

Abstract
The CpG Island Methylator Phenotype (CIMP) is fundamental to an important subset of colorectal cancer; however, its cause is unknown. CIMP is associated with microsatellite instability but is also found in BRAF mutant microsatellite stable cancers that are associated with poor prognosis. The isocitrate dehydrogenase 1 (IDH1) gene causes CIMP in glioma due to an activating mutation that produces the 2-hydroxyglutarate oncometabolite. We therefore examined IDH1 alteration as a potential cause of CIMP in colorectal cancer. The IDH1 mutational hotspot was screened in 86 CIMP-positive and 80 CIMP-negative cancers. The entire coding sequence was examined in 81 CIMP-positive colorectal cancers. Forty-seven cancers varying by CIMP-status and IDH1 mutation status were examined using Illumina 450K DNA methylation microarrays. The R132C IDH1 mutation was detected in 4/166 cancers. All IDH1 mutations were in CIMP cancers that were BRAF mutant and microsatellite stable (4/45, 8.9%). Unsupervised hierarchical cluster analysis identified an IDH1 mutation-like methylation signature in approximately half of the CIMP-positive cancers. IDH1 mutation appears to cause CIMP in a small proportion of BRAF mutant, microsatellite stable colorectal cancers. This study provides a precedent that a single gene mutation may cause CIMP in colorectal cancer, and that this will be associated with a specific epigenetic signature and clinicopathological features.
Keywords: BRAF, CIMP, colorectal cancer, IDH1, microsatellite
Abbreviations
- CIMP
CpG Island Methylator Phenotype
- MSI
microsatellite instability
- MSS
microsatellite stable
- IDH1
isocitrate dehydrogenase 1
Introduction
The CpG Island Methylator Phenotype (CIMP) describes the coordinate hypermethylation of CpG dinucleotides, often clustered in the promoters of genes silenced during colorectal tumorigenesis.1,2 CIMP is detected in approximately 20-30% of colorectal cancers and is highly associated with mutation of the BRAF oncogene.3 It is inversely associated with the global hypomethylation generally observed in the cancer genome.4 Approximately half of all CIMP cancers will hypermethylate the mismatch repair gene MLH1 and develop microsatellite instability (MSI), while the remainder will be microsatellite stable (MSS). This molecular phenotype is important because it characterizes a clinically distinct group of colorectal cancer and precursor serrated polyps.5,6 Importantly, while MSI confers an excellent prognosis, BRAF mutant cancers that are MSS have a particularly poor prognosis.7
Understanding of CIMP has been somewhat hampered by the lack of a consensus method for identifying the phenotype.8 Several marker panels have been used which results in different frequencies of CIMP.1,3,9-14 Genome wide studies may offer a more objective classification.15 A commonly used marker panel proposed by Weisenberger and colleagues in 2006 consistently and specifically identifies cancers with a high frequency BRAF mutation.3 CIMP classification may inform choice of therapy.16,17 Epigenetic modification is reversible and DNA methyltransferase inhibitors may be efficacious in CIMP-positive cancers. Recently, stage III CIMP-positive colorectal cancers have been shown to have a worse prognosis than CIMP-negative colorectal cancers, but be more sensitive to irinotecan-based chemotherapy.18
The cause of CIMP in colorectal cancer has been unknown. Association studies have revealed the possible influence of genetic and environmental factors on CIMP. For example, it has been suggested that variants in the methylenetetrahydrofolate reductase gene, in relation to low folate and high alcohol intake, may increase the risk of CIMP.19 Lifestyle factors such as level or alcohol and dietary folate intake, early life energy restriction and physical activity have also been associated with the phenotype.20-24 History of smoking has consistently been associated with CIMP in colorectal cancers.25-27
Somatic mutation of the IDH1 gene causes CIMP in the majority of grade II and II gliomas, which include astrocytomas and oligodendrogliomas, as well as a substantial proportion of secondary glioblastomas thought to arise from these tumors.28-30 This gene encodes cytosolic isocitrate dehydrogenase, which catalysis the conversion of isocitrate to α-ketoglutarate. The R132H mutation in the catalytic domain of the protein reduces the ability of IDH1 to decarboxylate isocitrate and results in a gain of enzymatic activity, causing conversion of α–ketoglutarate to 2-hydroxyglutarate (2HG).31,32 This 2HG ‘oncometabolite’ inhibits histone demethylation, resulting in the accumulation of histone H3K9 marks and subsequent increase in DNA methylation consistent with the glioma methylator phenotype.29,33,34 In addition to frequent mutation in brain cancers,28,30,35-37 approximately 10% of acute myeloid leukemias have an IDH1 mutation, which also produces 2HG and segregates with a distinct epigenetic signature.34,38-40 Mutation of the arginine at position 172 of the closely related IDH2 gene is thought to cause CIMP via a similar mechanism.33
A single R132C IDH1 mutation has been reported in colorectal cancers, from a series of 11 tumors.41 However, in 2 other series of 128 and 97 colorectal cancers, no mutations were identified.42,43 We hypothesized that IDH1 mutations would occur at increased frequency in the specific subset of colorectal cancers showing CIMP and that mutations may occur in the related gene IDH2.
