

Abstract
Kidney diseases, including chronic kidney disease (CKD) and acute kidney injury (AKI), are associated with inflammation. The mechanism that regulates inflammation in these renal injuries remains unclear. Here, we demonstrated that p300/CBP-associated factor (PCAF), a histone acetyltransferase, was overexpressed in the kidneys of db/db mice and lipopolysaccharide (LPS)-injected mice. Moreover, elevated histone acetylation, such as H3K18ac, and up-regulation of some inflammatory genes, such as ICAM-1, VCAM-1, and MCP-1, were found upon these renal injuries. Furthermore, increased H3K18ac was recruited to the promoters of ICAM-1, VCAM-1, and MCP-1 in the kidneys of LPS-injected mice. In vitro studies demonstrated that PCAF knockdown in human renal proximal tubule epithelial cells (HK-2) led to downregulation of inflammatory molecules, including VCAM-1, ICAM-1, p50 subunit of NF-κB (p50), and MCP-1 mRNA and protein levels, together with significantly decreased H3K18ac level. Consistent with these, overexpression of PCAF enhanced the expression of inflammatory molecules. Furthermore, PCAF deficiency reduced palmitate-induced recruitment of H3K18ac on the promoters of ICAM-1 and MCP-1, as well as inhibited palmitate-induced upregulation of these inflammatory molecules. In summary, the present work demonstrates that PCAF plays an essential role in the regulation of inflammatory molecules through H3K18ac, which provides a potential therapeutic target for inflammation-related renal diseases.
Keywords: acute kidney injury, diabetic nephropathy, histone acetylations, inflammation, PCAF
Abbreviations
- AKI
acute kidney injury
- CKD
chronic kidney disease
- COL4
type IV collagen
- GNAT
GCN5-related N-acetyltransferases
- HATs
histone acetyltransferases
- HDACs
histone deacetylases
- HL
hyperlipidemia
- ICAM-1
intercellular adhesion molecule-1
- LPS
lipopolysaccharide
- MCP-1
monocyte chemotactic protein 1
- MnSOD
manganese superoxide dismutase
- NF-κB
nuclear factor-κB
- PCAF
p300/CBP-associated factor
- TGFβ-1
transforming growth factor β-1
- VCAM-1
vascular cell adhesion molecule-1
Introduction
Renal disease is an enormous threat to human health. Chronic kidney disease (CKD) and acute kidney injury (AKI) are 2 major classes of renal disease. CKD is characterized by progressive loss of renal function over the years. Diabetic nephropathy is presently the leading cause of CKD and end stage renal disease in western countries.1 On the other hand, AKI can lead to a rapid loss of renal function. The etiologies of AKI include ischemia, acute tubular necrosis, and exposure to endotoxin, such as lipopolysaccharide (LPS).2-4
Inflammation is a biological response of tissues to harmful stimuli. Inflammation induces overexpression of various inflammatory molecules including adhesion molecules, chemokines, pro-inflammatory cytokines, as well as activation of some inflammatory transcription factors.5 Adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), enable leucocytes to adhere to endothelium, and these leucocytes then migrate into injured sites and initiate inflammatory responses.6,7 In addition, chemokines such as monocyte chemotactic protein 1 (MCP-1) can further recruit monocytes and memory T cells to the sites of inflammation.8 Moreover, NF-κB, a transcription factor activated by many inflammatory stimuli, regulates transcription of many inflammatory genes, including ICAM-1, VCAM-1, and MCP-1, which lead to a vicious cycle of inflammation.9 Accumulating evidences has suggested that chronic inflammation and unresolved inflammation play critical roles in diverse diseases, including renal diseases.10
Long-term activation of low-grade inflammation has been suggested to associate with hyperglycemia and oxidative stress, which eventually lead to diabetic nephropathy.11 Pharmacological or genetic inhibition of some inflammatory molecules has been shown to ameliorate diabetes-induced renal morphological changes.12,13 Similar to CKD, levels of proinflammatory cytokines and chemokines are also significantly elevated in the kidneys of LPS-induced AKI in mice.14 Inhibition of inflammation by nicotinic acetylcholine receptor agonists could effectively ameliorate LPS-induced renal failure.14 However, the exact mechanism that regulates elevation of these inflammatory molecules upon renal diseases remains unknown.
Histone modifications, including acetylation, methylation and phosphorylation, usually regulate gene transcription.15 Different histone modifications have distinct biological roles.16 Histone acetylation weakens the binding between histones and DNA, which makes transcription machinery more accessible for DNA; thus, histone hyperacetylation is often associated with transcription activation.17 Changes in histone acetylation have been reported also to associate with inflammation.18 For example, enriched acetylated histones on the promoters of pro-inflammatory cytokines, such as interleukin-1 (IL-1), IL-2, IL-8, and IL-12, can lead to transcriptional activation of these genes, and subsequent inflammation.18
In cells, the levels of histone acetylations are tightly controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs). There are 3 major families of HATs, including the GNAT family (GCN5 and PCAF), the p300/CBP family (p300 and CBP), and the MYST family (MOZ, MROF, HBO1 and MOF).19,20 Alteration in histone acetylation and their regulatory enzymes in rodent models of renal diseases have been reported. In the kidneys of Akita mice and db/db mice, classic models of type 1 and type 2 diabetes respectively, the levels of H3K9ac and H3K23ac were increased.21,22 Furthermore, increased recruitment of acetylated histones including H4K5/8/12/16ac, H2AK5ac, and H2BK4/7ac on the promoter of Tnf-α, a key inflammatory gene, are found in the kidneys of LPS-induced AKI.23 Additionally, curcumin, a potent antioxidant, prevents the development of early lesions of diabetic nephropathy in rats by inhibiting the interaction between p300 and NF-κB.24 Paradoxically, long-term administration of vorinostat, an inhibitor of HDACs, attenuates renal injury in diabetic rodents.25 Thus, the exact roles of histone acetylations and their regulatory enzymes in the development of renal diseases still await further investigation.
We hypothesized that histone hyperacetylation caused by overexpression of HATs in renal cells leads to elevation of inflammation that finally induces renal injury. In the present study, the levels of PCAF, histone acetylation, and inflammatory molecules were investigated in the kidneys of db/db mice and LPS-injected mice, respectively. Furthermore, renal proximal tubule epithelial cells that overexpress or knockdown PCAF were generated to investigate the critical role of PCAF in regulating inflammatory molecules. Our results demonstrate PCAF regulates the expression of inflammatory molecules and may contribute to the development of CKD and AKI, suggesting PCAF may be considered as a novel therapeutic target for renal diseases.
Results
Upregulation of histone acetylation, PCAF, and inflammatory genes in the kidneys of db/db mice
Multiple sites of increased histone acetylation in the kidneys of Akita mice, a classic type 1 diabetic mouse model, has been previously reported by us.21 We then asked whether altered histone acetylations also happened in the kidneys of db/db mice, a type 2 diabetic mouse model. Using enriched core histones extracted from different experimental groups, we found that total ac-K, H3K9ac, H3K9ac/S10p, H3K18ac, H3K23ac and H3K56ac were all significantly increased in the kidneys of db/db mice compared with wild type mice (1.5-fold, 3.3-fold, 2.5-fold, 4.9-fold, 1.8-fold, and 2.2-fold increase, respectively; Fig. 1A and B). However, the total renal histone methylation level was similar between these 2 groups (Fig. 1A and B).
Figure 1.
