

Abstract
Trihalomethanes (THM) are undesired disinfection byproducts (DBPs) formed during water treatment. Mice exposed to DBPs showed global DNA hypomethylation and c-myc and c-jun gene-specific hypomethylation, while evidence of epigenetic effects in humans is scarce. We explored the association between lifetime THM exposure and DNA methylation through an epigenome-wide association study. We selected 138 population-based controls from a case-control study of colorectal cancer conducted in Barcelona, Spain, exposed to average lifetime THM levels ≤85 μg/L vs. >85 μg/L (N = 68 and N = 70, respectively). Mean age of participants was 70 years, and 54% were male. Average lifetime THM level in the exposure groups was 64 and 130 µg/L, respectively. DNA was extracted from whole blood and was bisulphite converted to measure DNA methylation levels using the Illumina HumanMethylation450 BeadChip. Data preprocessing was performed using RnBeads. Methylation was compared between exposure groups using empirical Bayes moderated linear regression for CpG sites and Gaussian kernel for CpG regions. ConsensusPathDB was used for gene set enrichment. Statistically significant differences in methylation between exposure groups was found in 140 CpG sites and 30 gene-related regions, after false discovery rate <0.05 and adjustment for age, sex, methylation first principal component, and blood cell proportion. The annotated genes were localized to several cancer pathways. Among them, 29 CpGs had methylation levels associated with THM levels (|Δβ|≥0.05) located in 11 genes associated with cancer in other studies. Our results suggest that THM exposure may affect DNA methylation in genes related to tumors, including colorectal and bladder cancers. Future confirmation studies are required.
Keywords: drinking water, DNA methylation, environmental epidemiology, illumina humanMethylation450K, MYNN, RB1, SMAD3, SOX2, trihalomethanes
Introduction
Disinfection byproducts (DBPs) appear during water treatment when organic matter reacts with the disinfectant. Trihalometanes (THMs) are one of the most abundant classes in chlorinated waters; in the past, Spain has shown high levels of THMs compared to Northern Europe.1 Lifetime exposure to DBPs has been related to bladder cancer2,3 and, possibly, colorectal cancer4 in epidemiological studies. However, the mechanisms involved in these deleterious effects are not clear.
Some mechanisms of action have been proposed for DBPs from experimental models. Cytotoxicity, defined as an alteration of the cell integrity with or without direct DNA damage, has been shown by some compounds (i.e., chloroform and trichloroacetic acid), probably dependent on the release of oxidative free radicals during liver reductive metabolism through cytochromes.5,6 For some brominated compounds, it has been proposed that in vivo bioactivation through glutathione S transferases (GSTT1) generates carbonyl radicals, which could be genotoxic (as they attach as adducts to DNA).7 However, these 2 mechanisms do not completely explain the effects observed when doses are low and exposure is chronic. In chronic low doses, toxicants may generate non-mutational changes as a consequence of repetitive cytotoxicity/regeneration cycles that may produce somatic changes in the methylation of DNA in the DNA daughter strands, leading to DNA instability and induction of apoptotic pathways.8 In mice, the exposure to several trihalometanes and haloacetic acids (the second most abundant DBP in chlorinated waters) reduced global DNA methylation levels. Specifically, hypomethylation of some protooncogenes (c-myc and c-jun) associated with increased mRNA expression has been documented. The exposed mice had higher than expected rates of kidney and liver tumors.8-11
The mechanistic evidence in humans is limited. Epidemiological studies have shown an interaction with polymorphisms of GSTT1 and CYP2E1 for bladder cancer in subjects chronically exposed to THM in drinking water.12 Furthermore, levels of DNA methylation at some transposons (LINE-1) in granulocytes increased with lifetime THM exposure in controls.13 To our knowledge, there are no studies exploring THM chronic exposure and changes in genome-wide DNA methylation. In this context, we aim to explore the association between lifetime exposure to THM and DNA methylation levels through an epigenome-wide association study.
Results
Mean age and sex ratio of study participants was similar between exposure groups for the 138 analyzed subjects (Table 1). Only 6 subjects in the low-exposure group and 10 in the high-exposure group were current smokers. Most of the subjects (>70%) had elementary studies or less. The estimated blood cell proportions of CD8 + T-, CD4 + T-, NK-, B-lymphocytes and monocytes were not significantly different between exposure groups, while the proportion of granulocytes was significantly higher in the low-exposure group. In the high-exposure group, most of THM exposure was due to brominated THM, while chloroform predominated in the low-exposure group.
Table 1.
Characteristics of the study subjects by exposure groups
| THM* ≤ 85 µg/L | THM* > 85µg/L | ||
|---|---|---|---|
| Variables | n = 68 | n = 70 | P-value |
| Sex, n(%) | |||
| Male | 34 (50%) | 41 (59%) | 0.3 |
| Female | 34 (50%) | 29 (41%) | |
| Age years, mean (SD) | 70.5 (5.9) | 70.1 (5.6) | 0.7 |
| Trihalomethane levels (µg/L), median (IQR) | |||
| Chloroform | 21.7 (18.7, 24.1) | 17.3 (16.4, 17.6) | <0.001 |
| Bromodichloromethane | 21.0 (19.1, 23.2) | 35.1 (32.2, 35.6) | <0.001 |
| Dibromochloromethane | 11.7 (10.6, 13.1) | 28.1 (25.5, 29.3) | <0.001 |
| Bromoform | 12.0 (9.8, 14.1) | 49.2 (45.6, 51.9) | <0.001 |
| Total THM | 64.0 (58.8, 72.7) | 129.9 (118.4, 134.1) | <0.001 |
| Smoking status, n(%) | |||
| Never smoker | 37 (54%) | 35 (50%) | 0.6 |
| Former smoker | 25 (37%) | 25 (36%) | |
| Current smoker | 6 (9%) | 10 (14%) | |
| Municipality, n(%) | |||
| A | 10 (15%) | 0 (0%) | <0.001 |
| B | 57 (84%) | 3 (4%) | |
| C | 1 (1%) | 17 (24%) | |
| D | 0 (0%) | 50 (71%) | |
| Education level, n(%) | |||
| Elementary or less | 54 (79%) | 52 (74%) | 0.6 |
| High school | 11 (16%) | 12 (17%) | |
| Universitary or more | 3 (4%) | 6 (9%) | |
| Estimated proportions of blood cell types, median (IQR) | |||
| CD8 T-lymphocytes | 3.27 (1.32, 5.67) | 4.80 (1.89, 6.87) | 0.07 |
| CD4 T-lymphocytes | 13.36 (10.73, 16.79) | 14.46 (9.60, 17.31) | 0.6 |
| NK lymphocytes | 8.62 (5.98, 11.39) | 10.33 (6.51, 13.35) | 0.06 |
| B lymphocytes | 3.17 (2.25, 4.14) | 3.61 (2.30, 5.23) | 0.08 |
| Monocytes | 8.23 (7.00, 9.75) | 8.26 (6.56, 9.57) | 0.8 |
| Granulocytes | 61.63 (57.35, 69.07) | 59.74 (54.56, 64.41) | 0.01 |
average lifetime trihalomethanes level
The methylation intensities in both Infinium I and II assays showed the expected bimodal distribution when all probes were plotted together, but approximately normal distribution for most of the CpGs when considered individually. From the original 485,577 probes, we excluded potentially unreliable probes leaving 252,156 for further analyses (Fig. 1). Among these, crude mean methylation levels were significantly different between exposure groups in 23,302 CpG sites after multiple comparison correction (FDR < 0.05, λ = 1.207). Using the selectModel command, the variables that best adjusted all the models were age, sex, the first principal component (surrogate variable), and the estimated blood cell proportions. After adjustment for these covariables, 1840 CpG sites showed different mean methylation levels between exposure groups after FDR correction (FDR < 0.05, λ = 1.048). Using the observed mean methylation values, 140 CpG sites had an absolute observed Δβ > 0.05 between exposure groups (Table S1). Among them, in 29 CpG sites, the covariate adjusted β coefficient showed differences > 0.05 due to THM exposure (absolute expected Δβ between 0.051 and 0.150 Table 2). A cluster of high-exposure subjects was associated with differential methylation of these 29 CpG sites (Fig. 2).
Figure 1.
