

Abstract
Clinical trials with SRC family kinases (SFKs) inhibitors used alone or in a combination with anti-CD20 monoclonal antibodies (mAbs) are currently underway in the treatment of B-cell tumors. However, molecular interactions between these therapeutics have not been studied so far. A transcriptional profiling of tumor cells incubated with SFKs inhibitors revealed strong downregulation of MS4A1 gene encoding CD20 antigen. In a panel of primary and established B-cell tumors we observed that SFKs inhibitors strongly affect CD20 expression at the transcriptional level, leading to inhibition of anti-CD20 mAbs binding and increased resistance of tumor cells to complement-dependent cytotoxicity. Activation of the AKT signaling pathway significantly protected cells from dasatinib-triggered CD20 downregulation. Additionally, SFKs inhibitors suppressed antibody-dependent cell-mediated cytotoxicity by direct inhibition of natural killer cells. Abrogation of antitumor activity of rituximab was also observed in vivo in a mouse model. Noteworthy, the effects of SFKs inhibitors on NK cell function are largely reversible. The results of our studies indicate that development of optimal combinations of novel treatment modalities with anti-CD20 mAbs should be preceded by detailed preclinical evaluation of their effects on target cells.
Keywords: CD20, dasatinib, SRC family kinases, rituximab, ofatumumab
Abbreviations
- ADCC
antibody-dependent cell-mediated cytotoxicity
- BCR
B-cell receptor
- CDC
complement-dependent cytotoxicity
- CFSE
carboxyfluorescein succinimidyl ester
- CLL
chronic lymphocytic leukemia
- DLBCL
diffuse large B-cell lymphoma
- NHL
non-Hodgkin lymphoma
- PBMC
peripheral blood mononuclear cells
- SFKs
SRC family kinases
Introduction
Members of the SRC family kinases (SFKs) play a critical role in the pathogenesis of B-cell tumors by promoting survival and clonal expansion of malignant B-cells. Some of the SFKs including BLK, FYN and LYN have been reported to be uniquely expressed at high levels in various hematopoietic lineages and carry out highly specialized functions in the hematopoietic system. SFKs expressed in normal and malignant B-cells are found in association with the B-cell receptor (BCR) complex and are implicated in signal transduction events promoting tumor growth and progression.1,2 Both non-Hodgkin's lymphoma (NHL) and chronic lymphocytic leukemia (CLL) cells are characterized by anomalously high levels of constitutively active LYN, which plays a key role in initiating BCR signaling via tyrosine phosphorylation of BCR signal transduction components (Igα/Igβ) and BCR co-receptor (CD19).3,4 BCR signaling is further propagated by spleen tyrosine kinase (SYK) through association with the adaptor molecule B-cell linker protein (BLNK) and its downstream signaling components, namely Bruton's tyrosine kinase (BTK) and phospholipase Cγ2 (PLCγ2). Moreover, LYN-dependent phosphorylation of cytoplasmic domain of CD19 recruits p85 subunit of phosphoinositide 3-kinase (PI3Kδ). Recruitment of PI3Kδ to the plasma membrane leads to the production of phosphatidylinositol 3,4,5-triphosphate (PIP3), which is required for robust activation of BTK and AKT.5
Multiple experimental studies indicate that inhibition of SFKs blocks cell proliferation and induces apoptosis, thereby implicating SFKs as attractive molecular targets for anti-lymphoma/leukemia therapy.6 Clinical trials are currently underway to investigate the effects of bafetinib (BCR-ABL/LYN inhibitor) and dasatinib (BCR-ABL/SFKs inhibitor) in relapsed and refractory CLL patients. Moreover, dasatinib is being investigated in diffuse large B-cell lymphoma (DLBCL) and other NHL, multiple myeloma (MM) and in CLL in combination with chemotherapy (fludarabine) or anti-CD20 mAb (rituximab) (Phase 1/2 clinical trials NCT00949988 and NCT01173679, respectively).
CD20 is a membrane protein expressed on the surface of normal and malignant B-cells. It is an excellent molecular target for mAbs that are widely used in the treatment of NHL and CLL. Anti-CD20 mAbs are successfully and widely used therapeutic mAbs. They are routinely incorporated in cyclophosphamide- or fludarabine-containing combination regimens into all phases of conventional treatments, including first-line, maintenance and salvage treatment. Nonetheless, the overall response rates to rituximab have been reported to be 46–67%, and are even lower in patients relapsing after initial rituximab treatment.7 A number of mechanisms have been proposed to account for this inefficiency, including modulation of surface CD20 levels, occurring due to both transcriptional and posttranscriptional regulations.8-10 Therefore, identification of more effective therapeutic schemes is constantly being pursued. Before using them in cancer patients, extensive pre-clinical studies should be performed to better define interactions between drugs at the molecular level. We have recently reported that targeted therapeutics inhibiting BCR signaling pathways, including SYK, BTK or PI3K inhibitors downregulate CD20 expression resulting in impaired antitumor activity of anti-CD20 mAbs.11 Furthermore, SFKs being important downstream signaling molecules of Fc receptors are crucial for tumor cell killing via antibody-dependent cell-mediated cytotoxicity (ADCC). It has been shown that ADCC and FcR signaling represents one of the essential mechanisms of action for anti-CD20 therapy in vivo.12 These results, and the recent initiation of clinical trials of dasatinib with rituximab, a combination for which no pre-clinical studies have been published, prompted us to investigate the influence of SFKs inhibitors on antitumor activity of CD20 immunotherapy in B-cell tumors.
Here, we demonstrate that inhibition of SFKs results in increased resistance of tumor cells to the antitumor activity of anti-CD20 mAbs, both in vitro and in vivo. While impaired complement-dependent cytotoxicity (CDC) is a direct result of decreased CD20 levels, inhibition of ADCC is caused by impairment of the cytolytic activity of natural killer (NK) cells. Our observations strongly imply that CD20 downregulation relies on transcriptional mechanisms and highlight the role of AKT in SFKs-dependent transcriptional regulation of CD20. Moreover, we also delineate new perspectives in the field of CD20 regulation and identify PI3K/AKT signaling pathway as an important regulator of transcription factor(s) involved in CD20 transcription in tumor B-cells.
Results
Global gene expression analysis upon chemical inhibition of SFKs
A transcriptional profiling of Raji cells incubated with 100 nM dasatinib (a clinically-available multi-target tyrosine kinase inhibitor) or 10 μM PP2 (a more selective inhibitor of BCR-associated SFKs) was performed to characterize molecular changes in lymphoma cells incubated with SFKs inhibitors. The unpaired t test with Benjamin-Hochberg FDR <5% (false discovery rate) correction (with P value cut-off <0.01) revealed 28 upregulated and 86 downregulated (with at least 3-fold change) genes in cells incubated with either dasatinib or PP2 (Fig. 1A). The analysis of microarray data has been deposited in NCBI's Gene Expression Omnibus and is accessible via GEO Series accession number GSE50929. Compared with untreated cells, these up/downregulated genes were common for each treatment examined individually. A statistically-significant (with P value of 0.00109) downregulation of MS4A1 (CD20) gene was identified (fold change −6.22) when dasatinib-treated cells were analyzed along with PP2-treated cells and compared collectively with untreated cells (Fig. 1B). These results were further confirmed by quantitative PCR (Fig. S1). Since CD20 is a therapeutic target in B-cell malignancies and an increasing number of anti-CD20 monoclonal antibodies are approved for clinical use, we decided to further focus on the outcomes and mechanisms of CD20 expression regulation.
Figure 1.
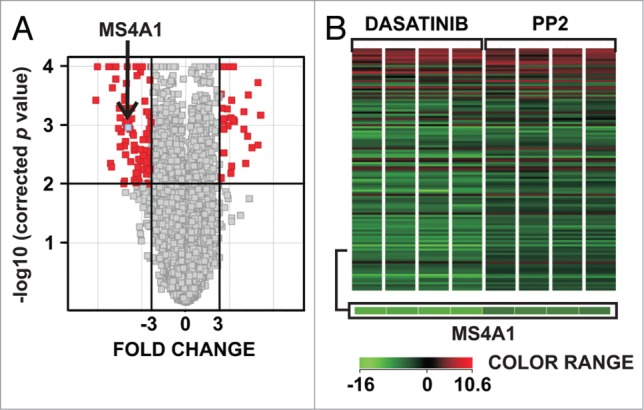
Transcriptional profiling of Raji cells incubated for 24 h with dasatinib or PP2. (A) Total RNA from control Raji cells or from cells incubated for 24 h with either 100 nM dasatinib or 10 μM PP2 was used to generate cRNA for hybridization to human-specific AMADID Release GE 8x60K microarrays. The volcano plot shows changes in expression of all the Agilent microarray transcripts in cells incubated with dasatinib or PP2. Red spots represent genes for which expression changed significantly (p < 0.01, Fold Change > 3, unpaired t test and Benjamin-Hochberg FDR < 5% correction) due to dasatinib or PP2 treatment, gray spots represent genes for which expression was not significantly changed. Arrow indicates MS4A1 gene (blue spot). The plot was made using GeneSpring (Agilent) software. (B) GeneSpring (Agilent, USA) cluster of genes (containing MS4A1 gene) for which expression changed similarly in cells upon incubation with dasatinib or PP2. All genes with a fold change cut-off > 3.0 (p value < 0.01 in unpaired t test and Benjamin-Hochberg FDR < 5% correction) were considered as significantly regulated and are presented in a matrix format: each row represents a single gene, and each column represents an experimental sample. In each sample, the ratio of the abundance of transcripts of each gene to the median abundance of the gene's transcript across all sample, is represented by the color of the corresponding cell in the matrix.
