

Abstract
Adoptive T-cell therapy of cancer often fails due to the tumor cells' immune escape mechanisms, like antigen loss or down-regulation. To anticipate immune escape by loss of a single antigen, it would be advantageous to equip T cells with multiple specificities. To study the possible interference of 2 T-cell receptors (TCRs) in one cell, and to examine how to counteract competing effects, we generated TETARs, CD8+ T cells expressing two additional T-cell receptors by simultaneous transient transfection with 2 TCRs using RNA electroporation. The TETARs were equipped with one TCR specific for the common melanoma antigen gp100 and one TCR recognizing a patient-specific, individual mutation of CCT6A (chaperonin containing TCP1, subunit 6A) termed “CCT6Am TCR.” These CD8+ T cells proved functional in cytokine secretion and lytic activity upon stimulation with each of their cognate antigens, although some reciprocal inhibition was observed. Murinisation of the CCT6Am TCR increased and prolonged its expression and increased the lytic capacity of the dual-specific T cells. Taken together, we generated functional, dual-specific CD8+ T cells directed against a common melanoma-antigen and an individually mutated antigen for the use in personalised adoptive T-cell therapy of melanoma. The intended therapy would involve repetitive injections of the RNA-transfected cells to overcome the transiency of TCR expression. In case of autoimmunity-related side effects, a cessation of treatment would result in a disappearance of the introduced receptors, which increases the safety of this approach.
Keywords: Adoptive T-cell therapy, anti-tumor activity, immune escape, immunotherapy, melanoma, neo-antigen, TCR transfer
Abbreviations
- ACT
Adoptive cell transfer
- DC
Dendritic cell
- CTL
Cytotoxic T lymphocytes
- TCR
T-cell receptor
- TETARs
T cells expressing two additional T-cell receptors.
Introduction
Cytotoxic T lymphocytes (CTL) specific for tumor antigens are crucial for cancer immunotherapy. As a patient's peripheral T-cell repertoire often lacks high-avidity, tumor-specific CTL due to thymic selection1 and because tumor-specific CTL are often anergic or show a limited life span, their expansion for therapeutic applications is laborious and often not feasible.1-3
Generating CTL of a desired specificity by equipping bulk T cells with new TCRs can solve these problems. Adoptive cell transfer4 (ACT) of CD8+ T cells reprogrammed in this way is a new experimental therapy for virus-induced, life-threatening diseases, like HIV-1, or different types of cancer. This therapeutic approach is in general performed using retroviruses or lentiviruses encoding T-cell receptors (TCRs) specific for the targeted antigens (reviewed by Hawkins et al.5 and Schumacher et al.6). Alternatively, these new specificities can be introduced into the CTL by mRNA-electroporation.7,8 This technique represents a safe and feasible method to generate tumor-specific CTL, as production and validation are more cost-effective than virus-production. These cells can easily be produced in therapeutic doses,9 and the risk of inducing autoimmunity is diminished by the transient expression of the new TCRs.
In the case of melanoma immunotherapy, targeting common melanoma-associated tumor antigens with CTL often leads to initial success with remission of the tumor,4 but also relapse occurs due to immunoediting mechanisms triggered in the tumor.10 Targeting individually mutated antigens in cancer patients might be a more promising approach in cancer immunotherapy,11 as T cells recognizing these mutated tumor antigens are not restricted by tolerance mechanisms,12 and as these mutations might also contribute to promote the cancer phenotype.13 Hence, a personalised therapy simultaneously targeting a common melanocytic antigen, and an individually mutated tumor antigen might represent a new strategy in melanoma therapy, as for immune escape, the tumor would have to lose both targeted antigens at the same time.