Results
IDH1 mutation
The IDH1 R132C mutation was detected in 4 of the 166 colorectal cancers sequenced (Fig. 1). Clinicopathological data was available for 3 of the 4 R132C mutant samples. One tumor was located in the distal colon (TNM stage T3N2M0, splenic flexure) of a 79 year old male. The other 2 tumors arose in the proximal colon at the hepatic flexure (T3N0M0, 74.9 year old female) or ascending colon (T3N0M0, 82.4 year old female). The 4 cases with the R132C somatic mutation were all CIMP-positive, had an activating BRAF mutation and were microsatellite stable. Of the 45 CIMP-positive, microsatellite stable cancers, the R132C mutation rate was 8.9% (4/45). A serine to proline mutation was also observed at position 326 in a microsatellite stable, CIMP-positive cancer. Although the functional significance of this mutation is unclear, it is unlikely to contribute to the same gain of function as the R132 mutation. Two frameshift mutations (one insertion, one deletion) were identified in the A7 repeat tract occurring at the third amino acid of the predicted protein sequence. These were both observed in CIMP-positive, microsatellite unstable cancers, likely as a consequence of mismatch repair deficiency in a cancer that had already established CIMP. These frameshift mutations early in the coding sequence of the gene would inactivate that allele, which would reduce normal IDH1 function, rather than produce a gain of function as is seen with the R132 mutation.
Figure 1.
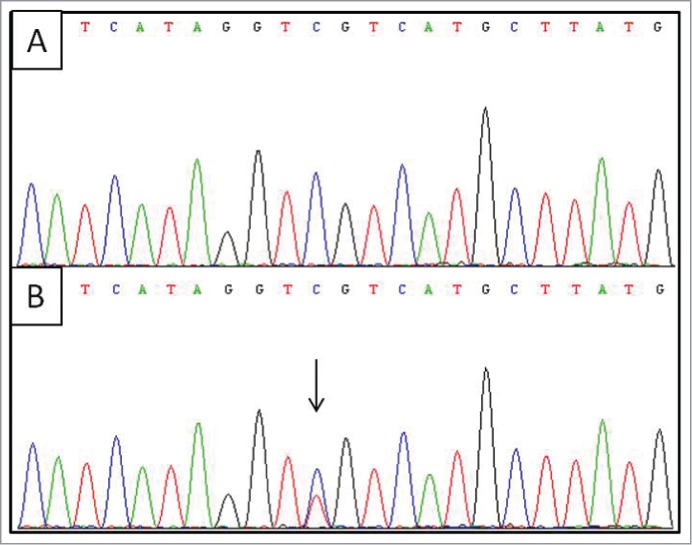
Sequence analysis for the IDH1 R132C somatic mutation. Upper panel (A) shows the wild-type sequence while the lower panel (B) shows the mutant sequence, indicated by an arrow.
IDH2 analysis
The previously reported R140Q and R172K mutations in IDH2 were not detected in any of 86 CIMP-positive or 80 CIMP-negative cancers assessed. Somatic frameshift mutation of the G7 repeat tract at nucleotide position 1248 was observed in 3 cancers with microsatellite instability. Again, this is likely the result of microsatellite instability in a cancer with established CIMP.
Methylation microarray analysis
Using a subset of the 3000 most variable probes, unsupervised hierarchical cluster analysis highlighted 4 distinct subgroups based on methylation profile. The CIMP-positive, BRAF mutant cancers segregated into 2 distinct subgroups, with all R132C mutants falling within CIMP subgroup 1 (Fig. 2). The clinical and molecular features of the 2 CIMP subclusters in comparison to the CIMP-negative cancers (RPMM clusters 3 and 4) is shown in Table 1.
Figure 2.

DNA methylation microarray analysis. Unsupervised hierarchical cluster analysis of CIMP-negative and CIMP-positive cancers, including 4 with the R132C IDH1 mutation (A). The recursively partitioned mixture model (RPMM) clusters are represented in red (CIMP-positive cluster 1), blue (CIMP-positive cluster 2), and CIMP negative clusters shown in yellow and green. Cancers with IDH1 mutation, CIMP or BRAF mutation are indicated by pink boxes below the heatmap. The overlap between the genes specifically methylated in defined subgroups of CIMP-positive cancers and the glioma methylator phenotype are shown in (B).
Table 1.