Inhibitors of SRC family kinases downregulate CD20 levels and impair antitumor activity of anti-CD20 mAbs in Raji cells
Dasatinib and more selective compounds targeting SFKs (bafetinib and PP2) were studied in more detail to determine their influence on CD20 levels. Flow cytometry revealed a severely impaired binding of anti-CD20 (clone L27) mAb to Raji cells pre-incubated for 48 h with increasing non-toxic concentrations of all tested SFKs inhibitors (Fig. 2A, Fig. S2). Likewise, binding of ofatumumab and rituximab was impaired in Raji cells pre-incubated with dasatinib (Fig. S3). Neither imatinib, an inhibitor of BCR-ABL, c-KIT and platelet-derived growth factor receptor (PDGFR), nor tandutinib, a Fms-like tyrosine kinase 3 receptor (FLT3), PDGFR and c-KIT inhibitor, exerted significant effects on CD20 levels and antitumor activity of rituximab in Raji cells (Fig. S4). To investigate whether modulation of CD20 levels results from specific inhibition of SFKs activity, we used shRNA to knock-down FYN, LCK and LYN expression (Fig. S5A). Using flow cytometry, we observed that SFKs knock-down significantly decreased surface CD20 levels (Fig. S5B).
Figure 2.

For figure legend, see next page.Figure 2 (See previous page). SFKs inhibitors downregulate surface CD20 levels and impair antitumor activity of rituximab and ofatumumab. (A) The surface CD20 level was determined with FITC-conjugated anti-CD20 antibody (clone L27, BD) in Raji cells pre-incubated for 48 h with increasing concentrations of multi-SRC family kinases inhibitors. Results are presented as a percentage of MFI of control cells (± SD). (B–C) Raji cells pre-incubated for 48 h with increasing concentrations of multi-SRC kinases inhibitors were washed and incubated for 1 h with rituximab (B) or ofatumumab (C) (1–100 μg/ml) and 10% human AB serum. Cell viability in CDC assay was measured with PI. The survival of cells is presented as a percentage of control cells without antibody (± SD). (D) CFSE-stained Raji cells were co-incubated for 4 h with either rituximab or ofatumumab (100 μg/ml) and NK cells at E:T ratio 6:1 in presence of dasatinib or PP2. Raji cell survival in ADCC assay was determined with flow cytometry after staining with PI and is presented as a percentage of control cells without rituximab/ofatumumab. (E) NK and Raji cells were incubated simultaneously at effector to target ratio 1:1 with rituximab (100 μg/ml) and either dasatinib (100 nM) or PP2 (10 μM) for 4 h at 37°C. For degranulation assay NK cells were co-incubated with GolgiStop and anti-CD107a antibody. Cells were washed, stained with anti-CD56, anti-CD3 and Fixable Viability Dye. To determine cytokines production, cells were washed, permeabilized with Cytoperm/Cytofix and stained with anti-interferon (IFN)-γ or anti-tumor necrosis factor (TNF) antibodies followed by flow cytometry analysis. Results are presented as a percentage of CD107a, TNF or IFN-γ positive NK cells (± SD) within the whole NK cell population. Shown is one representative of at least 3 independent experiments.
To determine the functional consequences of decreased CD20 levels on effector mechanisms of anti-CD20 mAbs, their ability to induce complement-dependent cytotoxicity (CDC) was examined. A dose-dependent impairment of rituximab- (R-CDC) and ofatumumab-induced complement-dependent cytotoxicity (O-CDC) was observed in Raji cells pre-incubated for 48 h with all tested SFKs inhibitors compared with control cells (Fig. 2B, C). Although binding of rituximab and ofatumumab was decreased to a similar extent, the highest tested concentrations of SFKs inhibitors almost completely abrogated R-CDC, while O-CDC was affected to a lesser extent. Collectively, flow cytometry measurements of relative surface antigens expression in Raji cells pre-incubated with SFKs inhibitors show a substantial decrease in CD20 levels, significant reduction in binding of therapeutic anti-CD20 mAbs and induction of strong resistance to antitumor activity of anti-CD20 mAbs in CDC assay. Moreover, the results of experiments using shRNA reveal that downmodulation of CD20 occurs selectively due to inhibition of SFKs.
Influence of dasatinib on other B-cell surface molecules
Surface levels of complement regulatory proteins (CD46 and CD55) and other B-cell molecules (HLA-DR, ICAM-1, CD52, CD38) remained unchanged in dasatinib-treated Raji cells (Fig. S6). Conspicuously, some of the BCR complex components were affected by dasatinib. While CD19 expression in cells pre-incubated with dasatinib remained roughly unchanged (a 20% decrease compared with controls), both CD21 and CD22 levels were decreased by approximately 50%, but still to a lesser extent compared with CD20. The changes in proteins levels correlated well with the changes in gene expression from microarray data. The expression of genes encoding CD20 and CD22 appeared to be strongly downregulated with log fold change (Log FC) below −1.584 and P < 0.01 in cells treated with either dasatinib or PP2. The downregulation of the gene encoding CD21 was less distinct with Log FC < −1.0 (Table S2). In line with previous findings by Davis et al. we also observed increased surface IgM expression upon dasatinib treatment likely resulting from decreased BCR internalization.13 Altogether, our results indicate that although dasatinib decreases CD20, CD21 and CD22 levels and modulates various surface antigens, it does not substantially influence other targets for mAbs-mediated therapy, such as CD38, CD52 and CD19 (Fig. S6).
Dasatinib impairs rituximab-mediated NK cell cytotoxicity
To determine the effects of SFKs inhibitors on NK effector cell function we used a model where both target Raji cells and effector NK cells were co-incubated for 4 h in presence of SFKs inhibitors and antibodies. Both rituximab-mediated ADCC (R-ADCC) and ofatumumab-mediated ADCC (O-ADCC) (Fig. 2D), as well as NK cells activation and degranulation (Fig. 2E), were severely impaired or even completely blocked at the highest concentrations of tested SFKs inhibitors. Also, in a spontaneous NK cell cytotoxicity assay against K562 cells in a co-incubation model with dasatinib or PP2, significant inhibition of NK cytotoxic effects (Fig. S7A) and NK cell degranulation (Fig. S7B) were observed. Dasatinib and PP2 were not toxic to NK cells within a tested range of concentrations. It can be concluded that anti-CD20 mAbs-mediated ADCC is inhibited by SFKs inhibitors due to suppression of NK cytotoxicity.
Dasatinib impairs antitumor activity of rituximab in vivo
Since dasatinib strongly impairs rituximab- and ofatumumab-mediated CDC and ADCC in cell culture studies we further aimed to assess antitumor efficacy of the combination treatment in vivo using a model of EL4 lymphoma cells stably expressing human CD20 (EL4-hCD20). In this cell line model CD20 expression, controlled by CMV promoter, remained unchanged upon treatment with increasing concentrations of dasatinib for 48 h (Fig. S8). Noteworthy, we observed in vivo that while treatment with rituximab alone significantly inhibited tumor growth compared with controls, dasatinib significantly decreased its antitumor activity (P = 0.0045 vs rituximab-treated group, Figure 3). These results suggest that the decrease in anti-tumor activity of rituximab in vivo could be attributed to the inhibition of ADCC mechanism.
Figure 3.
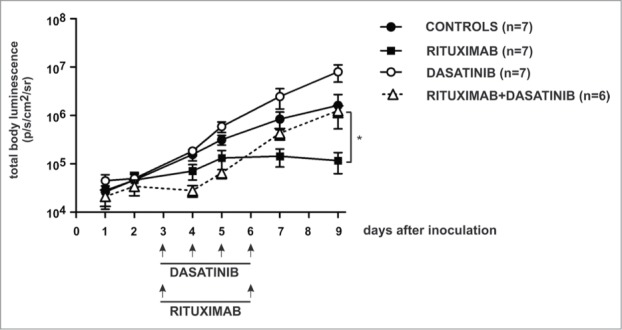
Dasatinib impairs antitumor activity of rituximab in a murine model. Mice were inoculated with 5x105 of EL4-hCD20 cells intravenously and randomized to groups of 6–8. Dasatinib was given once a day i.p. (50 mg/kg) for 4 consecutive days. Rituximab was given i.p. (10 mg/kg) at days 3 and 6. Control mice were injected with equal volume of PBS. Luminescence was followed from day 1 until termination of the experiment at day 9. Data are presented as mean ± SEM, statistical significance was reached by comparison between groups rituximab vs rituximab+dasatinib, p = 0.0045, t test. Shown is one representative of 2 independent experiments.
