in the cytosol with treatment of IC488743 or ZSTK474 relative to the controls all cells consistent with cell cycle analysis and lack of cleaved PARP. Fractionation of the nuclear and cytosolic compartments was confirmed by histone H3 and HSP90 localization.
From these data, only pan-PI3K inhibition or an isoform-selective inhibitor utilized at non-selective concentrations diminished PI3K signaling downstream to AKT and S6 molecules and resulted in transient loss of proliferation. We have observed that each of the Class IA PI3K isoforms is necessary for growth and proliferation and PI3K/AKT and RAS/MAPK activity.
Combined treatment of ZSTK474 and erlotinib inhibits proliferation of erlotinib-resistant NSCLC cells
Our data suggest that non-squamous NSCLC cell lines seem to require class IA PI3K activity to ensure proper cell growth and proliferation through both PI3K/AKT and Ras/MAPK pathways, we investigated whether pharmacological inhibition of all class IA enzymatic activity sensitized cells to the EGFR inhibitor, erlotinib. Ihle and colleagues demonstrated that PX-866, an inhibitor of PI3KCA, inhibited growth of xenografted A549 cells when administered with gefitinib, an EGFR inhibitor, but there are apparent adverse metabolic events that coincide with treatment.32 A549 and H460 cells are inherently insensitive to EGFR inhibition largely due to KRAS activation. These cell lines have inactivating mutations in LKB1 (Fig. 1) and have been shown to possess sensitivity to MAPK and mTOR inhibitors.23 Thus, treatment with ZSTK474, to ablate the activity of each class I enzyme, with the addition of erlotinib, to silence the MAPK pathway, may prove efficacious in these cells, especially if apoptotic activity is demonstrated at low levels of drug.33
The growth inhibitory activity of erlotinib and ZSTK474 was measured in the erlotinib-resistant NSCLC cell lines A549 and H460 (wt EGFR/KRAS) by Alamar Blue assay (Fig. 7A). We found that both A549 and H460 were similarly insensitive to single-agent exposure to erlotinib, only reaching 50% growth inhibition at concentrations greater than 3μM. The pan-PI3K inhibitor was markedly more potent, achieving half maximal growth inhibitory concentrations in the nanomolar range.
Figure 7.
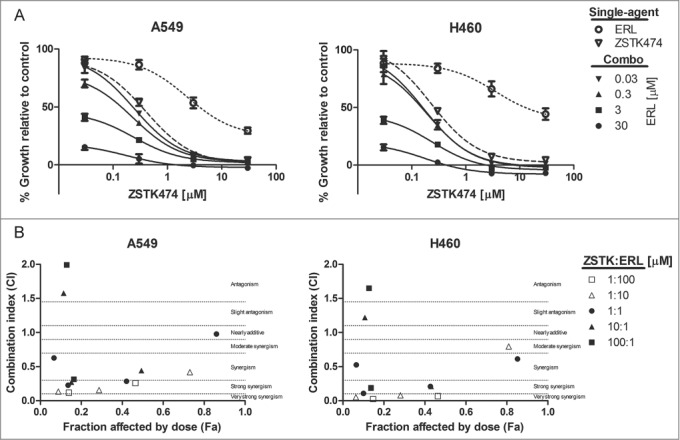
Combined ZSTK474 and erlotinib can synergistically reduce proliferation in KRAS active NSCLC. (A) A549 and H460 cells were treated with the indicated concentrations of each inhibitor, as a single-agent, or in combination. Combination data are presented as individual curves at fixed erlotinib concentrations (0.03–30μM) plotted against increasing ZSTK474 concentrations (0.03–30μM). Points represent the mean of 3 independent experiments +/− SD, and lines were fitted using non-linear regression analysis. (B) Median effect analysis was performed by CompuSyn software to study the interaction between the inhibitor combinations at the concentrations indicated in (A). Points represent the mean of 3 independent experiments. Dotted lines mark range of interaction classification.
We then determined the effect of combined EGFR and pan-PI3K inhibitor treatment on cell proliferation over 72 hr. Each inhibitor combination was tested at concentrations spanning GI50 values. We found that the combinations of erlotinib and ZSTK474 were more growth inhibitory than either compound alone. Although modest, H460 cells demonstrated greater growth inhibition in response to the drug combinations. Growth inhibition was evaluated by median effect analysis (CompuSyn, Memorial Sloan-Kettering Cancer Center and MIT) to determine if the improved growth inhibitory activity of the combinations was additive or synergistic (Fig. 7B). Combinations were synergistic at 1:1, 1:10, and 1:100 ratios (ZSTK474: erlotinib), but antagonistic effects were evident at 10:1 and 100:1 ratios (ZSTK474: erlotinib). Overall, while a synergistic interaction was observed with pan-PI3K and EGFR inhibitors that impaired cell proliferation, we identified modest cytotoxic effects in the H460 cell line occurring at highest tandem drug concentrations.
