

Abstract
O-GlcNAcylation is a posttranslational modification catalyzed by the O-Linked N-acetylglucosamine (O-GlcNAc) transferase (OGT) and reversed by O-GlcNAcase (OGA). Numerous transcriptional regulators, including chromatin modifying enzymes, transcription factors, and co-factors, are targeted by O-GlcNAcylation, indicating that this modification is central for chromatin-associated processes. Recently, OGT-mediated O-GlcNAcylation was reported to be a novel histone modification, suggesting a potential role in directly coordinating chromatin structure and function. In contrast, using multiple biochemical approaches, we report here that histone O-GlcNAcylation is undetectable in mammalian cells. Conversely, O-GlcNAcylation of the transcription regulators Host Cell Factor-1 (HCF-1) and Ten-Eleven Translocation protein 2 (TET2) could be readily observed. Our study raises questions on the occurrence and abundance of O-GlcNAcylation as a histone modification in mammalian cells and reveals technical complications regarding the detection of genuine protein O-GlcNAcylation. Therefore, the identification of the specific contexts in which histone O-GlcNAcylation might occur is still to be established.
Keywords: Chromatin, Epigenetics, Histone, HCF-1, OGT, O-GlcNAc, O-GlcNAcylation, Polycomb, posttranslational modification, TET2
Abbreviations
- HCF-1
Host Cell Factor-1
- H2B K120ub
Histone H2B lysine 120 monoubiquitination
- H2B S112 O-GlcNAc
Histone H2B serine 112 O-GlcNAc
- O-GlcNAc
O-Linked N-acetylglucosamine
- OGT
O-Linked N-acetylglucosamine transferase
- OGA
O-GlcNAcase
- PUGNAc
O-(2-acetamido-2-deoxyglucopyranosylidene) amino N-phenylcarbamate
- TET2
Ten-Eleven Translocation protein 2
- UDP-GlcNAc
Uridine Diphosphate N-Acetylglucosamine
- WGA
Wheat Germ Agglutinin
Introduction
O-GlcNAcylation is a widespread posttranslational modification corresponding to the addition of a single O-Linked N-acetylglucosamine (O-GlcNAc) moiety to nuclear and cytosolic proteins.1-3 Similar to other posttranslational modifications, O-GlcNAcylation regulates protein function by influencing protein-protein interactions, enzymatic activity, and sub-cellular localization.4,5 In mammals, this modification is coordinated by 2 enzymes, the O-Linked β-N-acetylglucosamine transferase (OGT), which catalyzes the attachment of the O-GlcNAc moiety on serine and threonine residues of target proteins, and the O-GlcNAcase (OGA), which ensures its removal through hydrolysis.6,7 O-GlcNAcylation signaling is dependent on the availability of the donor substrate, uridine diphosphate N-Acetylglucosamine (UDP-GlcNAc), which is produced via the hexosamine biosynthetic pathway.8 O-GlcNAcylation is a highly dynamic modification, being regulated by a plethora of intracellular and extracellular cues, including growth factor signaling, fluctuation of nutrient levels, and stress responses.9-11 Indeed, O-GlcNAcylation signaling acts as a metabolic sensor that links changes in the cellular metabolism to downstream regulation of numerous cellular pathways.5,12,13 Moreover, recent studies have shown that direct competition between phosphorylation and O-GlcNAcylation can occur for the same amino acid residue, thus adding another layer of complexity to the outcome and regulation of this posttranslational modification.14,15 The physiological importance of O-GlcNAcylation is further emphasized by the fact that defects in its regulation have been associated with human pathologies, such as diabetes, neurodegenerative diseases, and cancer.16-20
O-GlcNAcylation signaling was proposed to play important roles in regulating the epigenome.1,21 Indeed, several transcriptional regulators and chromatin-modifying enzymes are modified by O-GlcNAcylation, thus impacting their recruitment to chromatin, assembly into functional transcription regulatory complexes, stability, and activity.22-25 For instance, others and we have identified a non-canonical mechanism of OGT-mediated transcriptional regulation, which involves the O-GlcNAcylation of the Host Cell Factor 1 (HCF-1) transcriptional regulator, inducing its proteolytic maturation.26-28
Recent studies have also reported that histones are modified by O-GlcNAcylation, suggesting an interesting possibility of crosstalk with other well-established histone marks.29-31 Moreover, it was suggested that the methylcytosine dioxygenase Ten Eleven Translocation 2 (TET2) enzyme directly interacts with OGT to stimulate histone H2B S112 O-GlcNAcylation (H2B S112 O-GlcNAc) and gene expression.32 Several methods were used to detect histone O-GlcNAcylation, including mass spectrometry, immunodetection with O-GlcNAc-specific antibodies, and affinity binding to wheat germ agglutinin (WGA) lectin. 14,30, 33 However, discrepancies regarding the occurrence and the identity of the histones being modified were also reported. For instance, some studies suggested that histones H2A and H2B might be the principal targets for O-GlcNAcylation, while others have shown that histone H3 would be the main substrate.14,15, 31,33 Upon further characterization, it was reported that histone H2B S112 O-GlcNAcylation promotes H2B monoubiquitination on lysine 120 (H2B K120ub), an event associated with transcriptional activation.33,34 On the other hand, histone H3 serine 10 was also reported to be O-GlcNAcylated, and this appears to compete with the phosphorylation of this site as well as modulate the transcriptional state of chromatin.14,15 Other O-GlcNAcylation sites were reported within the globular domains of histones suggesting that they may function in maintaining higher-order chromatin structure.31 Strikingly, during our investigation on the role of OGT in chromatin function, we were unable to reproduce the previous findings regarding histone O-GlcNAcylation in mammalian cells, whereas modification of other known OGT substrates was readily detected. Our results raise questions about the occurrence of histone O-GlcNAcylation and its proposed function in chromatin regulation.
Results and Discussion
Histone O-GlcNAcylation was undetectable using various extraction techniques
OGT interacts with the TET family of methylcytosine dioxygenase enzymes, notably TET2, which appear to be required for the chromatin association of OGT and this was suggested to promote histone O-GlcNAcylation.32,35 To further investigate the potential biological significance of histone O-GlcNAcylation, we initially sought to reproduce previously published results on the modification of histones H2B and H2A.14,30,32-34 We co-expressed Flag-H2B or Flag-H2A with either Myc-OGT or the D925A catalytic inactive mutant (Myc-OGT CD).36 Co-expression of Myc-TET2 with Myc-OGT was also included, since their association was expected to significantly increase OGT-mediated histone O-GlcNAcylation.32,37 We conducted an immunoprecipitation of Flag-H2B or Flag-H2A under denaturing conditions to determine their potential O-GlcNAcylation levels by using the widely employed anti-O-GlcNAc antibodies RL2 and CTD110.6 (Fig. 1 and Fig. S1). We did not detect a specific signal at the molecular weight region corresponding to histones Flag-H2B or Flag-H2A using the 2 anti-O-GlcNAc antibodies. However, using similar conditions, we were able to detect HCF-1 and TET2 O-GlcNAcylation, 2 known substrates of OGT (Fig. S2).24,26 Of note, as expected, OGT-mediated HCF-1 proteolytic cleavage also confirmed the activity of OGT in our co-transfection conditions (Fig. S2).26-28 Next, using the same transfection conditions indicated above, we performed several established extraction methods in order to enrich endogenous histones for O-GlcNAcylation detection. Chromatin and histones were isolated using both high salt/detergent (300 mM NaCl, 1% NP-40) extraction (Fig. 2A and Fig. S3A) and acid extraction (0.2 N HCl) (Fig. 2B and Fig. S3B) methods, respectively. First, immunoblotting with RL2 or CTD110.6 antibodies was conducted on the soluble fractions to detect global O-GlcNAcylation levels (Fig. 2 and Fig. S3, Left panels). As expected, we observed an increase of cellular O-GlcNAcylation levels following OGT overexpression. However, chromatin fractions revealed faint signals between 10 and 20 kDa when the blots probed with RL2 antibody were overexposed. The CTD110.6 antibody occasionally produced more pronounced signals at the levels of histones (Fig. 2 and Fig. S3, Right panels). Overall, we did not observe increasing signals following overexpression of OGT or TET2, regardless of the O-GlcNAc antibody used, the quantity of proteins loaded or the method of extraction performed (Fig. 2 and Fig. S3). On the other hand, upon OGT overexpression, we detected increased O-GlcNAcylation of certain chromatin-associated proteins corresponding to bands ≥37 kDa. Based on these results, we concluded that the faint and inconsistent signals produced between 10 and 20 kDa by the RL2 or CTD110.6 antibodies are not indicative of histone O-GlcNAcylation.
Figure 1.
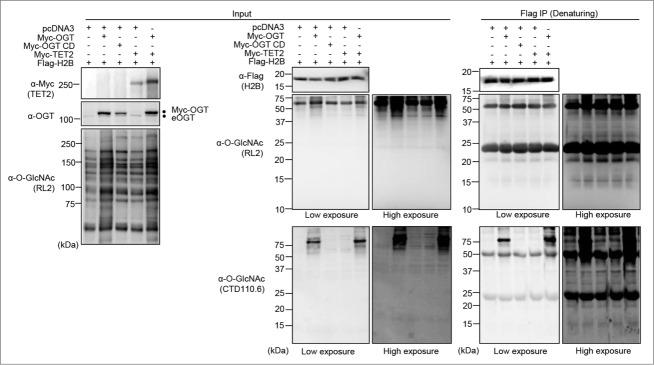
Undetectable Histone O-GlcNAcylation following OGT and TET2 overexpression. (A) HEK293T cells were transfected with Flag-H2B along with pcDNA3 empty vector, Myc-OGT or Myc-OGT catalytic dead (CD), as well as Myc-TET2 alone or in combination with Myc-OGT. Three days posttransfection, cells pellets were harvested and immunoprecipitation (Flag-IP) following protein denaturation was conducted to obtain purified Flag-H2B. The immunopurified histones were subjected to western blotting analysis using the indicated antibodies. Dots indicate Myc-OGT and endogenous OGT (eOGT).