Dasatinib downregulates CD20 levels and impairs activity of anti-CD20 mAbs in primary tumor cells and in various CD20 positive cell lines
To extend the observations in Raji cells we have used both primary cells from patients with B-cell tumors and established CD20 positive tumor cell lines. In a set of 30 primary tumor samples dasatinib decreased surface CD20 levels (P < 0.0001 vs control groups) as confirmed with Wilcoxon signed-rank test (Fig. 4, MFIs from each sample listed in Table S1). In normal B-cells dasatinib downregulated surface CD20 levels by 30%. In these B-cells, relatively resistant to R-CDC, dasatinib only modestly impaired O-CDC (Fig. S9A). Although the majority of primary cells isolated from patients were resistant to mAbs-CDC, in some sensitive patients we observed inhibited R-CDC and O-CDC after incubation with dasatinib (Fig. S9B). Dasatinib strongly decreased surface CD20 levels in the majority of established tumor B-cell lines (Fig. S10A). A strong downregulation of surface CD20 levels was observed in Ly-1, Ly-7, DHL-4, DHL-6 cells that were reported to be BCR-dependent,14 while CD20 modulation was relatively modest in Ly-4, Ly-19, Karpas-422, Pfeiffer and Toledo cells reported to be BCR-independent14 as well as in a single BCR-dependent Ly-10 cell line (Fig. S10A). These effects correlated with an impaired R-CDC and O-CDC (Fig. S10B, for cell lines resistant to R-CDC data with O-CDC is included). Together, these data confirm that CD20 expression is modulated by SFKs in most CD20 positive tumor cell lines and primary cells isolated from patients with B-cell malignancies.
Figure 4.
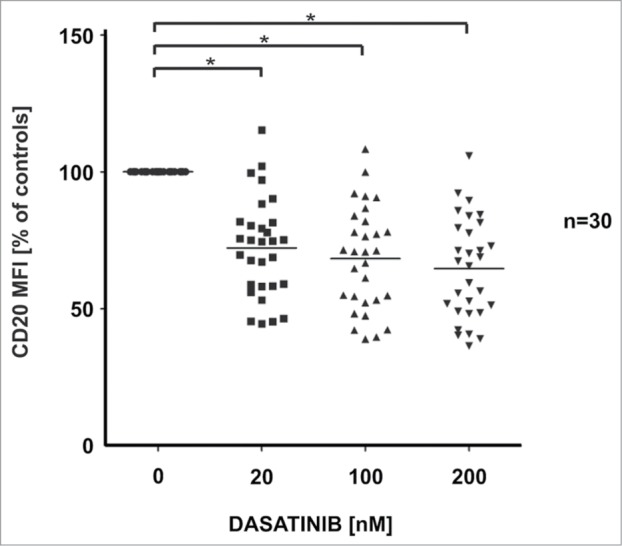
Influence of dasatinib on CD20 levels in primary CD20-positive leukemia/lymphoma cells. Primary human CD20-positive B-CLL/lymphoma cells (total n = 30; CLL n = 25, MCL n = 4, FL n = 1) were incubated for 48 h with increasing concentrations of dasatinib. After washing, cells were incubated with saturating amount of FITC-conjugated anti-CD20 mAb (clone L27, BD) for 30 min at room temperature in the dark. Prior to flow cytometry analysis, cells were washed with PBS and resuspended in PBS with PI. Results are presented as a percentage of MFI of control cells (± SD). The differences in CD20 MFI between control and drug-treated groups were statistically significant (p < 0.0001) as measured using Wilcoxon signed-rank test.
Dasatinib modulates CD20 at a transcriptional level
Immunoblotting of protein extracts from Raji cells confirmed that dasatinib downregulates total CD20 levels (Fig. 5A). A nearly 10-fold decrease in CD20 mRNA was also observed in qRT-PCR experiments with 100 and 200 nM dasatinib (Fig. 5B). Since incubation with cycloheximide, a protein synthesis inhibitor had no influence on dasatinib-induced CD20 mRNA downregulation as determined with qRT-PCR (Fig. 5C), dasatinib appears to mediate its effects on CD20 mRNA levels independently of translation. Since protein levels can also be regulated by the modulation of mRNA turnover, a conventional actinomycin D chase mRNA decay assay was done and revealed that in the context of transcriptional blockade dasatinib does not influence CD20 mRNA half-life (Fig. 5D). Therefore, it can be concluded that dasatinib-induced decrease in CD20 expression occurs mainly at the transcriptional level.
Figure 5.
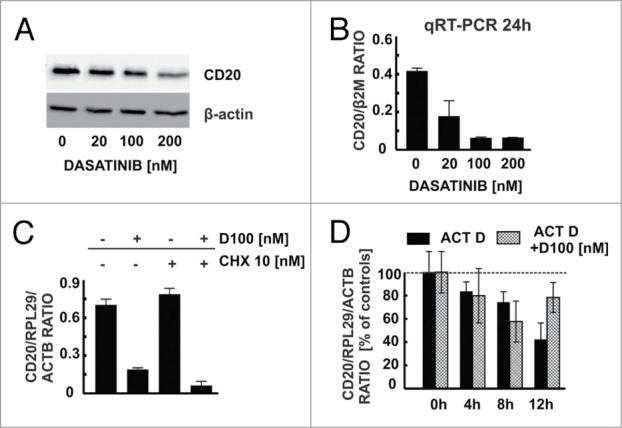
Dasatinib modulates CD20 levels at a transcriptional level. (A) Western blotting analysis of CD20 and β-actin in protein extracts from Raji cells pre-incubated for 48 h with increasing concentrations of dasatinib. (B) cDNA from Raji cells pre-incubated for 48 h with increasing concentrations of dasatinib was used for qRT-PCR amplification of CD20 and B2M products with corresponding probes labeled with FAM and DABCYL. (C) cDNA from Raji cells pre-incubated for 24 h with dasatinib (100 nM) and/or cycloheximide (10 nM) was used for qRT-PCR amplification of CD20, RPL29 and ACTB products. (D) In actinomycin D chase mRNA decay assay cDNA from Raji cells pre-incubated for 24 h with dasatinib (100 nM) and/or actinomycin (10 μg/ml) was used for qRT-PCR amplification of CD20, RPL29 and ACTB products. Shown is one representative of at least 3 independent experiments.
Modulation of CD20 expression by dasatinib requires CD20 promoter
To see whether dasatinib requires an endogenous promoter of CD20 gene to mediate its effects, HeLa cells were transduced with a vector containing CD20 gene driven by a constitutive CMV promoter. Although pre-incubation of Raji cells with dasatinib decreased endogenous CD20 levels, a pre-incubation of HeLa cells with dasatinib did not modulate exogenous surface CD20 expression (Fig. 6A). To further corroborate this observation Raji cells were stably transduced with a vector encoding CD20 His-tagged fusion protein under the CMV promoter (Raji His-CD20). While incubation of these cells for 48 h with increasing concentrations of dasatinib strongly downregulated endogenous CD20 in total cellular lysates, expression of CD20 His-tagged fusion protein remained unchanged (Fig. 6B). The activity of luciferase in Raji cells nucleofected with pGL4-CD20 promoter reporter plasmid (−431/+52 bp) decreased approximately 2-fold after incubation with 100 nM dasatinib (Fig. 6C, left). To get more detailed insight into the mechanism of CD20 promoter modulation, a series of truncated CD20 promoter constructs deprived of binding sites for selected transcription factors was generated (Fig. 6D). Incubation with dasatinib decreased the activity of del3 (−313/+52 bp) truncated promoter, whereas lack of the −313/−198 bp region made the promoter resistant to dasatinib treatment (Fig. 6C, right). We calculated the fold change (dasatinib-treated/controls) for each truncation from 4–7 independent experiments. The results clearly show that both wild-type and truncated del3 promoter respond to dasatinib treatment to a similar extent (fold changes 0.516 ± 0.048 and 0.548 ± 0.034, respectively), while the truncated del2, del1 and del0 promoters are much less susceptible to dasatinib treatment (fold changes 0.843 ± 0.105, 0.939 ± 0.075 and 1.173 ± 0.232, respectively). Since the -313/-198 bp region is known to contain a putative Oct-1/Oct-2 transcription factor binding site (BAT-box), we introduced mutations in the BAT-box sequence reported to abolish binding of transcription factors.15 Although the basal promoter activity of luciferase in Raji cells transfected with this construct was decreased when compared with wild type CD20 promoter (Fig. 6E), somewhat unexpectedly dasatinib reduced the activity of both wild type and BAT-box mutated CD20 promoter. Also in EMSA assay we observed no differences in the migration of protein-DNA complexes between nuclear extracts from control and dasatinib-treated Raji cells (Fig. 6F). Collectively, our results indicate that SFKs inhibitors decrease CD20 expression through inhibition of CD20 promoter activity. Moreover, we confirmed that transcription factors bound to BAT-box are important regulators of basal CD20 expression, but they are not the major mediators of the CD20 downregulation caused by SFKs inhibitors. The results of our experiments indicate that dasatinib mediates its effects on CD20 promoter and regulates the activity of the -313/-198 bp region of the promoter via novel, yet to be identified, transcription factor(s).
Figure 6.