These data suggest that ablation of both the PI3K/AKT and RAS/MAPK axes are essential for cytotoxicity in the KRAS active, non-squamous NSCLC cells. Utilizing these agents in combination may lead to effective cytotoxicity in KRAS active cells at concentrations of ZSTK474 that minimize side-effects.33-35 Pre-clinical in vivo experiments that test the combination as single agents will be important as will efforts to formulate the drugs as a combination particle to prevent off-target effects.
Discussion
Lung cancer is a disease characterized by extensive genomic changes that unfortunately lead to millions of deaths from the disease each year worldwide because patients do not achieve a sustained response to therapy.36 Only recently have actionable mutations and mutated signaling pathways been identified and targeted therapeutically.37-40 Our interests converge on the PI3K/AKT/mTOR signaling axis because it represents one of the most commonly activated pathways in cancer for which few targeted therapies have resulted in clinical use in lung cancer.41
In lung cancers, mutations have been reported in multiple genes that control PI3K/AKT pathway activation, including EGFR, KRAS, HER3 and BRAF,42-44 even though few adenocarcinomas of the lung demonstrate mutations in PIK3CA.45 Together, these observations make the effector molecules of the PI3K pathway alluring targets for the cancer therapy. First generation PI3K inhibitors targeted 3 class IA PI3K isoforms (α, β, and δ) and were not suitable for clinical use due largely to toxicity and poor bioavailability.46,47 Although class IA PI3K isoforms possess similar protein structure, control of expression, and regulation of activity, recent literature reports non-redundant cellular functions that appear to be isoform specific.48-54 Importantly, to our knowledge, PI3K isoform-specific activities have not been thoroughly dissected in lung cancers of non-squamous histology. Therefore, we chose to investigate the intersection of therapeutically-actionable mutations, isoform-specific inhibitory compounds, and deregulated activities of the PI3K/AKT signaling cascade in NSCLC cell lines.
The activities of a panel of PI3K inhibitory compounds were tested in vitro and in cell lines. The IC50 values for PI3K isoform specificity and selectivity have been previously published and further validated by this laboratory. Using these compounds as tools and mutational status of cell lines as potential biomarkers of response, we intended to evaluate the activity of each compound for inhibition of growth and/or cell killing in a panel of NSCLC cells and to independently assess the necessity of each PI3K class IA enzymes in NSCLC.
Importantly, we found poor anti-proliferative activity among the isoform-selective PI3K compounds using a range of treatment concentrations overlapping our in vitro estimation of IC50 for each isoform. Several compounds demonstrated anti-proliferative activity against the cell lines when tested at micromolar concentrations, thus non-selective. Even though we sought biomarkers of response in anticipation of moving these agents toward clinical use, the mutational analysis was mostly observational given poor activity of the compounds. We found that cell lines containing mutated PIK3CA were most sensitive to A66. Specifically, H460 bears an activating mutation in PIK3CA (E545K) as does H1975 (G188D) which apparently sensitizes the cells to A66 (GI50 8.1 µM vs 1.59 µM, respectively). CAL-101 (GS-1101) is a small molecule inhibitor of p110δ isoform that has been demonstrated to having promising activity against chronic lymphocytic leukemia (CLL).21,55,56 We found that CAL-101 has similar anti-proliferative activity as the other p110δ inhibitor tested, IC488743. IC488743 interestingly demonstrates the best activity in H1975 and H460 cell lines that retain KRAS and LKB1 mutations. Using the work of Ihle and others as a foundation, these results were contrary to our expectations.23,57
CAL-101 and IC488743 treatment differed most in the EGFR mutant lines PC9 and H1650. It is important to note that the GI50 values for even the most sensitive cell lines were well above those estimated IC50 values for isoform selectivity, and likely, physiologically unachievable. However, the data also suggest that dual inhibition of p110β and p110δ enzymes may be observed at micromolar concentrations while sparing p110α. These treatment conditions resulted in diminished cellular proliferation for cells of multiple genotypes. Having similar potency for individual class IA isoforms as the isoform-selective inhibitors, the pan-PI3K inhibitor (ZSTK474) was far more effective at blocking PI3K signaling and proliferation than any of the isoform selective inhibitors tested demonstrating nanomolar GI50 for all NSCLC cell lines tested.