As the equipment of T cells with 2 additional TCRs bears the risk of competition for signaling molecules or for mispairing of the TCR chains, proper mRNA quantities for transfection have to be found and preferential pairing of the matching chains has to be forced. To prevent competitive effects between both introduced TCRs, we titrated the quantities of TCR-encoding mRNAs to obtain suitable ratios for transfection. Further, we wanted to enhance the correct pairing of the introduced TCR chains. This can be done by murinisation of one of the TCRs, a method we found to be reliable and effective in the context of virus-specific TETARs.14 Murinisation of one TCR was performed to prevent competitive mechanisms, like surface dilution of the introduced TCRs,15 between the transfected and the endogenous TCR. Murinisation improves preferential pairing of the introduced TCR chains and diminishes the risk of formation of low-affinity or autoreactive TCR-chain dimers.16,17 As tumor-associated antigen-specific TCRs are actually self-reactive and underlie thymic selection, it was unclear whether this approach is also applicable for tumor-specific TCRs. To figure out whether this strategy can be extended to the field of tumor therapy, we generated tumor-specific TETARs. Therefore, we simultaneously transfected CD8+ T cells by mRNA electroporation with one TCR specific for a common melanoma-associated antigen, gp100 (also called PMEL), and a second TCR specific for the individually mutated tumor antigen CCT6A. The mutated CCT6A peptide was chosen as a model for an individually mutated, patient-specific neo-antigen. The CCT6A gene encodes the chaperonin subunit TCP 1-zeta-6A (CCT6A) and this protein has been described to take part in drug resistance to chemotherapy of malignant melanoma.18 The CCT6Am antigen had previously been identified and proved to be immunogenic in a melanoma patient (unpublished data). We chose this specific antigen, as we intended to generate T cells specific for antigens of different categories, namely one individually mutated and one common melanoma antigen.
The generated TETARs showed antigen-specific cytokine secretion and cytotoxic activity upon recognition of both cognate peptides, and proved to be activated superiorly upon antigen contact compared to a mixture of CTL transfected with either the gp100-specific TCR or the CCT6Am-specific TCR by both peptides.
Results
Our aim was to generate CD8+ T cells which express two additional T-cell receptors (TETARs), one specific for a common melanoma antigen, gp100, and one specific for an individually mutated tumor antigen, CCT6Am, by RNA electroporation.
Tumor-specific TETARs can be generated with 2 human TCRs or with a human TCR combined with a partially murinised TCR
The generation of TETARs requires the finding of proper quantities and ratios of TCR-RNAs for transfection to minimise competitive effects between the transfected TCRs. Therefore, we electroporated CD8+ T cells with RNA encoding the human gp100 TCR, the human (h)CCT6Am TCR, or the murinised (m)CCT6Am TCR, either alone or in combination (i.e. hTETARs were transfected with gp100 TCR and hCCT6Am TCR, whereas mTETARs were transfected with gp100 TCR and mCCT6Am TCR), and stimulated these TCR-transfected CD8+ T cells with HLA-A*0201+ HLA-B*27+ EBV-transformed B cells (D05-EBV) either loaded with the gp100 peptide or the CCT6Am peptide. The effector cells were used for stimulation 4, 24, and 48 h after electroporation (Fig. 1, Fig. S1). T cells transfected with only one type of TCR secreted IL-2 (Fig. 1), TNF (Fig. S1A), and IFNγ (Fig. S1B) upon antigen-specific stimulation. The amount of secreted cytokines decreased over time after electroporation (Fig. 1, Fig. S1A and B). Thereby, T cells transfected with the mCCT6Am TCR displayed a longer-lasting response to the CCT6Am peptide than T cells transfected with the hCCT6Am TCR. Similarly, mTETARs also showed cytokine secretion for a prolonged time compared to hTETARs upon stimulation with CCT6A peptide (Fig. 1, Fig. S1A). Four hours after electroporation, the T cells equipped only with gp100 TCR, as well as the mTETARs and the hTETARs, secreted high quantities of cytokines upon stimulation with the gp100 peptide, and cytokine secretion decreased at later time points. (Fig. 1, Fig. S1A). The transfection with different ratios of RNA encoding the gp100 TCR and the CCT6Am TCRs revealed a ratio of 10 µg gp100 TCR RNA and 20 µg hCCT6Am TCR RNA or 20 µg mCCT6Am TCR to be the most suitable combination for generating TETARs. Both, the hTETARs and the mTETARs, recognized the gp100 antigen specifically and secreted similar amounts of IL-2, TNF, and IFNγ, and this secretion sustained for a similar time compared to T cells only transfected with the gp100 TCR (Fig. 1, Fig. S1A and B). Upon stimulation with the CCT6Am peptide, the T cells transfected only with the CCT6Am TCR secreted similar amount of IL-2 as TETARs after 4 h after transfection, but not at the later time points (Fig. 1). The same effect was seen for TNF, but to a lesser extent (Fig. S1A). IFNγ secretion stayed similar for TETARs and T cells transfected with only one TCR upon stimulation with both cognate peptides at all time points (Fig. S1B). At the other indicated RNA ratios, competitive effects between the 2 TCRs inhibited proper cytokine secretion, especially after stimulation with CCT6Am peptide-loaded target cells, indicating that the gp100 TCR is dominant over the CCT6Am TCR (Fig. 1, Fig. S1A and B). These competitive effects refer to the hCCT6Am TCR and, although to a lower extent, also to the mCCT6Am TCR (Fig. 1, Fig. S1A and B). For these reasons, we decided to use TETARs generated by transfection with 10 µg gp100 TCR RNA and 20 µg CCT6Am TCR RNA for all further experiments. These results show that tumor-specific TETARs can be generated by RNA electroporation and secrete cytokines antigen-specifically upon stimulation with both cognate antigens.