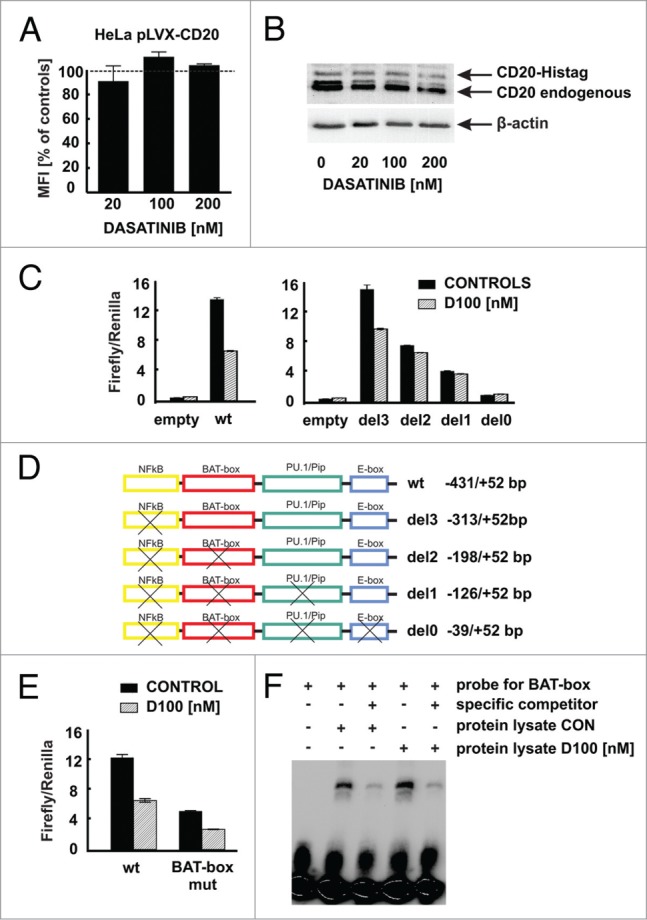
Modulation of CD20 expression by dasatinib requires CD20 promoter. (A) HeLa cells stably transduced to express CD20 (HeLa pLVX-CD20-IRES-PURO) were incubated for 48 h with increasing concentrations of dasatinib. Surface CD20 levels were determined as described earlier. (B) Protein lysates from Raji cells modified to express CD20-Histag fusion protein pre-incubated for 48 h with increasing concentrations of dasatinib were separated in polyacrylamide gel. CD20 and CD20-Histag proteins were detected with anti-CD20 antibody. (C) Relative luciferase activity was measured in lysates from Raji cells transfected with either empty vector or pGL4-wild type CD20 promoter or with pGL4-truncated CD20 promoters and further incubated with dasatinib (100 nM) for subsequent 24 h. (D) Scheme of truncated CD20 promoters used for reporter assays. (E) Relative luciferase activity was measured in lysates from Raji cells transfected with either pGL4-wild type or mutated (BAT-box mut) CD20 promoters incubated with dasatinib (100 nM) for subsequent 24 h. (F) Nuclear lysates from Raji cells either control or incubated for 24 h with dasatinib (100 nM) were mixed with biotinylated BAT-box probe in presence or absence of specific competitor. Formed DNA-protein complexes were analyzed for the binding of the proteins to the putative Oct-2 binding site in the CD20 promoter. Shown is one representative of at least 2 independent experiments.
Effects of SFKs inhibitors on CD20 levels is reversible and could be overcome by activation of AKT
Flow cytometry of Raji cells incubated for 48 h with dasatinib followed by washout periods of 0 to 96 h revealed that downregulation of CD20 levels is reversible (Fig. 7A). Since SFKs are reported to regulate multiple kinases we assessed the influence of selected signaling pathways inhibitors on the surface CD20 levels, including salirasib (RAS inhibitor), U0126 (MEK1/2 inhibitor), Ro 32–0432 (PKC inhibitor), BAY 11–7085 (NF-κB inhibitor), rapamycin (mTOR inhibitor) (Fig. S11) and MK-2206 (AKT inhibitor) (Fig. 7B). Interestingly, of all inhibitors tested only MK-2206 downregulated surface CD20 levels to the extent similar to dasatinib (Fig. 7B). We further confirmed that short incubation with dasatinib apart from inhibiting phosphorylation of SFKs at Tyr416 also inhibited AKT phosphorylation at Ser473 (Fig. 7C). Upon incubation with dasatinib the activity of CD20 promoter in Raji cells with a constitutively active AKT1 was almost 3-fold higher than in Raji cells co-transfected with control plasmid (Fig. 7D). PTEN, a negative regulator of AKT, decreased the activity of CD20 promoter, while expression of C124S-mutated form of PTEN had no influence on CD20 promoter activity (Fig. 7E). Given the crucial role of AKT in regulation of CD20 levels we have overexpressed a constitutively active AKT1 (myrAKT) in Raji cells (Fig. 7F). Raji pMIG-GFP cells exhibited baseline AKT phosphorylation that was inhibited by dasatinib, while in Raji pMIG-myrAKT cells AKT phosphorylation was high and not affected by dasatinib. Moreover, in Raji pMIG-myrAKT cells dasatinib failed to downregulate total CD20 levels (Fig. 7G). The same effects were observed at surface protein levels (Fig. 7H) as well as mRNA levels (Fig. 7I). Consistently, myrAKT expression sensitized Raji cells to R-CDC (Fig. 7J). Collectively, these data indicate that dasatinib causes a reversible, concentration-dependent decrease in CD20 levels, which is mitigated by stimulation of AKT signaling pathway.
Figure 7.

Effect of SFKs inhibitors on CD20 levels is rescued by activation of SFKs and BCR downstream signaling pathways. (A) Raji cells pre-incubated with 100 nM dasatinib for 48 h were washed and subsequently incubated without dasatinib for indicated times. CD20 levels were determined as described earlier. (B) Raji cells pre-incubated for 48 h with increasing concentrations of AKT inhibitor (MK-2206) were analyzed for surface CD20 levels as previously described. (C) p-AKT (Ser473), pan AKT and p-SRC (Tyr416) were detected with corresponding antibodies in protein lysates from Raji cells pre-incubated with dasatinib. (D) Relative luciferase activity was measured in protein lysates of Raji cells co-transfected with pGL4-wt CD20 promoter and either pMIG or pMIG-myrAKT and incubated with dasatinib for 48 h. (E) Relative luciferase activity was measured in protein lysates of Raji cells co-transfected with pGL4-wt CD20 promoter and either empty vector, wt PTEN or C124S PTEN and incubated with dasatinib for 48 h. (F) Raji cells stably transduced with either pMIG or pMIG-myrAKT were sorted based on GFP expression. AKT, p-AKT (Thr308, Ser473), CD20, β-actin were detected with corresponding antibodies in protein lysates from unsorted and sorted cells. (G) p-AKT, AKT, CD20 and β-actin were detected with corresponding antibodies in protein lysates from sorted Raji cells (Raji pMIG or Raji pMIG-myrAKT) pre-incubated with dasatinib for 24 h. (H) Surface CD20 levels in Raji pMIG or Raji pMIG-myrAKT cells pre-incubated with dasatinib for 48 h were determined as described earlier. Results are presented as PE MFI (± SD) of GFP-positive cells. (I) cDNA from Raji pMIG and Raji pMIG mAkt cells pre-incubated for 24 h with dasatinib was used for qRT-PCR amplification of CD20, ACTB and RPL29 products. (J) CDC assay was performed using Raji pMIG or Raji pMIG-myrAKT cells as previously described. Shown is one representative of at least 2 independent experiments.
Discussion
The most important finding of this study is that SFKs inhibitors strongly downregulate CD20 levels in tumor cells, leading to decreased binding of anti-CD20 mAbs to the surface CD20 and to impaired activation of antitumor effector mechanisms of the immune system. While impaired CDC mostly results from decreased CD20 levels, inhibition of ADCC is caused by impairment of cytolytic activity of NK cells.
Specificity of small-molecule inhibitors usually turns out to be rather limited, and sometimes even post-registration studies reveal their interactions with multiple kinases.16,17 This promiscuity is not necessarily a disadvantage as multi-kinase inhibitors were frequently shown to outperform selective compounds in terms of their antitumor activity. However, lack of specificity necessitates additional studies that can reveal unexpected interactions with approved therapeutic approaches, either positive or antagonistic, or even aggravating toxicity. SFKs have become particularly promising therapeutic targets for small-molecule inhibitors. Several studies revealed that SFKs are important for lymphoma growth in vitro18 and in vivo in a mouse model.19 LYN accounts for the majority of cellular SFKs activity in CLL cells,3 and constitutive BCR signaling in B-cell lymphoma cells is likely due to sustained activation of LYN. Accordingly, various SFKs inhibitors are currently being tested in clinical trials. For example, bafetinib (a dual BCR-ABL/LYN inhibitor) and dasatinib (BCR/ABL and SFKs inhibitor) are evaluated as single agents in relapsed and refractory CLL, DLBCL, NHL and MM patients.
Surprisingly, although CD20 has been reported to indirectly interact with LYN, LCK and FYN,20 the role of SFKs in CD20 regulation has not been investigated so far. To better characterize molecular mechanisms of SFKs inhibitors we used microarray analyses of gene expression in lymphoma cells and observed a strong downregulation of CD20 mRNA levels in cells incubated with either dasatinib or PP2. Since dasatinib is currently evaluated in the treatment of relapsed/refractory CLL patients in combination with rituximab, we decided to analyze the effect of SFKs inhibitors on CD20 levels in more detail.
In our study dasatinib was used at concentrations detected in CML patients taking FDA-approved doses.18 In flow cytometry an impaired binding of all tested antibodies was observed irrespectively of the recognized epitope, including murine L27 antibody, chimeric rituximab and fully human ofatumumab. Intriguingly, although binding of rituximab and ofatumumab was diminished to a similar extent, CDC of ofatumumab was impaired to a lesser degree when compared with rituximab. Ofatumumab has been widely reported to activate CDC more efficiently than rituximab, and a recent study indicates that triggering of CDC is a major mechanism of ofatumumab-mediated antitumor effects.21,22 Our data support the enhanced potential of ofatumumab to mediate CDC, when compared with rituximab.