The dose-dependent pharmacodynamics evaluation of each inhibitor largely mirrors the proliferation results. Treatment with ZSTK474 completely abrogated pAKT signaling at 0.5μM for A549, H1650 and PC9 cell lines (Fig. 4, top, and Fig. S2). Each isoform-selective inhibitor impaired PI3K/AKT pathway signaling at micromolar concentrations and only elicited an anti-proliferative response at those concentrations. Treatment at concentrations suitable for single isoform selectivity had little effect on proliferative signaling, with the exception of A66 treatment in PIK3CA –mutated H1975 and H460 cells. Importantly, time-dependent inhibition of PI3K/AKT signaling was achieved with partial off-target activity but showed differential effects among the NSCLC cell lines. Together, these data suggest that there may be inherent resistance mechanisms in place in NSCLC cells to preserve PI3K activity for cell proliferation and survival signaling. To this point, in the preclinical development of CAL-101, drug concentrations of 5-10 µM were needed to achieve cytotoxic activity. This represents at least 1000x greater concentrations than the predicted IC50 for p110δ, likely inhibiting both p110β and p110γ PI3K isoforms. Furthermore, clinical exposure to CAL-101 is greater than 2 µM at lowest doses, suggesting that partial inhibition of other PI3K isoforms such as p110β may contribute in part to clinical cytotoxic effects observed in patients.28,29,58,59
Because there is little literature that evaluates the roles of the class IA PI3K enzymes specifically in lung cancer, we also investigated the possibility of compensating PI3K activities in NSCLC with respect to resistance to the PIK3CD inhibitors, CAL-101 and IC488743, because each compound possesses a different selectivity profile for PIK3CA, PIK3CB and PIK3CD.10,60 Using siRNA-mediated silencing of p110β, we tested the activity of CAL-101 and IC488743 in 3 cell lines having variable sensitivity to either drug. From this experiment, it seemed likely that ablation of 2 of the 3 enzymes of interest (PI3KA/B/D) is necessary to eliminate the majority of the pAKT activity. However, to prove the hypothesis, silencing of PIK3CA followed with PIK3CB/D pharmacological inhibition would be necessary.
Interestingly, we observed reduced pERK1/2 signaling following treatment with IC488743 or ablation of p110β in the H1650 cell line, a PTEN null line. We expected that MAPK signaling might be up-regulated to compensate for loss of pAKT activity. This observation suggests that in some cellular contexts, PI3K enzymes may retain catalytic activities in multiple pathways or serve non-catalytic activities as observed by others.22 We will pursue experiments to assess protein-protein interactions to test the latter hypothesis.
From these data, we observed that non-selective concentrations of IC488743 and nanomolar concentrations of a pan-inhibitor slowed proliferation. We wanted to understand how proliferation is slowed in the presence of these agents. We found that GI75 concentrations of either inhibitor impaired PI3K/AKT pathway signaling and led to what appears to be a transient G1 cell cycle arrest. However, there was no observable induction of apoptosis for either drug suggesting that targeted inhibition the PI3K pathway. We suspect that these non-squamous NSCLC cells exploit pathway cross-talk and feedback mechanisms between PI3K/AKT and RAS/MAPK pathways that allow for cell cycle re-entry and escape from apoptosis. To determine this convincingly, treated cells would undergo drug washout and incubation with complete media after which cell cycle position would be re-evaluated.
In cells retaining wild-type EGFR and harboring KRAS mutations, co-incubation of ZSTK474 and erlotinib lead to reduced proliferation and, under some conditions, cell death in these cells resistant to erlotinib alone, suggesting that ablating upstream signals from receptor tyrosine kinases and activity of all class I PI3K enzymes may be an effective combination as has been postulated for other cancers.61 We did not test the combination on the EGFR single-mutant cells (PC9 and H1650) because we know that erlotinib is effective in cell killing, and we would not expect to observe notable improvement. Utilizing the combination of pan-PI3K and EGFR inhibitors in the clinic faces scheduling and dosing hurdles to reduce the possibility of adverse events in patients.
Finally, we hypothesized that isoform-selective inhibition of PI3K class IA enzymes was a rational choice for lung cancer patient therapy and might be able to be personalized. Importantly, we found that PIK3CA mutated cells responded to p110α inhibition, as demonstrated by other.26 Inhibition of multiple isoforms or pathways was more effective than inhibition of individual p110β and p110δ isoforms alone, and efficacy of single-agent treatment cannot be predicted from the genotypes of the NSCLC lines used in this study. Overall, our findings provide evidence that redundancy among class IA isoforms contributes to inherent drug resistance, and that pan-class I PI3K inhibitors, especially in combination with EGFR inhibitors, may hold therapeutic promise in erlotinib-resistant NSCLC.
Materials and Methods
Cell lines and culture conditions
Human NSCLC cell lines obtained from American Type Culture Collection were A549, H460, H1650, H1975, and PC9. The lines were frozen at low passage for future use and subsequently were confirmed by STR testing (Bio-synthesis Inc., Lewisville, TX.). All cell lines were propagated as monolayer cultures at 37°C in 5% CO2 using RPMI 1640 growth media supplemented with 10% fetal bovine serum (FBS), glucose, sodium pyruvate, HEPES buffer and penicillin-streptomycin (Life Technologies, Grand Island, NY USA). The mutation status of each cell line was determined from the Cosmic database (http://www.sanger.ac.uk/genetics/CGP/cosmic/) and is found in Fig. 1A.