Molecular and Clinical Features of DNA Methylation Array Cancer Subgroups
| CIMP+ RPMM Group 1 | CIMP+ RPMM Group 2 | CIMP−RPMM Group 3/4 | |
|---|---|---|---|
| Total | 17 | 12 | 18 |
| IDH1 R132C Mutation | 4/17 (24%) | 0 | 0 |
| BRAF Mutation | 17/17 | 17/17 | 0 |
| CIMP | 16/17 (94%) | 17/17 | 0 |
| Age (range, average years) | 51.2 − 93.0 (74.1) | 50.9 − 87.6 (69.0) | 30.5 − 93.4 (70.2) |
| Sex | |||
| Male | 6/17 (35%) | 4/12 (33%) | 8/18 (44%) |
| Female | 11/17 (65%) | 8/12 (67%) | 10/18 (56%) |
| Site | |||
| Cecum | 5/13 (38%) | 5/11 (45%) | 3/14 (21%) |
| Proximal Colon | 5/13 (38%) | 3/11 (27%) | 2/14 (14%) |
| Distal Colon | 3/13 (23%) | 3/11 (27%) | 5/14 (36%) |
| Rectum | 0 | 0 | 4/14 (29%) |
Samples within RPMM clusters were ordered in terms of similarity using the function “seriate,” in the seriation package. Within Cluster 1, the R132C mutants were positioned together, suggesting the profiles of the 4 R132C mutants were more closely related to each other than other CIMP-positive cancers without the R132C mutation (Fig. 2). In order to assess the stability of the clusters a variable number of probes (n = 1000, 2000, 4000, 5000) were clustered and compared to the original clustering pattern based on 3000 probes. The clusters were largely independent of the number of probes used with only one sample (CIMP-) changing cluster. In addition, to ensure that no one sample influenced the clustering, a leave-one-out resampling method was implemented. Once again, this indicated that the clusters were not sensitive to changes in the complete data set.
We next sought to assess the overlap between the ‘IDH1 mutant-like methylation signature’ in colorectal cancer (Cluster 1, Fig. 2) and the glioma methylator phenotype (G-CIMP+).29 Of the 50 G-CIMP+ tumors in this study, 46 had an IDH1 mutation (45 R132H, 1 R132L) and of the remaining 4 IDH1 wild type samples, 3 had an IDH2 mutation (172K). Turcan et al. identified 16455 unique genes differentially methylated in G-CIMP+ compared to G-CIMP- gliomas. This includes all genes hypo- and hypermethylated with probes associated with promoter regions, CpG islands or CpG island flanking regions (shores and shelves). We followed the same algorithm to identify hypermethylated gene promoter CpG islands in Cluster 1 cancers, the 4 R132C mutants, Cluster 1 without the 4 R132C mutants and Cluster 2 cancers, compared to CIMP-negative colorectal cancers (Fig. 2). Unique genes were then compared to unique G-CIMP+ genes to provide the percentage overlap between the G-CIMP+ phenotype and the IDH1 mutation-like methylation signature observed here. A high level of overlap was observed for Cluster 1 and G-CIMP+ target genes (13573/16445, 82.5%). When the 4 IDH1 mutants were removed from the analysis, 10355/16445 (63.0%) of genes remained in common with G-CIMP+, compared to only 3164/16445 (19.2%) of Cluster 2 genes, supporting the concept that even in the absence of the R132C mutation, the remaining Cluster 1 cancers arose via a similar mechanism.
Discussion
This study provides evidence that a single mutation can cause the development of CIMP in colorectal cancer. Direct mutation of the catalytic domain of IDH1 is uncommon, but may explain the phenotype in a proportion of CIMP-positive microsatellite stable colorectal cancers, and provides a precedent that a single mutation may underlie the development of CIMP in colorectal cancer.
The data presented here strongly support the hypothesis that the R132C IDH1 somatic mutation causes CIMP in a proportion of colorectal cancers. Functional data from glioma studies suggest the mechanism of this is through oncogenic gain of function to produce the 2-hydroxyglutarate oncometabolite, which inhibits histone demethylation to increase DNA methylation.29,33 Unsupervised hierarchical cluster analysis of colorectal data showed a similar global DNA hypermethylation pattern for all IDH1 R132C mutants, where the profile for each R132C mutant was more similar to other R132C mutants than any other cancers. This is consistent with the mechanism of CIMP development being due to the same cause in these 4 cancers. Furthermore, the 4 mutants were positioned within a subcluster of CIMP-positive cancers with what we term and IDH1 mutation-like methylation signature’, which clearly segregated from a second CIMP-positive subcluster.
The Cancer Genome Atlas public data set examined the exomes and methylation profiles of 224 colorectal cancers.44 A single IDH1 R132C mutation was identified and, consistent with the present study, this occurred in a microsatellite stable tumor. On methylation array analysis the sample segregated with other CIMP cancers (CIMP-low). Interestingly, the mucinous cancer arose at the hepatic flexure of an 81-year-old female, which is consistent with the clinicopathological profile expected of CIMP cancers.45,46 Two of the 3 tumors in the present study arose in the proximal colon, in the ascending colon and hepatic flexure. This is consistent with the finding of Yamauchi et al., who reported an increasing gradient of CIMP from the rectum to ascending colon.47
While the majority of mutations observed in gliomas are R132H, other mutation types have been reported. The R132C mutation was first identified in acute myeloid leukemia38 while R132C, R132G, and R132L mutations have been found in high grade gliomas.42 Andersson et al. have shown using recombinant mutant proteins that the R132C mutant has greater catalytic activity in the reduction of α-ketoglutarate to 2-hydroxyglutarate compared to the R132H mutant.48 Remarkable overlap of differentially methylated regions (82%) was seen between the colorectal IDH1 mutation-like cluster and the glioma methylator phenotype, which is caused by IDH1 mutation.29 We therefore propose that alteration of other genes in this pathway may contribute to the phenotype observed in Cluster 1 samples. We examined one other gene in this pathway (IDH2) but did not find strong evidence of its involvement.