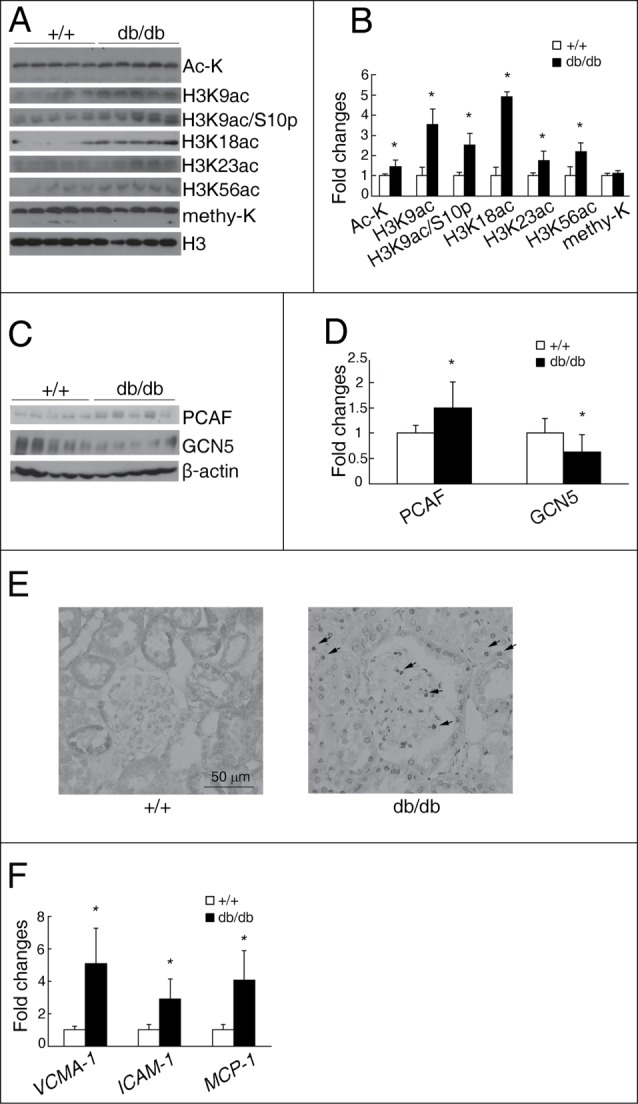
Increased histone acetylation, PCAF, and inflammatory genes in the kidneys of db/db mice. Representative western blots for Ac-K, H3K9ac, H3K9ac/S10p, H3K18ac, H3K23ac, H3K56ac, methy-K and H3 in the kidneys of db/db and wild type (+/+) mice shown in (A), with quantitative densitometric results summarized in (B). Representative western blots for PCAF, GCN5, and β-actin in the kidneys of db/db and wild type (+/+) mice (C), with quantitative densitometric results (D). (E) Representative PCAF immunostaining on kidney sections. Grey or black color indicates positive staining. Bar = 50 μm. (F) qPCR analysis of VCAM-1, ICAM-1, and MCP-1 in the kidneys of db/db and wild type (+/+) mice. (n = 5 in each group; *P < 0.05 compared with wild type mice).
The levels of some HATs were further investigated in these 2 groups. There was a significant increased level of PCAF (1.5-fold; Fig. 1C and D) and an unexpected decreased level of GCN5 (Fig. 1C and D) in the kidneys of db/db mice, when compared with wild type mice. However, similar PCAF and p300 mRNA levels, together with a slight downregulated GCN5 level, were found in the kidneys of db/db mice when compared with wild type mice (Supplemental data, Fig. S1). Immunohistochemistry confirmed that compared to the light cytoplasmic staining of PCAF on some renal tubular cells of wild type mice, a dramatically increased nuclear PCAF staining was found on the renal tubular cells of db/db mice (Fig. 1E). There was also a significant increase in nuclear PCAF staining on glomeruli of db/db mice, with almost no PCAF staining on glomeruli of wild type mice (Fig. 1E).
Since diabetic nephropathy is associated with chronic inflammation, we also investigated the mRNA levels of several inflammatory molecules in the kidneys of these 2 groups. qPCR analysis displayed significant increases of VCAM-1, ICAM-1, and MCP-1 in the kidneys of db/db mice compared with wild type mice (5.1-fold, 2.9-fold, and 4.1-fold increase, respectively; Fig. 1F).
Upregulation of histone acetylation, PCAF, and inflammatory genes in the kidneys of LPS-injected mice
We next examined whether changes of PCAF and histone acetylations can also be found in acute renal inflammatory diseases such as LPS-induced AKI. One hour after LPS injection, the protein level of PCAF was significantly increased in the kidneys of LPS-injected mice compared with PBS-injected mice (Fig. 2A), while GCN5 protein level was similar between these 2 groups (Supplemental data, Fig. S2). Consistent with the increased PCAF level, significantly increased levels of total ac-K, H3K9ac, and a dramatic raise of H3K18ac were found in the kidneys of LPS-injected mice (1.6-fold, 1.3-fold, and 3.5-fold, respectively) when compared with PBS-injected mice (Fig. 2B and C). Furthermore, compared with PBS injection, LPS injection led to significantly increased levels of VCAM-1, ICAM-1, and MCP-1 in the kidneys (6.9-fold, 13.5-fold, and 148.8-fold, respectively; Fig. 2D).
Figure 2.

Increased histone acetylation, PCAF, and inflammatory genes in the kidneys of LPS-injected mice. (A) Representative western blots for PCAF and β-actin in the kidneys of LPS-injected and PBS-treated mice. (B–C) Representative western blots and quantitative densitometric results for Ac-K, H3K9ac, H3K18ac, and H3 in the kidneys of LPS-injected and PBS-treated mice. (D) qPCR analysis for VCAM-1, ICAM-,1 and MCP-1 in the kidneys of LPS-injected and PBS-treated mice. (E) Quantitative results of H3K18ac ChIP assay in the kidneys of LPS-injected and PBS-treated mice. (n = 4–5 in each group; *P < 0.05 compared with PBS-treated mice).
To investigate whether H3K18ac is involved in transcriptional regulation of these inflammatory genes in this AKI model, CHIP assays were performed to examine the binding affinity of H3K18ac on the promoters of VCAM-1, ICAM-1, and MCP-1. The results demonstrated in the kidney, LPS injection enhanced the recruitment of H3K18ac on the promoters of VCAM-1, ICAM-1, and MCP-1 of all designed primer sets (Fig. 2E), suggesting an important role of H3K18ac on the regulation of inflammatory genes in AKI.
PCAF knockdown induces alteration of histone acetylation in cultured renal tubular cells
To investigate the role of PCAF in renal diseases, stable PCAF knockdown human proximal tubule epithelial cells were generated, which was named as shPCAF HK-2. Compared with wild type cells, both protein (Fig. 3A) and mRNA (Fig. 3B) levels of PCAF were significantly decreased in shPCAF HK-2 cells (43% and 47% of wild type cells, respectively). Whereas the mRNA levels of p300 and GCN5 were similar between PCAF knockdown and wild type cells, indicating PCAF knockdown has no significant impact on other HATs (Fig. 3C).
Figure 3.