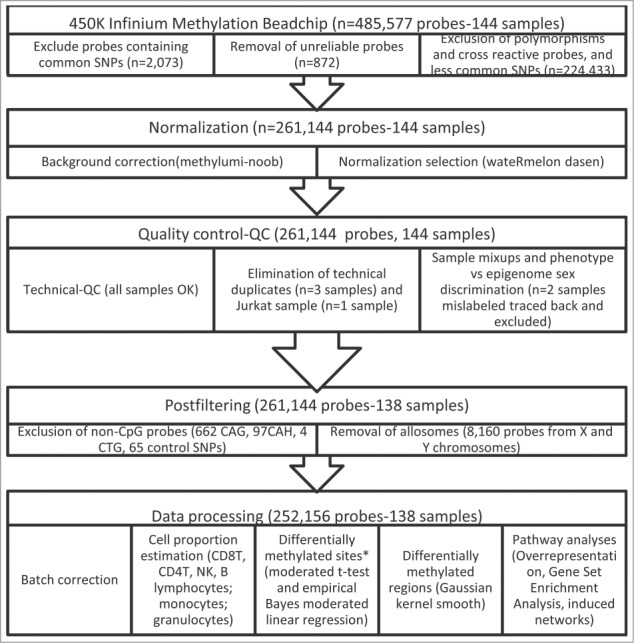
Flowchart of 450K Infinium Methylation BeadChip sample analyses. Note: *Differential methylation was defined as an |Δβ|>0.05 and false discovery rate-FDR<0.05.
Table 2.
Top CpG sites (n = 29) with methylation levels associated with THM exposure (|Δβ|>0.05 and FDR<0.05), after adjusting for covariables (age, sex, first principal component and blood cells proportion)
| IllumID | Gene symbola | Gene name | Gene features | EntrezID | Mean methylation if THM <85 | Observed Δβ (95%CI)b | Expected Δβ (95%CI)c | FDR | Associated diseases-functiond |
|---|---|---|---|---|---|---|---|---|---|
| cg12949141 | PCBD2 | pterin-4 α-carbinolamine dehydratase | Body_none | 84105 | 0.556 | 0.095 (0.07,0.12) | 0.113 (0.09,0.136) | 3.72E-11 | Cluster of genes of Colorectal cancer |
| cg07613278 | API5 | apoptosis inhibitor 5 | TSS200_shore | 8539 | 0.116 | 0.067 (0.051,0.082) | 0.054 (0.039,0.07) | 2.34E-06 | Cell cycle activation of protooncogenes, Hepatocellular carcinoma |
| cg27143246 | MYNN | myoneurin | TSS1500_shore | 55892 | 0.145 | 0.096 (0.073,0.119) | 0.071 (0.051,0.091) | 4.55E-06 | Colorectal cancer, Bladder cancer, ovarian cancer |
| cg08538416 | IGR_shelf | 0.881 | −0.119 (−0.15,−0.089) | −0.083 (−0.108,−0.059) | 7.35E-06 | ||||
| cg24866706 | IGR_none | 0.908 | −0.08 (−0.099,−0.06) | −0.061 (−0.079,−0.043) | 7.88E-06 | ||||
| cg14162627 | ZNF322B | zinc finger protein 322 pseudogene 1 | TSS1500_shore | 387328 | 0.489 | 0.065 (0.047,0.083) | 0.054 (0.038,0.07) | 9.61E-06 | Pseudogenes, duplications, MHC |
| cg05757007 | ASCC3 | activating signal cointegrator 1 complex subunit 3 | Body_none | 10973 | 0.787 | −0.127 (−0.161,−0.093) | −0.085 (−0.11,−0.059) | 9.74E-06 | Repair of N-Alkylated nucleotides, DNA demethylation |
| cg17558214 | PHF14 | PHD finger protein 14 | Body_none | 9678 | 0.884 | −0.105 (−0.133,−0.077) | −0.075 (−0.098,−0.052) | 1.17E-05 | Biliary tract cancer, colorectal cancer |
| cg26435481 | SNORD114–9 | small nucleolar RNA, C/D box 114−9 | TSS1500_none | 767585 | 0.866 | −0.083 (−0.105,−0.061) | −0.055 (−0.072,−0.038) | 1.22E-05 | Acute promyelocytic leukemia |
| cg22838357 | IGR_shelf | 0.838 | −0.117 (−0.148,−0.085) | −0.076 (−0.1,-0.052) | 2.17E-05 | ||||
| cg15739904 | IGR_island | 0.562 | −0.055 (−0.07,−0.039) | −0.051 (−0.067,−0.034) | 6.23E-05 | ||||
| cg27447696 | IGR_none | 0.831 | −0.069 (−0.087,−0.051) | −0.053 (−0.071,−0.035) | 9.73E-05 | ||||
| cg00338852 | ARL13B | ADP-ribosylation factor-like 13B | 3′UTR_none | 200894 | 0.893 | −0.083 (−0.107,−0.059) | −0.054 (−0.072,−0.036) | 1.01E-04 | Ciliopathies, Joubert syndrome |
| cg13325919 | SOX2 | SRY (sex determining region Y)-box 2 | 3′UTR_shore | 6657 | 0.778 | −0.071 (−0.092,−0.051) | −0.06 (−0.081,−0.04) | 1.09E-04 | Gastric cancer, cervical squamous cell carcinoma, non-small lung cell carcinomas, colorectal cancer, non-invasive bladder cancer, head and neck squamous cell, ovarian cancer, invasive gliomas, glioblastomas, esophageal carcinomas, testis and germ cell neoplasia, hepatocellular carcinoma, anaplastic thyroid carcinoma, osteosarcomas, upper urinary tract transitional carcinomas |
| cg02279953 | SNORD114–11 | small nucleolar RNA, C/D box 114-11 | TSS1500_none | 767589 | 0.915 | −0.084 (−0.109,−0.059) | −0.055 (−0.075,−0.036) | 2.53E-04 | No information available |
| cg16896134 | ZNF714 | zinc finger protein 714 | 3′UTR_none | 148206 | 0.851 | −0.085 (−0.111,−0.06) | −0.052 (−0.07,−0.033) | 4.20E-04 | non-syndromic cleft lip with or without cleft palate |
| cg17256697 | IGR_shore | 0.743 | −0.084 (−0.113,−0.055) | −0.069 (−0.095,−0.044) | 7.34E-04 | ||||
| cg21653586 | IGR_none | 0.330 | 0.103 (0.068,0.138) | 0.099 (0.062,0.136) | 8.75E-04 | ||||
| cg04141141 | PPAP2A | phosphatidic acid phosphatase type 2A | Body_none | 8611 | 0.866 | −0.092 (−0.12,−0.064) | −0.055 (−0.076,−0.034) | 8.90E-04 | Prostate cancer |
| cg22840583 | EMR4P | egf-like module containing, mucin-like, hormone receptor-like 4 pseudogene | TSS1500_none | 326342 | 0.850 | −0.096 (−0.125,−0.067) | −0.063 (−0.088,−0.037) | 2.80E-03 | Pseudogene, leukocyte migration in non human species |
| cg27103937 | IGR_shelf | 0.722 | 0.068 (0.04,0.096) | 0.075 (0.044,0.106) | 3.09E-03 | ||||
| cg19637330 | IGR_island | 0.577 | −0.11 (−0.173,−0.046) | −0.15 (−0.216,−0.084) | 7.61E-03 | ||||
| cg02698886 | IGR_shore | 0.591 | 0.072 (0.049,0.095) | 0.052 (0.028,0.075) | 1.08E-02 | ||||
| cg23048036 | CEP97 | centrosomal protein 97kDa | TSS1500_shore | 79598 | 0.788 | −0.078 (−0.105,−0.052) | −0.052 (−0.076,−0.028) | 1.21E-02 | Ciliogenesis, centromere migration |
| cg00389785 | IGR_shelf | 0.646 | −0.071 (−0.099,−0.044) | −0.056 (−0.083,−0.029) | 1.63E-02 | ||||
| cg21004684 | IGHMBP2 | immunoglobulin mu binding protein 2 | Body_none | 3508 | 0.673 | −0.027 (−0.053,−0.0004) | −0.051 (−0.076,−0.026) | 2.12E-02 | Breast cancer |
| cg22244039 | IGR_shore | 0.780 | −0.068 (−0.109,−0.026) | −0.088 (−0.131,−0.045) | 2.23E-02 | ||||
| cg25592910 | PCDH15 | protocadherin-related 15 | TSS200_none | 65217 | 0.512 | 0.056 (0.03,0.082) | 0.054 (0.027,0.08) | 2.37E-02 | Usher syndrome I, NK/T cell lymphomas |
| cg07707039 | COX11 | cytochrome c oxidase assembly homolog 11 (yeast) | Body_shelf | 1353 | 0.612 | 0.057 (0.033,0.082) | 0.051 (0.026,0.076) | 2.40E-02 | Breast cancer, type 2 diabetes mellitus |
Genes were sorted by false discovery rate-FDR from the regression models adjusted for age, sex, the first principal component and the estimated white blood cell proportion.