While CD20 levels appear to play an important role in CDC, they seem to be less relevant for ADCC.23 In clinically relevant conditions, when both NK and tumor cells were co-incubated with dasatinib, we observed a completely blocked NK cell cytotoxicity, cytokines release and impaired NK cell function in ADCC mechanism. Indeed, inhibition of SFKs has been already shown to impair immune function via suppression of T-cells, neutrophils and spontaneous NK cytotoxicity, both in vitro and in vivo.24,25 We also confirmed previous observations that the effect of dasatinib on NK function is largely reversible.24 Pre-incubation of NK cells for 24 h with dasatinib followed by intense washing almost completely restored cytotoxicity of NK cells (unpublished observations). In the same vein, it has been reported that pretreatment of NK cells with dasatinib followed by a washout of the drug leads to amplification of cytokine production and NK cell degranulation.26 Moreover, a significant expansion of large granular lymphocytes (LGL) (both CD8+ cells and NK cells) has been observed during dasatinib therapy in CML patients. Clearly, to better understand the effects of dasatinib, it seems necessary to study its influence on NK cell functions in animal models and clinical trials. Since both CDC and ADCC mechanisms are crucial to mediate antitumor effects of anti-CD20 mAbs in vivo, we employed a murine model of EL4 lymphoma cells expressing human CD20 and observed that a 4-d dasatinib treatment significantly impaired antitumor activity of rituximab. Considering constitutive expression of CD20 in this model it can be surmised that dasatinib-mediated attenuation of antitumor effects of rituximab results from impaired activity of NK cells.
Flow cytometry clearly demonstrated that levels of various surface antigens remained roughly unchanged upon dasatinib treatment. Importantly, complement inhibitors CD46 and CD55, as well as other B-cell markers (HLA-DR, CD38, CD52, CD19) were only slightly downregulated. However, we observed downregulation of CD21 and CD22, of which CD22 is currently used as a target for toxin-conjugated anti-CD22 mAbs (inotuzumab ozogamicin). Taken together, our results suggest that SFKs inhibitors could potentially reduce the activity of therapeutic schemes employing anti-CD38, anti-CD52 or anti-CD19 mAbs through impairment of ADCC mechanism. Moreover, activity of anti-CD22 immunotoxins could be reduced due to CD22 downregulation. The significance and mechanisms of these phenomena require further studies.
Proteomic approaches have demonstrated that dasatinib, developed initially to target BCR/ABL, can inhibit the activity of other tyrosine kinases including BTK, CSK, as well as Ser/Thr kinases like p38 MAPK.27,28 Therefore, we used shRNA to specifically knock-down LYN, FYN or LCK and demonstrated that these kinases are important regulators of CD20 levels in B-cell tumors. Moreover, the results of experiments performed with a large set of malignant B-cell tumors indicated that dasatinib downregulated CD20 in a series of Burkitt's lymphoma (BL) cell lines, as well as in majority of tested DLBCL cell lines and primary tumor cells.
Using a series of truncated and mutated CD20 promoters we revealed that the −313 to −198 bp region of CD20 promoter is indispensable for dasatinib effects on CD20 mRNA levels. Surprisingly, none of the transcription factors known to regulate CD20 expression seemed to mediate dasatinib effects. Given the role of SFKs in the regulation of PI3K signaling pathway we focused on AKT and observed that constitutively active myrAKT largely protected Raji cells from dasatinib-triggered CD20 downregulation. Our results highlight the role of AKT in SFKs-dependent transcriptional regulation of CD20. Moreover, we also hypothesize that PI3K/AKT signaling pathway is implicated in the regulation of transcription factor(s) important in CD20 transcription in tumor B-cells.
Although anti-CD20 directed therapies constitute a substantial improvement in the treatment of B-cell tumors, further attempts to improve rituximab efficacy depend on thorough studies of CD20 regulation to allow for translation of preclinical findings into the clinic. An increasing number of studies reveals that treatment with anti-CD20 mAbs may lead to an antigen-negative relapses.29-36 Transcriptional37,38 and epigenetics-dependent36,39,40 downregulation of CD20 has been observed in B-cell malignancies. Also mutations in MS4A1 gene can lead to antigen-loss variants in patients treated with rituximab-containing therapies.36,41,42 Other mechanisms of CD20 regulation include internalization43 or “shaving”.44 Finally, conformational changes or altered reorganization of CD20 within lipid rafts can be associated with impaired binding and reduced antitumor activity of anti-CD20 mAbs.9,45 All processes that cause CD20 downregulation could potentially impair antitumor activity of anti-CD20 mAbs and lead to resistance to rituximab. Here, we add another mechanism that seems to be of clinical significance, i.e., a drug-mediated transcriptional downregulation of CD20 levels.
The outcomes of our experiments shed new light on the regulation of CD20 level in B-cell malignancies. Moreover, in accordance with our previous study, we report here that inhibition of BCR signaling pathway on nearly every step of signal initiation and propagation considerably impairs antitumor activity of anti-CD20 mAbs.11 Our observations are supported by recently published data of Kohrt et al., where the combination of ibrutinib (BTK inhibitor) and rituximab was shown to be antagonistic in both in vitro and in vivo models.46 Notably, the combination of idelalisib (PI3Kδ inhibitor) and rituximab has been recently shown to improve progression-free survival, response rate and overall survival among patients with relapsed CLL in a Phase 3 study.47 However, this combination was compared only with placebo and rituximab group and antitumor activity of idelalisib as a single agent treatment was not assessed. Moreover, in this study a surprising finding of a reduction in rates of infusion-related toxicity from rituximab in the idelalisib plus rituximab group was reported. It has been already shown that infusion-related side-effects during rituximab treatment result from complement activation followed by macrophages and mast cells activation and cytokine release. Thus, lower frequency of infusion-related cytotoxicity may result from CD20 downregulation and impaired activation of R-CDC by idelalisib as we described earlier.11
In conclusion, our data presented here, together with results from other groups,46,47 suggest that it is utterly important to elucidate the molecular mechanisms of CD20 regulation and combine new agents with already used anti-CD20 mAbs in a planned manner tailored for individual patients. These interactions are particularly important considering an increasing number of small-molecule inhibitors of signaling pathways undergoing pre-clinical or clinical development. Many of those new therapeutic agents will be used in various combinations, pursued to optimize the therapeutic efficacy and to limit concomitant toxicities. However, optimal combinations of novel treatment modalities with already existing ones should take into account potential and yet to be identified “off-target” activity that could unexpectedly result in high toxicity or impaired outcomes of treatment.48
Materials and Methods
Cell culture
All cells were cultured at 37°C in a humidified atmosphere of 5% CO2 in appropriate media (Table S3). Primary cells from patients with B-cell tumors (CLL, n = 25; mantle cell lymphoma (MCL), n = 4; follicular lymphoma (FL), n = 1) or healthy donors PBMC were isolated from blood or bone marrow using Histopaque-1077 (Sigma Aldrich). Approval for the study was obtained from the Institutional Review Board of the Medical University of Warsaw and was conducted according to the Declaration of Helsinki. Each patient signed an informed consent for the procedures.
Reagents
Rituximab was purchased from Roche. Alemtuzumab (Genzyme) and ofatumumab (GlaxoSmithKline) were purchased from the hospital pharmacy of the Medical University of Warsaw, Poland. Dasatinib (LC Laboratories), bafetinib (Adooq Bioscience), PP2 (Calbiochem), U0126, MK-2206 and everolimus (SelleckChem), Ro 32–0432 and BAY 11–7085 (TOCRIS Bioscience), Salirasib (Caymanchem) were all dissolved in DMSO. Propidium iodide (PI), puromycin, mitomycin C and cycloheximide (Sigma Aldrich) were dissolved in water. Carboxyfluorescein succinimidyl ester (CFSE) (Invitrogen) and actinomycin D (Sigma Aldrich) were dissolved in DMSO.
Gene expression microarray
RNA was isolated using a Universal RNA Purification Kit (EurX) according to the manufacturer's protocol. The samples were treated with DNaseI to eliminate a possibility of DNA contamination and subsequently purified using RNeasy MiniElute Cleanup Kit (Qiagen). Labeling, hybridization, acquisition and analysis were performed as described in Supplementary Information. The unpaired t test with Benjamin-Hochberg FDR <5% (false discovery rate) correction was applied (with P value cut-off <0.01). Only genes with at least a 3-fold change in the expression level in all the examined samples were further analyzed. The data has been deposited in NCBI's Gene Expression Omnibus and is accessible via GEO Series accession number GSE50929.
Cytotoxicity assay
Propidium iodide (PI) staining in the CDC assay was performed as described earlier.11 Cell viability was analyzed using a flow cytometer (FACScan, BD Biosciences, La Jolla, CA, USA). ADCC, NK cells degranulation and cytokine production was evaluated in a co-incubation model as described earlier11 and in Supplementary Information.
Staining of surface antigens
Cells were incubated with saturating amounts of fluorochrome-conjugated Abs (Table S4) for 30 min. Binding of unconjugated mAbs (rituximab, ofatumumab and alemtuzumab - final concentration of 100 μg/ml and anti-IgM - final concentration of 10 μg/ml) for 30 min was followed by 30 min incubation with Alexa Fluor® 488-conjugated AffiniPure F(ab’)2 Goat anti-Human IgG (H+L) (Jackson ImmunoResearch). Prior to analysis cells were washed and resuspended in PBS. Cells were analyzed on a FACScan, BD. The MFI of PI-negative cells served as a measure for mAb binding on a per-cell basis.