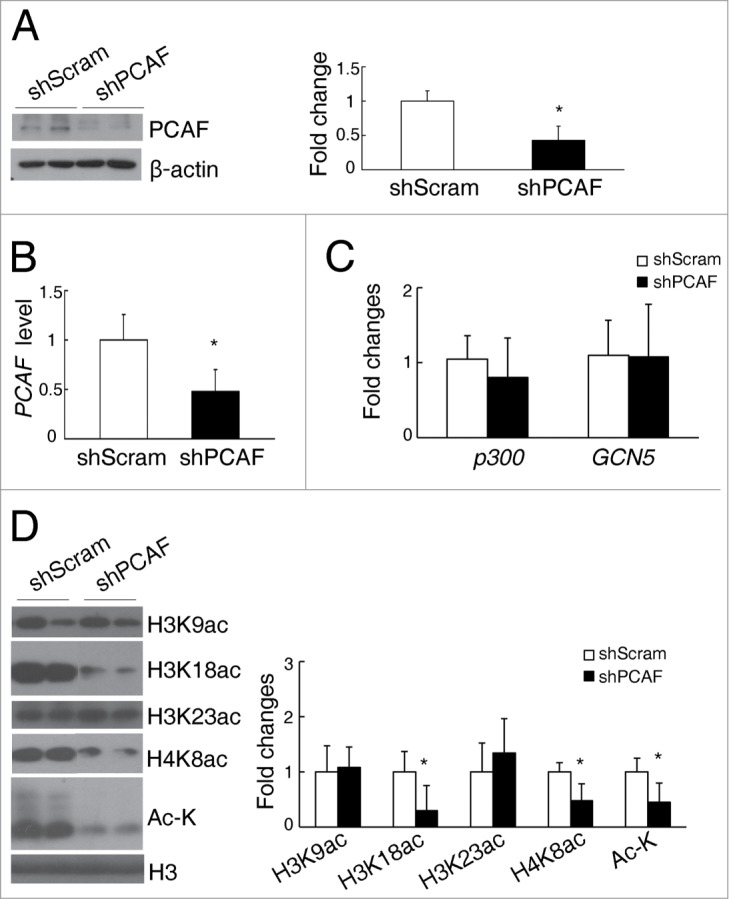
PCAF deficiency induces alteration of histone acetylation in cultured renal tubular cells. (A) Representative western blots for PCAF and β-actin in shScram and shPCAF cells (left panel), and their quantitative densitometric results (right panel). qPCR analysis for PCAF (B), p300, and GCN5 (C) in shScram and shPCAF cells. (D) Representative western blots for H3K9ac, H3K18ac, H3K23ac, H4K8ac, Ac-K, and H3 (left panel) with their quantitative densitometric results (right panel) in shScram and shPCAF cells. (shScram, cells stably transfected with pSUPER-shScram; shPCAF, cells stably transfected with pSUPER-shPCAF. Each experiment was performed in duplicates for 3 times with a representative result shown. *P < 0.05 compared with cells transfected with shScram).
Consistent with the decreased PCAF level, total histone acetylation level was impaired in shPCAF HK-2 cells (45% of wild type cells, Fig. 3D). Using antibodies that detect different specific histone acetylation sites, we demonstrated that the levels of H3K18ac and H4K8ac were significantly downregulated in shPCAF HK-2 cells (29% and 48% of wild type cells, respectively; Fig. 3D), whereas the levels of H3K9ac and H3K23ac were similar between the PCAF knockdown and wild type HK-2 cells (Fig. 3D).
PCAF knockdown reduces the levels of inflammatory molecules in cultured renal tubular cells
To further explore the effects of PCAF knockdown in renal tubular cells, the mRNA levels of several inflammatory, fibrotic and oxidative stress related genes were investigated. VCAM-1, ICAM-1, ICAM-2, NF-κB p50 (encoding the p50 subunit of NF-κB), and MCP-1 were significantly downregulated in shPCAF HK-2 cells (Fig. 4A). However, the mRNA levels of fibrotic and oxidative stress related genes, such as TGFβ−1, COL4 (Type IV collagen), and MnSOD, were similar between the PCAF knockdown and wild type cells (Fig. 4B). Consistent with their transcription levels, the protein levels of VCAM-1, ICAM-1, NF-κB p50, and MCP-1 were significantly decreased in shPCAF HK-2 cells (Fig. 4C).
Figure 4.
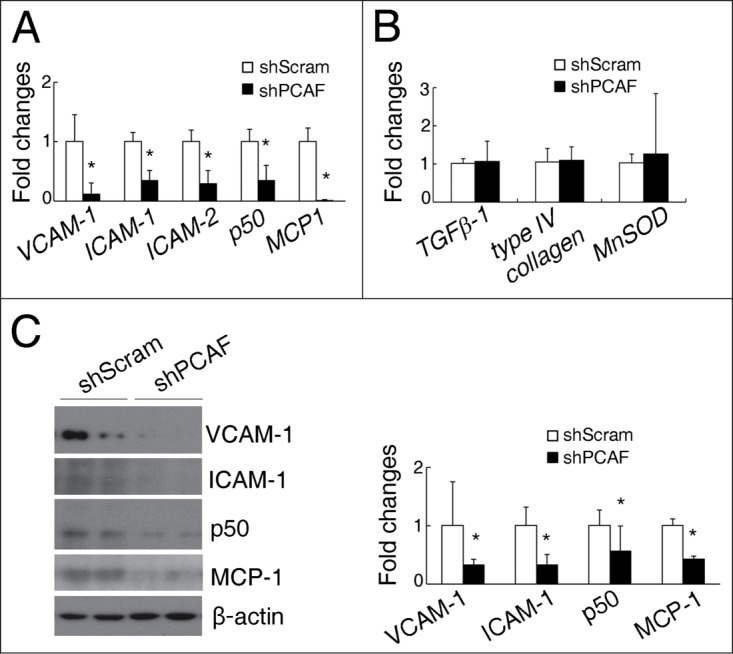
PCAF deficiency reduces the levels of inflammatory molecules in cultured renal tubular cells. (A) qPCR analysis for VCAM-1, ICAM-1, ICAM-2, NF-κB p50, and MCP-1 in shScram and shPCAF cells. (B) qPCR analysis for TGFβ-1, type IV collagen, and MnSOD in shScram and shPCAF cells. (C) Representative protein gel blots for VCAM-1, ICAM-1, NF-κB p50, MCP-1 and β-actin (left panel) with their quantitative densitometric results (right panel) in shScram and shPCAF cells. (shScram, cells stably transfected with pSUPER-shScram; shPCAF, cells stably transfected with pSUPER-shPCAF. Each experiment was performed in duplicates for 3 times with a representative result shown.*P < 0.05 compared with cells transfected with shScram).
PCAF induces upregulation of inflammatory molecules in cultured renal tubular cells
To test whether PCAF directly regulates the transcription of these inflammation-related genes, HK-2 cells were transfected with either a full-length PCAF plasmid (pCI-Flag-PCAF) or a control plasmid (pCI). Successful transfection was verified by a dramatic increase of PCAF mRNA level in the transfected cells (Supplemental data, Figure S3). Two days after transfection, significant increases in VCAM-1, ICAM-1, ICAM-2, and MCP-1 levels were found in the pCI-Flag-PCAF transfected HK-2 cells when compared with the pCI transfected HK-2 cells (Fig. 5A), whereas the level of NF-κB p50 was found unchanged. Furthermore, overexpression of PCAF significantly promoted the transcription of MCP-1, as demonstrated by luciferase assay in 293T cells (Supplemental data, Figure S4). To test the effects of other HATs, such as GCN5, on the transcription of these inflammation-related genes, HK-2 cells were also transfected with either a full-length GCN5 plasmid or a control plasmid. Although the mRNA level and protein level of GCN5 were significantly increased after transfection, the mRNA levels of VCAM-1, ICAM-1, ICAM-2, p50, and MCP-1 were similar between the HK-2 cells transfected with the GCN5 plasmid and the control plasmid (Supplemental data, Fig. S5A and B).
Figure 5.