The observed deltabeta corresponds to the mean difference of high exposed vs. low exposed group to THM, confidence interval is based on a empirical Bayes moderated t-test.
The expected deltabeta corresponds to the β coefficient of high exposed vs. low exposed group to THM adjusted for age, sex, the first principal component and the estimated white blood cell proportion, confidence interval is based on a empirical Bayes moderated linear regression.
From PubMed search (gene expression, protein levels or genetic polymorphisms associated with specific molecular mechanisms or diseases in humans).
Figure 2.
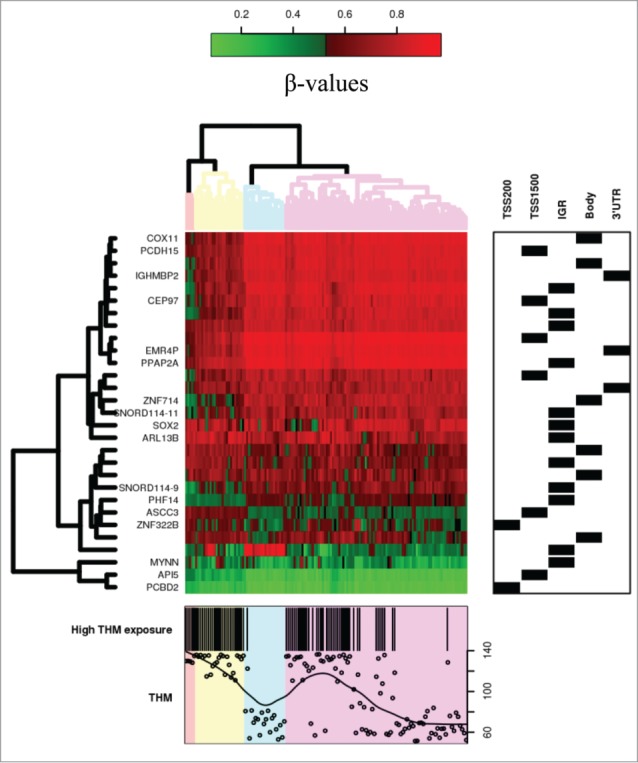
Heatmap of CpG sites with methylation levels associated with THM exposure (|Δβ|>0.05 and FDR<0.05). Note: DNA methylation heatmap of methylated genes passing FDR<0.05, in white blood cells DNA of persons exposed during lifetime to trihalomethanes. Each row represents a CpG site with columns representing each sample. The top dendrogram shows the results of an unsupervised hierarchical clustering of 138 samples based on 29 CpG sites, which separates those subjects flagged as highly exposed in average to lifetime THM levels >85 μg/L (marked as black in the bottom box), from those exposed to lower levels (the remainder columns). In the right box each site is marked to its corresponding region. A scatterplot of the actual lifetime THM levels is shown at the bottom box.
For pathway analyses, we included all the genes annotated by Illumina to specific CpG sites (intergenomic regions were not included, as they cannot be properly annotated to a contiguous gene). Using GSEA, 96 pathways were enriched using KeGG (P-value of the family wise error rate (FWER) < 0.001), and 128 using Reactome (Tables S2 and S3). Induced networks of the 140 CpG sites showed multiple interrelationships with SOX2 and RB1 pathways (Fig. 3) which are respectively an oncogene and a tumor suppressor that are associated with different classes of cancer, including bladder and colorectal cancer (Table 2).
Figure 3.
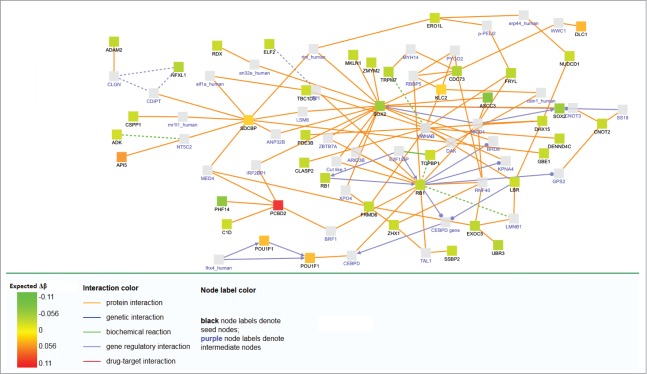
Network analysis of differentially methylated genes after adjustement by covariables. Expected Δβ is the methylation change expected after adjusting for age, sex, the first principal component and the estimated white blood cell proportion.
A specific evaluation of the 29 CpG sites with absolute β coefficients above 0.05 in the adjusted model was also explored. As the pathways were not enriched using this shortlist, a manual PubMed search of the related genes was performed. Eleven genes (PCBD2,14 API5,15 MYNN,16 ASCC3,17 PHF14,18 SNORD114–9,19 SOX2,20–24 PPAP2A,25 IGHMBP2,26 PCDH15,27 and COX1128) were related to different classes of cancer (colorectal and bladder cancer, among others) and 2 to ciliopathies (ARL13B29,30 and CEP9731) (see Table 2). Other diseases (type 2 diabetes mellitus, Usher syndrome, and non-syndromic cleft lip), and some cell specific pathological and physiological mechanisms (duplications, repair of N-alkylated nucleotides, leukocyte migration) were also found for some of these genes. Differentially methylated regions (Table 3) overlap several genes (CEP97, API5, SNORD114–9, SNORD114–11, MYNN, and PCDH15).
Table 3.