Quantitative Real-time PCR
Cells were washed with PBS, pelleted, and resuspended in 1 ml of TRIzol Reagent (Invitrogen) to extract total RNA according to the manufacturer protocol. cDNA synthesis and real-time PCR were done using either LightCycler® Fast Start DNA Master PLUS SYBRGreen I (Roche) and specific intron-spanning primers or LightCycler® Probes Master (Roche) supplemented with mix of primers (Tables S5–7) and probes labeled with FAM and DABCYL as described earlier.49 The results were analyzed after 40 cycles of amplification and normalized to the level of the housekeeping genes (B2M, RPL29 and ACTB for qRT-PCR using SYBRGreen and B2M for qRT-PCR using hydrolysis probes).
Western blotting
Cells were washed with PBS, pelleted, lysed and separated on 10% SDS-polyacrylamide gel as described elsewhere.11 Anti-CD20 (polyclonal, Abcam), anti-pSRC family Tyr416, anti-pAKT Ser473 (D9E) or Thr308 (C31E5E), anti-pan AKT (11E7) antibodies all from Cell Signaling at 1:1000 or 1:2000 dilutions were used for the overnight incubation at 4°C either in the presence of 5% BSA or 5% nonfat dry milk. After washing with TBST, membranes were incubated with anti-rabbit HRP-coupled secondary antibody (Jackson ImmunoResearch). The chemiluminescence reaction for HRP was developed using luminol-based chemiluminescence reagent and visualized with Stella 8300 bioimager (Raytest, Straubenhardt, Germany). The blots were re-probed with anti-β-actin-peroxidase purified immunoglobulin (clone AC-15, Sigma Aldrich) at 1:50000 dilution for 45 min.
Plasmids
Wild type CD20 promoter (-431/+52 bp) was synthesized by Epoch Life Science (Missouri City, TX, USA). The truncated promoters were generated by PCR amplification from the wild type promoter (Table S8). Wild type and truncated promoters were cloned into pGL4 promoter-less Firefly luciferase reporter plasmid (Promega). Mutated CD20 promoter and CD20-Histag constructs were generated as described in Supplementary Information. The EL4 lymphoma cell line was transduced with the pMX-hCD20 vector as described earlier.23 A retroviral vector containing the human CD20 cDNA under the control of the Moloney murine leukemia virus (Mo-MuLV) LTR was used to modify EL4 lymphoma cells.50 For introduction of constitutively active AKT1 isoform into Raji cells pMIG-GFP and pMIG-myrAKT1-IRES-GFP (kindly provided by Dr. Kira Gritsman from DFCI,51 thereafter named pMIG and pMIG-myrAKT, respectively) were used. The pMIG-myrAKT plasmid encodes a constitutively active AKT1 that is fused in-frame with SRC myristoylation domain and localized to cell membrane. The pGW1-myc-PTEN constructs encoding human PTEN (wild type or C124S mutant lacking the phosphatase activity) were kindly provided by Prof. Jacek Jaworski.52 The mutant PTEN (C124S) was generated by site-directed mutagenesis. The sequences of all constructs were confirmed by DNA sequencing.
Lentiviral/retroviral transduction of target cells
Lentiviruses and retroviruses were produced as described in Supplementary Information. To select gene of interest-containing cells either puromycin selection or sorting for GFP-positive cells on FACSAria III cell sorter (BD Biosciences, La Jolla, USA) was performed 5 d post transduction. HeLa cells modified with pLVX-CD20-IRES-PURO construct (HeLa pLVX-CD20) were cultured in DMEM medium supplemented with 8 μg/ml puromycin. Raji cells modified with pLVX-CD20-Histag-IRES-PURO (Raji CD20-Histag) were cultured in RPMI medium supplemented with 2 μg/ml puromycin.
Luciferase assay
Raji cells were nucleofected with the plasmid mix consisting of CD20 promoter Firefly luciferase reporter and Renilla luciferase reporter (pRL-TK) using the Cell Line Nucleofector Kit V (Lonza) and Amaxa Nucleofector Technology (Lonza, Basel, Switzerland). To introduce constitutively active AKT1 isoform the nucleofection mix was supplemented with pMIG-myrAKT or control pMIG-GFP vectors. For PTEN overexpression pGW1-myc-PTEN (wild type or C124S mutant) vectors were used. 12 h after transfection cells were treated with either dasatinib (100 nM) or DMSO and lysed with Passive Lysis Buffer (Promega) 24–48 h later. The relative luciferase activity was measured using a Dual-Luciferase Reporter Assay System (Promega) and a multilabel reader (VictorTM X4, PerkinElmer, Waltham, MA, USA).
EMSA
Nuclear proteins were extracted from Raji cells using NE-PER nuclear and cytoplasmic extraction kit (Pierce Chemical) with a Complete® protease inhibitor cocktail (Roche Diagnostics). An oligonucleotide probe containing the BAT-box consensus region in CD20 promoter (sequence 5′- GAA GTT CTT CTA ATT AAG GTA GC-3′) was 5′-labeled with biotin by Metabion International AG (Planegg-Martinsried, Germany). The biotin-labeled and unlabeled nucleotides were cooled down in the water bath from 95 to 25°C for 3 h to form double-stranded probes. 10 fmol of biotin-labeled probe was incubated with 6 μg of the nuclear extract for 20 min at room temperature. To confirm the specificity of binding, the extract-probe mixtures were further admixed with a 200-fold excess of the unlabeled, consensus probe. All the binding reactions were enriched with poly dI·dC nucleotides to eliminate any unspecific interactions between probe and protein lysate. After 25 min of incubation mixtures were resolved in 6% polyacrylamide/TBE gel and immediately transferred onto positively charged nylon membrane (Pierce Chemical) for 30 min at 380 mA. DNA on the membrane was then cross-linked using 312 nm UV lamp for 15 min. Detection of transcription factor-oligonucleotide complexes was performed using a LightShift chemiluminescent EMSA kit (Pierce Chemical), according to the manufacturer's protocol. The position of the DNA-protein complexes on the membrane was visualized using streptavidin-HRP conjugate and enhanced luminol substrate with Stella 8300 bioimager.
Mice, CD20 lymphoma model and therapy
For in vivo experiments 7–8-wk-old C57BL/6 female mice were used. Mice were obtained from the Animal Facility of the Mossakowski Medical Research Centre, Polish Academy of Sciences (Warsaw, Poland). On day 0 of the experiment, mice were injected into the tail vein with 5 × 105 of mouse EL4 lymphoma cells, stably expressing human CD20 and Firefly luciferase. Mice were randomized to groups of 6–8 animals that were treated with dasatinib and rituximab, or to control groups with single-agent treatment or no treatment (PBS). Mice were intraperitoneally (i.p.)-administered dasatinib (50 mg/kg) daily from day 3 until day 6 for a total of 4 doses; rituximab was administered i.p. (10 mg/kg), on days 3 and 6. Bioluminescence of EL4 cells was measured 4 times a week (starting from the day of injection). For in vivo imaging of EL4 growth, mice were injected (i.p.) with d-luciferin (150 mg/kg), and after 5 min were anaesthetized with isoflurane and visualized using IVIS® Imaging System (Xenogen, Alameda, CA, USA). Images were analyzed with the Living Image 4.2 software package (Caliper Life Science, Hopkinton, MA, USA). To quantify the bioluminescence (BLI) of mice, the regions of interest (ROIs) were drawn on the whole body and the results were used to generate the growth curve of EL4 lymphoma. Luminescence data were presented as average radiance (photons/sec/cm2/steradian). All in vivo experiments were performed in accordance with the guidelines and approved by the local Ethics Committee.
Disclosure of Potential Conflicts of Interest
The authors state they have no conflict of interest.
Author Contributions
MW, KB, BP designed, performed and analyzed experiments. JB, MS, MD, MB, NM, PZ, AS and AZ performed experiments. Microarray analysis was conducted by MK, sorting of transduced cells was performed by PP-B. In vivo experiments were performed by ZP with contribution of TPR and JHWL. AD-I, PJ, MS and TZ provided primary material and were involved in manuscript preparation. PJ and DGE provided expert advice and guidance throughout the study, ALR and DO contributed to the experiments using NK cells. JG supervised the project, designed experiments and analyzed the data. This manuscript was written by MW, KB and JG. All authors reviewed the manuscript.