Figure 1.
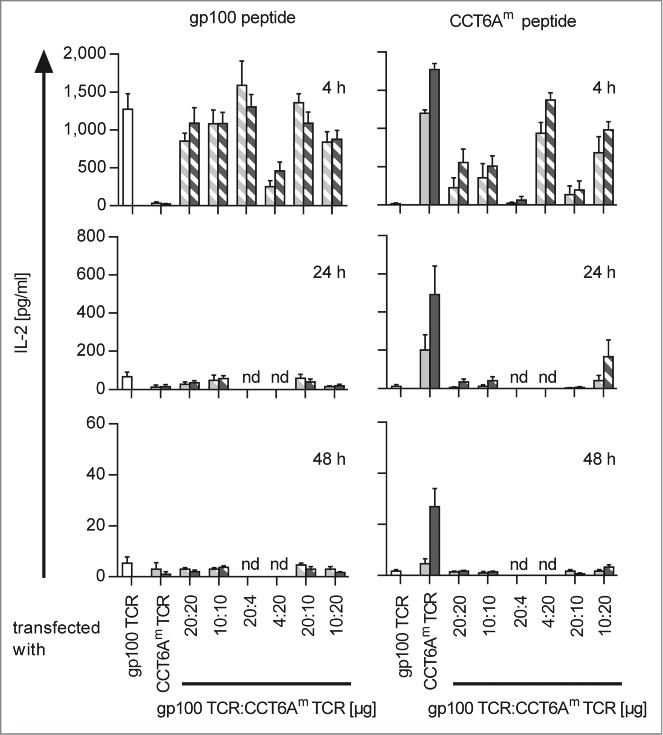
Antigen-specific IL-2 secretion efficiency of TETARs is dependent on the ratios of the 2 transfected TCRs. CD8+ T cells were equipped with single TCRs (plain-colored columns): the gp100 TCR (white columns), the human CCT6AmTCR (light gray columns), or the murinised CCT6Am TCR (dark gray columns). TETARs (hatched columns) were generated by transfecting CD8+ T cells with the gp100 TCR in combination with the human CCT6AmTCR (white and light gray), or with the murinised CCT6AmTCR (white and dark gray) by electroporation of the indicated quantities of RNA. IL-2 secretion was measured in a cytometric bead array (CBA) after stimulation with either gp100 peptide- or CCT6Am peptide-loaded D05-EBV cells for 4, 24, or 48 h. Average values of 3 (24 and 48 h) or 4 (4 h) independent experiments ± SEM are depicted. Some conditions were not determined at 24 h and 48 h (nd).