Differentially methylated regions (n = 30) identified using a Gaussian kernel (DMRcate) with methylation levels associated with trihalomethane exposure ((|Δβ|>0.05 and FDR<0.05) after adjusting for covariables (age, sex, first principal component and blood cells proportion)
| Associated gene symbol(s) | Genomic area(s) | Coordinates (hg19) | no. probes | Min P-value | Mean P-value | Max Δβ |
|---|---|---|---|---|---|---|
| chr8:125313573–125313940 | 3 | 3.06E-31 | 2.60E-28 | −0.05 | ||
| CEP97 | TSS1500,Body | chr3:101442954–101443851 | 4 | 5.44E-22 | 8.32E-13 | −0.05 |
| API5 | TSS1500,TSS200,1stExon,Body | chr11:43333145–43333782 | 7 | 1.13E-21 | 2.28E-19 | 0.05 |
| SNORD114–9,SNORD114–10,SNORD114–11 | TSS1500,TSS200 | chr14:101432120–101433601 | 4 | 3.67E-15 | 4.04E-12 | −0.06 |
| MYNN | TSS1500,TSS200,1stExon,5′UTR | chr3:169489583–169491183 | 8 | 1.23E-13 | 5.29E-08 | 0.07 |
| chr6:24360304–24360603 | 3 | 9.85E-06 | 1.77E-05 | 0.05 | ||
| chr1:19110734–19110922 | 2 | 1.28E-05 | 1.40E-05 | −0.15 | ||
| EXOC3L2 | 5′UTR,TSS200,TSS1500 | chr19:45737011–45738115 | 9 | 1.05E-04 | 5.98E-04 | 0.06 |
| PCDH15 | TSS200 | chr10:56561096–56561124 | 2 | 7.46E-04 | 7.48E-04 | 0.05 |
| DNHD1 | Body | chr11:6592066–6592745 | 3 | 1.22E-03 | 1.66E-03 | 0.06 |
| PCDHGA5,PCDHGA4,PCDHGA2,PCDHGB2,PCDHGA1,PCDHGB1,PCDHGA3 | TSS1500,Body,TSS200,1stExon | chr5:140743575–140744556 | 6 | 1.45E-03 | 7.12E-03 | 0.06 |
| C22orf27 | TSS1500,TSS200,Body | chr22:31317764–31318546 | 10 | 2.91E-03 | 4.95E-03 | −0.06 |
| GPX6 | 1stExon,5′UTR,TSS200 | chr6:28483537–28483691 | 2 | 3.66E-03 | 5.22E-03 | −0.07 |
| NXPH2 | TSS1500 | chr2:139538356–139539001 | 4 | 3.73E-03 | 1.13E-02 | 0.09 |
| SMAD3 | TSS1500 | chr15:67356310–67356942 | 3 | 5.66E-03 | 7.02E-03 | 0.09 |
| PRSS21 | TSS1500,Body | chr16:2866901–2868001 | 5 | 6.08E-03 | 1.30E-02 | −0.05 |
| chr8:599963–600488 | 4 | 6.68E-03 | 2.46E-02 | −0.07 | ||
| SLFN12 | 1stExon,5′UTR,TSS1500 | chr17:33759484–33759986 | 4 | 9.44E-03 | 1.97E-02 | −0.06 |
| LMTK3 | Body | chr19:49000743–49002477 | 6 | 1.18E-02 | 1.83E-02 | −0.06 |
| KCNMA1 | Body | chr10:79110632–79111034 | 2 | 1.19E-02 | 1.92E-02 | 0.06 |
| chr4:25090198–25090665 | 5 | 1.36E-02 | 1.93E-02 | −0.06 | ||
| chr2:47799165–47799268 | 2 | 1.44E-02 | 1.49E-02 | −0.05 | ||
| chr3:133502540–133503437 | 6 | 1.49E-02 | 2.07E-02 | 0.06 | ||
| KIF25 | Body | chr6:168435636–168435923 | 3 | 2.48E-02 | 2.54E-02 | −0.09 |
| DUSP22 | TSS200,1stExon,5′UTR,Body | chr6:291903–292596 | 5 | 2.89E-02 | 3.62E-02 | −0.08 |
| PPP4R2 | TSS1500 | chr3:73045556–73045686 | 2 | 2.94E-02 | 2.98E-02 | 0.07 |
| chr4:1512820–1513089 | 3 | 3.31E-02 | 3.78E-02 | −0.06 | ||
| C21orf56 | Body | chr21:47581558–47582049 | 2 | 3.33E-02 | 3.81E-02 | 0.07 |
| STK32C | Body | chr10:134045578–134046066 | 3 | 4.17E-02 | 4.66E-02 | 0.06 |
| chr8:125313573–125313940 | 3 | 3.06E-31 | 2.60E-28 | −0.05 |
All sensitivity analyses provided larger top lists with slightly different variations on the order of top hits. However, the differentially methylated top hits were preserved independently of the strategy used (normalization, robust modeling, or less stringent filtering of probes). We opted to keep the shortest most conservative list.
Discussion
This is the first DNA methylation epigenome-wide study linking chronic THM exposure to changes in human DNA methylation. We found that subjects with an average lifetime exposure to THM higher than 85 µg/L showed differential methylation of 29 CpG sites (FDR < 0.05 and absolute expected Δβ > 0.05) compared to those with less exposure. Some of the genes annotated to these sites are associated with different cancers, including bladder and colorectal.
Epidemiological studies have suggested that chronic exposure to DBPs increases bladder cancer risk.2,3 Experimental evidence has suggested that the mechanisms behind DBP carcinogenicity may partially be explained by epigenetic alterations that result from chronic cytotoxicity mediated by a mixture of oxidative metabolites release, reductive free radicals, and cell integrity disruption.5,32 For non-genotoxic compounds, cycles of mitotic regeneration in response to cytotoxicity may produce cumulative epigenetic changes.8 These cumulative epigenetic changes and, particularly, global DNA hypomethylation, may lead to genomic instability and cell apoptosis.33 In particular, global DNA hypomethylation and hypomethylation of several protooncogenes after exposure to several THM and HAA has been described in rodents.8–11 This specific derepression of protooncogenes may lead to overcome the apoptotic pathways and generate survival of transformed neoplastic cells.34 In our data, the induced networks and several individual CpG sites, annotated to specific genes, suggest a mechanism related to tumor suppressor release (RB1), or oncogene activation (SOX2), which may explain part of the THM mechanism in humans. Moreover, some of the differentially methylated sites and regions are located in genes related to both or, specifically, to either bladder cancer16,20 or colorectal cancer21,35 (MYNN, SOX2, RB1, and SMAD3), which supports this hypothesis. In addition, the cancer-related KeGG pathway was ranked 2nd in the GSEA analysis (colorectal cancer was ranked 22nd and bladder cancer 58th, both significant). Using Reactome, EGFR signaling in cancer was ranked 45th. Other cancer-related pathways that were enriched include p53, MAPK, and PPAR signaling. Other metabolic, immunological, and neurodevelopment pathways were also non-specifically enriched using both curated databases.
We used total THM as a DBP exposure proxy and classified subjects into 2 exposure categories. We followed a simplistic approach in order to optimize the statistical power that ignored potential differences attributable to THM composition and quantitative estimates of exposure. We found that THM exposure did not perfectly classify the subjects DNA methylation levels (Fig. S1). Limited sample size, exposure misclassification, and individual unmeasured characteristics may explain these classification differences. Given that exposure was assigned based on residence, unaccounted socioeconomic factors (other than educational level) might explain some of the observed differences.36 DBPs and, particularly, THM as proxy of the whole mixture may behave differentially according to the specific route of exposure. Given the different molecular weights and polarity of the DBPs, potential exposure routes include not only ingestion but also inhalation and/or dermal absorption during showering and water related activities (cleaning, swimming, flushing the toilet, etc.).37–41 These routes, plus the subject water handling and consumption behaviors, may produce differential internal doses and partially explain why subjects apparently exposed to similar THM levels had a wide range of changes in DNA methylation levels in our sample. Exposure misclassification due to unaccounted non-residential exposure (e.g., workplace) is expected to be low, only affecting the ingested THM fraction. On the other hand, individual DNA methyltransferases activity or specific protective genetic polymorphisms may enhance or reduce the epigenetic changes and reduce the potential for new DNA methylation changes. Unfortunately, we do not have data about these specific variables.42,43 Finally, as these processes may be perpetuated through inflammatory pathways, the role of chronic inflammation and its modification through anti-inflammatory medications (either intended or pleiotropic) is another potential target of future research in the area.44
We used microarrays as a cost-effective approach to interrogate epigenome-wide DNA methylation changes associated with THM exposure. However, there are several analytical gaps in the Infinium HumanMethylation450 BeadChip analyses, as there is no consensus for preprocessing and downstream analysis of the data.45 We decided to be as conservative as possible and compared different approaches to observe internal reproducibility of results. The results were concordant using different approaches and we are confident that we tried to maximize our results while reducing potential analytical bias. This is the main strength of our study. In addition, the design was intended to maximize the power even under the small sample size constrain. In this first exploratory study with limited sample size and absence of experimental validation, we generate some hypotheses that require replication in new studies with larger sample size.
The use of whole blood samples instead of the target organ is a limitation in our study. As DNA methylation is time and organ specific we cannot assure that the differences found can be replicated on the target organs. Second, the cross sectional blood sampling does not allow us to observe the evolution of changes in time, as the samples were collected from a case-control study. However, there is a limited amount of longitudinal studies of cancer and, to our knowledge, none has evaluated THM exposure, ours being the first study to provide some potential hypotheses about these mechanisms in humans. Third, the changes found in methylation levels are relatively small (utmost 15%), which increases the probability of positive results by chance, even after adjusting for multiple comparison. Fourth, differentially methylated regions methods and pathway analyses for DNA methylation data are active and rapidly evolving research areas and results may differ by method applied. Our differentially methylated region results seem to be driven by the most differentially methylated probes, even if other probe differences were modest (absolute Δβ < 0.01) or non-significant (FDR > 0.05) in the differential methylation site analysis. Pathway analyses try to summarize common pathways assuming large gene-specific interactions included on curated databases but do not account for unknown intergenomic regions without annotated genes. In addition, without gene expression data, DNA methylation pathway analyses only may suggest but cannot provide exact information about the gene network underlying the observed methylation changes. Finally, we have not performed a laboratory validation using a different technique. This and the replication of the results are future steps that should be performed for confirmation. Thus, we call for a cautious interpretation of the results, given that some of them may be the product of chance.