Reagents
IC488743 and CAL-101 (GS-1101) were provided by Gilead Sciences (Foster City, CA). A66, TGX-221, and ZSTK474 was purchased from Selleckchem (Houston, TX). All inhibitors were dissolved in DMSO (Sigma Aldrich Corp, St. Louis, MO; BP231-100) to a stock concentration of 10mM, stored at −20C, and diluted to indicated final concentration in RPMI 1640 containing 1% FBS at time of use.
PI3K activity ELISA
PI3K isoform-specific inhibitor concentration (IC) was determined in vitro using an ELISA assay (Millipore, Billerica, MA; 17-493) according to manufacturer's instructions. The values for biotinylated-PIP3 were set to 100. Experimental signals are divided by biotinylated-PIP3 then multiplied by 100 to give percent positive signal. IC50 values were calculated using Graphpad Prism software based upon the previous calculations for relative percent experimental to control for drug/isoform concentration as indicated. For detailed instructions, refer to the following document: http://www.millipore.com/userguides.nsf/a73664f9f981af8c852569b9005b4eee/ca49bfd0cd79e94b852577c3007bcebd/$FILE/PI3K.pdf
Cell proliferation assessment
The growth inhibitory activity (GI) of each compound was tested on cells using the Alamar Blue viability and Trypan Blue exclusion assays.62,63 In the Alamar Blue assay, 2 × 103 cells/well were plated in 96-well cell culture plates (USA Scientific, Ocala, FL USA; CC7682-7596). Cells were treated 24hr later with PI3K inhibitors for 72hr in RPMI 1640 containing 1% serum. For the Trypan Blue exclusion assay, 1 × 104 cells/well were plated in 24-well cell culture plate (USA Scientific, CC7682-7524). The cells were synchronized 24 hr later by changing media to RPMI containing 0.1% serum. After 24hr serum deprivation, cells were released in RPMI containing 1% serum with PI3K inhibitors for 72 hr. Cells were trypsinized, and mixed 1:1 with trypan blue for visual counting of both viable and dead cells. Experimental concentrations from (0.3 – 30 µM) of A66, TGX-221, IC488743 and CAL-101 were tested. GI50 values were calculated using Graphpad Prism software.
Cell cycle and apoptosis analysis
For the cell cycle study, NSCLC cell lines were plated in 60-mm dishes (Corning, Corning, NY; 25382-381). After 24hr, the cells were treated with ZSTK474 and IC488743 at the indicated GI75 concentrations for 24 and 48 hr. Following incubation, cells were trypsinized, quenched with serum, washed, resuspended in phosphate buffered saline (PBS), and fixed in cold 70% ethanol. Cell cycle distribution was assessed by propidium iodide (PI) staining then quantified by BD FACS Calibur flow cytometery or Life Technologies Attune flow cytometry. The data were analyzed with CellQuest software (BD Biosciences, San Jose, CA) or FlowJo (TreeStar software, Ashland OR USA), respectively. An analysis of apoptosis was performed using the same conditions as the cell cycle study, but whole-cell lysates were analyzed by western blot analysis for cleavage of PARP (Cell Signaling Technology, Danvers, MA USA).
Analysis of protein expression
NSCLC cells were plated in 60-mm dishes. After 24hr, plating media was removed, and cells were synchronized in starvation media (RPMI 1640 containing 0.1% FBS) for 24 hr. Concentration-dependent response experiments were carried out using indicated drug concentrations for 3 hr. Similarly, time-dependent response to drug exposure was undertaken at 1 µM concentration of each isoform-selective inhibitor at indicated time intervals. After drug treatment, cells were harvested, pelleted by centrifugation, and lysed with M-PER protein extraction reagent (Thermo Fisher Scientific, Waltham, MA USA; 78503).
Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes for antibody exposure. Signal was elicited using chemiluminescent reagents (ThermoScientific/Pierce). Primary antibodies used in these experiments were phospho-AKT (Ser473), total AKT, phospho-ERK1/2 (Thr302/Tyr204), total ERK1/2, phospho-S6 ribosomal protein (pS6RP) (Ser235/236), α-tubulin, PTEN, PARP, p85, cyclin D1, HSP90, Histone H3, PI3K p110α, p110β (Cell Signaling Technology, Danvers, MA; 9271, 9272, 9101, 4695, 4858, 2125, 9559, 9532, 9661, 4255, 3011, respectively) and PI3K p110δ (Santa Cruz Biotechnology, Santa Cruz, CA; sc-7176).
Cytosolic and nuclear protein extraction
A549, PC9 and H1650 NSCLC cell lines were treated with indicated concentrations of agents, representing the estimated GI75 concentrations for IC488743 and ZSTK474. After 48hr exposure, cells were trypsinized, quenched with serum, pelleted, and washed once with PBS. Nuclear and cytosolic compartments were separated according to NE-PER Nuclear and Cytoplasmic Extraction Kit protocol (ThermoScientific/Pierce, 78835). Western blot analysis for cyclin D1, and controls for cytosolic and nuclear fractionation, HSP90 and Histone H3, were performed as described above.