Funding
The work was supported by National Science Center grant 2012/07/B/NZ6/03498 (MW), Polish Ministry of Science grant NN402352938 (MW), the European Commission 7th Framework Programme FP7-REGPOT-2012-CT2012–316254-BASTION (JG) and the Medical University of Warsaw grant 1M19/PM12D/13 (KB).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Yamanashi Y, Kakiuchi T, Mizuguchi J, Yamamoto T, Toyoshima K. Association of B cell antigen receptor with protein tyrosine kinase Lyn. Science 1991; 251:192-4; PMID:1702903; http://dx.doi.org/ 10.1126/science.1702903 [DOI] [PubMed] [Google Scholar]
- 2. Burkhardt AL, Brunswick M, Bolen JB, Mond JJ. Anti-immunoglobulin stimulation of B lymphocytes activates src-related protein-tyrosine kinases. Proc Natl Acad Sci U S A 1991; 88:7410-4; PMID:1714601; http://dx.doi.org/ 10.1073/pnas.88.16.7410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Contri A, Brunati AM, Trentin L, Cabrelle A, Miorin M, Cesaro L, Pinna LA, Zambello R, Semenzato G, Donella-Deana A. Chronic lymphocytic leukemia B cells contain anomalous Lyn tyrosine kinase, a putative contribution to defective apoptosis. J Clin Invest 2005; 115:369-78; PMID:15650771; http://dx.doi.org/ 10.1172/JCI200522094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tauzin S, Ding H, Khatib K, Ahmad I, Burdevet D, van Echten-Deckert G, Lindquist JA, Schraven B, Din NU, Borisch B, et al. . Oncogenic association of the Cbp/PAG adaptor protein with the Lyn tyrosine kinase in human B-NHL rafts. Blood 2008; 111:2310-20; PMID:18070987; http://dx.doi.org/ 10.1182/blood-2007-05-090985 [DOI] [PubMed] [Google Scholar]
- 5. Packard TA, Cambier JC. B lymphocyte antigen receptor signaling: initiation, amplification, and regulation. F1000Prime Rep 2013; 5:40; PMID:24167721; http://dx.doi.org/ 10.12703/P5-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li S. Src kinases as targets for B cell acute lymphoblastic leukaemia therapy. Expert Opin Ther Targets 2005; 9:329-41; PMID:15934919; http://dx.doi.org/ 10.1517/14728222.9.2.329 [DOI] [PubMed] [Google Scholar]
- 7. McLaughlin P, Grillo-López AJ, Link BK, Levy R, Czuczman MS, Williams ME, Heyman MR, Bence-Bruckler I, White CA, Cabanillas F, et al. . Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol 1998; 16:2825-33; PMID:9704735 [DOI] [PubMed] [Google Scholar]
- 8. Bil J, Winiarska M, Nowis D, Bojarczuk K, Dabrowska-Iwanicka A, Basak GW, Sułek K, Jakobisiak M, Golab J. Bortezomib modulates surface CD20 in B-cell malignancies and affects rituximab-mediated complement-dependent cytotoxicity. Blood 2010; 115:3745-55; PMID:20200358; http://dx.doi.org/ 10.1182/blood-2009-09-244129 [DOI] [PubMed] [Google Scholar]
- 9. Winiarska M, Bil J, Wilczek E, Wilczynski GM, Lekka M, Engelberts PJ, Mackus WJ, Gorska E, Bojarski L, Stoklosa T, et al. . Statins impair antitumor effects of rituximab by inducing conformational changes of CD20. PLoS Med 2008; 5:e64; PMID:18366248; http://dx.doi.org/ 10.1371/journal.pmed.0050064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beum PV, Peek EM, Lindorfer MA, Beurskens FJ, Engelberts PJ, Parren PW, van de Winkel JG, Taylor RP. Loss of CD20 and bound CD20 antibody from opsonized B cells occurs more rapidly because of trogocytosis mediated by Fc receptor-expressing effector cells than direct internalization by the B cells. J Immunol 2011; 187:3438-47; PMID:21841127; http://dx.doi.org/ 10.4049/jimmunol.1101189 [DOI] [PubMed] [Google Scholar]
- 11. Bojarczuk K, Siernicka M, Dwojak M, Bobrowicz M, Pyrzynska B, Gaj P, Karp M, Giannopoulos K, Efremov DG, Fauriat C, et al. . B-cell receptor pathway inhibitors affect CD20 levels and impair antitumor activity of anti-CD20 monoclonal antibodies. Leukemia 2014; 28:1163-7; PMID:24492323; http://dx.doi.org/ 10.1038/leu.2014.12 [DOI] [PubMed] [Google Scholar]
- 12. de Haij S, Jansen JH, Boross P, Beurskens FJ, Bakema JE, Bos DL, Martens A, Verbeek JS, Parren PW, van de Winkel JG, et al. . In vivo cytotoxicity of type I CD20 antibodies critically depends on Fc receptor ITAM signaling. Cancer Res 2010; 70:3209-17; PMID:20354182; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-4109 [DOI] [PubMed] [Google Scholar]
- 13. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, et al. . Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010; 463:88-92; PMID:20054396; http://dx.doi.org/ 10.1038/nature08638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen L, Monti S, Juszczynski P, Daley J, Chen W, Witzig TE, Habermann TM, Kutok JL, Shipp MA. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood 2008; 111:2230-7; PMID:18006696; http://dx.doi.org/ 10.1182/blood-2007-07-100115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thévenin C, Lucas BP, Kozlow EJ, Kehrl JH. Cell type- and stage-specific expression of the CD20/B1 antigen correlates with the activity of a diverged octamer DNA motif present in its promoter. J Biol Chem 1993; 268:5949-56; PMID:7680653 [PubMed] [Google Scholar]
- 16. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009; 9:28-39; PMID:19104514; http://dx.doi.org/ 10.1038/nrc2559 [DOI] [PubMed] [Google Scholar]
- 17. Peters EC, Gray NS. Chemical proteomics identifies unanticipated targets of clinical kinase inhibitors. ACS Chem Biol 2007; 2:661-4; PMID:18041816; http://dx.doi.org/ 10.1021/cb700203j [DOI] [PubMed] [Google Scholar]
- 18. Yang C, Lu P, Lee FY, Chadburn A, Barrientos JC, Leonard JP, Ye F, Zhang D, Knowles DM, Wang YL. Tyrosine kinase inhibition in diffuse large B-cell lymphoma: molecular basis for antitumor activity and drug resistance of dasatinib. Leukemia 2008; 22:1755-66; PMID:18596745; http://dx.doi.org/ 10.1038/leu.2008.163 [DOI] [PubMed] [Google Scholar]
- 19. Ke J, Chelvarajan RL, Sindhava V, Robertson DA, Lekakis L, Jennings CD, Bondada S. Anomalous constitutive Src kinase activity promotes B lymphoma survival and growth. Mol Cancer 2009; 8:132; PMID:20043832; http://dx.doi.org/ 10.1186/1476-4598-8-132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deans JP, Kalt L, Ledbetter JA, Schieven GL, Bolen JB, Johnson P. Association of 75/80-kDa phosphoproteins and the tyrosine kinases Lyn, Fyn, and Lck with the B cell molecule CD20. Evidence against involvement of the cytoplasmic regions of CD20. J Biol Chem 1995; 270:22632-8; PMID:7545683; http://dx.doi.org/ 10.1074/jbc.270.38.22632 [DOI] [PubMed] [Google Scholar]
- 21. Pawluczkowycz AW, Beurskens FJ, Beum PV, Lindorfer MA, van de Winkel JG, Parren PW, Taylor RP. Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): considerably higher levels of CDC are induced by OFA than by RTX. J Immunol 2009; 183:749-58; PMID:19535640; http://dx.doi.org/ 10.4049/jimmunol.0900632 [DOI] [PubMed] [Google Scholar]
- 22. Bologna L, Gotti E, Da Roit F, Intermesoli T, Rambaldi A, Introna M, Golay J. Ofatumumab is more efficient than rituximab in lysing B chronic lymphocytic leukemia cells in whole blood and in combination with chemotherapy. J Immunol 2013; 190:231-9; PMID:23225880; http://dx.doi.org/ 10.4049/jimmunol.1202645 [DOI] [PubMed] [Google Scholar]
- 23. van Meerten T, van Rijn RS, Hol S, Hagenbeek A, Ebeling SB. Complement-induced cell death by rituximab depends on CD20 expression level and acts complementary to antibody-dependent cellular cytotoxicity. Clin Cancer Res 2006; 12:4027-35; PMID:16818702; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-0066 [DOI] [PubMed] [Google Scholar]
- 24. Blake SJ, Bruce Lyons A, Fraser CK, Hayball JD, Hughes TP. Dasatinib suppresses in vitro natural killer cell cytotoxicity. Blood 2008; 111:4415-6; PMID:18398058; http://dx.doi.org/ 10.1182/blood-2008-02-138701 [DOI] [PubMed] [Google Scholar]
- 25. Fraser CK, Blake SJ, Diener KR, Lyons AB, Brown MP, Hughes TP, Hayball JD. Dasatinib inhibits recombinant viral antigen-specific murine CD4+ and CD8 +T-cell responses and NK-cell cytolytic activity in vitro and in vivo. Exp Hematol 2009; 37:256-65; PMID:19056158; http://dx.doi.org/ 10.1016/j.exphem.2008.09.013 [DOI] [PubMed] [Google Scholar]
- 26. Hassold N, Seystahl K, Kempf K, Urlaub D, Zekl M, Einsele H, Watzl C, Wischhusen J, Seggewiss-Bernhardt R. Enhancement of natural killer cell effector functions against selected lymphoma and leukemia cell lines by dasatinib. Int J Cancer 2012; 131:E916-27; PMID:22419518; http://dx.doi.org/ 10.1002/ijc.27537 [DOI] [PubMed] [Google Scholar]