In summary, our results suggest that long-term THM exposure affect DNA methylation in genes related to tumors, including bladder and colorectal cancer. These findings, if confirmed or validated in other populations, may contribute to understanding the molecular mechanisms or THM pathogenesis.
Materials and Methods
Study population
This study is part of a population-based case-control study of colorectal cancer, which in turn is part of a larger multicase-control study (MCC-Sp) conducted in Spain between 2007 and 2012.46 A subset of population-based controls who provided a blood sample (81% among the recruited) were selected using a stratified random selection based on residence in Barcelona metropolitan area (4 municipalities), age at recruitment (>60 years), and availability of lifetime estimates of THM levels (see below). Study subjects were frequency matched by age group (60–70 vs. 70–80 y) and THM levels (<85 µg/L vs. ≥85 µg/L, the median). A total of 70 subjects (males and females) were selected in each exposure group (50% 60–70 y old, 50% 70–80 y old). The MCC-Sp project and the present study have been approved by the Investigation Review Boards of the different participant hospitals in Barcelona. All the participants had signed the informed consent of the main MCC study and agreed to the molecular analyses.
Questionnaires
Trained interviewers administered a comprehensive computer-assisted questionnaire to the study subjects in the primary care centers. The interview took around 90 min and included personal, socio-demographic, lifestyle, family history, and medical history variables. A validated self-administered food frequency questionnaire was also filled in. The questionnaire is available at the study website: www.mccspain.org.
Lifetime trihalomethane exposure
Exposure estimates were based on levels in the residence of the subject, as a proxy of exposure through all the routes (ingestion, inhalation, dermal contact) in different situations (drinking water and water-based fluids, showering, bathing, dish-washing, etc.). Residential history, including complete address of all residences where study subjects had lived at least during one year since age 18 was requested. Trihalomethane levels in the study municipalities were estimated back to 1940 using available measurements. Historical data on water source and the available THM measurements were used to estimate annual average THM levels for years when measurements were absent. Available THM measurements were averaged and imputed to the past when water source and treatment were unchanged. Proportion of surface water was used as a weight to this average in the event of changes in water source. Before chlorination started, THM levels were assumed to be zero. Residential histories and estimated THM levels were merged by zip code and year to estimate average THM levels from age 18 to the time of interview in study subjects, according to previously published methodology.47 Subjects included in this analysis had THM exposure data covering at least 70% of years from age 18 until the time of interview, with an average (range) of available data of 95.2% (74.1–100) of the years.
DNA extraction and genome-wide DNA methylation array
DNA was extracted from EDTA-treated blood using the Chemagic DNA Blood5k Kit (Perkin Elmer) following manufacturer's protocol at the Spanish National Genotyping Center (CEGEN-Barcelona) and using a manual DNA extraction kit (PROMEGA) at the Bellvitge Biomedical Research Institute (IDIBELL). None of the samples presented visual signs of DNA degradation (smear bands or bands below 10,000 bp) as observed after running 100 ng of DNA on a 1.3% agarose gel. The isolated genomic DNA was stored at −80°C until use. For every sample, 1 µg of DNA was bisulfite-converted using the Zymo EZ DNA methylation kit (ZYMO Research Corporation, Irvine, CA) according to manufacturer instructions. Converted DNA was eluted in 22 µL elution buffer. DNA methylation level was assessed using the Infinium HumanMethylation450 BeadChip (Illumina Inc., San Diego, CA)48 at the CEGEN facility at the Barcelona Biomedical Research Park (PRBB) according to manufacturer's instructions.49 Briefly, 4 µL of bisulfite-converted DNA was isothermally amplified overnight (20–24 h) and fragmented enzymatically. DNA was precipitated using isopropanol and collected after centrifugation at 4°C. Precipitated DNA was resuspended in hybridization buffer and dispensed onto the Infinium HumanMethylation450 BeadChips using a robot (Tecan Group Ltd., Männedorf, Switzerland). Samples were distributed in random blocks according to inclusion criteria to reduce potential chip associated batch effects. Two samples were run by duplicate and one in triplicate, plus a Jurkat DNA control. In total, 12 chips were run (12 samples/chip). Every Infinium slide has 8 chips, thus 2 slides were run simultaneously in a single laboratory batch. Hybridization was performed at 48°C overnight (16–20 h) using an Illumina hybridization oven. Amplified and fragmented DNA samples annealed to locus-specific 50mers (covalently linked to some of the bead types) during hybridization. After hybridization, free DNA was washed away and the BeadChips were processed through a single nucleotide extension following immunohistochemistry staining (ddNTP) in capillary flow through chambers (Tecan GenePaint automated slide processor) using the Freedom Evo robot. Fluorescence signal was captured as an image on an Illumina iScan system using Illumina iScan software. After background subtraction, 2 raw intensity data (idat) files were produced (one per channel) using Illumina GenomeStudio software.
Illumina 450K data preprocessing considerations
The 450K array combines 2 assays in one platform: the Infinium I (type I probes) and Infinium II (type II probes).48 The probes used to assess methylation levels are technically different. In consequence, preprocessing of 450K data should also control for potential differences between assays. In addition, 2 other technical biases are possible: probes that cross-hybridize, and probes mapping in polymorphic residues.50,51 Probes that cross-hybridize account for about 8.6% of the 450K array. Methylation levels measured in these probes likely reflect a combination of levels of methylation at the various locations to which these probes hybridize. On the other hand, polymorphic target probes (4.3% of 450K probes) are probes with polymorphisms at the target C or G at the extension point. Since the 450K platform quantitatively genotypes level of C/T SNPs after bisulfite conversion, these probes may assess a difference in genotype rather than a true difference in methylation levels.
Bioinformatics and statistical analysis
The array data were preprocessed and then processed using R version 3.1.252 and different Bioconductor packages.53 The pipeline used (adapted from RnBeads)54 included the following steps: (1) loading raw intensity data (idat);55 (2) prefiltering (removal of SNP-enriched probes, greedycut algorithm removal of unreliable measurements, and removal of predefined blacklisted probes—all crossreactive probes and polymorphisms as reported by Chen et al.51); (3) normalization (methylumi-noob for background correction56 and dasen normalization57); (4) quality control (detection and exclusion of technical failures during bisulfite conversion, hybridization, extension and staining, detection of potential sample mixups using the default 65 SNPs included in the array); (5) postfiltering (removal of non CpG probes, removal of sex chromosomes); (6) negative control batch effect correction58 and surrogate variable analysis-sva multidimensional reduction adjusting for residual confounding; (7) visualization of the general unadjusted methylation profile. Once data were clean, the downstream data analysis was performed using β values. The β value index was calculated using both intensities of DNA methylation fraction at a specific CpG site: β = M/(M + U + α), where M represents methylated and U unmethylated signal intensities and α is an arbitrary offset (100) to stabilize β values whether the intensities are low. β values are bounded between 0 and 1.59
Epigenome wide DNA methylation association analyses
In total, 144 samples were processed. From the original 140 study subjects, one was processed in duplicate and one in triplicate, and a Jurkat sample was added as a technical control. The duplicates correlation and triplicates correlation was between 0.995 and 0.998. One of the samples was selected at random to keep for further analyses. The other duplicates, the Jurkat sample, and 2 mislabeled (cancer cases) were discarded, leaving 138 samples for analyses. We used an empirical Bayes moderated t-test and/or empirical Bayes moderated linear regression models using limma58 to test the associations between THM levels and DNA methylation (measured as β values). We selected some potential adjustment covariables: age, sex, municipality, highest education reached (elementary or less, high school, and university or more), and tobacco smoking (classified as never, former, and current smokers). Using a surrogate variable approach, we detected a potential residual uncorrected batch effect and we included a surrogate variable in the adjusted models.58 Finally, we used the Houseman algorithm (included in minfi) to estimate the proportions of white blood cells in our samples. In brief, this algorithm uses ∼473 most informative CpG probes to estimate the proportions of T- (CD8, CD4), NK-, and B-lymphocytes, monocytes, and total granulocytes. Adjustment by cell mixtures control population cell stratification in the model due to specific cells populations DNA methylation epigenetic landmarks.55,60,61 We used the selectModel option from limma to reduce multiple comparisons. This command compares nested models using the Akaike Information Criteria-AIC. Genomic inflation factor (λ)62 was calculated for adjusted and unadjusted models. Statistical significance after multiple testing comparison was established using the Benjamini-Hochberg false discovery rate (FDR).63 Statistically significant differential methylation between groups was defined as an absolute Δβ ≥ 0.05. To define cut-offs, we defined the absolute observed Δβ as the difference between the average methylation of the comparison groups and the absolute expected Δβ as the β coefficient of the adjusted models. Finally, to capture differentially methylated regions we used DMRcate,64 which uses the limma empirical Bayes t-moderated statistic calculated per probe and a Gaussian kernel smooth using a 1000 window. The estimate is smoothed based on the varying density of CpG sites given the irregular spacing of the data on the genome interrogated by the Infinium HumanMethylation450 BeadChip.