Figure 2.
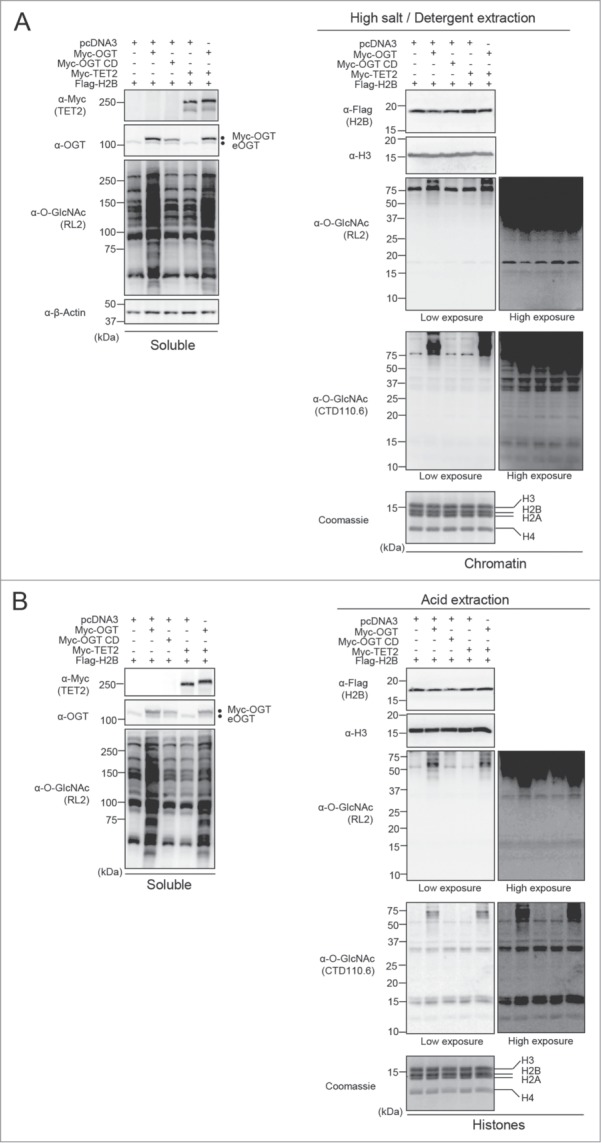
Undetectable histone O-GlcNAcylation following various extraction procedures. (A) HEK293T cells were transfected with Flag-H2B along with either pcDNA3 empty vector, Myc-OGT, Myc-OGT catalytic dead (CD) or Myc-TET2, as well as the combination of Myc-OGT with Myc-TET2. Three days posttransfection, cells pellets were collected for subsequent high salt/detergent extraction and cellular extracts were then analyzed by western blotting with the indicated antibodies. (Left panel) Soluble fraction showing global increase of O-GlcNAcylation following OGT overexpression. (Right panel) Immunodetection of histone O-GlcNAcylation by RL2 and CTD110.6 antibodies on chromatin fraction. β-Actin and histone H3 were used as loading controls. (B) HEK293T cells were transfected as in (A). Three days posttransfection, cells were harvested and histones were extracted. The samples were analyzed by western blotting with the indicated antibodies. (Left panel) Soluble fraction showing global O-GlcNAcylation levels. (Right panel) Histones fraction detected with both RL2 and CTD110.6 anti-O-GlcNAc antibodies. Coomassie brilliant blue staining indicates abundance of histones loaded. Histone H3 was use as a loading control. Dots indicate Myc-OGT and endogenous OGT (eOGT). kDa: Molecular weight marker in kilodaltons.
Modulation of O-GlcNAc levels does not result in the detection of specific histone O-GlcNAcylation
We reasoned that if the signals detected by RL2 or CTD110.6 around 10–20 kDa correspond to histone O-GlcNAcylation, then it might be possible to modulate these signals by depleting endogenous OGT. Thus, we conducted siRNA knockdown of OGT in U2OS cells and performed cellular fractionation to separate the soluble and histone-containing chromatin fractions. As shown in Figure 3A (Right panel), the signals detected with RL2 or CTD110.6 antibodies, around 10–20 kDa, did not decrease following OGT depletion suggesting that these signals are unspecific. In contrast, using both antibodies, we detected a significant decrease in global O-GlcNAcylation in the soluble fraction upon siRNA treatment (Fig. 3A, Left panel). Again, we noted that while the signal obtained with RL2 antibody in the 10–20 KDa region can be seen only upon overexposure of the membrane, the CTD110.6 antibody produced a much more readily detectable signal in this region. As RL2 and CTD110.6 are respectively IgG and IgM isotypes, we sought to determine if the signal detected at the level of histones with CTD110.6 could be due to the peroxidase-coupled secondary anti-IgM antibody. This is particularly relevant as high quantities of histones were probed with anti-O-GlcNAc antibodies. Thus, we incubated the blots with a non-relevant anti-rhodamine IgM antibody prior to incubation with the same anti-IgM HRP-conjugated antibody or with this secondary antibody alone (Fig. 3A, Right panel). Interestingly, both combinations displayed a similar signal pattern as that obtained using the CTD110.6, suggesting that the non-specific signal originates from the combination of secondary anti-IgM antibody and high protein density of histone bands. In addition, probing the membranes with a non-relevant anti-BAP1 antibody also resulted in background signals at molecular weights corresponding to histone bands (Fig. 3A, Right panel). These data indicate that the high abundance of histones present on membranes renders the detection of O-GlcNAcylation amenable to false-positive immunoblotting signals. Of note, previous studies questioned the specificity of CTD110.6 toward O-GlcNAc and revealed cross-reactivity with N-GlcNAc2-modified glycoprotein and GlcNAcylated O-mannose modified proteins.38,39 Consequently, we used the RL2 antibody to continue our investigation since it was not shown to cross react with other GlcNAc modifications. Nonetheless, our data suggested that the faint signal obtained with RL2 corresponds to a non-specific background caused by high amounts of histones. To further support our data, we extracted endogenous histones from HeLa cells for comparison with purified yeast H2B (yH2B) and recombinant human H2B (hH2B) produced in bacteria. We reasoned that if the faint signal produced by RL2 corresponds to histone O-GlcNAcylation, this signal should not be detected for histones purified from yeast or bacteria, which are not O-GlcNAcylated. We observed that the RL2 antibody produced low signals following overexposure of the blot, and these signals increased proportionally with the amount of histones loaded, irrespective of the species from which the histones were isolated (Fig. S4, panels A, B). Next, we conducted competition assays and found that, as expected, N-Acetylglucosamine (GlcNAc) inhibited RL2 binding to high molecular weight O-GlcNAcylated proteins present in the soluble fraction or associated with the chromatin fraction (Fig. S4C). However, GlcNAc also strongly reduced the signal produced by RL2, in the region of histone bands, for both mammalian chromatin and recombinant human H2B (Fig. S4C). These results are not surprising as a non-specific and low affinity binding of the RL2 antibody to histone fraction could also be potentially blocked by GlcNAc. Thus, these results further suggest that the RL2 antibody recognizes non-specifically antigenic determinants on histones through its paratope.
Figure 3.
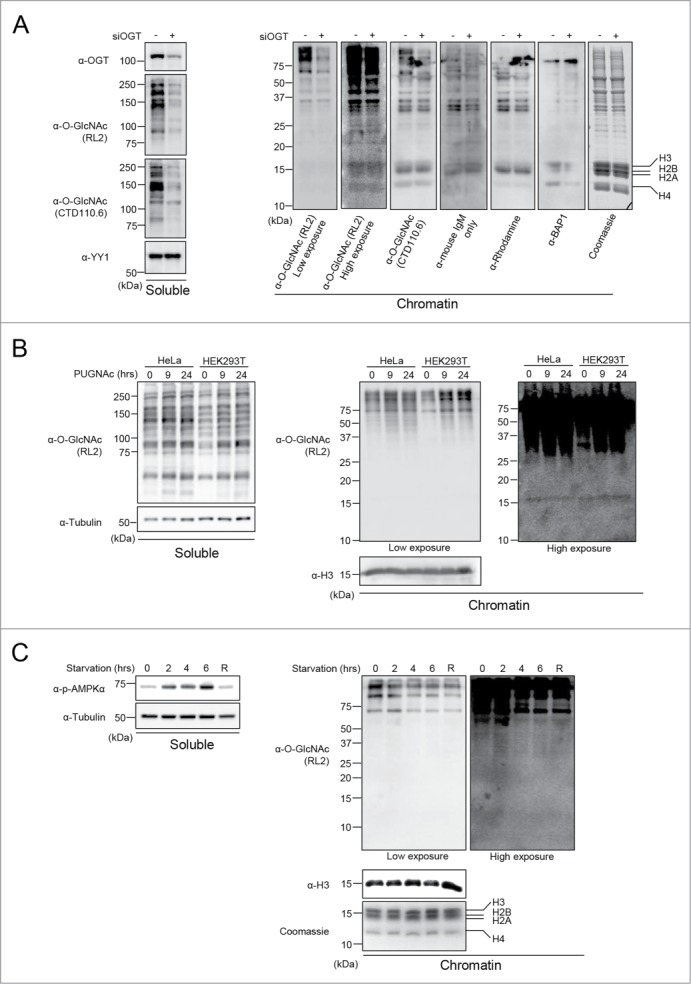
Modulation of O-GlcNAcylation does not result in detectable histone O-GlcNAcylation. (A) U2OS cells were transfected twice with OGT siRNA and three days posttransfection, cells were harvested. Chromatin fraction was isolated and protein levels were analyzed by western blotting with the indicated antibodies. (Left) U2OS soluble fraction showing OGT depletion and decrease of global O-GlcNAcylation levels. YY1 was used as a loading control. (Right) Chromatin fraction showing O-GlcNAcylation background detection of histones using various antibodies. (B) HeLa and HEK293T cells that were treated with 100 µM PUGNAc for 0, 9 and 24 hours. The soluble and chromatin fractions were blotted with RL2 antibody. Histone H3 and Tubulin were used as loading controls. (C) C2C12 mouse myoblast cell line were starved by incubation in HBSS buffer, and harvested at the indicated times for cellular fractionation. R: 4 hours of starvation followed by 2 hours of replenishment with complete culture medium. (Left) Western blot analysis of the soluble fraction showing increasing levels of phosphorylated AMPKα (α-p-AMPKα) as a control of the starvation treatment. Tubulin was used as a loading control. (Right) The chromatin fraction was analyzed by western blotting using the indicated antibodies. Histone H3 was used as a loading control and Coomassie brilliant blue staining indicates the abundance of histones loaded. kDa: Molecular weight marker in kilodaltons.