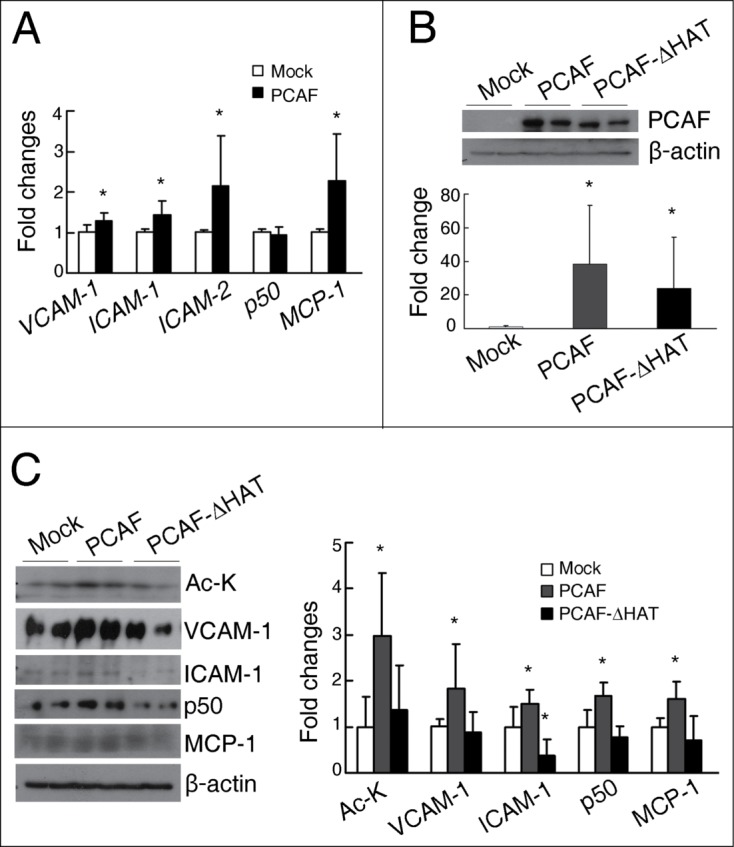
PCAF overexpression induces the expression of inflammatory molecules in cultured renal tubular cells. (A) qPCR analysis for VCAM-1, ICAM-1, ICAM-2, NF-κB p50, and MCP-1 in HK-2 cells transfected with either a pCI plasmid (Mock) or a pCI-Flag-PCAF plasmid (PCAF). (B) Representative western blots for PCAF and β-actin in the HK-2 cells transfected with a pCI plasmid (Mock) or a pCI-Flag-PCAF plasmid (PCAF) or a pCI-Flag-PCAF-ΔHAT plasmid (PCAF-ΔHAT) shown on the up panel, with their quantitative densitometric results shown on the bottom panel. (C) Representative western blots for Ac-K, VCAM-1, ICAM-1, NF-κB p50, MCP-1, and β-actin in HK-2 cells transfected with Mock, PCAF, or PCAF-ΔHAT shown on the left panel, with their quantitative densitometric results shown on the right panel. (Each experiment was performed in duplicates for 3 times with a representative result shown. *P < 0.05 compared with the Mock group).
To further test whether the HAT activity of PCAF is involved in the regulation of these inflammatory molecules, HK-2 cells were transfected with either the pCI-Flag-PCAF plasmid or the pCI-Flag-PCAF-ΔHAT plasmid, the latter plasmid codes a mutant PCAF with partial HAT domain deletion. Similar overexpressed PCAF level was found in cells transfected with either plasmid (Fig. 5B); however, total histone acetylation level was increased only in the pCI-Flag-PCAF transfected cells (Fig. 5C). Furthermore, compared with control cells, it was the overexpression of full-length PCAF, but not the mutant PCAF in HK-2 cells that induced upregulations of VCAM-1, ICAM-1, NF-κB p50 and MCP-1 (Fig. 5C). It has been reported that due to different regulatory mechanisms and synthesis/degradation rates, the correlation between the levels of mRNA and protein of certain genes are not linear.26 The absence of correlation between mRNA level and protein level of NF-κB p50 after transfection of the full length PCAF plasmid implicates PCAF may regulate the degradation of NF-κB p50, although further studies are necessary to verify this possibility. However, overexpression of GCN5 did not induce and increase in the protein levels of VCAM-1, ICAM-1 and NF-κB p50 in HK-2 cells (Supplemental data, Fig. S5C).
PCAF knockdown inhibits palmitate-induced upregulation of inflammatory molecules by decreasing recruitment of H3K18ac on their promoters
To investigate whether PCAF regulates the transcription of inflammatory genes by manipulating histone acetylation level, ChIP assays were performed. Two sets of primers were designed to target the distinct regions on the promoters of ICAM-1 and MCP-1. In normal cultured condition, PCAF knockdown induced significant decreases of H3K18ac recruitment on the promoters of ICAM-1-a/b and MCP-1-a/b in HK-2 cells (Fig. 6A). To further investigate whether PCAF knockdown can inhibit the hyperlipidemia-induced inflammation in the kidneys, HK-2 cells were treated with 300 μM palmitate to mimic the hyperlipidemic condition in vivo. The presence of palmitate led to significant upregulation of ICAM-1 and MCP-1 mRNA levels in HK-2 cells; however, PCAF knockdown significantly inhibited the palmitate-induced upregulation of these inflammatory genes (Fig. 6B). Furthermore, ChIP assays also showed that after palmitate treatment, increased recruitment of H3K18ac on the promoters of ICAM-1 and MCP-1 were observed, and PCAF knockdown attenuated such palmitate-induced increased recruitment of H3K18ac on these promoters (Fig. 6C and D), which was consistent with what observed on the transcription level.
Figure 6.