Pathway analyses
Pathways associated with differentially methylated sites were interrogated using ConsensusPathDB and GSEA65,66 Three different approaches were used: (1) Overrepresentation analyses (number of overrepresented selected genes per set); (2) Gene set enrichment analyses (GSEA) using the KeGG and Reactome curated databases (full list of genes pre-ranked using the t-statistic of the adjusted model); and (3) Induced networks (induced relationships among genes-proteins filling the gaps of unmeasured products not represented/included on the differentially methylated list).66,67 For overrepresentation analyses, the background gene pool was limited to those genes annotated by Illumina in the array. A manual PubMed search of the top CpG sites was performed to annotate the genes associated with specific diseases/conditions.
Sensitivity analyses
We tested our preprocessing approach and modeling compared to other potential approaches to test reproducibility of the top hits. The following analyses were performed: β values normalized using BMIQ,68 a longer probe list with a less conservative filtering excluding only those probes with MAF > 5% in European population, M-values (logit2 transformation of β values)59 used as the outcome, and robust regression instead of linear regression.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Authors thank our fieldworkers Lourdes Arjona and Estela Carrasco. We also thank the CEGEN and Magda Monfort for processing the blood samples and performing the microarray laboratory analyses. Thanks to Nadia Vilahur and Eva Morales for their initial feedback in some issues related to the microarray analyses. We thank the Parc de Salut Mar Biobank (MARBiobanc) and Núria Somoza for their support to the project.
Funding
This study was funded by the Spanish Health Ministry (Fondo de Investigaciones Sanitarias–FIS, Instituto de Salud Carlos III, Spain number FIS PI11–226). Lucas A Salas received a Colciencias PhD Scholarship, Colombia (Grant: 529/2011). Parc de Salut Mar Biobank (MARBiobanc) provided biobanking facilities for to the project. MARBiobanc is supported by Instituto de Salud Carlos III FEDER (RD09/0076/00036).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Ethics Committee Approval
The study was approved by the Ethics Committee of the Research Center (IMIM-IMAS).
References
- 1.Premazzi G, Cardoso C, Conio O, Palumbo F, Ziglio G, Borgioli A, Griffini O, Meucci L, Commission JRCE . Exposure of the European population to trihalomethanes (THMs) in drinking water: volume 2 Luxembourg: Office for Official Publications of the European Communities; 1997 [Google Scholar]
- 2.Villanueva CM, Cantor KP, Grimalt JO, Malats N, Silverman D, Tardon A, Garcia-Closas R, Serra C, Carrato A, Castaño-Vinyals G, et al.. Bladder cancer and exposure to water disinfection by-products through ingestion, bathing, showering, and swimming in pools. Am J Epidemiol 2007165:148-56; PMID:17079692; http://dx.doi.org/ 10.1093/aje/kwj364 [DOI] [PubMed] [Google Scholar]
- 3.Villanueva CM, Cantor KP, Cordier S, Jaakkola JJK, King WD, Lynch CF, Porru S, Kogevinas M. Disinfection byproducts and bladder cancer: a pooled analysis. Epidemiology 2004; 15:357-67; PMID:15097021; http://dx.doi.org/ 10.1097/01.ede.0000121380.02594.fc [DOI] [PubMed] [Google Scholar]
- 4.Rahman MB, Driscoll T, Cowie C, Armstrong BK. Disinfection by-products in drinking water and colorectal cancer: a meta-analysis. Int J Epidemiol 2010; 39:733-45; PMID:20139236; http://dx.doi.org/ 10.1093/ije/dyp371 [DOI] [PubMed] [Google Scholar]
- 5.Tomasi A, Albano E, Biasi F, Slater TF, Vannini V, Dianzani MU. Activation of chloroform and related trihalomethanes to free radical intermediates in isolated hepatocytes and in the rat in vivo as detected by the ESR-spin trapping technique. Chem Biol Interact 1985; 55:303-16; PMID:3000632; http://dx.doi.org/ 10.1016/S0009-2797(85)80137-X [DOI] [PubMed] [Google Scholar]
- 6.Ni YC, Wong TY, Lloyd R V, Heinze TM, Shelton S, Casciano D, Kadlubar FF, Fu PP. Mouse liver microsomal metabolism of chloral hydrate, trichloroacetic acid, and trichloroethanol leading to induction of lipid peroxidation via a free radical mechanism. Drug Metab Dispos 1996; 24:81-90; PMID:8825194 [PubMed] [Google Scholar]
- 7.DeMarini DM, Shelton ML, Warren SH, Ross TM, Shim JY, Richard AM, Pegram RA. Glutathione S-transferase-mediated induction of GC → AT transitions by halomethanes in salmonella. Environ Mol Mutagen 1997; 30:440-7; PMID:9435885 http://dx.doi.org/ 10.1002/(SICI)1098-2280(1997)30:4%3c440::AID-EM9%3e3.0.CO;2-M [DOI] [PubMed] [Google Scholar]
- 8.Coffin JC, Ge R, Yang S, Kramer PM, Tao L, Pereira MA. Effect of trihalomethanes on cell proliferation and DNA methylation in female B6C3F1 mouse liver. Toxicol Sci 2000; 58:243-52; PMID:11099637; http://dx.doi.org/ 10.1093/toxsci/58.2.243 [DOI] [PubMed] [Google Scholar]
- 9.Pereira MA, Kramer PM, Conran PB, Tao L. Effect of chloroform on dichloroacetic acid and trichloroacetic acid-induced hypomethylation and expression of the c-myc gene and on their promotion of liver and kidney tumors in mice. Carcinogenesis 2001; 22:1511-9; PMID:11532874; http://dx.doi.org/ 10.1093/carcin/22.9.1511 [DOI] [PubMed] [Google Scholar]
- 10.Ge R, Yang S, Kramer PM, Tao L, Pereira MA. The effect of dichloroacetic acid and trichloroacetic acid on DNA methylation and cell proliferation in B6C3F1 mice. J Biochem Mol Toxicol 2001; 15:100-6; PMID:11284051; http://dx.doi.org/ 10.1002/jbt.5 [DOI] [PubMed] [Google Scholar]
- 11.Tao L, Wang W, Li L, Kramer PK, Pereira MA. DNA hypomethylation induced by drinking water disinfection by-products in mouse and rat kidney. Toxicol Sci 2005; 87:344-52; PMID:16014735; http://dx.doi.org/ 10.1093/toxsci/kfi257 [DOI] [PubMed] [Google Scholar]
- 12.Cantor KP, Villanueva CM, Silverman DT, Figueroa JD, Real FX, Garcia-Closas M, Malats N, Chanock S, Yeager M, Tardon A, et al.. Polymorphisms in GSTT1, GSTZ1, and CYP2E1, disinfection by-products, and risk of bladder cancer in Spain. Environ Health Perspect 2010; 118:1545-50; PMID:20675267; http://dx.doi.org/ 10.1289/ehp.1002206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salas LA, Villanueva CM, Tajuddin SM, Amaral AFS, Fernandez AF, Moore LE, Carrato A, Tardón A, Serra C, García-Closas R, et al.. LINE-1 methylation in granulocyte DNA and trihalomethane exposure is associated with bladder cancer risk. Epigenetics 2014; 9:1532-9; PMID:25482586; http://dx.doi.org/ 10.4161/15592294.2014.983377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia W-H, Zhang B, Matsuo K, Shin A, Xiang Y-B, Jee SH, Kim D-H, Ren Z, Cai Q, Long J, et al.. Genome-wide association analyses in East Asians identify new susceptibility loci for colorectal cancer. Nat Genet 2013; 45:191-6; PMID:23263487; http://dx.doi.org/ 10.1038/ng.2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Jove Navarro M, Basset C, Arcondéguy T, Touriol C, Perez G, Prats H, Lacazette E. Api5 Contributes to E2F1 Control of the G1/S Cell Cycle Phase Transition. PLoS One 2013; 8:e71443; PMID:23940755; http://dx.doi.org/24163127 10.1371/journal.pone.0071443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Figueroa JD, Ye Y, Siddiq A, Garcia-Closas M, Chatterjee N, Prokunina-Olsson L, Cortessis VK, Kooperberg C, Cussenot O, Benhamou S, et al.. Genome-wide association study identifies multiple loci associated with bladder cancer risk. Hum Mol Genet 2014; 23:1387-98; PMID:24163127; http://dx.doi.org/ 10.1093/hmg/ddt519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dango S, Mosammaparast N, Sowa ME, Xiong LJ, Wu F, Park K, Rubin M, Gygi S, Harper JW, Shi Y. DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation. Mol Cell 2011; 44:373-84; PMID:22055184; http://dx.doi.org/ 10.1016/j.molcel.2011.