Short interfering RNA knockdown (siRNA)
Cells were plated in 6-well dishes in RPMI growth medium (10% FBS) without antibiotics. After 24hr and immediately prior to transfection, the media was changed to RPMI 1640 containing 1% FBS without antibiotics. Isoform-specific PI3K p110β siRNA (Smartpool- Dharmacon, Thermo Fisher Scientific, Waltham, MA USA; L-003019-00) (25 nM) was pre-incubated with RNAiMAX transfection reagent (Life Technologies, 13778-150) in Opti-Mem (Life Technologies) then added to cells for 24 hr. siRNA-containing media was aspirated then replaced with RPMI 1640 containing 1% serum with or without 1 µM CAL-101 or IC488743 for 3 hr. Cells were then harvested and extracts prepared for western blot analysis, as previously described for analysis of protein expression.
Statistical analyses
Cell proliferation assays were completed 3 times with 3 replicates per experiment (Fig. 2, Table 2). The technical replicate values were averaged for the 3 experiments and plotted, mean ± standard deviation (SD). CompuSyn software was used to evaluate drug combination median effects and combination indices (CI). CompuSyn algorithm simulating drug interactions utilize Chou-Talalay method, where additive effect (CI = 1), synergism (CI > 1), and antagonism (CI > 1). 64
ELISA experiments were completed once with technical replicate measures. IC50 values were calculated using Graphpad Prism.
Pharmacodynamic analysis represents 3 independent experiments quantified by densitometric analysis, pAKT (S473) normalized to tAKT, and averaged to reflect changes in pAKT levels (Fig. 3). Image J software was used for densitometric analysis of pAKT and tAKT levels from films. The error bars for each agent and cell line combination represents the SEM, n=3.
Cell cycle and apoptosis data represent the mean ± SD of 3 separate experiments, (Fig. 6). Figures were generated using Graphpad Prism. Pearson correlation calculation was performed using GraphPad Prism software to compare paired GI50 values for each inhibitor quantified in serum-synchronized cells vs. asynchronous cells. The r-value represents the correlation coefficient.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: Cancer J Clin 2014; 64:9-29; PMID:24399786 [DOI] [PubMed] [Google Scholar]
- 2.Stabile LP, Lyker JS, Gubish CT, Zhang W, Grandis JR, Siegfried JM. Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non-small cell lung cancer shows enhanced antiproliferative effects. Cancer Res 2005; 65:1459-70; PMID:15735034; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-1872 [DOI] [PubMed] [Google Scholar]
- 3.Pao W, Miller VA. Epidermal growth factor receptor mutations, small-molecule kinase inhibitors, and non-small-cell lung cancer: current knowledge and future directions. J Clin Oncol 2005; 23:2556-68; PMID:15767641; http://dx.doi.org/ 10.1200/JCO.2005.07.799 [DOI] [PubMed] [Google Scholar]
- 4.Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Bri J Cancer 2006; 94:455-9; PMID:16449998; http://dx.doi.org/ 10.1038/sj.bjc.6602970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts PJ, Stinchcombe TE, Der CJ, Socinski MA. Personalized medicine in non-small-cell lung cancer: is KRAS a useful marker in selecting patients for epidermal growth factor receptor-targeted therapy? J Clin Oncol 2010; 28:4769-77; PMID:20921461; http://dx.doi.org/ 10.1200/JCO.2009.27.4365 [DOI] [PubMed] [Google Scholar]
- 6.Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, et al.. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res 2009; 69:3256-61; PMID:19351834; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat 2008; 11:32-50; PMID:18166498; http://dx.doi.org/ 10.1016/j.drup.2007.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006; 441:424-30; PMID:16724053; http://dx.doi.org/ 10.1038/nature04869 [DOI] [PubMed] [Google Scholar]
- 9.Gustafson AM, Soldi R, Anderlind C, Scholand MB, Qian J, Zhang X, Cooper K, Walker D, McWilliams A, Liu G, et al.. Airway PI3K pathway activation is an early and reversible event in lung cancer development. Sci Translat Med 2010; 2:26ra5; http://dx.doi.org/ 10.1126/scitranslmed.3000251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Dis 2009; 8:627-44; PMID:19644473; http://dx.doi.org/ 10.1038/nrd2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blagden S, Olmin A, Josephs D, Stavraka C, Zivi A, Pinato DJ, Anthoney A, Decordova S, Swales K, Riisnaes R, et al.. First-in-Human Study of CH5132799, an Oral Class I PI3K Inhibitor, Studying Toxicity, Pharmacokinetics, and Pharmacodynamics, in Patients with Metastatic Cancer. Clin Cancer Res 2014; 20:5908-17; PMID:25231405; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia S, Roberts TM, Zhao JJ. Should individual PI3 kinase isoforms be targeted in cancer? Curr Opin Cell Biol 2009; 21:199-208; PMID:19200708; http://dx.doi.org/ 10.1016/j.ceb.2008.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol 2010; 11:329-41; PMID:20379207; http://dx.doi.org/ 10.1038/nrm2882 [DOI] [PubMed] [Google Scholar]