To further investigate potential histone O-GlcNAcylation, we sought to determine if inhibiting O-GlcNAcase (OGA), the enzyme responsible for O-GlcNAc removal, with O-(2-acetamido-2-deoxyglucopyranosylidene) amino N-phenylcarbamate (PUGNAc) would increase the signal of histone O-GlcNAcylation above background levels. Treatment of HeLa and HEK293T cells with PUGNAc promoted the accumulation of O-GlcNAcylated proteins but not of the background signal at the level of histones (Fig. 3B, Left and Right panels). We also conducted a nutrient starvation in C2C12 myoblasts, and analyzed global protein and potential histone O-GlcNAcylation. As expected, AMPK phosphorylation progressively increased and decreased with starvation and medium replenishment (R), respectively (Fig. 3C, Left panel).40 We observed that while chromatin-associated high molecular weight protein O-GlcNAcylation was reduced upon starvation, only weak and inconsistent background signals were detected for histones (Fig. 3C, Right panel).
Taken together, our results indicate that the immunoblot signal detected by RL2 in the region corresponding to histones is a non-specific O-GlcNAcylation-independent background signal.
Histone O-GlcNAcylation was undetectable in different cell lines and during cell cycle progression
Although our data did not reveal constitutive histone O-GlcNAcylation, we could not exclude that this posttranslational modification might occur on histones in specific cell types. Thus, we investigated potential histone O-GlcNAcylation in a variety of previously used cell lines including mouse Embryonic Fibroblast (MEF) as well as mouse Embryonic Stem Cells (mESCs).32,34,37 We also included the mouse pre-adipocyte 3T3-L1 cell line often used in differentiation studies as well as several multiple myeloma cell lines, RPMI-8226, JJN-3, and NCI-H292 cells, that we had in culture at the time of our investigation. We find that this cell line panel is representative of multiple tissue-origins, as well as primary and cancer cells. When immunoblotting the insoluble chromatin fraction for O-GlcNAcylation, we detected a very faint signal at the level of histones (Fig. 4A, Top panel). This signal, obtained only following extended exposure of the blot, does not correlate with the cell-specific levels of endogenous O-GlcNAcylation detected for high molecular weight proteins. Instead, by probing total H2A levels, it can be noticed that the RL2 signal follows the trend of histones abundance (Fig. 4A, Bottom blot).
Figure 4.
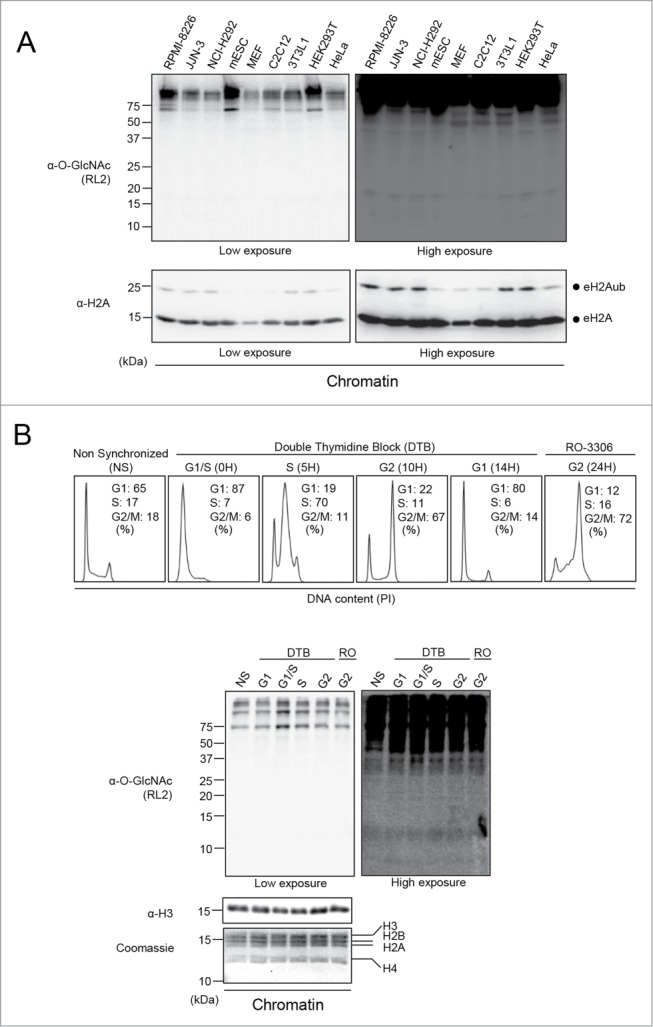
Unspecific signal detected at the level of histones in various cell lines and during cell cycle progression. (A) Various cell lines were cultured and harvested to perform chromatin extraction. The chromatin fraction was analyzed by western blotting with the indicated antibodies. Total histone H2A was used as a loading control. Dots indicate endogenous H2A ubiquitination (eH2Aub) and total H2A (eH2A). (B) Histones O-GlcNAcylation analysis of synchronized U2OS cells. Cells were blocked in G1/S boundary by double thymidine block (DTB) and then released to progress through the cell cycle. U2OS cells were also treated with the CDK1 inhibitor, RO-3306, for 24 hours to block cells in late G2. Cells were harvested at the indicated times for FACS analysis (Upper panel) and for chromatin extraction (Bottom panel). The chromatin fraction was analyzed by western blotting with the indicated antibodies and by Coomassie brilliant blue staining. Histone H3 was used as a loading control. NS: Non-Synchronized; DTB: Double Thymidine Block; kDa: Molecular weight marker in kilodaltons.
To further determine whether histone O-GlcNAcylation might be enriched in a specific phase of the cell cycle, U2OS cells were synchronized at the G1/S boundary with a double thymidine block and released to progress through the cell cycle.41 These cells were also treated with the CDK1 inhibitor (RO-3306) to enrich for late G2 cells (Fig. 4B, Top panel).42 The chromatin fraction was isolated from cells at different phases of the cell cycle and histone O-GlcNAcylation was monitored by western blotting using the RL2 antibody. We noticed the expected faint signal at the level of histones and that this signal did not change during cell cycle progression. However, high molecular weight chromatin-associated proteins show significantly increased O-GlcNAcylation at G1/S transition. Therefore, we concluded that fluctuating histone O-GlcNAcylation could not be observed during cell cycle. Moreover, along with our previous data, these results strengthen the notion that the signal detected by RL2 at the level of histones following blot overexposure corresponds to background.
Lack of evidence supporting H2B S112 O-GlcNAcylation and its link with H2B Lys120 monoubiquitination
It was reported that H2B S112 is O-GlcNAcylated (H2B S112 O-GlcNAc), an event that appears to promote the monoubiquitination of H2B Lys120 (H2B K120ub) thus coordinating gene expression.33 It was also described that AMPK-mediated OGT Thr444 phosphorylation hindered its ability to associate with chromatin, and this was shown to reduce the reported H2B S112 O-GlcNAc signal.9 In turn, this effect was proposed to inhibit monoubiquitination of H2B K120, thus repressing gene expression in MEF cells.34 As an anti-O-GlcNAcylated H2B S112 is commercially available,34 we first inquired about the specificity of this antibody. Since H2B S112 O-GlcNAc was shown to decrease dramatically following knockdown of OGT by RNAi, we used a similar approach to deplete OGT by siRNA in HeLa cells (Fig. 5A).33,34 As expected, OGT knockdown resulted in the accumulation of the precursor form of HCF-1, as its proteolytic maturation is O-GlcNAcylation-dependent (Fig. 5A, Left panel).26-28 However, neither the signal generated by the anti-H2B S112 O-GlcNAc nor that for anti-H2B K120ub changed following OGT depletion (Fig. 5A, Right panel). In addition, we expressed Flag-H2B WT and Flag-H2B S112A mutant in HEK293T cells and conducted anti-Flag immunoprecipitation under denaturing conditions (Fig. 5B). Unexpectedly, the anti-H2B S112 O-GlcNAc signal decreases by only ∼3-fold in the H2B S112A mutant, instead of completely disappearing. In addition, we observed no change in H2B K120ub levels. Our results could not validate the reported link between H2B S112 O-GlcNAc and H2B K120ub and, moreover, further suggest that the anti-H2B S112 O-GlcNAc antibody might not confer specific detection for H2B O-GlcNAcylation. Next, we sought to further investigate the anti-H2B S112 O-GlcNAc antibody specificity by producing recombinant human histones H2B (His-hH2B) and H2B S112A (His-hH2B S112A) purified from bacteria (Fig. 5C). We also used a GST-H2A construct as a negative control. Following relative quantification of recombinant histones shown by Coomassie brilliant blue staining (Fig. 5C, Top panel), we subsequently probed increasing quantities of these proteins by immunoblotting using the anti-H2B S112 O-GlcNAc antibody. We found that purified recombinant proteins are detectable using this antibody and that this signal decreased by about 2 to 3 folds when the serine was mutated to alanine (His-hH2B S112A) (Fig. 5C, Middle panel). Moreover, the H2B S112 O-GlcNAc signal detected for the recombinant His-hH2B protein is comparable to that detected for a similar amount of mammalian endogenous H2B in the chromatin fraction (Fig. 5C, Bottom panel). Thus, these results indicate that this antibody recognizes the H2B backbone itself rather than O-GlcNAc moiety and that unmodified S112 is a major determinant in epitope recognition by this antibody.