Recently, the chromatin regulator genes CHD7 and CHD8 were found to be mutated at an enriched frequency in CIMP-positive colorectal cancers compared to CIMP-negative.49 In this series, CIMP1 (high level CIMP) was classified based on a panel of 7 markers. A classification of CIMP1 required hypermethylation of the MLH1 gene promoter, as well as 4/6 of the remaining markers. By definition, CIMP1 cancers in this series were all MSI. The majority of the CHD mutations identified were therefore in MSI cancers and it is not clear that these were the cause of CIMP or may have arisen as a consequence of MSI. In fact, a high proportion of alterations identified were frameshift insertion or deletion mutations, which are typically observed in MSI cancers as a consequence of mismatch repair deficiency. In these cancers CIMP is established early in polyp development, therefore a mutation that causes CIMP would necessarily have to occur prior to the onset of MSI. It is possible that CHD gene family members contribute to a methylator phenotype and it will be important to study how these and other epigenetic regulator genes may further contribute to or modulate CIMP.
Interestingly, IDH1 mutations were exclusively observed in microsatellite stable cancers. Biologically, this suggests that mutation of this gene is sufficient to promote tumorigenesis without the necessity to compromise mismatch repair activity, which is required for the development of dysplasia in approximately half of all CIMP colorectal cancers. Furthermore, we hypothesize that mutation of IDH1 results in deregulation of DNA methylation for a defined group of gene promoters that does not include the MLH1 mismatch repair gene.
IDH1 mutation is associated with a favorable prognosis in glioma,37,50-52 increased sensitivity to temozolomide51 and to radiotherapy.53 It is possible that colorectal cancers with IDH1 mutation, or an IDH1 mutation-like methylation signature (Cluster 1 samples) may be similarly sensitive to particular therapeutics. Furthermore, direct inhibition of mutant IDH1 suppresses the production of 2HG54-56 and, therefore, may present a therapeutic option for treating CIMP cancers arising via this mechanism.
The present study shows that a modest proportion of CIMP-positive colorectal cancers are associated with an activating mutation of IDH1, which is known to cause DNA methylation. This is important “proof of principle” that specific causes of CIMP can be identified in colorectal cancer. All the IDH1 mutations occurred in one cancer subgroup (MSS BRAF mutant) and they were associated with an identifiable methylation signature. This suggests that different causes of CIMP lead to different patterns of methylation and, thus, different clinicopathological outcomes.
Methods
Patient samples
A series of 669 colorectal cancers was collected at the Royal Brisbane and Women's Hospital (RBWH), Australia, between 1992 and 2012. The cancers had previously been characterized for BRAF V600E mutation using allelic discrimination,57 the CpG Island Methylator Phenotype using the Weisenberger et al. 5 marker panel (CACNA1G, IGF2, NEUROG1, RUNX3, SOCS1)3 and microsatellite instability using the National Cancer Institute 5 marker panel (BAT25, BAT26, D5S346, D2S123 and D17S250).58 All patients provided written, informed consent and the study was approved by the RBWH and Bancroft Human Research Ethics Committees.
IDH1 mutation detection
A cohort of 86 CIMP-positive and 80 CIMP-negative cancers were screened for the IDH1 R132C mutation by automated sequencing. Amplification was performed by the Australian Genomic Research Facility using previously described primers,42 Applied Biosystems® AmpliTaq Gold® 360 Master Mix and a touchdown cycling profile of 95°C for 7 minutes, 12 cycles of 95°C for 30 seconds, 68°C (−1°C per cycle) for 30 seconds, 72°C for 1 minute, 29 cycles of 95°C for 30 seconds, 57°C for 15 seconds, 72°C for 1 minute followed by a 10 minute hold at 72°C. An additional internal forward sequencing primer (5′-GCC ATC ACT GCA GTT GTA GGT TA - 3′) was used to ensure specificity.
The entire coding region of IDH1 was screened for mutation in 81 CIMP+ colorectal cancers (but not the CIMP-negative cancers). Sequencing was performed by Macrogen USA using BigDye v3.1 (Life Technologies, Applied Biostystems) as per the manusfacturer's protocol. Thermal cycling conditions are as follows: 96°C hold for 2 minutes, followed by 25 cycles of 96°C for 15 seconds, 50°C for 5 seconds, 60°C for 2 minutes. Sequence detection was performed by capillary electrophoresis on a 3730xl Genetic Analyzer (Life Technologies, Applied Biosystems) using a 50 cm array, the Long DNA sequencing module (LongSeq50_POP7) and the KB analysis protocol (KB basecaller) with the default instrument settings. Post-detection, raw signal data was processed on the 3730xl Genetic Analyzer computer using Sequencing Analysis v5.3.1 (Life Technologies, Applied Biosystems). Primer sequences for IDH1 coding exons are shown in Table 2.