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ivanov I, Lo KC, Hawthorn L, Cowell JK, Ionov Y. Identifying candidate colon cancer tumor suppressor genes using inhibition of nonsense-mediated mRNA decay in colon cancer cells. Oncogene 2007; 26:2873-84; PMID:17086209 http://dx.doi.org/ 10.1038/sj.onc.1210098 [DOI] [PubMed] [Google Scholar]
- 19.Valleron W, Laprevotte E, Gautier E-F, Quelen C, Demur C, Delabesse E, Agirre X, Prósper F, Kiss T, Brousset P. Specific small nucleolar RNA expression profiles in acute leukemia. Leukemia 2012; 26:2052-60; PMID:22522792 http://dx.doi.org/ 10.1038/leu.2012.111 [DOI] [PubMed] [Google Scholar]
- 20.Tung CL, Hou PH, Kao YL, Huang YW, Shen CC, Cheng YH, Wu SF, Lee MS, Li C. SOX2 modulates alternative splicing in transitional cell carcinoma. Biochem Biophys Res Commun 2010; 393:420-5; PMID:20138825 http://dx.doi.org/ 10.1016/j.bbrc.2010.02.010 [DOI] [PubMed] [Google Scholar]
- 21.Liu H, Du L, Wen Z, Yang Y, Li J, Dong Z, Zheng G, Wang L, Zhang X, Wang C. Sex determining region Y-box 2 inhibits the proliferation of colorectal adenocarcinoma cells through the mTOR signaling pathway. Int J Mol Med 2013; 32:59-66.; PMID:23599173 [DOI] [PubMed] [Google Scholar]
- 22.Ruan J, Wei B, Xu Z, Yang S, Zhou Y, Yu M, Liang J, Jin K, Huang X, Lu P, et al.. Predictive value of Sox2 expression in transurethral resection specimens in patients with T1 bladder cancer. Med Oncol 2013; 30:445; PMID:23307254 http://dx.doi.org/ 10.1007/s12032-012-0445-z [DOI] [PubMed] [Google Scholar]
- 23.Fang X, Yu W, Li L, Shao J, Zhao N, Chen Q, Ye Z, Lin S-C, Zheng S, Lin B. ChIP-seq and functional analysis of the SOX2 gene in colorectal cancers. OMICS 2010; 14:369-84; PMID:20726797; http://dx.doi.org/ 10.1089/omi.2010.0053 [DOI] [PubMed] [Google Scholar]
- 24.Long KB, Hornick JL. SOX2 is highly expressed in squamous cell carcinomas of the gastrointestinal tract. Hum Pathol 2009; 40:1768-73; PMID:19716157 http://dx.doi.org/ 10.1016/j.humpath.2009.06.006 [DOI] [PubMed] [Google Scholar]
- 25.Ulrix W, Swinnen JV, Heyns W, Verhoeven G. Identification of the phosphatidic acid phosphatase type 2a isozyme as an androgen-regulated gene in the human prostatic adenocarcinoma cell line LNCaP. J Biol Chem 1998; 273:4660-5; PMID:9468526 http://dx.doi.org/ 10.1074/jbc.273.8.4660 [DOI] [PubMed] [Google Scholar]
- 26.Shen J, Terry MB, Gammon MD, Gaudet MM, Teitelbaum SL, Eng SM, Sagiv SK, Neugut AI, Santella RM. IGHMBP2 Thr671Ala polymorphism might be a modifier for the effects of cigarette smoking and PAH-DNA adducts to breast cancer risk. Breast Cancer Res Treat 2006; 99:1-7; PMID:16752224; http://dx.doi.org/ 10.1007/s10549-006-9174-3 [DOI] [PubMed] [Google Scholar]
- 27.Jaijo T, Oshima A, Aller E, Carney C, Usami S, Millán JM, Kimberling WJ. Mutation screening of the PCDH15 gene in Spanish patients with Usher syndrome type I. Mol Vis 2012; 18:1719-26; PMID:22815625 [PMC free article] [PubMed] [Google Scholar]
- 28.Tang L, Xu J, Wei F, Wang L, Nie WW, Chen LB, Guan XX. Association of STXBP4/COX11 rs6504950 (G>A) polymorphism with breast cancer risk: evidence from 17,960 cases and 22,713 controls. Arch Med Res 2012; 43:383-8; PMID:22863968; http://dx.doi.org/ 10.1016/j.arcmed.2012.07.008 [DOI] [PubMed] [Google Scholar]
- 29.Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, Audollent S, Attié-Bitach T, Holden KR, Dobyns WB, et al.. Mutations in the cilia gene ARL13B lead to the classical form of joubert syndrome. Am J Hum Genet 2008; 83:170-9; PMID:18674751; http://dx.doi.org/ 10.1016/j.ajhg.2008.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miertzschke M, Koerner C, Spoerner M, Wittinghofer A. Structural insights into the small G-protein Arl13B and implications for Joubert syndrome. Biochem J 2014; 457:301-11; PMID:24168557; http://dx.doi.org/ 10.1042/BJ20131097 [DOI] [PubMed] [Google Scholar]
- 31.Spektor A, Tsang WY, Khoo D, Dynlacht BD. Cep97 and CP110 suppress a cilia assembly program. Cell 2007; 130:678-90; PMID:17719545; http://dx.doi.org/ 10.1016/j.cell.2007.06.027 [DOI] [PubMed] [Google Scholar]
- 32.Amy GL, United Nations Environment Programme, International Labour Organisation, Organization WH, Inter-Organization Programme for the Sound Management of Chemicals, Safety IP on C, By-products WHOTG on EHC for D and D . Disinfectants and disinfectant by-products. Geneva: World Health Organization; 2000 [Google Scholar]
- 33.Moore LE, Huang WY, Chung J, Hayes RB. Epidemiologic considerations to assess altered DNA methylation from environmental exposures in cancer. Ann N Y Acad Sci 2003; 983:181-96; PMID:12724223; http://dx.doi.org/ 10.1111/j.1749-6632.2003.tb05973.x [DOI] [PubMed] [Google Scholar]
- 34.Cheung HH, Lee TL, Rennert OM, Chan WY. DNA methylation of cancer genome. Birth Defects Res C Embryo Today 2009; 87:335-50; PMID:19960550; http://dx.doi.org/ 10.1002/bdrc.20163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Houlston RS, Cheadle J, Dobbins SE, Tenesa A, Jones AM, Howarth K, Spain SL, Broderick P, Domingo E, Farrington S, et al.. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet 2010; 42:973-7; PMID:20972440; http://dx.doi.org/ 10.1038/ng.670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mcguinness D, Mcglynn LM, Johnson PCD, Macintyre A, Batty GD, Burns H, Cavanagh J, Deans KA, Ford I, Mcconnachie A, et al.. Socio-economic status is associated with epigenetic differences in the pSoBid cohort. Int J Epidemiol 2012; 41:151-60; PMID:22253320; http://dx.doi.org/ 10.1093/ije/dyr215 [DOI] [PubMed] [Google Scholar]
- 37.Leavens TL, Blount BC, DeMarini DM, Madden MC, Valentine JL, Case MW, Silva LK, Warren SH, Hanley NM, Pegram RA. Disposition of bromodichloromethane in humans following oral and dermal exposure. Toxicol Sci 2007; 99:432-45; PMID:17656487; http://dx.doi.org/ 10.1093/toxsci/kfm190 [DOI] [PubMed] [Google Scholar]
- 38.Lourencetti C, Grimalt JO, Marco E, Fernandez P, Font-Ribera L, Villanueva CM, Kogevinas M. Trihalomethanes in chlorine and bromine disinfected swimming pools: air-water distributions and human exposure. Environ Int 2012; 45:59-67; PMID:22572118; http://dx.doi.org/ 10.1016/j.envint.2012.03.009 [DOI] [PubMed] [Google Scholar]