- 27. Hantschel O, Rix U, Schmidt U, Bürckstümmer T, Kneidinger M, Schütze G, Colinge J, Bennett KL, Ellmeier W, Valent P, et al. . The Btk tyrosine kinase is a major target of the Bcr-Abl inhibitor dasatinib. Proc Natl Acad Sci U S A 2007; 104:13283-8; PMID:17684099; http://dx.doi.org/ 10.1073/pnas.0702654104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kneidinger M, Schmidt U, Rix U, Gleixner KV, Vales A, Baumgartner C, Lupinek C, Weghofer M, Bennett KL, Herrmann H, et al. . The effects of dasatinib on IgE receptor-dependent activation and histamine release in human basophils. Blood 2008; 111:3097-107; PMID:18180381; http://dx.doi.org/ 10.1182/blood-2007-08-104372 [DOI] [PubMed] [Google Scholar]
- 29. Seliem RM, Freeman JK, Steingart RH, Hasserjian RP. Immunophenotypic changes and clinical outcome in B-cell lymphomas treated with rituximab. Appl Immunohistochem Mol Morphol 2006; 14:18-23; PMID:16540725; http://dx.doi.org/ 10.1097/01.pai.0000145130.02931.74 [DOI] [PubMed] [Google Scholar]
- 30. Davis TA, Czerwinski DK, Levy R. Therapy of B-cell lymphoma with anti-CD20 antibodies can result in the loss of CD20 antigen expression. Clin Cancer Res 1999; 5:611-5; PMID:10100713 [PubMed] [Google Scholar]
- 31. Foran JM, Norton AJ, Micallef IN, Taussig DC, Amess JA, Rohatiner AZ, Lister TA. Loss of CD20 expression following treatment with rituximab (chimaeric monoclonal anti-CD20): a retrospective cohort analysis. Br J Haematol 2001; 114:881-3; PMID:11564080; http://dx.doi.org/ 10.1046/j.1365-2141.2001.03019.x [DOI] [PubMed] [Google Scholar]
- 32. Schmitz K, Brugger W, Weiss B, Kaiserling E, Kanz L. Clonal selection of CD20-negative non-Hodgkin's lymphoma cells after treatment with anti-CD20 antibody rituximab. Br J Haematol 1999; 106:571-2; PMID:10460626; http://dx.doi.org/ 10.1046/j.1365-2141.1999.01608.x [DOI] [PubMed] [Google Scholar]
- 33. Kinoshita T, Nagai H, Murate T, Saito H. CD20-negative relapse in B-cell lymphoma after treatment with Rituximab. J Clin Oncol 1998; 16:3916; PMID:9850038 [PubMed] [Google Scholar]
- 34. Haidar JH, Shamseddine A, Salem Z, Mrad YA, Nasr MR, Zaatari G, Bazarbachi A. Loss of CD20 expression in relapsed lymphomas after rituximab therapy. Eur J Haematol 2003; 70:330-2; PMID:12694172; http://dx.doi.org/ 10.1034/j.1600-0609.2003.00007.x [DOI] [PubMed] [Google Scholar]
- 35. Maeshima AM, Taniguchi H, Nomoto J, Maruyama D, Kim SW, Watanabe T, Kobayashi Y, Tobinai K, Matsuno Y. Histological and immunophenotypic changes in 59 cases of B-cell non-Hodgkin's lymphoma after rituximab therapy. Cancer Sci 2009; 100:54-61; PMID:19038008; http://dx.doi.org/ 10.1111/j.1349-7006.2008.01005.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hiraga J, Tomita A, Sugimoto T, Shimada K, Ito M, Nakamura S, Kiyoi H, Kinoshita T, Naoe T. Down-regulation of CD20 expression in B-cell lymphoma cells after treatment with rituximab-containing combination chemotherapies: its prevalence and clinical significance. Blood 2009; 113:4885-93; PMID:19246561; http://dx.doi.org/ 10.1182/blood-2008-08-175208 [DOI] [PubMed] [Google Scholar]
- 37. Jilani I, O’Brien S, Manshuri T, Thomas DA, Thomazy VA, Imam M, Naeem S, Verstovsek S, Kantarjian H, Giles F, et al. . Transient down-modulation of CD20 by rituximab in patients with chronic lymphocytic leukemia. Blood 2003; 102:3514-20; PMID:12893761; http://dx.doi.org/ 10.1182/blood-2003-01-0055 [DOI] [PubMed] [Google Scholar]
- 38. Pickartz T, Ringel F, Wedde M, Renz H, Klein A, von Neuhoff N, Dreger P, Kreuzer KA, Schmidt CA, Srock S, et al. . Selection of B-cell chronic lymphocytic leukemia cell variants by therapy with anti-CD20 monoclonal antibody rituximab. Exp Hematol 2001; 29:1410-6; PMID:11750099; http://dx.doi.org/ 10.1016/S0301-472X(01)00753-6 [DOI] [PubMed] [Google Scholar]
- 39. Ushmorov A, Leithäuser F, Sakk O, Weinhaüsel A, Popov SW, Möller P, Wirth T. Epigenetic processes play a major role in B-cell-specific gene silencing in classical Hodgkin lymphoma. Blood 2006; 107:2493-500; PMID:16304050; http://dx.doi.org/ 10.1182/blood-2005-09-3765 [DOI] [PubMed] [Google Scholar]
- 40. Tomita A, Hiraga J, Kiyoi H, Ninomiya M, Sugimoto T, Ito M, Kinoshita T, Naoe T. Epigenetic regulation of CD20 protein expression in a novel B-cell lymphoma cell line, RRBL1, established from a patient treated repeatedly with rituximab-containing chemotherapy. Int J Hematol 2007; 86:49-57; PMID:17675267; http://dx.doi.org/ 10.1532/IJH97.07028 [DOI] [PubMed] [Google Scholar]
- 41. Terui Y, Mishima Y, Sugimura N, Kojima K, Sakurai T, Mishima Y, Kuniyoshi R, Taniyama A, Yokoyama M, Sakajiri S, et al. . Identification of CD20 C-terminal deletion mutations associated with loss of CD20 expression in non-Hodgkin's lymphoma. Clin Cancer Res 2009; 15:2523-30; PMID:19276251; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-1403 [DOI] [PubMed] [Google Scholar]
- 42. Sar A, Perizzolo M, Stewart D, Mansoor A, Difrancesco LM, Demetrick DJ. Mutation or polymorphism of the CD20 gene is not associated with the response to R-CHOP in diffuse large B cell lymphoma patients. Leuk Res 2009; 33:792-7; PMID:19054557; http://dx.doi.org/ 10.1016/j.leukres.2008.10.013 [DOI] [PubMed] [Google Scholar]
- 43. Ivanov A, Beers SA, Walshe CA, Honeychurch J, Alduaij W, Cox KL, Potter KN, Murray S, Chan CH, Klymenko T, et al. . Monoclonal antibodies directed to CD20 and HLA-DR can elicit homotypic adhesion followed by lysosome-mediated cell death in human lymphoma and leukemia cells. J Clin Invest 2009; 119:2143-59; PMID:19620786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beum PV, Kennedy AD, Williams ME, Lindorfer MA, Taylor RP. The shaving reaction: rituximab/CD20 complexes are removed from mantle cell lymphoma and chronic lymphocytic leukemia cells by THP-1 monocytes. J Immunol 2006; 176:2600-9; PMID:16456022; http://dx.doi.org/ 10.4049/jimmunol.176.4.2600 [DOI] [PubMed] [Google Scholar]
- 45. Ringshausen I, Feuerstacke Y, Krainz P, den Hollander J, Hermann K, Buck A, Peschel C, Meyer Zum Bueschenfelde C. Antifungal therapy with itraconazole impairs the anti-lymphoma effects of rituximab by inhibiting recruitment of CD20 to cell surface lipid rafts. Cancer Res 2010; 70:4292-6; PMID:20460536; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-0259 [DOI] [PubMed] [Google Scholar]
- 46. Kohrt HE, Sagiv-Barfi I, Rafiq S, Herman SE, Butchar JP, Cheney C, Zhang X, Buggy JJ, Muthusamy N, Levy R, et al. . Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood 2014; 123:1957-60; PMID:24652965; http://dx.doi.org/ 10.1182/blood-2014-01-547869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, et al. . Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 2014; 370:997-1007; PMID:24450857; http://dx.doi.org/ 10.1056/NEJMoa1315226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reeder CB, Ansell SM. Novel therapeutic agents for B-cell lymphoma: developing rational combinations. Blood 2011; 117:1453-62; PMID:20978267; http://dx.doi.org/ 10.1182/blood-2010-06-255067 [DOI] [PubMed] [Google Scholar]
- 49. Winiarska M, Nowis D, Bil J, Glodkowska-Mrowka E, Muchowicz A, Wanczyk M, Bojarczuk K, Dwojak M, Firczuk M, Wilczek E, et al. . Prenyltransferases regulate CD20 protein levels and influence anti-CD20 monoclonal antibody-mediated activation of complement-dependent cytotoxicity. J Biol Chem 2012; 287:31983-93; PMID:22843692; http://dx.doi.org/ 10.1074/jbc.M112.374751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Di Gaetano N, Cittera E, Nota R, Vecchi A, Grieco V, Scanziani E, Botto M, Introna M, Golay J. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol 2003; 171:1581-7; PMID:12874252; http://dx.doi.org/ 10.4049/jimmunol.171.3.1581 [DOI] [PubMed] [Google Scholar]
- 51. Kharas MG, Okabe R, Ganis JJ, Gozo M, Khandan T, Paktinat M, Gilliland DG, Gritsman K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 2010; 115:1406-15; PMID:20008787; http://dx.doi.org/ 10.1182/blood-2009-06-229443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci 2005; 25:11300-12; PMID:16339025; http://dx.doi.org/ 10.1523/JNEUROSCI.2270-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.