Table 2.
Primer Sequences for IDH1 Mutation Detection
| Forward Primer | Reverse Primer | Product Size (bp) | |
|---|---|---|---|
| Exon 3 | 5’ – TTG TTC AGA GAA GAT ACT CAA TTC – 3’ | 5’ – TAA GTA GCT GGG ACT TCA CG – 3’ | 422 |
| Exon 4 | 5’ – CTG TGT TTA GGG TGT GCC AG – 3’ | 5’ – AAT TTC ATA CCT TGC TTA ATG GG – 3’ | 573 |
| Exon 5 | 5’ – TGT CTG GAC CTC TTC ATC CC – 3’ | 5’ – TGT CAA GTT TCG GGT TTT GC – 3’ | 418 |
| Exon 6 | 5’ – GTT GGA AAC CTG TCT GGG AC – 3’ | 5’ – TTT TGT TTC ACT CCT GCT AAA CC – 3’ | 497 |
| Exon 7 | 5’ – CTG TTT GGG ACA AGC AGA TG – 3’ | 5’ – GGA CTA CAA AAC TCC CCT TCC – 3’ | 312 |
| Exon 8 | 5’ – CCT ATC AAG ATT GAG TCA TTT ATG C – 3’ | 5’ – CCA AGG GAA CAC ATC TGG G – 3’ | 305 |
| Exon 9 | 5’ – GGG GAA CTA TGA GAC ATT TGG – 3’ | 5’ – ACC GAT GCT CTG AGC CC – 3’ | 307 |
| Exon 10 | 5’ – TTT CTA GGA CTT TAC CAC TAC CTG C – 3’ | 5’ – TTT TGC CTT TAT CCT TGA GTG – 3’ | 359 |
IDH2 mutation detection
The IDH2 mutational hotspot was assessed in 86 CIMP-positive and 80 CIMP-negative cancers. Automated sequencing was performed by the Australian Genomic Research Facility using the same protocol developed for IDH1, with primers previously described by Chotirat et al.59
DNA methylation array profiling
The potential impact of the R132C IDH1 mutation on genome-wide DNA methylation profiles was assessed using Infinium HumanMethylation450 BeadChips (Illumina Inc., California, USA). Bisulfite conversion of 500 ng of DNA for each sample was performed using the EZ-96 DNA Methylation™ kit (Zymo Research, Irvine, CA, USA) then half the resulting yield was labeled and hybridized to the arrays according to the Illumina Infinium methylation protocol. BeadChips were scanned on an iScan (Illumina). Twenty-nine CIMP-positive and 18 CIMP-negative cancers were chosen for hybridization. Of the CIMP-positive samples, 4 had the R132C mutation.
For DNA methylation analysis IDAT files were loaded into the R (2.15) environment using the Bioconductor (http://www.bioconductor.org) minfi package.60 The arrays were then background and control normalized using the minfi package. Technical differences between Infinium I and Infinium II probes were removed using Subset-quantile Within Array Normalization (SWAN), developed by Maksimovic et al 61 and available in the minfi package. The methylation status for each probe was recorded as a β value that ranged between 0 and 1, where values close to 1 represent high levels of methylation and where values close to 0 represent low levels of methylation. A detection P-value was calculated for all probes on all arrays, with P > 0.05 being considered significantly different from background measurements. Probes were removed from analysis if greater than 50% of the samples had a detection P-value greater than 0.05 (n = 173). Probes on the X- and Y-chromosomes were removed, together with probes with SNPs present, leaving a total of 385183 probes for analysis.
For cluster analysis, the top 3000 most variable probes were selected (based on the standard deviation of the β value across the sample set). These probes were selected based solely on variability and not genomic classification (CpG island, promoter, shore, shelf). A recursively partitioned mixture model (RPMM) was used to cluster the β scores. RPMM is a model-based unsupervised clustering algorithm developed for measurements that lie between 0 and 1. This algorithm was implemented using the RPMM Bioconductor package.62 The implementation of RPMM was identical to Hinoue et al.15 who used a fuzzy clustering algorithm for initialization and level-weighted version of BIC as a split criterion.
In order to asses differences in methylation between groups, the original n = 385183 β values were converted to M values via the logit transformation as recommended by Du et al.63 Differentially methylated probes were detected using minfi, which utilises an F-test in order to detect differences between categorical groups. In addition sample estimates of the variances were estimated using the limma package. Probes were considered to be differentially methylated if the resulting q-value was less than 0.05. Selection of all probes with q-value less than 0.05 ensures that the false discovery rate (FDR) is less than 0.05.64
Statistical analysis
Categorical variables were analyzed for significance using Fisher's Exact Test and continuous variables were assessed by Student's T-test (SPSS version 19; Graphpad software). Odds ratios and 95% confidence intervals were calculated in SPSS version 19. P-values ≤0.05 were considered statistically significant.