Tumor-specific TETARs show increased activity compared to mixed T cells transfected with a single TCR after stimulation with their cognate antigens
The next step was to show that one individual TETAR cell is indeed able to recognize the epitopes with both receptors and that TETARs are hence activated more efficiently than a mix of T cells, with each T cell transfected with only one of the TCRs. This was done by determination of the antigen-specific induction of CD25, an activation marker on T cells. Additionally, the lytic capacities of TETARs and a mix of T cells expressing only one TCR were compared. Therefore, hTETARs and mTETARs were compared with T cells transfected with only one type of TCR, and with 1:1-mixtures of these T cells (hT-cell mix containing T cells transfected with the gp100 TCR and T cells transfected with the hCCT6Am TCR, and mT-cell mix containing T cells transfected with the gp100 TCR and T cells transfected with the mCCT6Am TCR). In all cases we investigated CD25 expression after stimulation with target cells loaded with gp100 peptide (Fig. 2A) or CCT6Am peptide (Fig. 2B). CD8+ T cells transfected with only one type of TCR showed an increase in CD25 expression after activation with their cognate antigen (Fig. 2A and B), with a slightly higher percentage of mCCT6Am TCR-transfected T cells being activated after stimulation with CCT6Am peptide compared to T cells transfected with the hCCT6Am TCR (Fig. 2B). Compared to the hT-cell mix, the hTETARs revealed a significantly higher percentage of CD25-positive cells upon activation by gp100 peptide-loaded target cells (Fig. 2A). The same was true for the mTETARs and the mT-cell mix (Fig. 2A). Upon stimulation with CCT6Am peptide, mTETARs showed a higher percentage of CD25-positive cells than hTETARs (Fig. 2B). Although scarcely missing significance, the mTETARs showed a higher percentage of CD25-positive cells than the mT-cell mix (p = 0.0712) (Figure 2B). The difference between hTETARs compared with hT-cell mix was less distinctive (p = 0.2340) (Fig. 2B).
Figure 2.
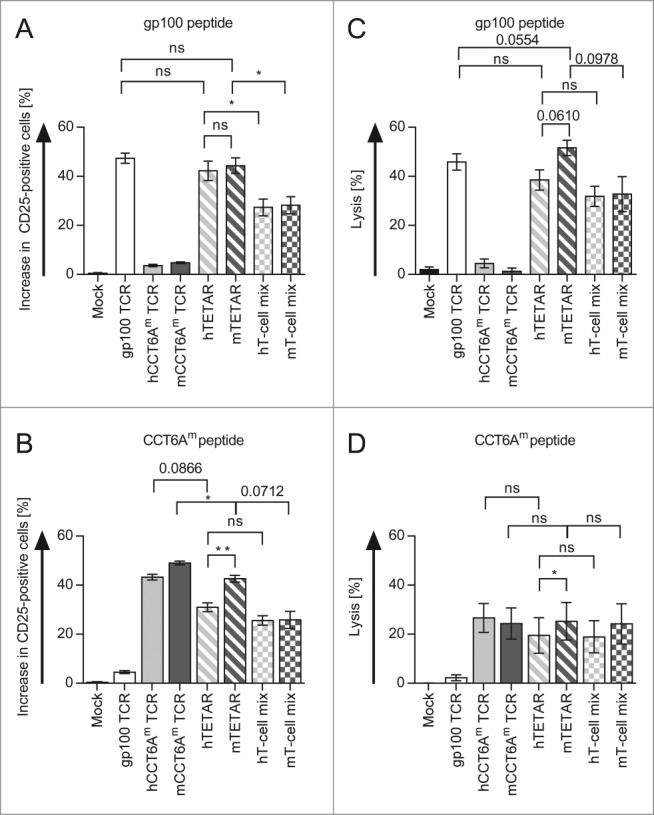
TETARs can be activated more efficiently than a mixture of 2 pools of CD8+ T cells each reprogrammed with only one specificity. CD8+ T cells were RNA-transfected with the single TCRs (plain-colored columns): the gp100 TCR (white columns), the human CCT6Am (hCCT6Am) TCR (light gray column), or the murinised CCT6Am (mCCT6Am) TCR (dark gray columns). TETARs (hatched columns) were generated by transfecting CD8+ T cells with the gp100 TCR in combination with the human CCT6Am TCR (hTETAR; white and light gray), or the murinised CCT6Am TCR (mTETAR; white and dark gray). As a control, T cells were mock electroporated. Additionally, T cells transfected with gp100 TCR were mixed in a 1:1 ratio with T cells transfected with either the hCCT6Am TCR (white and light gray chequered columns) or the mCCT6Am TCR (white and dark gray chequered columns). Cells were stimulated with gp100 peptide (A) - or CCT6Am peptide-loaded (B) D05-Mel#6 tumor cells overnight, and CD25-expression was measured by flow cytometry. The specific increase in the percentage of CD25-positive T cells was determined by subtraction of non-peptide-stimulated T cells. Cytotoxicity assays were performed with gp100 peptide-loaded (C), or CCT6Am peptide-loaded (D) D05-EBV cells as target cells with a target-to-effector cell ratio of 1:60. Average values of 3 (A and B) or 5 (C and D) independent experiments ± SEM are shown. p-values were calculated with the Student's t-test (*p ≤ 0.05, **p ≤ 0.01, ns = not significant).