Figure 5.

(See previous page). H2B S112 O-GlcNAc antibody is not specific and S112 O-GlcNAcylation is not linked to H2B K120 monoubiquitination. (A) HeLa cells were transfected twice with OGT siRNA and three days posttransfection, cells were harvested to perform cellular fractionation. Protein levels were analyzed by western blotting with the indicated antibodies. HeLa soluble fraction was analyzed for HCF-1 proteolytic cleavage. YY1 was used as a loading control (Left panel). Chromatin fraction was analyzed for H2B S112 O-GlcNAc and H2B K120ub levels (Right panel). (B) HEK293T cells were transfected with Flag-H2B or Flag-H2B S112A. Three days posttransfection, cells were harvested and total cell lysates were subjected to protein denaturation and immunoprecipitation (IP) using α-Flag antibody. Input as well as IP fractions were subjected to immunoblotting analysis using the indicated antibodies. Arrows indicate Flag-H2B and endogenous H2B (eH2B). Tubulin was used as a loading control for the input fraction. (C) Relative quantification of eluted purified recombinant histones detected by Coomassie brilliant blue staining. Known amounts of BSA protein were used as standards for relative quantification (Top panel). Increasing amounts of recombinant His-hH2B, His-hH2B S112A mutant, GST-H2A as well as chromatin extract from HEK293T cells were analyzed by western blot using the indicated antibodies. Red Ponceau S staining of the membrane used for subsequent western blotting showing the loading of purified proteins (Bottom panel). Arrows and lines indicate the position of recombinant and endogenous histones respectively. kDa: Molecular weight marker in kilodalton.
Undetectable binding of mammalian histones to the wheat germ agglutinin (WGA) lectin
It was previously reported that HeLa cell nucleosomes obtained by micrococcal digestion can be enriched with a WGA lectin resin, which is known to strongly bind O-GlcNAcylated proteins.33 However, the extraction procedure does not preclude histone binding to the WGA column as a consequence of the interaction of histones with O-GlcNAcylated proteins associated with nucleosomes. Thus, we sought to use an acid extraction procedure to separate the soluble histones from all other proteins that remain in the insoluble fraction. Moreover, acid treatment ensures denaturation of proteins, thereby preventing potential interactions of histones with O-GlcNAcylated proteins associated with chromatin. We cultured HEK293T cells with PUGNAc for 24 hours and also included this inhibitor during the extraction steps to exclude the possibility of losing protein O-GlcNAc modification.14 Moreover, to ensure that acid treatment does not influence detection of O-GlcNAc modification, we also analyzed the ability of chromatin-associated-proteins in the acid extraction mixture to bind the WGA column (Fig. 6). Importantly, HCF-1 and several chromatin-associated proteins were enriched approximately 5 to 10 times compared to the input signal, thus confirming that the WGA resin strongly and efficiently binds O-GlcNAcylated proteins (Fig. 6, Left panel). This result also indicates that acid treatment did not covalently modify or disrupt the O-GlcNAc moiety. In contrast, immunoblotting with RL2, anti-H2B S112 O-GlcNAc, or anti-H2A revealed that histones were found only in the input and the flow through fractions (Fig. 6, Right panel). Consistently, staining of histones with Coomassie brilliant blue did not reveal histones binding to WGA-agarose. Note that the background signal produced by the RL2 antibody at the level of histones was observed in the input and the flow through fractions. To further exclude the possibility that only a small fraction of histones are modified in proliferating human cells, and that such limited amounts of modified histones are below immunoblotting detection thresholds, we carried out a WGA pull-down experiment in conditions that would allow for the total depletion of HCF-1 from the cell lysate (Fig. S5, Left panel). Even in these conditions, histones were again only detected in the inputs and the flow through fractions by immunodetection and silver staining (Fig. S5, Right panels). All together, these data support our findings that histone O-GlcNAcylation is undetectable in mammalian cells.
Figure 6.

HCF-1 and several chromatin-associated proteins but not histones bind to WGA lectin. HEK293T cells were treated with 100 µM PUGNAc or equal volume of DMSO for 24 hours as indicated and harvested to perform acid extraction of histones. The indicated soluble and chromatin-associated-proteins fractions (Left panels), and histone fraction (Right panels) were incubated for 2 hours with agarose bound WGA lectin or with agarose resin to control for non-specific binding. Two elutions (E1 and E2) of agarose and WGA bound proteins as well as the flow through (FT) fractions were analyzed by western blot using the indicated antibodies. The Coomassie Brilliant Blue staining and the anti-H2A antibody indicate that histones are found in the input and the FT fractions. Arrows indicate endogenous H2A ubiquitination (eH2Aub) and total H2A (eH2A). kDa: Molecular weight marker in kilodalton.
Histone O-GlcNAcylation is not detected by Click-iT biotin-alkyl chemistry, mass spectrometry, or following in vitro O-GlcNAcylation reactions
To further corroborate our observations, we sought to use other methods for O-GlcNAcylation detection. We first used the commercially available in vitro Click-iT chemistry system. This procedure consists in modifying O-GlcNAcylated proteins with an azido sugar (GalNAz), which can then react with biotin-alkyne thus becoming detectable via streptavidin-conjugated HRP.44 This approach is highly sensitive since it allows for the detection of the O-GlcNAcylation of α-crystallin, a lens protein that was reported to be O-GlcNAcylated at a very low stoichiometric ratio.44,45 Thus, we performed the Click-iT chemistry reaction on histones extracted from HeLa cells, as well as on α-crystallin, used as the positive control. As shown in Figure 7A, although similar amount of both purified histones and α-crystallin were used (Fig. 7A, Right panel), we could only detect O-GlcNAcylation of α-crystallin in these conditions (Fig. 7A, Left panel). Moreover, O-GlcNAcylation of co-purified high molecular weight proteins could be readily observed, thus demonstrating the efficiency of GalNAz labeling in the same reaction conditions as histones.
Figure 7.

(See previous page). Histones are not modified by Click-iT biotin-alkyl chemistry or by in vitro OGT-mediated O-GlcNAcylation. (A) The poorly O-GlcNAcylated α-crystallin is detected with the Click-iT biotin-alkyl chemistry but not histones. HeLa cells were harvested and acid extraction followed by acetone precipitation was performed to purify histones. The GalNAz labeling reaction was carried out for a minimum of 16 hours and the biotin-alkyl reaction was performed in the presence or absence of the GalT1 enzyme. (Left panel) O-GlcNAcylation was analyzed by blotting with streptavidin-HRP. (Right panel) Coomassie brilliant blue staining shows the indicated amounts of used purified proteins. (B–D) Histones are not O-GlcNAcylated in vitro by OGT. (B) Purified proteins analyzed by Coomassie brilliant blue or silver staining as indicated. (Left panel) Relative quantification of purified recombinant His-OGT and GST-HCF-1-N to known amounts of BSA. (Middle and Right panels) Integrity of Flag-H2A-purified mammalian nucleosomes and recombinant His-hH2B. (C) In vitro O-GlcNAcylation reaction of the purified recombinant His-hH2B. GST-HCF-1-N and recombinant His-hH2B were incubated with increasing amounts of recombinant His-OGT for 4 hours. O-GlcNAcylation was detected by western blotting using the anti-O-GlcNAc antibody (RL2). (D) In vitro O-GlcNAcylation reaction of the purified mammalian nucleosomes by OGT. GST-HCF-1-N and nucleosomes were incubated in the absence or presence of His-OGT for 11 hours. The O-GlcNAcylation was detected as in (C). kDa: Molecular weight marker in kilodalton.
Next, we sought to determine, by performing ETD-MS/MS analysis, if we could recapitulate the reported histone O-GlcNAcylation sites using acid extracted histones from HeLa cells. We also purified HCF-1 from HEK293T cells and included it as a positive control. Mass spectrometry analysis by HCD and ETD for proper glycan localization was conducted.46 As shown in Figure S6 and Table S1, in addition to several novel sites, we were able to identify 6 of the reported HCF-1 O-GlcNAcylation sites.26,27, 47-51 However, we were unable to detect O-GlcNAcylation of the core histones. Indeed, even though the MS analysis revealed the presence of histone peptides that were reported to be modified (e.g., H2B S112, H2A T101, H3 S10, and H4 S47), we did not detect their corresponding O-GlcNAcylated forms (Figs. S7 and S8).