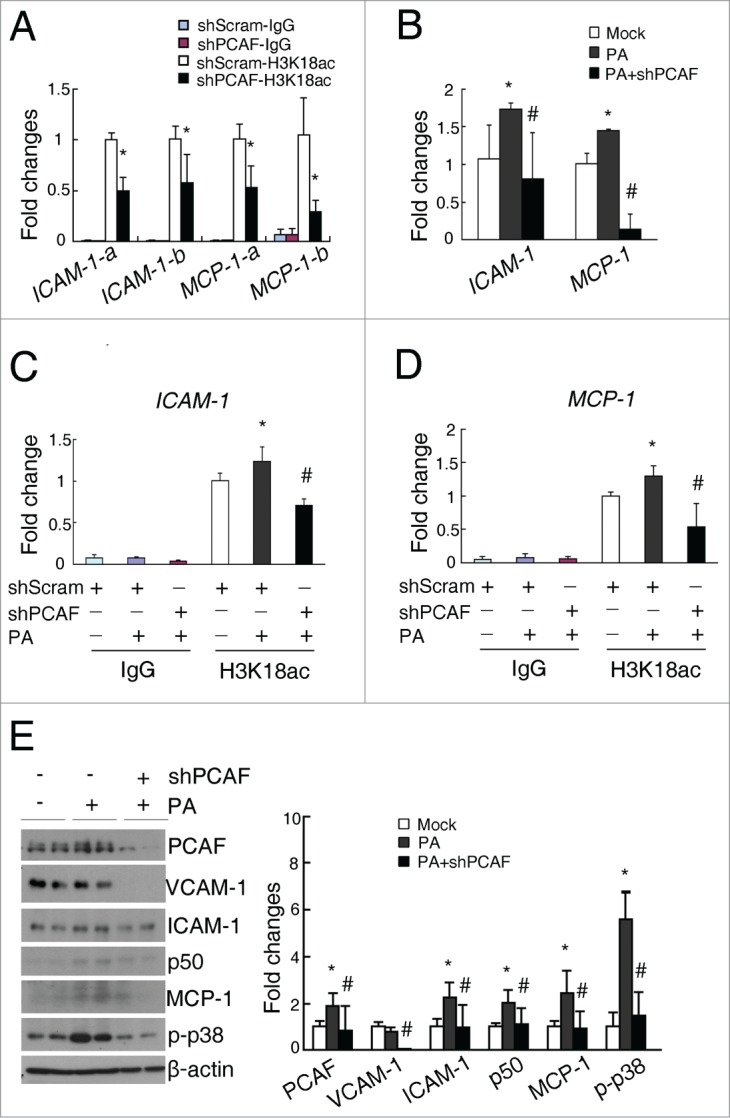
PCAF deficiency inhibits the palmitate-induced recruitment of H3K18ac on the promoters of ICAM-1 and MCP-1 and upregulations of these inflammatory molecules. (A) Quantitative results for the H3K18ac ChIP assay in shScram and shPCAF cells. (B) qPCR analysis for ICAM-1 and MCP-1 in HK-2 cells treated with 300 μM PA. Quantitative results of the H3K18ac ChIP assay on the promoters of ICAM-1 (C) and MCP-1 (D) in shScram and shPCAF cells treated with 300 μM palmitate. (E) Representative western blots for PCAF, VCAM-1, ICAM-1, NF-κB p50, MCP-1, p-p38, and β-actin in the shScram and shPCAF cells treated with 300 μM palmitate (left panel), with their densitometric quantitative results (right panel). (shScram, cells stably transfected with pSUPER-shScram; shPCAF, cells stably transfected with pSUPER-shPCAF; Mock, cells treated with vehicle; PA, palmitate treatment. Each experiment was performed in duplicates for 3 times with a representative result shown. * P < 0.05 compared with Mock group. # P < 0.05 compared with PA-treated cells)
Meanwhile, palmitate induced a 1.9-fold increase of PCAF in HK-2 cells, as well as upregulations of ICAM-1, NF-κB p50, MCP-1 and phosphorylated p38 levels (2.2-fold, 2.0-fold, 2.4-fold, and 5.6-fold, respectively; Fig. 6E). PCAF knockdown in HK-2 cells not only normalized palmitate-induced increase in PCAF level, but also inhibited all these palmitate-induced increases in the inflammatory molecules (Fig. 6E). Also, PCAF knockdown significantly downregulated VCAM-1 level in palmitate-treated HK-2 cells, although no changes of VCAM-1 were found between palmitate-treated and un-treated cells (Fig. 6E).
Discussion
Renal diseases severely threat human health. Both CKD and AKI contribute to the development of high mortality end stage renal diseases. Therefore, it is urgent to identify the common mechanisms underlying pathogenesis of these renal diseases, and seek for potential therapeutic targets. In this study, we have demonstrated that PCAF, a histone acetyltransferase, regulates the expression of inflammatory molecules in renal tubular epithelial cells, which contribute to the development of CKD and AKI.
It has been reported that alterations in histone modifications are associated with renal diseases. For example, increased levels of H3K9ac, H3K23ac, and H3S10p have been observed in the kidneys of db/db mice after uninephrectomy surgery.22 Furthermore, in the kidneys of Akita mice and high glucose treated mesangial cells, acetylation on multiple histone H3 lysine residues, including H3K9, H3K18, and H3K23, are increased.21 Consistent with these observations, in the present study, we demonstrated that increased histone acetylation also happened in the kidneys of db/db mice and LPS-injected mice (Fig. 1 and 2), suggesting such accumulation of histone acetylation exists in multiple rodent models of renal diseases.
In addition to histone acetylation, alterations in histone methylation have also been reported to contribute to the development of some renal diseases. For example, increased level of H3K4me2 has been observed in the kidneys of db/db mice after uninephrectomy surgery.22 In renal ischemia and reperfusion injury, accumulated H3K4me3 has been found on the promoters of TGFβ−1, MCP-1, and Collagen III.27 In the present study, although total methylation level was not changed in the kidneys of db/db mice compared with wild type mice (Fig. 1), we still cannot rule out the possibility that some specific histone methylation sites are altered in the kidneys of db/db mice.
Among all HATs, p300 and CBP have been reported to be involved in the development of renal diseases. The interaction between p300 and NF-κB has been reported to be inhibited by curcumin, a potent antioxidant, to prevent the development of diabetic nephropathy.24 In cultured rat mesangial cells, overexpression of p300 or CBP has been demonstrated to significantly enhance TGFβ−1-induced PAL-1 and p21 levels, with increased recruitment of H3K9ac and H3K14ac on the promoters of PAL-1 and p21.28 However, these changes are not observed when PCAF is overexpressed.28 Consistent with this, our study also demonstrated that PCAF overexpression did not induce increased transcription of TGFβ-1 in cultured renal tubular epithelial cells (Fig. 4). In this study, we have demonstrated that PCAF regulates the transcription of inflammatory genes (Fig. 4 and 5). We found that PCAF, but not GCN5, is overexpressed in the kidneys of db/db mice and LPS-injected mice. Moreover, we have previously demonstrated that PCAF, but not GCN5, is overexpressed in the kidneys of Akita mice.21 Although GCN5 and PCAF both act as a HAT, increasing evidence has suggested that they have different biological functions. For example, mice lacking Pcaf develop normally without a distinct phenotype; however, mice lacking Gcn5 die in the gestation stage, suggesting the distinct roles of GCN5 and PCAF in embryogenesis.29 In vitro study has demonstrated that GCN5 deficiency, but not PCAF deficiency, inhibits the growth rate of chicken B cells.30 In addition, specifically inhibition of PCAF, but not p300 or GCN5, suppresses the β-amyloid induced inflammatory cytokine production and neuronal cell death.31 All these observations may be due to the fact that GCN5 and PCAF bind to distinct transcriptional factors and regulators; moreover, they also acetylate different sites of histones.32 Thus, it is possible that p300, PCAF, and GCN5, although all are HATs, regulate different sets of genes upon same stimulation. The different roles of HATs, such as p300, PCAF, and GCN5, in the development of renal diseases await further investigation.
Previous studies have found that PCAF is involved in the development of pancreatitis, arteriogenesis, and liver injury, suggesting PCAF plays an important role in inflammation.33-35 PCAF is recruited to the promoter of TNFα and causes increased histone H3K9 acetylation in acute necrotic pancreatitis, where inflammation is taken place.33 A microarray study further demonstrates that in a hindlimb ischemia model, proinflammatory genes are profoundly reduced in the surrounding skeletal muscle of PCAF knockout mice compared to wild type mice.34 Impaired blood flow recovery has also been found in PCAF deficient mice, either genetic or therapeutic, after hindlimb ischemia injury,34 as arteriogenesis is a classic inflammation-driven process after this injury. In cultured leukocytes and endothelial cells, PCAF deficient inhibits ischemia and reperfusion-induced inflammatory response.34 Moreover, downregulation of PCAF by miR-181a/b provides a negative feedback regulation to TNFα-induced inflammatory reaction in liver epithelial cells.35 In the present study, we also demonstrated that PCAF specifically regulated some inflammatory molecules in renal tubule epithelial cells (Fig. 4 and 5). Furthermore, knockdown PCAF inhibited palmitate-induced increases of inflammatory molecules by decreasing recruitment of H3K18ac on the promoters of these genes (Fig. 6), which prevents the palmitate-induced damage in renal tubular epithelial cells.