- 14.Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup OV, Mikami A, Roberts TM. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci U S A 2006; 103:16296-300; PMID:17060635; http://dx.doi.org/ 10.1073/pnas.0607899103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E, Sancho S, Smith AJ, Withers DJ, Vanhaesebroeck B. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 2006; 441:366-70; PMID:16625210; http://dx.doi.org/ 10.1038/nature04694 [DOI] [PubMed] [Google Scholar]
- 16.Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 2007; 129:957-68; PMID:17540175; http://dx.doi.org/ 10.1016/j.cell.2007.03.051 [DOI] [PubMed] [Google Scholar]
- 17.Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A 2008; 105:13057-62; PMID:18755892; http://dx.doi.org/ 10.1073/pnas.0802655105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, et al.. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008; 454:776-9; PMID:18594509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A, Meek S, Smith AJ, Okkenhaug K, Vanhaesebroeck B. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc Natl Acad Sci U S A 2008; 105:8292-7; PMID:18544649; http://dx.doi.org/ 10.1073/pnas.0707761105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puri KD, Gold MR. Selective inhibitors of phosphoinositide 3-kinase delta: modulators of B-cell function with potential for treating autoimmune inflammatory diseases and B-cell malignancies. Front Immunol 2012; 3:256; PMID:22936933; http://dx.doi.org/ 10.3389/fimmu.2012.00256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, Jones J, Andritsos L, Puri KD, Lannutti BJ, et al.. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010; 116:2078-88; PMID:20522708; http://dx.doi.org/ 10.1182/blood-2010-02-271171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, Russo G, Azzolino O, Rybalkin SD, et al.. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 2004; 118:375-87; PMID:15294162; http://dx.doi.org/ 10.1016/j.cell.2004.07.017 [DOI] [PubMed] [Google Scholar]
- 23.Mahoney CL, Choudhury B, Davies H, Edkins S, Greenman C, Haaften G, Mironenko T, Santarius T, Stevens C, Stratton MR, et al.. LKB1/KRAS mutant lung cancers constitute a genetic subset of NSCLC with increased sensitivity to MAPK and mTOR signalling inhibition. Bri J Cancer 2009; 100:370-5; PMID:19165201; http://dx.doi.org/ 10.1038/sj.bjc.6604886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koivunen JP, Kim J, Lee J, Rogers AM, Park JO, Zhao X, Naoki K, Okamoto I, Nakagawa K, Yeap BY, et al.. Mutations in the LKB1 tumour suppressor are frequently detected in tumours from Caucasian but not Asian lung cancer patients. Bri J Cancer 2008; 99:245-52; PMID:18594528; http://dx.doi.org/ 10.1038/sj.bjc.6604469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balko JM, Potti A, Saunders C, Stromberg A, Haura EB, Black EP. Gene expression patterns that predict sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer cell lines and human lung tumors. BMC genomics 2006; 7:289; PMID:17096850; http://dx.doi.org/ 10.1186/1471-2164-7-289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jamieson S, Flanagan JU, Kolekar S, Buchanan C, Kendall JD, Lee WJ, Rewcastle GW, Denny WA, Singh R, Dickson J, et al.. A drug targeting only p110alpha can block phosphoinositide 3-kinase signalling and tumour growth in certain cell types. Biochem J 2011; 438:53-62; PMID:21668414; http://dx.doi.org/ 10.1042/BJ20110502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, Kenche V, Anderson KE, Dopheide SM, Yuan Y, et al.. PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat Med 2005; 11:507-14; PMID:15834429; http://dx.doi.org/ 10.1038/nm1232 [DOI] [PubMed] [Google Scholar]
- 28.Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, et al.. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 2011; 117:591-4; PMID:20959606; http://dx.doi.org/ 10.1182/blood-2010-03-275305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ikeda H, Hideshima T, Fulciniti M, Perrone G, Miura N, Yasui H, Okawa Y, Kiziltepe T, Santo L, Vallet S, et al.. PI3K/p110{delta} is a novel therapeutic target in multiple myeloma. Blood 2010; 116:1460-8; PMID:20505158; http://dx.doi.org/ 10.1182/blood-2009-06-222943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yaguchi S, Fukui Y, Koshimizu I, Yoshimi H, Matsuno T, Gouda H, Hirono S, Yamazaki K, Yamori T. Antitumor activity of ZSTK474, a new phosphatidylinositol 3-kinase inhibitor. J Natl Cancer Inst 2006; 98:545-56; PMID:16622124; http://dx.doi.org/ 10.1093/jnci/djj133 [DOI] [PubMed] [Google Scholar]