- 39.Xu X, Weisel CP. Human respiratory uptake of chloroform and haloketones during showering. J Expo Anal Environ Epidemiol 2005; 15:6-16; PMID:15138448; http://dx.doi.org/ 10.1038/sj.jea.7500374 [DOI] [PubMed] [Google Scholar]
- 40.Xu X, Weisel CP. Dermal uptake of chloroform and haloketones during bathing. J Expo Anal Environ Epidemiol 2005; 15:289-96; PMID:15316574; http://dx.doi.org/ 10.1038/sj.jea.7500404 [DOI] [PubMed] [Google Scholar]
- 41.Weisel CP, Kim H, Haltmeier P, Klotz JB. Exposure estimates to disinfection by-products of chlorinated drinking water. Environ Health Perspect 1999; 107:103-10; PMID:9924004; http://dx.doi.org/ 10.1289/ehp.99107103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luczak MW, Jagodziński PP. The role of DNA methylation in cancer development. Folia Histochem Cytobiol 2006; 44:143-54; PMID:16977793 [PubMed] [Google Scholar]
- 43.Weisenberger DJ, Velicescu M, Cheng JC, Gonzales FA, Liang G, Jones PA. Role of the DNA methyltransferase variant DNMT3b3 in DNA methylation. Mol Cancer Res 2004; 2:62-72; PMID:14757847 [PubMed] [Google Scholar]
- 44.Bayarsaihan D. Epigenetic mechanisms in inflammation. J Dent Res 2011; 90:9-17; PMID:21178119; http://dx.doi.org/ 10.1177/0022034510378683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilhelm-Benartzi CS, Koestler DC, Karagas MR, Flanagan JM, Christensen BC, Kelsey KT, Marsit CJ, Houseman EA, Brown R. Review of processing and analysis methods for DNA methylation array data. Br J Cancer 2013; 109:1394-402; PMID:23982603; http://dx.doi.org/ 10.1038/bjc.2013.496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castano-Vinyals G, Aragones N, Perez-Gomez B, Martin V, Llorca J, Moreno V, Altzibar JM, Ardanaz E, de Sanjose S, Jimenez-Moleon JJ, et al.. Population-based multicase-control study in common tumors in Spain (MCC-Spain): rationale and study design. Gac Sanit 2015; S0213-9111:00288-X; PMID:25613680; http://dx.doi.org/16556748 10.1016/j.gaceta.2014.12.003 [DOI] [PubMed] [Google Scholar]
- 47.Villanueva CM, Cantor KP, Grimalt JO, Castaño-Vinyals G, Malats N, Silverman D, Tardon A, Garcia-Closas R, Serra C, Carrato A, et al.. Assessment of lifetime exposure to trihalomethanes through different routes. Occup Environ Med 2006; 63:273-7; PMID:16556748; http://dx.doi.org/ 10.1136/oem.2005.023069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, Gunderson KL. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics 2009; 1:177-200; PMID:22122642; http://dx.doi.org/ 10.2217/epi.09.14 [DOI] [PubMed] [Google Scholar]
- 49.Infinium HD Assay Methylation Protocol Guide. Catalog # WG-914-1001. Part # 15019519 Rev A [Internet]. San Diego (CA): Illumina, Inc (US); [modified: December 2010; cited: June 2015]. Available from: http://bit.ly/1B36xzy. [Google Scholar]
- 50.Price ME, Cotton AM, Lam LL, Farré P, Emberly E, Brown CJ, Robinson WP, Kobor MS. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin 2013; 6:4; PMID:23452981; http://dx.doi.org/ 10.1186/1756-8935-6-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Y, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013; 8:203-9; PMID:23314698; http://dx.doi.org/ 10.4161/epi.23470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.R Core Team R: A language and environment for statistical computing. 2014 [Google Scholar]
- 53.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al.. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5(10):R80; PMID:15461798; http://dx.doi.org/23476028 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Assenov Y, Müller F, Lutsik P, Walter J, Lengauer T, Bock C. Comprehensive analysis of DNA methylation data with RnBeads. Nat Methods 2014; 11(11):1138-40; PMID:25262207; http://dx.doi.org/23476028 10.1038/nmeth.3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hansen KD, Aryee M. minfi: analyze Illumina's 450k methylation arrays. R package version 1.8.8. 2013 [Google Scholar]
- 56.Triche TJ, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low-level processing of Illumina infinium DNA methylation BeadArrays. Nucleic Acids Res 2013; 41:e90; PMID:23476028; http://dx.doi.org/ 10.1093/nar/gkt090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pidsley R, Wong CCCY, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics 2013; 14:293; PMID:23631413; http://dx.doi.org/ 10.1186/1471-2164-14-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012; 28:882-3; PMID:22257669;http://dx.doi.org/ 10.1093/bioinformatics/bts034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 2010; 11:587; PMID:21118553;http://dx.doi.org/ 10.1186/1471-2105-11-587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koestler DC, Marsit CJ, Christensen BC, Accomando WP, Langevin SM, Houseman EA, Nelson HH, Karagas MR, Wiencke JK, Kelsey KT. Peripheral blood immune cell methylation profiles are associated with nonhematopoietic cancers. Cancer Epidemiol Biomarkers Prev 2012; 21:1293-302; PMID:22714737; http://dx.doi.org/ 10.1158/1055-9965.EPI-12-0361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012; 13:86; PMID:22568884; http://dx.doi.org/ 10.1186/1471-2105-13-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barfield RT, Almli LM, Kilaru V, Smith AK, Mercer KB, Duncan R, Klengel T, Mehta D, Binder EB, Epstein MP, et al.. Accounting for population stratification in DNA methylation studies. Genet Epidemiol 2014; 38:231-41; PMID:24478250 http://dx.doi.org/ 10.1002/gepi.21789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 1995; 57:289-300 [Google Scholar]
- 64.Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, Lord R V, Clark SJ, Molloy PL. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015; 8:6; PMID: 25972926; http://dx.doi.org/ 10.1186/1756-8935-8-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102:15545-50; PMID:16199517; http://dx.doi.org/ 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kamburov A, Stelzl U, Lehrach H, Herwig R. The ConsensusPathDB interaction database: 2013 Update. Nucleic Acids Res 2013; 41:D793-800; PMID: 23143270; http://dx.doi.org/17916244 10.1093/nar/gks1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berger SI, Posner JM, Ma'ayan A. Genes2Networks: connecting lists of gene symbols using mammalian protein interactions databases. BMC Bioinformatics 2007; 8:372; PMID:17916244; http://dx.doi.org/ 10.1186/1471-2105-8-372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marabita F, Almgren M, Lindholm ME, Ruhrmann S, Fagerström-Billai F, Jagodic M, Sundberg CJ, Ekström TJ, Teschendorff AE, Tegnér J, et al.. An evaluation of analysis pipelines for DNA methylation profiling using the Illumina HumanMethylation450 BeadChip platform. Epigenetics 2013; 8:333-46; PMID:23422812; http://dx.doi.org/ 10.4161/epi.24008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.