The cytotoxicity assay revealed no significant differences in the lytic capacity toward gp100-peptide-loaded target cells between hTETARs and hT-cell mix and between mTETARs and mT-cell mix (p = 0.0978), although there was a tendency toward better lysis by TETARs (Fig. 2C). Target cells loaded with the CCT6Am peptide were lysed similar by the TETARs and their corresponding T-cell mixes, with the mTETARs and the mT-cell mix showing a slightly higher lytic capacity than the T cells containing the murinised TCRs only (Fig. 2D). Concerning the differences in the lytic capacity of hTETARs and mTETARs, the mTETARs showed increased lysis, which was significant for lysis of CCT6Am-peptide-loaded target cells, but just not for gp100-peptide-loaded cells (i.e., p = 0.0610; Figure 2C and D). Additionally, the mTETARs showed slightly better lysis of gp100-peptide-loaded target cells, than T cells transfected with only the gp100 TCR (p = 0.0554) (Fig. 2C). We observed that the mTETARs were activated by each target epitope to the same extent as the T-cells transfected with one individual receptor were activated by their cognate epitope, and better than the 1:1 mixtures. Compared to the T-cell mixtures, the TETARs showed increased lytic capacity of target cells loaded with the gp100 peptide, and similar lytic capacity of target cells loaded with the CCT6Am peptide. From these results, we conclude that the TETARs indeed contained T cells capable of recognizing both epitopes, and were not merely a mixture of T cells with only one additional specificity.
Tumor-specific TETARs effectively kill target cells loaded with the cognate peptides for each TCR
To test the functionality of the generated tumor-specific TETARs, we further investigated the lytic capacity of the TETARs in cytotoxicity assays. Therefore, CD8+ T cells were equipped with the gp100 TCR, the hCCT6Am TCR, or the mCCT6Am TCR alone or in combination. T cells electroporated without RNA were used as negative control. The antigen-specific lytic capacity of the transfected T cells was tested in a 51chromium-release assay. The T cells were co-incubated with the EBV-transformed B cell line D05-EBV, which was loaded with gp100 peptide or CCT6Am peptide, or was left unloaded as control. The cytolytic capacity of the TCR-transfected T cells was tested in different target to effector ratios, as indicated, and the percentage of lysis was determined (Fig. 3). The statistical comparisons between the different conditions are shown in Table S1. Only a slight background lysis of the target cells without peptide was observed (Fig. 3). The gp100 peptide-loaded cells were lysed in an antigen-specific manner at every effector to target ratio with similar efficiency (Fig. 3). The lytic capacity of the TETARs was similar to that of T cells only equipped with the gp100-TCR, using gp100 peptide-loaded target cells (Fig. 3). Target cells loaded with CCT6Am peptide were lysed antigen-specifically, with the highest lytic capacity displayed by TETARs containing the hCCT6Am TCR (Fig. 3). Comparing the lytic capacity of CCT6Am-specific T cells on D05-EBV target cells, hTETARs revealed a slightly higher lytic capacity than mTETARs and T cells equipped with only one type of TCR (Fig. 3).
Figure 3.