- 31.Weigelt B, Warne PH, Lambros MB, Reis-Filho JS, Downward J. PI3K pathway dependencies in endometrioid endometrial cancer cell lines. Clin Cancer Res 2013; 19:3533-44; PMID:23674493; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ihle NT, Paine-Murrieta G, Berggren MI, Baker A, Tate WR, Wipf P, Abraham RT, Kirkpatrick DL, Powis G. The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A-549 human non-small cell lung cancer xenografts. Mol Cancer Ther 2005; 4:1349-57; PMID:16170026; http://dx.doi.org/ 10.1158/1535-7163.MCT-05-0149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garassino MC, Martelli O, Broggini M, Farina G, Veronese S, Rulli E, Bianchi F, Bettini A, Longo F, Moscetti L, et al.. Erlotinib versus docetaxel as second-line treatment of patients with advanced non-small-cell lung cancer and wild-type EGFR tumours (TAILOR): a randomised controlled trial. Lancet Oncol 2013; 14:981-8; PMID:23883922; http://dx.doi.org/ 10.1016/S1470-2045(13)70310-3 [DOI] [PubMed] [Google Scholar]
- 34.Kong DX, Yamori T. ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor identified using the JFCR39 drug discovery system. Acta pharmacologica Sinica 2010; 31:1189-97; PMID:20729870; http://dx.doi.org/ 10.1038/aps.2010.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith GC, Ong WK, Costa JL, Watson M, Cornish J, Grey A, Gamble GD, Dickinson M, Leung S, Rewcastle GW, et al.. Extended treatment with selective phosphatidylinositol 3-kinase and mTOR inhibitors has effects on metabolism, growth, behaviour and bone strength. FEBS J 2013; 280:5337-49; PMID:23837532; http://dx.doi.org/ 10.1111/febs.12428 [DOI] [PubMed] [Google Scholar]
- 36.Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012; 489:519-25; PMID:22960745; http://dx.doi.org/ 10.1038/nature11404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al.. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304:1497-500; PMID:15118125; http://dx.doi.org/ 10.1126/science.1099314 [DOI] [PubMed] [Google Scholar]
- 38.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al.. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129-39; PMID:15118073; http://dx.doi.org/ 10.1056/NEJMoa040938 [DOI] [PubMed] [Google Scholar]
- 39.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, et al.. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007; 448:561-6; PMID:17625570; http://dx.doi.org/ 10.1038/nature05945 [DOI] [PubMed] [Google Scholar]
- 40.Hammerman PS, Janne PA, Johnson BE. Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. Clin Cancer Res 2009; 15:7502-9; PMID:20008850; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-0189 [DOI] [PubMed] [Google Scholar]
- 41.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol 2010; 28:1075-83; PMID:20085938; http://dx.doi.org/ 10.1200/JCO.2009.25.3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Faber AC, Li D, Song Y, Liang MC, Yeap BY, Bronson RT, Lifshits E, Chen Z, Maira SM, Garcia-Echeverria C, et al.. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci U S A 2009; 106:19503-8; PMID:19850869; http://dx.doi.org/ 10.1073/pnas.0905056106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev 2008; 18:73-9; PMID:18325754; http://dx.doi.org/ 10.1016/j.gde.2008.01.004 [DOI] [PubMed] [Google Scholar]
- 44.Yamamoto H, Shigematsu H, Nomura M, Lockwood WW, Sato M, Okumura N, Soh J, Suzuki M, Wistuba II, Fong KM, et al.. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res 2008; 68:6913-21; PMID:18757405; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang L, Hu H, Pan Y, Wang R, Li Y, Shen L, Yu Y, Li H, Cai D, Sun Y, et al.. PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PloS one 2014; 9:e88291; PMID:24533074; http://dx.doi.org/ 10.1371/journal.pone.0088291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maira SM, Stauffer F, Schnell C, Garcia-Echeverria C. PI3K inhibitors for cancer treatment: where do we stand? Biochem Soc Trans 2009; 37:265-72; PMID:19143644; http://dx.doi.org/ 10.1042/BST0370265 [DOI] [PubMed] [Google Scholar]
- 47.Hu L, Zaloudek C, Mills GB, Gray J, Jaffe RB. In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3-kinase inhibitor (LY294002). Clin Cancer Res 2000; 6:880-6; PMID:10741711 [PubMed] [Google Scholar]