Disclosure of Potential Conflicts of Interest
No conflicts of interest were disclosed.
Acknowledgments
We are grateful to Professor Nick Hayward for helpful discussion.
Funding
This work was funded by the National Health and Medical Research Council project grants 442965 and 1050455; Cancer Council Queensland grant 1025268; Pathology Queensland, Clinical and Statewide Services, Queensland, Australia; an Australian Postgraduate Award and a Cancer Council Queensland Postgraduate Award.
References
- 1. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999;96:8681–6; PMID:10411935; http://dx.doi.org/ 10.1073/pnas.96.15.8681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zoratto F, Rossi L, Verrico M, Papa A, Basso E, Zullo A, Tomao L, Romiti A, Lo Russo G, Tomao S. Focus on genetic and epigenetic events of colorectal cancer pathogenesis: implications for molecular diagnosis. Tumour Biol 2014;35:6195–206; PMID:25051912; http://dx.doi.org/ 10.1007/s13277-014-1845-9 [DOI] [PubMed] [Google Scholar]
- 3. Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006;38:787–93; PMID:16804544; http://dx.doi.org/ 10.1038/ng1834 [DOI] [PubMed] [Google Scholar]
- 4. Ogino S, Kawasaki T, Nosho K, Ohnishi M, Suemoto Y, Kirkner GJ, Fuchs CS. LINE-1 hypomethylation is inversely associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Int J Cancer 2008;122:2767–73; PMID:18366060; http://dx.doi.org/ 10.1002/ijc.23470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology 2010;138:2088–100; PMID:20420948; http://dx.doi.org/ 10.1053/j.gastro.2009.12.066 [DOI] [PubMed] [Google Scholar]
- 6. Bettington M, Walker N, Clouston A, Brown I, Leggett B, Whitehall V. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology 2013;62:367–86; PMID:23339363; http://dx.doi.org/ 10.1111/his.12055 [DOI] [PubMed] [Google Scholar]
- 7. Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, Wolff RK, Slattery ML. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res 2005;65:6063–9; PMID:16024606; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-0404 [DOI] [PubMed] [Google Scholar]
- 8. Hughes LA, Khalid-de Bakker CA, Smits KM, van den Brandt PA, Jonkers D, Ahuja N, Herman JG, Weijenberg MP, van Engeland M. The CpG island methylator phenotype in colorectal cancer: progress and problems. Biochim Biophys Acta 2012;1825:77–85; PMID:22056543 [DOI] [PubMed] [Google Scholar]
- 9. Ogino S, Kawasaki T, Kirkner GJ, Loda M, Fuchs CS. CpG island methylator phenotype-low (CIMP-low) in colorectal cancer: possible associations with male sex and KRAS mutations. J Mol Diagn 2006;8:582–8; PMID:17065427; http://dx.doi.org/ 10.2353/jmoldx.2006.060082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, Hernandez NS, Chen X, Ahmed S, Konishi K, et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A 2007;104:18654–9; PMID:18003927; http://dx.doi.org/ 10.1073/pnas.0704652104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yagi K, Akagi K, Hayashi H, Nagae G, Tsuji S, Isagawa T, Midorikawa Y, Nishimura Y, Sakamoto H, Seto Y, et al. Three DNA methylation epigenotypes in human colorectal cancer. Clin Cancer Res 2010;16:21–33; PMID:20028768; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-2006 [DOI] [PubMed] [Google Scholar]
- 12. Whitehall VL, Wynter CV, Walsh MD, Simms LA, Purdie D, Pandeya N, Young J, Meltzer SJ, Leggett BA, Jass JR. Morphological and molecular heterogeneity within nonmicrosatellite instability-high colorectal cancer. Cancer Res 2002;62:6011–4; PMID:12414620 [PubMed] [Google Scholar]
- 13. Dahlin AM, Palmqvist R, Henriksson ML, et al. The role of the CpG island methylator phenotype in colorectal cancer prognosis depends on microsatellite instability screening status. Clin Cancer Res 2010;16:1845–55; PMID:20197478; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-2594 [DOI] [PubMed] [Google Scholar]
- 14. Ogino S, Kawasaki T, Kirkner GJ, Kraft P, Loda M, Fuchs CS. Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J Mol Diagn 2007;9:305–14; PMID:17591929; http://dx.doi.org/ 10.2353/jmoldx.2007.060170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D, Malik S, Pan F, Noushmehr H, van Dijk CM, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012;22:271–82; PMID:21659424; http://dx.doi.org/ 10.1101/gr.117523.