In addition to its role in the regulation of inflammation, PCAF also has multiple other functions, including regulation of endoplasmic reticulum (ER) stress, mitosis, and cell growth. It has been shown that PCAF binds to XBP-1S (X-box binding protein 1S), an isoform of XBP-1 and important regulator of cellular unfolded protein response, to mediate XBP-1S-related gene transcription and regulates ER stress.36 During mitosis, the acetyltransferase activity of PCAF can temporally regulate the interaction of plus-end tracking protein 150 (TIP150) and end-binding protein 1 (EB1), 2 mitosis related proteins, to ensure chromosome stability.37 In several types of cancer, PCAF has also been shown to acetylate ATP-citrate lyase (ACLY) under high glucose to stabilize the protein level of ACLY, thus promoting lipid synthesis, cell proliferation, and tumor growth.38 In summary, in the present study, upregulation of PCAF was found in the kidney of db/db mice and LPS-injected mice. PCAF knockdown and overexpression specifically regulates the inflammatory molecules in cultured renal tubular epithelial cells. Furthermore, PCAF knockdown reduced the palmitate-induced upregulation of these inflammatory molecules by reducing H3K18ac level on their gene promoters. These evidences suggest a critical role for PCAF in the regulation of inflammation in renal diseases (Fig. 7).
Figure 7.
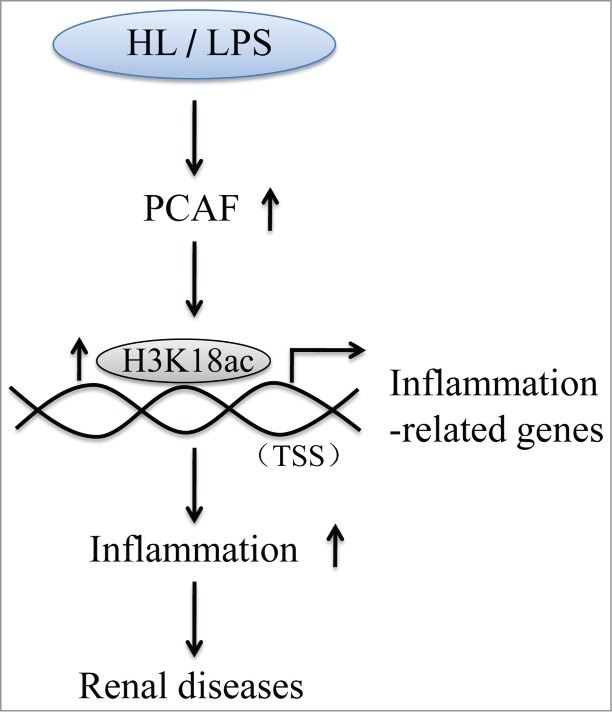
A possible molecular mechanism of how PCAF regulates inflammation in renal diseases. HL, hyperlipidemia; LPS, lipopolysaccharide; TSS, transcription start site.
Material and Methods
Animals
Seven-eight weeks old male db/db (LepR−/−) mice and their sex- and age-matched wild type controls were obtained from the Model Animal Research Center of Nanjing University. Male C57BL/6 mice were obtained from the ABSL-III laboratory of Wuhan University. The mice were housed in ventilated microisolator cages with free access to water and food. For the study on diabetic nephropathy, body weight (BW) of each mouse was measured weekly. Non-fasted blood glucose level of each mouse was measured by an OneTouch Ultra Glucometer (Lifescan, Inc., Milpitas, California) at time of sacrifice. The physiological characteristics of db/db mice and their controls are shown in Supplemental Table S1, indicating syndromes of diabetes, obesity, and renal hypertrophy for db/db mice at 3 months of age. For the LPS injection experiment, male C57BL/6 mice were intraperitoneally injected with LPS (3 mg/kg BW) or same volume of PBS. The mice were sacrificed one hour after LPS or PBS injection, and whole kidneys were collected. Mice were handled according to the Guidelines of China Animal Welfare Legislation, as approved by the Committee on Ethics in the Care and Use of Laboratory Animals of College of Life Sciences, Wuhan University. Kidney samples were either frozen at −80°C until use (for histone enrichment and western blotting) or fixed with formaldehyde solution (for immunohistochemistry study).
Plasmids
Plasmids pCI-Flag-PCAF and pCI-Flag-PCAF-ΔHAT (a deletion of amino acids 497–526) were gifts from Dr. Costas Demonacos (School of Pharmacy and Pharmaceutical Sciences, Manchester, UK). Plasmids pCI and myc-GCN5 were obtained from Dr. Xiaodong Zhang (Wuhan University) and Dr. Haining Du (Wuhan University), respectively. Transfections were carried out using FuGENE HD Transfection Reagent (Promega, Madison, WI) following the manufacturer's instruction.
Cell culture and stable PCAF knockdown cell line
Human renal proximal tubule epithelial cell line, HK-2, was cultured in RPMI-1640 media (Hyclone, South Logan, UT) supplemented with 10% FBS (Gibco, Grand Island, NY), 1 mM sodium pyruvate (Hyclone) and 5 ng/μl hEGF (Peprotech, Inc., Rocky Hill, CT). pSUPER vector system (Oligoengine, Seattle, WA) was used to establish stable PCAF knockdown cells. PCAF shRNA (shPCAF) was designed using Thermo Scientific siDESIGN Center (http://rnaidesigner.lifetechnologies.com/rnaiexpress). HK-2 cells were transfected with shPCAF, and stable clones were selected with puromycin (1 μg/μl). Palmitate stock solution was prepared as previously reported.39 Cells were treated with palmitate solution at a final concentration of 300 μM for 3 d.
Histone extraction
Acid extraction was performed as we described previously.40 Briefly, tissue was homogenized in fresh made hypotonic lysis buffer (10 mM Tris-HCl, 1 mM KCl, 1.5 mM MgCl2, 1 mM PMSF, 1.3 mM DTT, pH 8.0) by a Dounce homogenizer. The sample was lysed in hypotonic lysis buffer with 10% NP-40. Nuclei was precipitated and then incubated with 0.2 M H2SO4 at 4°C for 2 hours. After centrifuging at 16,000 g, 100% TCA was added to precipitate histones, followed by washing twice with ice-cold acetone. Concentration of enriched histones was quantitated with the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA), aliquoted and stored at −80°C until use.
Western blotting analysis
Western blots were performed as described previously.41 Equal amount of proteins were separated by SDS-PAGE (15% for extracted histones and 8–10% for whole cell lysate) and electroblotted onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA). A list of primary antibodies used is provided in Supplemental Table S2. Corresponding secondary antibodies (Bio-Rad) were used at a 1:5000 dilution. The blots were visualized by ECL reagent (Thermo Scientific, Rockland, MA) after exposure of X-OMAT autoradiograph films. The developed films were subsequently scanned, and band intensities were quantified with the Quantity One Software (Bio-Rad). Targeted protein expression levels were quantitated relative to histone H3 or β-actin in the same sample, and normalized to the respective control group, which was arbitrarily set as one-fold.
Immunohistochemistry staining
Kidney samples were routinely embedded in paraffin and sectioned. After being deparaffinized in xylene and rehydrated in decreasing concentrations of ethanol, sections were incubated with 3% H2O2 for 5 min to quench endogenous superoxidase activity followed by blocking with 2% goat serum in PBS and immunostained with an anti-PCAF antibody (1: 200 dilution, Santa Cruz Biotechnology, Dallas, TX) at 4°C overnight. After extensive washing, sections were incubated with a biotinylated anti-rabbit antibody (Vector Laboratories, Burlingame, CA) for 1 h at room temperature. Positive staining was visualized by DAB substrate reaction (Vector laboratories) following the ABC kit (Vector laboratories). High resolution pictures (× 400 power) were taken under an Olympus BX60 microscope equipped with a digital CCD.