Previous studies reported that histones could be modified by OGT in vitro.33 Thus, we performed in vitro O-GlcNAcylation reactions using purified recombinant human His-hH2B and purified mammalian nucleosomes. As a positive control, we also used purified recombinant GST-HCF-1N (for N-Terminal HCF-1),43 as this domain is a well-established substrate of OGT.26,27, 43 The relative quantity and integrity of the purified proteins were assessed by Coomassie brilliant blue staining or silver staining as indicated (Fig. 7B). As shown in Figures 7C and 7D, we detected a strong O-GlcNAcylation of HCF-1-N, but not of the recombinant His-hH2B (Fig. 7C) or nucleosomal histones (Fig. 7D).
In summary, using various approaches, we were unable to detect histone O-GlcNAcylation. Thus, our study provides strong evidence that histone O-GlcNAcylation, if occurring, must be present at levels below detection limits of commonly available tools, while O-GlcNAcylation of other known proteins including HCF-1 and TET2 can be observed. We emphasize that histones are hundred- to thousand-folds more abundant than the majority of cellular proteins and their modifications, even in relatively low abundance, are in general easily monitored. On the other hand, detection of histone modifications can be prone to false-positive signals, especially when analyzing a large amount of proteins by immunoblotting.
Material and Methods
Plasmids and mutagenesis
The cDNA of human OGT and TET2 were cloned from HeLa total RNA by reverse transcription and inserted into pENTR D-Topo plasmid (Life Technologies). Expression construct of Myc-TET2 was generated by recombination using LR clonase kit (Life Technologies) into pDEST-Myc construct. pCGN-HCF-1 FL was previously described.52,53 Myc-OGT, Myc-OGT D925A catalytic inactive mutant (Myc-OGT CD) were also previously described.26 GFP-OGT and GFP-OGT CD were generated by recombination into pDEST-GFP expression construct. pcDNA3-Flag-H2A and pcDNA3-Flag-H2B were obtained from Dr. Moshe Oren.54 H2B and H2A were generated using gene synthesis (BioBasic) and then subcloned into modified pENTR D-Topo plasmid. H2B S112A construct was generated by site direct mutagenesis using Q5 High-Fidelity DNA Polymerase. The Primers used are: Forward primer: CACGCCGTGGCGGAGGGCACCAAGGCCGTCA; Reverse primer: TGCCCTCCGCCACGGCGTGCTTGGCCAGCTC. HCF-1N was amplified from pCGN-HCF-1 FL and inserted into pENTR D-Topo plasmid. The T antigen NLS coding sequence was added in the primers. His-OGT was generated by subcloning the OGT cDNA into pET30a+ vector. Expression constructs of H2B and H2B S112A were generated by recombination using LR clonase kit into pDEST-Flag or pDEST-6xHis constructs. Expression constructs of H2B and HCF-1N were generated by recombination into pDEST-Flag or pDEST-GST constructs. All DNA constructs were sequenced.
Immunoblotting and antibodies
Total cell lysates were prepared by harvesting cells with buffer containing 25 mM Tris-HCl pH 7.3 and 1% sodium dodecyl sulfate (SDS). Cell extracts were boiled at 95°C for 10 min and sonicated. Quantification of total proteins was conducted using the bicinchoninic acid (BCA) assay.55 Total cell extracts as well as chromatin fractions and immunoprecipitation samples were diluted in 2X or 4X Laemmli buffer. SDS-PAGE and immunoblotting were conducted according to standard procedures. The band signals were acquired with a LAS-3000 LCD camera coupled to MultiGauge software (Fuji, Stamford, CT, USA).
Mouse monoclonal anti-BAP1 (C4, sc-28383), rabbit polyclonal anti-YY1 (H414, sc-1703), rabbit polyclonal anti-OGT (H300, sc-32921), mouse monoclonal anti-Tubulin (B-5–1–2, sc-SC-23948), were from Santa Cruz. Rabbit polyclonal anti-HCF-1 (A301–400A) was from Bethyl Laboratories. Mouse monoclonal anti-Flag (M2) was from Sigma-Aldrich. Mouse monoclonal anti-MYC (9E10) was from Covance. Rabbit polyclonal anti-H2B K120ub (D11 XP) was from Cell Signaling. Rabbit polyclonal anti-H2B S112 O-GlcNAc (ab130951), rabbit polyclonal anti-H2A (ab18255) Rabbit polyclonal anti-H3 (ab1791) and mouse monoclonal anti-O-Linked N-acetylglucosamine (RL2, ab2739) were from Abcam. The mouse monoclonal anti-O-Linked N-acetylglucosamine (CTD110.6) was kindly provided by Dr. Gerald Hart.56 Mouse monoclonal anti-β-Actin (MAB1501, clone C4) was from Millipore. Anti-rhodamine (IgM) was a generous gift from Dr. Li-Huei Tsai. Peroxidase Affini-Pure Goat anti-mouse IgM µ chain specific secondary antibody was from Jackson Laboratories.
Cell culture and cell transfection
U2OS osteosarcoma, HeLa, human embryonic kidney HEK293T, 3T3-L1 mouse preadipocytes, C2C12 mouse myoblasts, mouse embryonic fibroblasts (MEF) cell lines were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% of fetal bovine serum (FBS), L-glutamine and penicillin/streptomycin. Multiple myeloma cell lines (JJN-3, RPMI-8226, NCI-H292) were cultured in RPMI-1640 medium supplemented with 10% of fetal bovine serum (FBS), L-glutamine and penicillin/streptomycin. Mouse embryonic stem cells (mESCs) were maintained in DMEM medium supplemented with 15% of embryonic stem cells qualified FBS (Gibco), L-glutamine, penicillin/streptomycin, 0.1 mM β-mercaptoethanol, 0.1 mM MEM (Non-essential amino acids), 1 mM sodium pyruvate and 1,000 U/ml of leukemia inhibitory factor (LIF) (Life technologies).
HeLa or HEK293T cells were treated with 100 µM of PUGNAc or equal volume of DMSO for 0, 9 and 24 hours and were subjected to sub-cellular fractionation (soluble and chromatin) or histone extraction. HEK293T cells were transfected with mammalian expressing vectors using polyethylenimine (PEI) (Sigma-Aldrich). Three days posttransfection, cells were harvested for immunoblotting using total cell extracts or following cellular fractionation and histones extraction. Prior to immunoblotting, histones were also immunoprecipitated using anti-Flag antibody following denaturation of cell extracts. HeLa or U2OS cells were transfected using Lipofectamine 2000 (Life technologies) with 200 pmol of either ON-TARGET plus Non-targeting pool (D-001810–10–50) or ON-TARGET plus SMARTpool OGT (L-019111–00–0050) (Thermo Scientific, Dharmacon). Cells were transfected in a serum-free DMEM medium for 16 hours, then changed with DMEM complemented with 5% FBS, 1% Glutamine and 1% Penicillin-Streptomycin and 8 hours later, cells were transfected again as described above. Three days following the first transfection, cells were harvested in PBS and soluble and chromatin extractions were conducted and used for immunoblotting.
Histone and chromatin extraction
For histone extraction, HEK293T cells were transfected with Flag-H2A or Flag-H2B with or without Myc-OGT, Myc-OGT CD, Myc-TET2 or with the combination of Myc-OGT and Myc-TET2 and harvested three days posttransfection. For high salt/detergent chromatin extraction, cells were lysed in 300 mM NaCl, 1% NP-40, 2 µM PUGNAc, 1 mM PMSF and 1 X protease inhibitors (Sigma). Samples were kept on ice for 15 min and then centrifuged at 6,000 rpm for 10 min. The supernatant was kept as the soluble fraction for western blotting analysis. The pellet was washed 2 times with the previous buffer and resuspended in 1% SDS for protein quantification. For the histones acid extraction, cells were lysed in 50 mM Tris-HCl pH 7.4, 300 mM NaCl, 2 mM EDTA and 2 µM PUGNAc. The lysate was centrifuged at 6,000 rpm for 10 min and the supernatant was kept as the soluble fraction. The pellet was washed 2 times with the same buffer and treated with 0.2 N HCl (1 volume of buffer for 1 volume 0.4 HCl) for 1 hour on ice. After centrifugation at 14,000 rpm for 5 min, the supernatant containing histones was neutralized by adding equal volume of 100 mM Tris-HCl pH 8.8. The pellet was also resuspended in 1% SDS. To quantify proteins, part of the histones fraction was precipitated using 100% acetone at −20°C for 2 hours, centrifuged at 14,000 rpm for 10 min and resuspended in 1% SDS. Proteins were then quantified using the BCA assay. For chromatin extraction, the cell pellet was resuspended in 50 mM Tris-HCl pH 7.3, 300 mM NaCl, 5 mM EDTA, 1% Triton, 2 µM PUGNAc, 1X protease inhibitors (Sigma) and 1 mM PMSF and kept on ice for 30 min. The chromatin was pelleted by centrifugation at 10,000 rpm for 10 min at 4°C, the supernatant was kept (soluble fraction) and the chromatin pellet was resuspended in 25 mM Tris-HCl pH 7.3 and 1% SDS. Both fractions were quantified using the BCA protein quantification method.