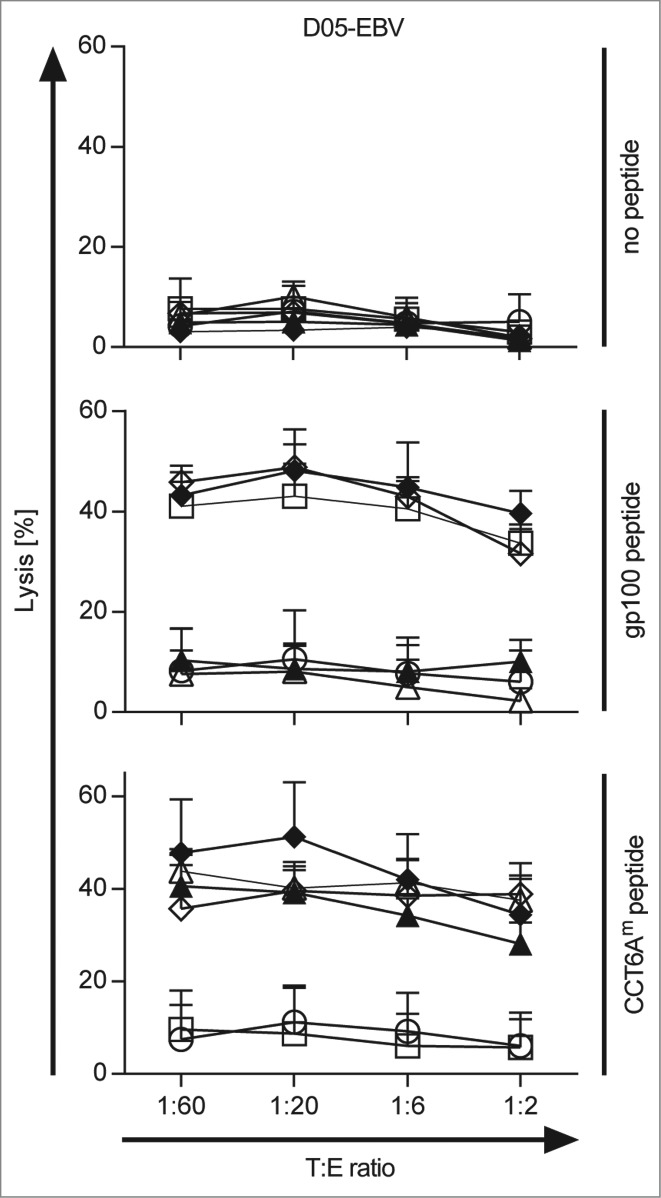
TETARs antigen-specifically lyse target cells loaded with each cognate peptide. CD8+ T cells were transfected with RNA encoding the single TCRs: the gp100 TCR (open squares), the human CCT6Am TCR (closed triangles), or the murinised CCT6Am TCR (open triangles), or were mock-electroporated (white circle) as control. TETARs were generated by transfecting CD8+ T cells with the gp100 TCR in combination with the human CCT6AmTCR (closed diamonds) or the murinised CCT6Am TCR (open diamonds). These cells were then used in 51chromium-release assays to determine their lytic capacity. EBV-immortalised B cells (D05-EBV) were loaded with either the gp100 peptide, or the CCT6Am peptide, or were left unloaded as control, and were used as target cells at the indicated target to effector cell (T:E) ratios. The percentage of lysed cells was determined and the average values of 3 independent experiments ± SEM are shown. The statistical significances between the different conditions are summarised in Table S1.
As the lytic capacity of both TETARs was at least similar to that of T cells transfected with only one TCR, these results clearly show that both TETARs, especially the mTETARs, are able to effectively recognize both of their cognate antigens and that they are able to lyse target cells loaded with these peptides antigen-specifically.
Murinisation of the TCR results in a higher and more stable expression
To investigate the influence of the murinisation on the expression and stability of the CCT6Am TCR, we determined the surface expression of the TCR Vβ14 chain, which is shared by both the gp100 and the CCT6Am TCRs. Therefore, we transfected CD8+ T cells either with the gp100 TCR, the hCCT6Am TCR, or the mCCT6Am TCR, or mock-electroporated them as a control, and determined the Vβ14 expression 4, 24, and 48 h after transfection. Although the total mean fluorescence intensities (MFI) were weak, a clear shift of the TCR-RNA-transfected T cells, especially of mCCT6Am TCR-transfected T cells, compared to mock-electroporated cells was visible in the histograms (Fig. S2). Compared to the control, all TCRs showed significant expression of the Vβ14 chain 4 h and 24 h after electroporation, whereas 48 h after transfection only the mCCT6Am TCR was still detectable (Fig. 4). While the expression of the 2 human TCRs, i.e. gp100 TCR and hCCT6Am TCR, was similar at all time points, the mCCT6Am TCR was expressed significantly higher than the hCCT6Am TCR (Fig. 4). These results show that murinisation of the CCT6Am TCR made it more stable over time and increased its expression on the surface of the TCR-transfected CD8+ T cells.
Figure 4.