- 48.Edgar KA, Wallin JJ, Berry M, Lee LB, Prior WW, Sampath D, Friedman LS, Belvin M. Isoform-specific phosphoinositide 3-kinase inhibitors exert distinct effects in solid tumors. Cancer Res 2010; 70:1164-72; PMID:20103642; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-2525 [DOI] [PubMed] [Google Scholar]
- 49.Marques M, Kumar A, Cortes I, Gonzalez-Garcia A, Hernandez C, Moreno-Ortiz MC, Carrera AC. Phosphoinositide 3-kinases p110alpha and p110beta regulate cell cycle entry, exhibiting distinct activation kinetics in G1 phase. Mol Cell Biol 2008; 28:2803-14; PMID:18285463; http://dx.doi.org/ 10.1128/MCB.01786-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marques M, Kumar A, Poveda AM, Zuluaga S, Hernandez C, Jackson S, Pasero P, Carrera AC. Specific function of phosphoinositide 3-kinase beta in the control of DNA replication. Proc Natl Acad Sci U S A 2009; 106:7525-30; PMID:19416922; http://dx.doi.org/ 10.1073/pnas.0812000106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murga C, Fukuhara S, Gutkind JS. A novel role for phosphatidylinositol 3-kinase beta in signaling from G protein-coupled receptors to Akt. J Biol Chem 2000; 275:12069-73; PMID:10766839; http://dx.doi.org/ 10.1074/jbc.275.16.12069 [DOI] [PubMed] [Google Scholar]
- 52.Berenjeno IM, Guillermet-Guibert J, Pearce W, Gray A, Fleming S, Vanhaesebroeck B. Both p110alpha and p110beta isoforms of PI3K can modulate the impact of loss-of-function of the PTEN tumour suppressor. Biochem J 2012; 442:151-9; PMID:22150431; http://dx.doi.org/ 10.1042/BJ20111741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Silio V, Redondo-Munoz J, Carrera AC. Phosphoinositide 3-kinase beta regulates chromosome segregation in mitosis. Mol Biol Cell 2012; 23(23):4526-42; PMID:23051731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Utermark T, Rao T, Cheng H, Wang Q, Lee SH, Wang ZC, Iglehart JD, Roberts TM, Muller WJ, Zhao JJ. The p110alpha and p110beta isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Gen Dev 2012; 26:1573-86; PMID:22802530; http://dx.doi.org/ 10.1101/gad.191973.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, Higashi K, Volinia S, Downward J, Waterfield MD. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci U S A 1997; 94:4330-5; PMID:9113989; http://dx.doi.org/ 10.1073/pnas.94.9.4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chantry D, Vojtek A, Kashishian A, Holtzman DA, Wood C, Gray PW, Cooper JA, Hoekstra MF. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem 1997; 272:19236-41; PMID:9235916; http://dx.doi.org/ 10.1074/jbc.272.31.19236 [DOI] [PubMed] [Google Scholar]
- 57.Ihle NT, Lemos R, Jr., Wipf P, Yacoub A, Mitchell C, Siwak D, Mills GB, Dent P, Kirkpatrick DL, Powis G. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res 2009; 69:143-50; PMID:19117997; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-6656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Herman SE, Lapalombella R, Gordon AL, Ramanunni A, Blum KA, Jones J, Zhang X, Lannutti BJ, Puri KD, Muthusamy N, et al.. The role of phosphatidylinositol 3-kinase-delta in the immunomodulatory effects of lenalidomide in chronic lymphocytic leukemia. Blood 2011; 117:4323-7; PMID:21378270; http://dx.doi.org/ 10.1182/blood-2010-11-315705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fruman DA, Rommel C. PI3Kdelta inhibitors in cancer: rationale and serendipity merge in the clinic. Cancer Dis 2011; 1:562-72; PMID:22586681; http://dx.doi.org/ 10.1158/2159-8290.CD-11-0249 [DOI] [PubMed] [Google Scholar]
- 60.Foukas LC, Berenjeno IM, Gray A, Khwaja A, Vanhaesebroeck B. Activity of any class IA PI3K isoform can sustain cell proliferation and survival. Proc Natl Acad Sci U S A 2010; 107:11381-6; PMID:20534549; http://dx.doi.org/ 10.1073/pnas.0906461107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Glaysher S, Bolton LM, Johnson P, Torrance C, Cree IA. Activity of EGFR, mTOR and PI3K inhibitors in an isogenic breast cell line model. BMC Res Notes 2014; 7:397; PMID:24964744; http://dx.doi.org/ 10.1186/1756-0500-7-397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Altman SA, Randers L, Rao G. Comparison of trypan blue dye exclusion and fluorometric assays for mammalian cell viability determinations. Biotechnol Prog 1993; 9:671-4; PMID:7764357; http://dx.doi.org/ 10.1021/bp00024a017 [DOI] [PubMed] [Google Scholar]
- 63.O'Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem 2000; 267:5421-6; PMID:10951200; http://dx.doi.org/ 10.1046/j.1432-1327.2000.01606.x [DOI] [PubMed] [Google Scholar]
- 64.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22:27-55; PMID:6382953; http://dx.doi.org/ 10.1016/0065-2571(84)90007-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.