Quantitative real-time PCR (qPCR)
Total RNA was extracted using RNAiso (TaKaRa Biotechnology, Japan), and converted to cDNA by M-MLV cDNA synthetic system (Invitrogen, Carlsbad, CA). qPCR was performed with SsoFastTM EvaGreenR Supermix (Invitrogen) using Bio-Rad iQ5 Real Time PCR System. β−actin was used as an internal control. The relative difference was expressed as the fold change calculated by the 2-ΔΔCT method. Primer list for qPCR is provided in Supplemental Table S3.
Chromatin Immunoprecipitation (CHIP) assay
For cultured cells, cells were fixed with 1% formaldehyde for 10 min at room temperature. For tissue, kidneys were minced first and fixed with 1% formaldehyde for 10 min at room temperature. The cross-linking was then quenched with 125 mM glycine. Lysis buffer (400 μl) containing protease inhibitor cocktail (Roche, Basel, CH) was added. The samples were sonicated, and an aliquot of each sample was saved as the input. Chromatin was immunoprecipitated with an anti-H3K18ac antibody (Abcam, Cambridge, MA) or an anti-rabbit IgG. The immune complexes were captured with Dynabeads Protein G (Thermo Scientific). Input DNA and CHIP-enriched DNA were analyzed by qPCR. The input samples were used as the internal control for comparison between samples. Primer list for CHIP assays is provided in Supplemental Table S4.
Statistical analysis
All results were expressed as the mean ± SD. Statistical significance was evaluated with the Kruskal-Wallis test followed by the Mann-Whitney test for 3 groups comparison, and the Mann-Whitney test only for 2 groups comparison. Differences were considered statistically significant at P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Basic Research Program of China (2012CB524901), the Natural Science Foundation of China (Nos. 81222043, 31271370, 81172971, and 31471208), and the Fundamental Research Funding of Central University. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Soldatos G, Cooper ME. Diabetic nephropathy: important pathophysiologic mechanisms. Diabetes Res Clin Pract 2008; 82 Suppl 1:S75-9; PMID:18994672; http://dx.doi.org/ 10.1016/j.diabres.2008.09.042 [DOI] [PubMed] [Google Scholar]
- 2. Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 2011; 121:4210-21; PMID:22045571; http://dx.doi.org/ 10.1172/JCI45161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Soares TJ, Costa RS, Volpini RA, Da Silva CG, Coimbra TM. Long-term evolution of the acute tubular necrosis (ATN) induced by glycerol: role of myofibroblasts and macrophages. Int J Exp Pathol 2002; 83:165-72; PMID:12485461; http://dx.doi.org/ 10.1046/j.1365-2613.2002.00223.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nath KA. Renal response to repeated exposure to endotoxin: implications for acute kidney injury. Kidney Int 2007; 71:477-9; PMID:17344894; http://dx.doi.org/ 10.1038/sj.ki.5002150 [DOI] [PubMed] [Google Scholar]
- 5. Medzhitov R. Origin and physiological roles of inflammation. Nature 2008; 454:428-35; PMID:18650913; http://dx.doi.org/ 10.1038/nature07201 [DOI] [PubMed] [Google Scholar]
- 6. Jones SC, Banks RE, Haidar A, Gearing AJ, Hemingway IK, Ibbotson SH, Dixon MF, Axon AT. Adhesion molecules in inflammatory bowel disease. Gut 1995; 36:724-30; PMID:7541009; http://dx.doi.org/ 10.1136/gut.36.5.724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frank PG, Lisanti MP. ICAM-1: role in inflammation and in the regulation of vascular permeability. Am J Physiol Heart Circ Physiol 2008; 295:H926-H7; PMID:18689494; http://dx.doi.org/ 10.1152/ajpheart.00779.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rovin BH, Lu L, Saxena R. A novel polymorphism in the MCP-1 gene regulatory region that influences MCP-1 expression. Biochem Biophys Res Commun 1999; 259:344-8; PMID:10362511; http://dx.doi.org/ 10.1006/bbrc.1999.0796 [DOI] [PubMed] [Google Scholar]
- 9. Ha H, Yu MR, Choi YJ, Kitamura M, Lee HB. Role of high glucose-induced nuclear factor-kappaB activation in monocyte chemoattractant protein-1 expression by mesangial cells. J Am Soc Nephrol 2002; 13:894-902; PMID:11912248 [DOI] [PubMed] [Google Scholar]
- 10. Duran-Salgado MB, Rubio-Guerra AF. Diabetic nephropathy and inflammation. World J Diabetes 2014; 5:393-8; PMID:24936261; http://dx.doi.org/ 10.4239/wjd.v5.i3.393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rivero A, Mora C, Muros M, Garcia J, Herrera H, Navarro-Gonzalez JF. Pathogenic perspectives for the role of inflammation in diabetic nephropathy. Clin Sci (Lond) 2009; 116:479-92; PMID:19200057; http://dx.doi.org/ 10.1042/CS20080394 [DOI] [PubMed] [Google Scholar]
- 12. Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Tesch GH. Intercellular adhesion molecule-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. J Am Soc Nephrol 2005; 16:1711-22; PMID:15857924; http://dx.doi.org/ 10.1681/ASN.2004070612 [DOI] [PubMed] [Google Scholar]
- 13. Kanamori H, Matsubara T, Mima A, Sumi E, Nagai K, Takahashi T, Abe H, Iehara N, Fukatsu A, Okamoto H, et al. . Inhibition of MCP-1/CCR2 pathway ameliorates the development of diabetic nephropathy. Biochem Biophys Res Commun 2007; 360:772-7; PMID:17631861; http://dx.doi.org/ 10.1016/j.bbrc.2007.06.148 [DOI] [PubMed] [Google Scholar]
- 14. Lai PF, Cheng CF, Lin H, Tseng TL, Chen HH, Chen SH. ATF3 Protects against LPS-Induced Inflammation in Mice via Inhibiting HMGB1 Expression. Evid Based Complement Alternat Med 2013; 2013:716481; PMID:24062788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Richards EJ. Inherited epigenetic variation–revisiting soft inheritance. Nat Rev Genet 2006; 7:395-401; PMID:16534512; http://dx.doi.org/ 10.1038/nrg1834 [DOI] [PubMed] [Google Scholar]
- 16. Lennartsson A, Ekwall K. Histone modification patterns and epigenetic codes. Biochim Biophys Acta 2009; 1790:863-8; PMID:19168116; http://dx.doi.org/ 10.1016/j.bbagen.2008.12.006 [DOI] [PubMed] [Google Scholar]
- 17. Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev 2002; 12:142-8; PMID:11893486; http://dx.doi.org/ 10.1016/S0959-437X(02)00279-4 [DOI] [PubMed] [Google Scholar]
- 18. Bayarsaihan D. Epigenetic mechanisms in inflammation. J Dent Res 2011; 90:9-17; PMID:21178119; http://dx.doi.org/ 10.1177/0022034510378683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun WJ, Zhou X, Zheng JH, Lu MD, Nie JY, Yang XJ, Zheng ZQ. Histone acetyltransferases and deacetylases: molecular and clinical implications to gastrointestinal carcinogenesis. Acta Biochim Biophys Sin (Shanghai) 2012; 44:80-91; PMID:22194016; http://dx.doi.org/ 10.1093/abbs/gmr113 [DOI] [PubMed] [Google Scholar]