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Colussi D, Brandi G, Bazzoli F, Ricciardiello L. Molecular pathways involved in colorectal cancer: implications for disease behavior and prevention. Int J Mol Sci 2013;14:16365–85; PMID:23965959; http://dx.doi.org/ 10.3390/ijms140816365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bardhan K, Liu K. Epigenetics and colorectal cancer pathogenesis. Cancers (Basel) 2013;5:676–713; PMID:24216997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shiovitz S, Bertagnolli MM, Renfro LA, et al. CpG Island Methylator Phenotype is Associated With Response to Adjuvant Irinotecan-Based Therapy for Stage 3 Colon Cancer. Gastroenterology 2014; 147:637–45; PMID:24859205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Curtin K, Slattery ML, Ulrich CM, et al. Genetic polymorphisms in one-carbon metabolism: associations with CpG island methylator phenotype (CIMP) in colon cancer and the modifying effects of diet. Carcinogenesis 2007;28:1672–9; PMID:17449906; http://dx.doi.org/ 10.1093/carcin/bgm089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Slattery ML, Curtin K, Wolff RK, Herrick JS, Caan BJ, Samowitz W. Diet, physical activity, and body size associations with rectal tumor mutations and epigenetic changes. Cancer Causes Control 2010;21:1237–45; PMID:20383576; http://dx.doi.org/ 10.1007/s10552-010-9551-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hughes LA, Simons CC, van den Brandt PA, et al. Body size, physical activity and risk of colorectal cancer with or without the CpG island methylator phenotype (CIMP). PLoS One 2011; 6:e18571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Slattery ML, Curtin K, Sweeney C, Levin TR, Potter J, Wolff RK, Albertsen H, Samowitz WS. Diet and lifestyle factor associations with CpG island methylator phenotype and BRAF mutations in colon cancer. Int J Cancer 2007;120:656–63; PMID:17096326; http://dx.doi.org/ 10.1002/ijc.22342 [DOI] [PubMed] [Google Scholar]
- 23. Slattery ML, Wolff RK, Herrick JS, Curtin K, Caan BJ, Samowitz W. Alcohol consumption and rectal tumor mutations and epigenetic changes. Dis Colon Rectum 2010;53:1182–9; PMID:20628283; http://dx.doi.org/ 10.1007/DCR.0b013e3181d325db [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hughes LA, van den Brandt PA, de Bruine AP, Wouters KA, Hulsmans S, Spiertz A, Goldbohm RA, de Goeij AF, Herman JG, Weijenberg MP, et al. Early life exposure to famine and colorectal cancer risk: a role for epigenetic mechanisms. PLoS One 2009;4:e7951; PMID:19956740; http://dx.doi.org/ 10.1371/journal.pone.0007951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Samowitz WS, Albertsen H, Sweeney C, Herrick J, Caan BJ, Anderson KE, Wolff RK, Slattery ML. Association of smoking, CpG island methylator phenotype, and V600E BRAF mutations in colon cancer. J Natl Cancer Inst 2006;98:1731–8; PMID:17148775; http://dx.doi.org/ 10.1093/jnci/djj468 [DOI] [PubMed] [Google Scholar]
- 26. Walker RG, Landmann JK, Hewett DG, et al. Hyperplastic polyposis syndrome is associated with cigarette smoking, which may be a modifiable risk factor. Am J Gastroenterol 2010;105:1642–7; PMID:20125129; http://dx.doi.org/ 10.1038/ajg.2009.757 [DOI] [PubMed] [Google Scholar]
- 27. Limsui D, Vierkant RA, Tillmans LS, et al. Cigarette smoking and colorectal cancer risk by molecularly defined subtypes. J Natl Cancer Inst 2010;102:1012–22; PMID:20587792; http://dx.doi.org/ 10.1093/jnci/djq201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008;321:1807–12; PMID:18772396; http://dx.doi.org/ 10.1126/science.1164382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012;483:479–83; PMID:22343889; http://dx.doi.org/ 10.1038/nature10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360:765–73; PMID:19228619; http://dx.doi.org/ 10.1056/NEJMoa0808710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462:739–44; PMID:19935646; http://dx.doi.org/ 10.1038/nature08617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010;17:225–34; PMID:20171147; http://dx.doi.org/ 10.1016/j.ccr.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012;483:474–8; PMID:22343901; http://dx.doi.org/ 10.1038/nature10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010;18:553–67; PMID:21130701; http://dx.doi.org/ 10.1016/j.ccr.2010.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Balss J, Meyer J, Mueller W, et al. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 2008;116:597–602; PMID:18985363; http://dx.doi.org/ 10.1007/s00401-008-0455-2 [DOI] [PubMed] [Google Scholar]
- 36. Watanabe T, Nobusawa S, Kleihues P, et al. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 2009;174:1149–53; PMID:19246647; http://dx.doi.org/ 10.2353/ajpath.2009.080958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sanson M, Marie Y, Paris S, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 2009;27:4150–4; PMID:19636000; http://dx.doi.org/ 10.1200/JCO.2009.21.9832 [DOI] [PubMed] [Google Scholar]
- 38. Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009;361:1058–66; PMID:19657110; http://dx.doi.org/ 10.1056/NEJMoa0903840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ward PS, Cross JR, Lu C, et al. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene 2012;31:2491–8; PMID:21996744; http://dx.doi.org/ 10.1038/onc.2011.416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wagner K, Damm F, Gohring G, et al. Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol 2010;28:2356–64; PMID:20368538; http://dx.doi.org/ 10.1200/JCO.2009.27.6899 [DOI] [PubMed] [Google Scholar]
- 41. Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006;314:268–74; PMID:16959974; http://dx.doi.org/ 10.1126/science.1133427 [DOI] [PubMed] [Google Scholar]