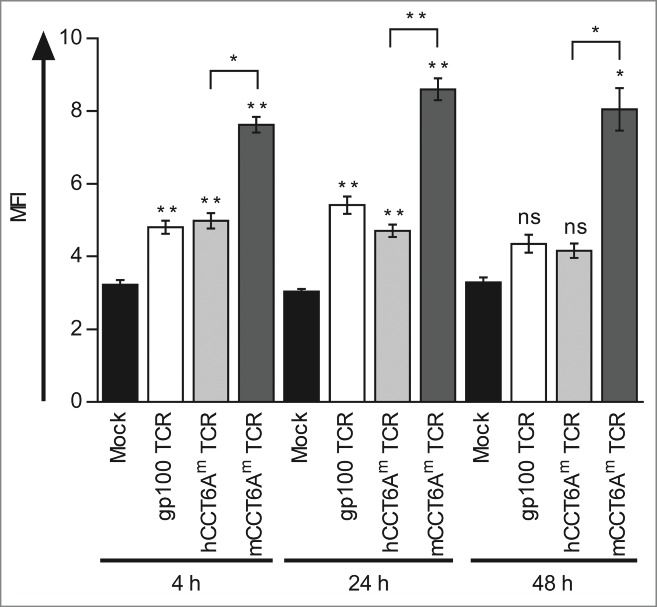
The murinised CCT6Am TCR is higher and more stably expressed than the gp100 TCR and the human CCT6Am TCR. CD8+ T cells were transfected with RNA encoding the gp100 TCR (white columns), the human CCT6Am (hCCT6Am) TCR (light gray columns), or the murinised mCCT6Am TCR (dark gray columns) by RNA electroporation, or were mock-electroporated (black columns) as control. The surface expression of the TCR Vβ14-chain, shared between the 3 TCRs, was determined 4, 24, and 48 h after electroporation by flow cytometry. Average values of 3 independent experiments ± SEM are shown. p-values were calculated with the Student's t-test (*p ≤ 0.05, **p ≤ 0.01).
Discussion
In the current study we explore a novel strategy that can be used for adoptive T-cell therapy of melanoma. Personalised cancer immunotherapy currently emphasizes on adoptive T-cell therapy,19 which means either TIL therapy or adoptive T-cell transfer. Current ACT using monospecific T cells often results in initial therapeutic success, but patients frequently relapse due to the immune escape mechanisms induced by the tumors (reviewed by Dunn et al.10). Melanomas escape immune surveillance by altering the gene expression of the tumor cells, resulting in removal or editing of epitopes presented by these cells, for example by downregulation or loss of tumor-associated antigens20-24 due to the phenotypic plasticity of melanoma cells.23 Many T-cell transfer approaches include only T cells with monoclonal specificity, as often no TILs are available for tumor therapy. Thus, they represent only a narrow attack on the tumor, thereby facilitating the development of escape variants, as concluded by Restifo et al.4. The TETARs we generated by RNA electroporation and that are presented in this study were able to target 2 melanoma-associated antigens simultaneously, thus increasing the immune pressure on the tumors. Our approach reduces the risk of immune escape, as it is unlikely that both epitopes recognized by the TETARs will be lost or down-regulated at the same time.
As several common tumor antigens that serve as targets for current immunotherapeutic approaches, like the melanocytic differentiation antigen gp100, are over-expressed self-antigens, they are often subject to tolerance mechanisms.11 In contrast, neo-antigens like our model antigen, the mutated CCT6A, are exclusively expressed in the tumor, so they are foreign to the patient's immune system and thus ideal targets for immunotherapy, as T cells specific for these antigens are not affected by central tolerance mechanisms.12 The T cells transfected with the CCT6Am TCR did not recognize the endogenously processed antigen on the tumor cell line D05-MEL#6, although the original T-cell clone did. The observation that RNA-reprogrammed T cells displayed a slightly lower functional avidity than the T-cell clone from which the TCR was derived, has been reported before.7 We nevertheless used this TCR and the CCT6Am antigen as a model system for TETAR generation.
In this study, we engineered T cells that target the common melanoma antigen gp100 and the individually mutated neo-antigen CCT6A simultaneously, and present a new kind of personalised ACT, which can easily be adjusted to many different patients by exchange of the TCR specific for the individually mutated neo-antigen.
We showed that our TETARs were able to recognize both cognate antigens and, in response, secrete cytokines and lysed target cells loaded with these peptides antigen-specifically. However, the cytokine production by TETARs stimulated with the CCT6Am peptide ceased more rapidly than that of T cells transfected with a single TCR. One reason for this timely limitation could be that the competition between the 2 TCRs occurs in a delayed fashion, only affecting the “weaker” CCT6Am TCR.