Wheat germ agglutinin lectin pull-down
HEK293T cells were either treated with 100 µM PUGNAc or DMSO for 24 hours. Cells were then harvested in 1X PBS and the cell pellet was lysed with 0.25 M sucrose, 3 mM CaCl2, 1 mM Tris-HCl pH 8.0, 0.5% triton, 2 µM PUGNAc and 1X protease inhibitors (Sigma). The chromatin was pelleted by centrifugation at 3,900 rpm for 5 minutes at 4°C and the supernatant was kept (soluble fraction). Next, the pellet was washed with 300 mM NaCl, 5 mM MgCl2, 1% Triton, 50 mM Tris-HCl pH 8.0, 5 mM DTT and 2 µM PUGNAc and centrifuged at 3900 rpm for 5 min at 4°C. Supernatant was discarded and the pellet was very quickly resuspended in 3 volumes of acid extraction buffer containing 0.5 M HCl, 10% glycerol and 0.1 M β-mercaptoethanol and left on ice for 30 min. The sample was centrifuged at 14,000 rpm for 5 min at 4°C, the supernatant containing the histones was transferred in a new tube and 10 volumes of acetone were added to both the pellet (chromatin-associated proteins) and the supernatant (histones) fractions and were left at −20 °C overnight. The next day, protein precipitates were pelleted at 14,000 rpm for 1 hour at 4°C, resuspended in 25 mM Tris-HCl pH7.3 and 1 % SDS, sonicated and diluted in 10 volumes of EB300 Buffer containing 50 mM Tris-HCl pH7.5, 300 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1X protease inhibitors (Sigma), 1 mM PMSF, 1 mM DTT and 2 µM PUGNAc. The samples were incubated for 2 hours with WGA lectin resin (Vector Laboratories, #AL-1023) or agarose beads, washed with EB300 buffer and eluted with 500 mM N-acetylglucosamine. Sample were then analyzed by western blotting.
Immunoprecipitation
For histone immunoprecipitation following denaturation, HEK293T cells were transfected with Flag-H2A or Flag-H2B with pcDNA3 empty vector, Myc-OGT or Myc-OGt CD, Myc-TET2 or the combination of Myc-OGT with Myc-TET2 using Polyethylenimine (PEI). Three days posttransfection, cells were harvested and the cell pellets were lysed in 20 mM Tris-HCl pH 8.0, 600 mM NaCl, 0.5% NP-40, 0.5% SDS, 0.5% sodium deoxycholate, 1 mM EDTA, and 2 µM PUGNAc. Samples were sonicated and centrifuged at 14,000 rpm for 10 min. The lysate was then diluted in 5 volumes of 50 mM Tris-HCl pH 7.4, 2 mM EDTA, and 100 mM NaCl. For TET2 and HCF-1 immunoprecipitation, HEK293T cells were transfected with Myc-TET2 or HA-HCF-1 FL with and without GFP-OGT or GFP-OGT CD. Three days posttransfection, cells were harvested to perform denaturing immunoprecipitation. Briefly cells pellets were lysed using 300 mM NaCl containing buffer (5 mM Tris-HCl pH 7.5, 300 mM NaCl, 1% Triton, 1% SDS, 10 mM NaF, 5 mM EDTA 1 mM PMSF, 2 µM PUGNAc, and 1X protease inhibitors (Sigma)). After boiling for 3 min in the lysis buffer, cell lysate was sonicated and samples were diluted 10 folds with the same buffer but without SDS prior to immunoprecipitation using anti-Myc or anti-HA antibodies. For mutant histones, HEK293T cells were transfected with PEI with either FLAG-H2B or FLAG-H2B S112A. Three days posttransfection, cells were harvested for denaturing immunoprecipitation in 25 mM Tris-HCl pH 7.3 and 1.5% SDS. Next, suspensions were diluted 10 times with dilution buffer containing 50 mM Tris-HCl pH7.5, 100 mM NaCl, 1% Triton, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, 1X protease inhibitors (Sigma), 2 µM PUGNAc, and 20 mM N-Ethylmaleimide (NEM, Sigma). Suspensions were mixed with anti-FLAG M2 resin (Sigma Aldrich #A2220) overnight at 4 °C. The next day, beads were washed 5 times with the dilution buffer. Proteins were then eluted from the beads by adding Laemmli Buffer 2X and analyzed by western blotting.
Recombinant proteins
pDEST-6xHis-H2B, pDEST-6xHis-H2BS112A, pDEST-GST-HCF-1-N and pET30a+ OGT were transformed into RIL bacteria. Following induction with 400 µM IPTG, recombinant proteins were purified either under native or denaturing conditions. For the denaturing immunopurification, the bacterial pellets were lysed in 50 mM Tris-HCl pH 8, 8 M urea, and 3 mM DTT and left on ice for 30 min. After incubation, suspensions were sonicated and centrifuged at 16,000 rpm for 20 min. Supernatants were incubated with Ni-NTA Agarose resin (Invitrogen #R901–15) overnight at 4°C and the resin was then washed with 50 mM Tris-HCl pH 8.0, 500 mM NaCl, 3 mM DTT and 20 mM imidazole and transferred into a Bio-Spin Disposable Chromatography columns (Bio Rad #732–6008). Proteins were eluted with 200 mM imidazole. For the native purification of His-H2B, His-H2B S112A and His-OGT, cell pellets were lysed in 50 mM Tris-HCl pH 8, 500 mM NaCl and 3 mM DTT, 1 mM PMSF, 1X protease inhibitors (Sigma), and left on ice for 30 min. After incubation and sonication, the purified proteins were obtained as described above except for His-OGT, which was kept on the Ni-NTA Agarose resin in order to use it for the in vitro O-GlcNAcylation reaction. The OGT Ni-NTA Agarose resin beads were washed 6 times and kept in 50 mM Tris-HCl pH 7.5, 12.5 mM MgCl2, 3 mM DTT, 10% glycerol, 1 mM PMSF, 1X protease inhibitors (Sigma). The induction of GST-H2A expression in bacteria was done in the same manner as indicated above. Pellets of cells were lysed on ice in 50 mM Tris-HCl pH 8, 150 mM NaCl, 1 mM EDTA, 1% Triton, and 1 mM PMSF, 1 mM DTT, 1X protease inhibitors (Sigma). The lysates were sonicated, centrifuged at 16,000 rpm for 20 min and supernatants were incubated with GSH-Agarose (Sigma Aldrich #A8580) overnight at 4 °C. Then, the resin was washed with 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% Tween20, 1 mM PMSF, 200 µM DTT, and 1X protease inhibitors (Sigma). Proteins were eluted with the same buffer containing 250 mM reduced glutathione. GST-HCF-1-N was purified in the same manner as GST-H2A. Elutions of His-H2B, His-H2B S112A, GST-H2A and GST-HCF-1N were loaded on SDS-PAGE for Coomassie brilliant blue staining using BSA for relative quantification.
Purification of nucleosomes
For mammalian nucleosomes purification, HEK293T cells were transfected with 7 μg of pCDNA-Flag-H2A using PEI in serum free media. Three days posttransfection, cells were harvested and chromatin fraction extraction and nucleosomes were purified using anti-Flag beads as previously described.57 For yeast Flag-H2B nucleosomes purification, 2 liters of cells expressing Flag-H2B were grown under standard conditions and 4 g of cell pellet were used for the purification. Briefly, cells were resuspended in the SP Buffer (20 mM HEPES 7.4, 1.2 M sorbitol, 10 mM DTT) supplemented with 5 mg/ml of Zymolyase 20T. When the spheroplasting is completed, the cell pellet is washed again with the SP buffer and then resuspended in the Buffer L (20 mM HEPES 7.4, 18% Ficoll 400, 20 mM KCl, 5 mM MgCl2, 1 mM EDTA, protease inhibitors (1 μg/ml leupeptin, pepstatin, aprotinin), 3 mM DTT, 1 mM PMSF) prior to dounce homogenization. The extract was then diluted with the Buffer S (buffer L supplemented with 2.4 M sorbitol instead of Ficoll) and chromatin fraction was recovered by centrifugation at 11,000 rpm for 20 min. The chromatin pellet was washed with the IP buffer (20 mM HEPES 7.6, 150 mM KCl, 5% glycerol, 5 mM MgCl2, 1 mM CaCl2, 0.1% NP-40, protease inhibitors, 1 mM PMSF prior to the MNase treatment. After MNase treatment (15KU/ml for 30 min at room temperature), the reaction was stopped with 1 mM EDTA and 5 mM EGTA. Following centrifugation at 20,000g for 5 min at 4°C, the soluble chromatin fraction was incubated 1 hour at 4°C with anti-Flag M2 beads. The beads were washed 4 times with the IP2 buffer (same as IP buffer but with 1 mM EDTA, 1 mM EGTA and without CaCl2). Bound nucleosomes were then eluted with 200 μg/ml of Flag peptides (Biobasic Inc).
Click-iT chemistry
HeLa cells were harvested and acid extraction was performed to purify histones as described for the WGA pull-down. Following acetone precipitation the histones were resuspended in 1% SDS, 20 mM HEPES pH 7.9 and quantified using the BCA assay. The GalNAz labeling reaction was performed following the manufacturer's instructions using the Click-iT® GalNAz metabolic glycoprotein labeling reagent kit (Life Technologies). α-crystalline was included as a positive control. Following labeling of histones and α-crystalline, the detection was carried out with the Click-iT® Biotin Glycoprotein Detection Kit (Life Technologies) and the reaction was loaded on SDS-PAGE for blotting analysis using streptavidin-HRP (Cell Signaling #3999S) affinity detection.