- 42. Bleeker FE, Lamba S, Leenstra S, et al. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat 2009;30:7–11; PMID:19117336; http://dx.doi.org/ 10.1002/humu.20937 [DOI] [PubMed] [Google Scholar]
- 43. Kang MR, Kim MS, Oh JE, et al. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer 2009;125:353–5; PMID:19378339; http://dx.doi.org/ 10.1002/ijc.24379 [DOI] [PubMed] [Google Scholar]
- 44. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7; PMID:22810696; http://dx.doi.org/ 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen D, Huang JF, Liu K, et al. BRAFV600E mutation and its association with clinicopathological features of colorectal cancer: a systematic review and meta-analysis. PLoS One 2014; 9:e90607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nosho K, Irahara N, Shima K, et al. Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS One 2008; 3:e3698; PMID:19002263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yamauchi M, Morikawa T, Kuchiba A, et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut 2012;61:847–54; PMID:22427238; http://dx.doi.org/ 10.1136/gutjnl-2011-300865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Andersson AK, Miller DW, Lynch JA, et al. IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia 2011;25:1570–7; PMID:21647154; http://dx.doi.org/ 10.1038/leu.2011.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tahara T, Yamamoto E, Madireddi P, et al. Colorectal carcinomas with CpG island methylator phenotype 1 frequently contain mutations in chromatin regulators. Gastroenterology 2014;146:530-38 e5; PMID:24211491; http://dx.doi.org/ 10.1053/j.gastro.2013.10.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shibahara I, Sonoda Y, Kanamori M, et al. IDH1/2 gene status defines the prognosis and molecular profiles in patients with grade III gliomas. Int J Clin Oncol 2011; PMID:21971842 [DOI] [PubMed] [Google Scholar]
- 51. Houillier C, Wang X, Kaloshi G, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology 2010;75:1560–6; PMID:20975057; http://dx.doi.org/ 10.1212/WNL.0b013e3181f96282 [DOI] [PubMed] [Google Scholar]
- 52. van den Bent MJ, Dubbink HJ, Marie Y, et al. IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res 2010;16:1597–604; PMID:20160062; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-2902 [DOI] [PubMed] [Google Scholar]
- 53. Li S, Chou AP, Chen W, et al. Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro Oncol 2013;15:57–68; PMID:23115158; http://dx.doi.org/ 10.1093/neuonc/nos261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Popovici-Muller J, Saunders JO, Salituro FG, et al. Discovery of the First Potent Inhibitors of Mutant IDH1 That Lower Tumor 2-HG in Vivo. ACS Med Chem Lett 2012;3:850–5; PMID:24900389; http://dx.doi.org/ 10.1021/ml300225h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Losman JA, Looper RE, Koivunen P, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013;339:1621–5; PMID:23393090; http://dx.doi.org/ 10.1126/science.1231677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013;340:626–30; PMID:23558169; http://dx.doi.org/ 10.1126/science.1236062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Young J, Barker MA, Simms LA, et al. Evidence for BRAF mutation and variable levels of microsatellite instability in a syndrome of familial colorectal cancer. Clin Gastroenterol Hepatol 2005;3:254–63; PMID:15765445; http://dx.doi.org/ 10.1016/S1542-3565(04)00673-1 [DOI] [PubMed] [Google Scholar]
- 58. Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998;58:5248–57; PMID:9823339 [PubMed] [Google Scholar]
- 59. Chotirat S, Thongnoppakhun W, Promsuwicha O, et al. Molecular alterations of isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) metabolic genes and additional genetic mutations in newly diagnosed acute myeloid leukemia patients. J Hematol Oncol 2012; 5:5; PMID:22397365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hansen KD, Aryee M. The minfi User's Guide: Analyzing Illumina 450K Methylation Arrays. R Package 2012; Version 1.4.0:1–21. [Google Scholar]
- 61. Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile Within Array Normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol 2012;13:R44; PMID:22703947; http://dx.doi.org/ 10.1186/gb-2012-13-6-r44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Houseman EA. RPMM: Recursively Partitioned Mixture Model. R Package 2012;Version 1.10:http://CRAN.R-project.org/package=RPMM. [Google Scholar]
- 63. Du P, Zhang X, Huang CC, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 2010;11:587; PMID:21118553; http://dx.doi.org/ 10.1186/1471-2105-11-587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Storey JD. The positive false discovery rate: a Bayesian interpretation and the q-value. The Annals of Statistics. 2003;31:2013–2035; http://dx.doi.org/ 10.1214/aos/1074290335 [DOI] [Google Scholar]