- 20. Graff J, Tsai LH. Histone acetylation: molecular mnemonics on the chromatin. Nat Rev Neurosci 2013; 14:97-111; PMID:23324667; http://dx.doi.org/ 10.1038/nrn3427 [DOI] [PubMed] [Google Scholar]
- 21. Chen H, Li J, Jiao L, Petersen RB, Peng A, Zheng L, Huang K. Apelin inhibits the development of diabetic nephropathy by regulating histone acetylation in Akita mouse. J Physiol 2014; 592:505-21; PMID:24247978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sayyed SG, Gaikwad AB, Lichtnekert J, Kulkarni O, Eulberg D, Klussmann S, Tikoo K, Anders HJ. Progressive glomerulosclerosis in type 2 diabetes is associated with renal histone H3K9 and H3K23 acetylation, H3K4 dimethylation and phosphorylation at serine 10. Nephrol Dial Transplant 2010; 25:1811-7; PMID:20067909; http://dx.doi.org/ 10.1093/ndt/gfp730 [DOI] [PubMed] [Google Scholar]
- 23. Bomsztyk K, Flanagin S, Mar D, Mikula M, Johnson A, Zager R, Denisenko O. Synchronous recruitment of epigenetic modifiers to endotoxin synergistically activated Tnf-α gene in acute kidney injury. PLoS One 2013; 8:e70322; PMID:23936185; http://dx.doi.org/ 10.1371/journal.pone.0070322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chiu J, Khan ZA, Farhangkhoee H, Chakrabarti S. Curcumin prevents diabetes-associated abnormalities in the kidneys by inhibiting p300 and nuclear factor-kappaB. Nutrition 2009; 25:964-72; PMID:19268536; http://dx.doi.org/ 10.1016/j.nut.2008.12.007 [DOI] [PubMed] [Google Scholar]
- 25. Advani A, Huang Q, Thai K, Advani SL, White KE, Kelly DJ, Yuen DA, Connelly KA, Marsden PA, Gilbert RE. Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am J Pathol 2011; 178:2205-14; PMID:21514434; http://dx.doi.org/ 10.1016/j.ajpath.2011.01.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Sousa Abreu R, Penalva LO, Marcotte EM, Vogel C. Global signatures of protein and mRNA expression levels. Mol Biosyst 2009; 5:1512-26; PMID:20023718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zager RA, Johnson AC. Renal ischemia-reperfusion injury upregulates histone-modifying enzyme systems and alters histone expression at proinflammatory/profibrotic genes. Am J Physiol Renal Physiol 2009; 296:F1032-41; PMID:19261745; http://dx.doi.org/ 10.1152/ajprenal.00061.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yuan H, Reddy MA, Sun G, Lanting L, Wang M, Kato M, Natarajan R. Involvement of p300/CBP and epigenetic histone acetylation in TGF-beta1-mediated gene transcription in mesangial cells. Am J Physiol Renal Physiol 2013; 304:F601-13; PMID:23235480; http://dx.doi.org/ 10.1152/ajprenal.00523.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamauchi T, Yamauchi J, Kuwata T, Tamura T, Yamashita T, Bae N, Westphal H, Ozato K, Nakatani Y. Distinct but overlapping roles of histone acetylase PCAF and of the closely related PCAF-B/GCN5 in mouse embryogenesis. Proc Natl Acad Sci U S A 2000; 97:11303-6; PMID:11027331; http://dx.doi.org/ 10.1073/pnas.97.21.11303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kikuchi H, Takami Y, Nakayama T. GCN5: a supervisor in all-inclusive control of vertebrate cell cycle progression through transcription regulation of various cell cycle-related genes. Gene 2005; 347:83-97; PMID:15715965; http://dx.doi.org/ 10.1016/j.gene.2004.12.007 [DOI] [PubMed] [Google Scholar]
- 31. Park SY, Lee YH, Seong AR, Lee J, Jun W, Yoon HG. Selective inhibition of PCAF suppresses microglial-mediated β-amyloid neurotoxicity. Int J Mol Med 2013; 32:469-75; PMID:23740527 [DOI] [PubMed] [Google Scholar]
- 32. Nagy Z, Tora L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 2007; 26:5341-57; PMID:17694077; http://dx.doi.org/ 10.1038/sj.onc.1210604 [DOI] [PubMed] [Google Scholar]
- 33. Sandoval J, Pereda J, Rodriguez JL, Escobar J, Hidalgo J, Joosten LA, Franco L, Sastre J, Lopez-Rodas G. Ordered transcriptional factor recruitment and epigenetic regulation of tnf-α in necrotizing acute pancreatitis. Cell Mol Life Sci 2010; 67:1687-97; PMID:20130956; http://dx.doi.org/ 10.1007/s00018-010-0272-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bastiaansen AJ, Ewing MM, de Boer HC, van der Pouw Kraan TC, de Vries MR, Peters EA, Welten SM, Arens R, Moore SM, Faber JE, et al. . Lysine acetyltransferase PCAF is a key regulator of arteriogenesis. Arterioscler Thromb Vasc Biol 2013; 33:1902-10; PMID:23788761; http://dx.doi.org/ 10.1161/ATVBAHA.113.301579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao J, Gong AY, Zhou R, Liu J, Eischeid AN, Chen XM. Downregulation of PCAF by miR-181a/b provides feedback regulation to TNF-α-induced transcription of proinflammatory genes in liver epithelial cells. J Immunol 2012; 188:1266-74; PMID:22219331; http://dx.doi.org/ 10.4049/jimmunol.1101976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lew QJ, Chu KL, Lee J, Koh PL, Rajasegaran V, Teo JY, Chao SH. PCAF interacts with XBP-1S and mediates XBP-1S-dependent transcription. Nucleic Acids Res 2011; 39:429-39; PMID:20817929; http://dx.doi.org/ 10.1093/nar/gkq785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ward T, Wang M, Liu X, Wang Z, Xia P, Chu Y, Wang X, Liu L, Jiang K, Yu H, et al. . Regulation of a dynamic interaction between two microtubule-binding proteins, EB1 and TIP150, by the mitotic p300/CBP-associated factor (PCAF) orchestrates kinetochore microtubule plasticity and chromosome stability during mitosis. J Biol Chem 2013; 288:15771-85; PMID:23595990; http://dx.doi.org/ 10.1074/jbc.M112.448886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin R, Tao R, Gao X, Li T, Zhou X, Guan KL, Xiong Y, Lei QY. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol Cell 2013; 51:506-18; PMID:23932781; http://dx.doi.org/ 10.1016/j.molcel.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cousin SP, Hugl SR, Wrede CE, Kajio H, Myers MG, Jr., Rhodes CJ. Free fatty acid-induced inhibition of glucose and insulin-like growth factor I-induced deoxyribonucleic acid synthesis in the pancreatic β-cell line INS-1. Endocrinology 2001; 142:229-40; PMID:11145586 [DOI] [PubMed] [Google Scholar]
- 40. Li J, Huang J, Li JS, Chen H, Huang K, Zheng L. Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J Hepatol 2012; 56:900-7; PMID:22173165; http://dx.doi.org/ 10.1016/j.jhep.2011.10.018 [DOI] [PubMed] [Google Scholar]
- 41. Wang LL, Chen H, Huang K, Zheng L. Elevated histone acetylations in Muller cells contribute to inflammation: a novel inhibitory effect of minocycline. Glia 2012; 60:1896-905; PMID:22915469; http://dx.doi.org/ 10.1002/glia.22405 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.