In vitro O-GlcNAcylation reaction
Purified GST-HCF-1-N (0.2 µg) and recombinant His-H2B (0.4 µg) were incubated with purified His-OGT (0.2 to 0.5 µg) in the presence of UDP-GlcNAc (1 mM) at 37°C overnight for 4 or 11 hours. The reaction was carried out in 50 mM Tris-HCl pH 7.5 containing 12.5 mM MgCl2 and 3 mM DTT. Purified nucleosomes were also included in the O-GlcNAcylation reaction as described above. The reaction was stopped by adding Laemmli buffer and analyzed by immunoblotting.
Synchronization and cell cycle analysis
U2OS cells were synchronized at the G1/S border using the method of thymidine (2 mM) double block as previously described.41 Cells were then released into new media to follow the progression through S and G2/M phases. U2OS cells were also arrested in late G2 by treating them with 10 µM of the CDK1 inhibitor RO-3306 for 24 hours.42 Cell cycle analysis was carried out as described.41
Cell starvation
C2C12 cells were incubated for 6 hours in the Hanks Balanced Salt Solution (HBSS) medium completed with 10 mM HEPES pH 7.5 and penicillin/streptomycin. After 4 hours of treatment, a separate plate dish of cells was replenished with fresh media and released for another 2 hours. Cells pellets were harvested at the indicated times for chromatin extraction.
Mass spectrometry analysis
Reduction of histone and HCF-1 samples was performed by adding 5 mM DTT in 50 mM ammonium bicarbonate. Alkylation was performed with chloroacetamide 50 mM with ammonium bicarbonate 50 mM. The digestion with trypsin was performed for 8 h at 37°C. Samples were loaded and separated on a homemade reversed-phase column (150 µm i.d. × 150 mm) with a 106-min gradient from 0–40% acetonitrile (0.2% FA) and a 600 nl/min flow rate on an Easy nLC-1000 (Thermo Fisher Scientific) connected to an LTQ-Orbitrap Fusion (Thermo Fisher Scientific). Each full MS spectrum acquired with a 70,000 resolution was followed by 10 MS/MS spectra, where the 10 most abundant multiply charged ions were selected for MS/MS sequencing. Tandem MS experiments were performed using high-energy C-trap dissociation (HCD) and electron transfer dissociation (ETD) acquired in the Orbitrap. Peaks were identified using a Peaks 7.0 (Bioinformatics Solution Inc.) and peptide sequences were blasted against the human Uniprot database (74,530 sequences). Tolerance was set at 10 ppm for precursor and 0.01 Da for fragment ions during data processing and with carbamidomethylation (C), oxidation (M), deamidation (NQ), and Hex-N-acylation (ST) as variable modifications.
Funding
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) (MOP-115132) and the Natural Sciences and Engineering Research Council of Canada (NSERC) (355814–2010) to EBA and (435636–2013) to HW. EBA is a scholar of the Fonds de la Recherche du Québec - Santé (FRQ-S) and the CIHR. HW is a scholar of the FRQ-S, JG has a M.Sc. scholarship from the FRQ-S. The Proteomics facility at The Institute for Research in Immunology and Cancer (IRIC) receives infrastructure support from IRICoR, the Canadian Foundation for Innovation, and the Fonds de Recherche du Québec- Santé (FRQS).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank Haider Dar and Diana Adjaoud for technical assistance.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Lewis BA, Hanover JA. O-GlcNAc and the epigenetic regulation of gene expression. J Biol Chem 2014; 289(50):34440-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janetzko J, Walker S. The Making of a Sweet Modification: Structure and Function of O-GlcNAc Transferase. J Biol Chem 2014; 289(50):34424-32; PMID:25336649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hart GW, Housley MP, Slawson C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 2007; 446:1017-22; PMID:17460662; http://dx.doi.org/ 10.1038/nature05815 [DOI] [PubMed] [Google Scholar]
- 4.Hart GW. Three decades of research on o-glcnacylation - A major nutrient sensor that regulates signaling, transcription and cellular metabolism. Front Endocrinol 2014; 5:183; PMID:25386167; http://dx.doi.org/ 10.3389/fendo.2014.00183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruan HB, Singh JP, Li MD, Wu J, Yang X. Cracking the O-GlcNAc code in metabolism. Trends Endocrinol Metab 2013; 24:301-9; PMID:23647930; http://dx.doi.org/ 10.1016/j.tem.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagel AK, Ball LE. O-GlcNAc transferase and O-GlcNAcase: achieving target substrate specificity. Amino acids 2014; 46:2305-16; PMID:25173736; http://dx.doi.org/ 10.1007/s00726-014-1827-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vocadlo DJ. O-GlcNAc processing enzymes: catalytic mechanisms, substrate specificity, and enzyme regulation. Curr Opin Chem Biol 2012; 16:488-97; PMID:23146438; http://dx.doi.org/ 10.1016/j.cbpa.2012.10.021 [DOI] [PubMed] [Google Scholar]
- 8.Hanover JA, Krause MW, Love DC. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim Biophys Acta 2010; 1800:80-95; PMID:19647043; http://dx.doi.org/ 10.1016/j.bbagen.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bullen JW, Balsbaugh JL, Chanda D, Shabanowitz J, Hunt DF, Neumann D, Hart GW. Cross-talk between two essential nutrient-sensitive enzymes: O-GlcNAc transferase (OGT) and AMP-activated protein kinase (AMPK). J Biol Chem 2014; 289:10592-606; PMID:24563466; http://dx.doi.org/ 10.1074/jbc.M113.523068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanover JA, Krause MW, Love DC. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol 2012; 13:312-21; PMID:22522719; http://dx.doi.org/ 10.1038/nrm3334 [DOI] [PubMed] [Google Scholar]
- 11.Harwood KR, Hanover JA. Nutrient-driven O-GlcNAc cycling - think globally but act locally. J Cell Sci 2014; 127:1857-67; PMID:24762810; http://dx.doi.org/ 10.1242/jcs.113233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caldwell SA, Jackson SR, Shahriari KS, Lynch TP, Sethi G, Walker S, Vosseller K, Reginato MJ. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene 2010; 29:2831-42; PMID:20190804; http://dx.doi.org/ 10.1038/onc.2010.41 [DOI] [PubMed] [Google Scholar]
- 13.Fardini Y, Dehennaut V, Lefebvre T, Issad T. O-GlcNAcylation: A New Cancer Hallmark? Front Endocrinol 2013; 4:99; PMID:23964270; http://dx.doi.org/ 10.3389/fendo.2013.00099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang S, Roche K, Nasheuer HP, Lowndes NF. Modification of histones by sugar β-N-acetylglucosamine (GlcNAc) occurs on multiple residues, including histone H3 serine 10, and is cell cycle-regulated. J Biol Chem 2011; 286:37483-95; PMID:21896475; http://dx.doi.org/ 10.1074/jbc.M111.284885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fong JJ, Nguyen BL, Bridger R, Medrano EE, Wells L, Pan S, Sifers RN. β-N-Acetylglucosamine (O-GlcNAc) is a novel regulator of mitosis-specific phosphorylations on histone H3. J Biol Chem 2012; 287:12195-203; PMID:22371497; http://dx.doi.org/ 10.1074/jbc.M111.315804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lazarus BD, Love DC, Hanover JA. O-GlcNAc cycling: implications for neurodegenerative disorders. Int J Biochem Cell Biol 2009; 41:2134-46; PMID:19782947; http://dx.doi.org/ 10.1016/j.biocel.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jozwiak P, Forma E, Brys M, Krzeslak A. O-GlcNAcylation and Metabolic Reprograming in Cancer. Front Endocrinol 2014; 5:145; PMID:25250015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forma E, Jozwiak P, Brys M, Krzeslak A. The potential role of O-GlcNAc modification in cancer epigenetics. Cell Mol Biol Lett 2014; 19:438-60; PMID:25141978; http://dx.doi.org/ 10.2478/s11658-014-0204-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Issad T, Pagesy P. [Protein O-GlcNAcylation and regulation of cell signalling: involvement in pathophysiology]. Biol Aujourdhui 2014; 208:109-17; PMID:25190571; http://dx.doi.org/ 10.1051/jbio/2014015 [DOI] [PubMed] [Google Scholar]
- 20.Slawson C, Copeland RJ, Hart GW. O-GlcNAc signaling: a metabolic link between diabetes and cancer? Trends Biochem Sci 2010; 35:547-55; PMID:20466550; http://dx.doi.org/ 10.1016/j.tibs.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dehennaut V, Leprince D, Lefebvre T. O-GlcNAcylation, an Epigenetic Mark. Focus on the Histone Code, TET Family Proteins, and Polycomb Group Proteins. Front Endocrinol 2014; 5:155; PMID:25309514; http://dx.doi.org/ 10.3389/fendo.2014.00155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng RP, He X, Guo SJ, Liu WF, Tao Y, Tao SC. Global identification of O-GlcNAc transferase (OGT) interactors by a human proteome microarray and the construction of an OGT interactome. Proteomics 2014; 14:1020-30; PMID:24536041; http://dx.doi.org/ 10.1002/pmic.201300144 [DOI] [PubMed] [Google Scholar]
- 23.Ozcan S, Andrali SS, Cantrell JE. Modulation of transcription factor function by O-GlcNAc modification. Biochim Biophys Acta 2010; 1799:353-64; PMID:20202486; http://dx.doi.org/ 10.1016/j.bbagrm.2010.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Q, Liu X, Gao W, Li P, Hou J, Li J, Wong J. Differential regulation of the ten-eleven translocation (TET) family of dioxygenases by O-linked β-N-acetylglucosamine transferase (OGT). J Biol Chem 2014; 289:5986-96; PMID:24394411; http://dx.doi.org/ 10.1074/jbc.M113.524140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang X, Zhang F, Kudlow JE. Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell 2002; 110:69-80; PMID:12150998; http://dx.doi.org/ 10.1016/S0092-8674(02)00810-3 [DOI] [PubMed] [Google Scholar]
- 26.Daou S, Mashtalir N, Hammond-Martel I, Pak H, Yu H, Sui G, Vogel JL, Kristie TM, Affar el B. Crosstalk between O-GlcNAcylation and proteolytic cleavage regulates the host cell factor-1 maturation pathway. Proc Natl Acad Sci U S A 2011; 108:2747-52; PMID:21285374; http://dx.doi.org/ 10.1073/pnas.1013822108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Capotosti F, Guernier S, Lammers F, Waridel P, Cai Y, Jin J, Conaway JW, Conaway RC, Herr W. O-GlcNAc transferase catalyzes site-specific proteolysis of HCF-1. Cell 2011; 144:376-88; PMID:21295698; http://dx.doi.org/ 10.1016/j.cell.2010.12.030 [DOI] [PubMed] [Google Scholar]
- 28.Lazarus MB, Jiang J, Kapuria V, Bhuiyan T, Janetzko J, Zandberg WF, Vocadlo DJ, Herr W, Walker S. HCF-1 is cleaved in the active site of O-GlcNAc transferase. Science 2013; 342:1235-9; http://dx.doi.org/ 10.1126/science.1243990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanover JA. Epigenetics gets sweeter: O-GlcNAc joins the “histone code.” Chem Biol 2010; 17:1272-4; PMID:21168762; http://dx.doi.org/ 10.1016/j.chembiol.2010.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakabe K, Wang Z, Hart GW. Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci U S A 2010; 107:19915-20; PMID:21045127; http://dx.doi.org/ 10.1073/pnas.1009023107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnaudo AM, Garcia BA. Proteomic characterization of novel histone post-translational modifications. Epigenetics Chromatin 2013; 6:24; PMID:23916056; http://dx.doi.org/ 10.1186/1756-8935-6-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 2013; 493:561-4; PMID:23222540; http://dx.doi.org/ 10.1038/nature11742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S, Imai Y, Kim J, He HH, Igarashi K, et al.. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature 2011; 480:557-60; PMID:22121020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Q, Yang C, Du Y, Chen Y, Liu H, Deng M, Zhang H, Zhang L, Liu T, Liu Q, et al.. AMPK regulates histone H2B O-GlcNAcylation. Nucleic Acids Res 2014; 42:5594-604; PMID:24692660; http://dx.doi.org/ 10.1093/nar/gku236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ito R, Katsura S, Shimada H, Tsuchiya H, Hada M, Okumura T, Sugawara A, Yokoyama A. TET3-OGT interaction increases the stability and the presence of OGT in chromatin. Genes Cells 2014; 19:52-65; http://dx.doi.org/ 10.1111/gtc.12107 [DOI] [PubMed] [Google Scholar]
- 36.Clarke AJ, Hurtado-Guerrero R, Pathak S, Schuttelkopf AW, Borodkin V, Shepherd SM, Ibrahim AF, van Aalten DM. Structural insights into mechanism and specificity of O-GlcNAc transferase. EMBO J 2008; 27:2780-8; PMID:18818698; http://dx.doi.org/ 10.1038/emboj.2008.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vella P, Scelfo A, Jammula S, Chiacchiera F, Williams K, Cuomo A, Roberto A, Christensen J, Bonaldi T, Helin K, et al.. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol Cell 2013; 49:645-56; PMID:23352454; http://dx.doi.org/ 10.1016/j.molcel.2012.12.019 [DOI] [PubMed] [Google Scholar]
- 38.Isono T. O-GlcNAc-specific antibody CTD110.6 cross-reacts with N-GlcNAc2-modified proteins induced under glucose deprivation. PloS One 2011; 6:e18959; PMID:21526146; http://dx.doi.org/ 10.1371/journal.pone.0018959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ogawa M, Nakamura N, Nakayama Y, Kurosaka A, Manya H, Kanagawa M, Endo T, Furukawa K, Okajima T. GTDC2 modifies O-mannosylated α-dystroglycan in the endoplasmic reticulum to generate N-acetyl glucosamine epitopes reactive with CTD110.6 antibody. Biochem Biophys Res Commun 2013; 440:88-93; PMID:24041696; http://dx.doi.org/ 10.1016/j.bbrc.2013.09.022 [DOI] [PubMed] [Google Scholar]
- 40.Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metabol 2009; 9:407-16; PMID:19416711; http://dx.doi.org/ 10.1016/j.cmet.2009.03.012 [DOI] [PubMed] [Google Scholar]
- 41.Hammond-Martel I, Pak H, Yu H, Rouget R, Horwitz AA, Parvin JD, Drobetsky EA, Affar el B. PI 3 kinase related kinases-independent proteolysis of BRCA1 regulates Rad51 recruitment during genotoxic stress in human cells. PloS One 2010; 5:e14027; PMID:21103343; http://dx.doi.org/ 10.1371/journal.pone.0014027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci U S A 2006; 103:10660-5; PMID:16818887; http://dx.doi.org/ 10.1073/pnas.0600447103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W. Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev 2003; 17:896-911; PMID:12670868; http://dx.doi.org/ 10.1101/gad.252103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hart C, Chase LG, Hajivandi M, Agnew B. Metabolic labeling and click chemistry detection of glycoprotein markers of mesenchymal stem cell differentiation. Methods Mol Biol 2011; 698:459-84. [DOI] [PubMed] [Google Scholar]
- 45.Haynes PA, Aebersold R. Simultaneous detection and identification of O-GlcNAc-modified glycoproteins using liquid chromatography-tandem mass spectrometry. Anal Chem 2000; 72:5402-10; PMID:11080893; http://dx.doi.org/ 10.1021/ac000512w [DOI] [PubMed] [Google Scholar]
- 46.Vester-Christensen MB, Halim A, Joshi HJ, Steentoft C, Bennett EP, Levery SB, Vakhrushev SY, Clausen H. Mining the O-mannose glycoproteome reveals cadherins as major O-mannosylated glycoproteins. Proc Natl Acad Sci U S A 2013; 110:21018-23; PMID:24101494; http://dx.doi.org/ 10.1073/pnas.1313446110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alfaro JF, Gong CX, Monroe ME, Aldrich JT, Clauss TR, Purvine SO, Wang Z, Camp DG 2nd, Shabanowitz J, Stanley P, et al.. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc Natl Acad Sci U S A 2012; 109:7280-5; PMID:22517741; http://dx.doi.org/ 10.1073/pnas.1200425109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Myers SA, Panning B, Burlingame AL. Polycomb repressive complex 2 is necessary for the normal site-specific O-GlcNAc distribution in mouse embryonic stem cells. Proc Natl Acad Sci U S A 2011; 108:9490-5; PMID:21606357; http://dx.doi.org/ 10.1073/pnas.1019289108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vosseller K, Trinidad JC, Chalkley RJ, Specht CG, Thalhammer A, Lynn AJ, Snedecor JO, Guan S, Medzihradszky KF, Maltby DA, et al.. O-linked N-acetylglucosamine proteomics of postsynaptic density preparations using lectin weak affinity chromatography and mass spectrometry. Mol Cell Proteomics 2006; 5:923-34; http://dx.doi.org/ 10.1074/mcp.T500040-MCP200 [DOI] [PubMed] [Google Scholar]
- 50.Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, Schoepfer R, Burlingame AL. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol Cell Proteomics 2012; 11:215-29; http://dx.doi.org/ 10.1074/mcp.O112.018366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Myers SA, Daou S, Affar el B, Burlingame A. Electron transfer dissociation (ETD): the mass spectrometric breakthrough essential for O-GlcNAc protein site assignments-a study of the O-GlcNAcylated protein host cell factor C1. Proteomics 2013; 13:982-91; PMID:23335398; http://dx.doi.org/ 10.1002/pmic.201200332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson AC, LaMarco K, Peterson MG, Herr W. The VP16 accessory protein HCF is a family of polypeptides processed from a large precursor protein. Cell 1993; 74:115-25; PMID:8392914; http://dx.doi.org/ 10.1016/0092-8674(93)90299-6 [DOI] [PubMed] [Google Scholar]
- 53.Wilson AC, Boutros M, Johnson KM, Herr W. HCF-1 amino- and carboxy-terminal subunit association through two separate sets of interaction modules: involvement of fibronectin type 3 repeats. Mol Cell Biol 2000; 20:6721-30; PMID:10958670; http://dx.doi.org/ 10.1128/MCB.20.18.6721-6730.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Minsky N, Oren M. The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Mol Cell 2004; 16:631-9; PMID:15546622; http://dx.doi.org/ 10.1016/j.molcel.2004.10.016 [DOI] [PubMed] [Google Scholar]
- 55.Stoscheck CM. Quantitation of protein. Methods Enzymol 1990; 182:50-68; PMID:2314256 [DOI] [PubMed] [Google Scholar]
- 56.Comer FI, Vosseller K, Wells L, Accavitti MA, Hart GW. Characterization of a mouse monoclonal antibody specific for O-linked N-acetylglucosamine. Anal Biochem 2001; 293:169-77; PMID:11399029; http://dx.doi.org/ 10.1006/abio.2001.5132 [DOI] [PubMed] [Google Scholar]
- 57.Yu H, Pak H, Hammond-Martel I, Ghram M, Rodrigue A, Daou S, Barbour H, Corbeil L, Hebert J, Drobetsky E, et al.. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci U S A 2014; 111:285-90; PMID:24347639; http://dx.doi.org/ 10.1073/pnas.1309085110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.