This dual-specific T cells may circumvent the current obstacle of ACT, as mentioned above, by targeting both epitopes simultaneously. Additionally, we could show that mTETARs were activated by both target epitopes obviously better than the 1:1 mixtures. This leads to the conclusion that TETARs are not merely a mixture of T cells with only one additional specificity, but indeed contain T cells capable of recognizing both epitopes.
Another possibility of immune evasion, the loss or down-regulation of MHC molecules,25 might nonetheless have negative impact on the TETARs engineered and tested in this study, as the target epitopes are not presented by the tumor cells anymore. One possible attempt to overcome this problem is to equip T cells with receptor-combinations that include chimeric antigen receptors (CARs), thus enabling the recognition of unprocessed tumor antigens independent of MHC-presentation.26 Additionally, TCRs restricted by different HLA haplotypes could be used for the generation of TETARs, thus being able to compensate for loss of single alleles.
The difficulties in the generation of TETARs, i.e alternative pairing of the TCR chains and competition for cellular factors, could be resolved. Titration of the TCR RNAs, thus adjusting the expression levels of the TCRs, strongly reduced the competitive effects between both TCRs. For future approaches, such titrations could be performed to require proper ratios of the introduced TCRs to engineer viral vectors without the obstacle of competitive effects.
Mispairing of the TCR chains was reduced by murinisation, but seemed to be less relevant for this TCR combination than for others that were examined before.14
Murinisation of human TCRs can be used to improve proper pairing of the TCR chains and thus improvement of TCR expression and function.16,27 In a previous study investigating TETARs in the context of HIV-1, we showed that murinisation was necessary to prevent competitive effects between the 2 introduced HIV-1-specific TCRs.14 In the current study, mispairing seemed to be less relevant as 2 human TCRs could also be used to generate TETARs with little competitive effects, when RNA quantities were properly adapted. However, murinisation resulted in increased and more stable expression of the CCT6Am TCR, and in an increased lysis of antigen-loaded target cells. Therefore, in this study, murinisation of the CCT6Am TCR prolonged the time of potential antigen-recognition and thus increases the probability of T-cell activation. For future TETAR approaches using other TCRs for transfection, the impact and benefits of murinisation have to be investigated depending on potentially upcoming competitive effects. As the induction of xenogeneic TCR regions might induce immune reactions, which might weaken T-cell function, another approach would be to replace not the whole C-regions of the TCR chains, but to exchange only selected murine residues in the C-regions of the TCR chains.15
The equipment of T cells with additional TCRs bears the general risk of inducing autoimmunity. The exogenous TCR itself may mediate autoimmunity or mixed dimers may be formed which can be self-reactive. These risks exponentially increase if 2 exogenous TCRs are introduced. If the TCRs are introduced retrovirally, the consequences for the patients could be very severe and the induced autoimmunity would be long-lasting. Especially a selective expansion of the autoimmune T cells by constant activation in the patient would be a threatening consequence. In our approach, this risk is reduced but not completely obliterated by murinisation of one TCR, which results in preferred formation of correctly paired dimers. Also for this reason, we avoided stable transfection by using RNA electroporation, resulting in only transient expression and a short half-life of the exogenous TCRs in the T cells. Autoimmune reactions would hence be limited to a few days, and an expansion of the autoreactive T cells in the patient is prevented. This gain in safety would, however, be at the cost of memory formation, as the transiency of expression would prevent this. Therefore, the transfected T cells need to be injected repetitively. This was shown to overcome the lack of memory formation in different murine models,28,29 and even in human case studies with mRNA-transfected T cells.30 As an alternative approach, a well characterized murinised TCR could be introduced into the T cells by retroviruses, thus by stable transfection, while the human TCR, which might more likely form mixed dimers with the endogenous TCR, could be introduced by RNA transfection.
Taken together, we could show that it is possible to generate CD8+ T cells with 2 additional TCRs specific for a common melanoma antigen and a patient-specific neo-antigen by RNA electroporation. These dual-specific T cells were able to secrete cytokines upon recognition of their cognate antigens and to lyse target cells antigen-specifically. To our knowledge, this is the first time that melanoma-specific dual-specific T cells were engineered. We believe that tumor-specific TETARs could be used as efficient new tools in the T-cell therapy of cancer.
Materials and Methods
Isolation and cultivation of CD8+ T cells
All human material from healthy donors was obtained after informed consent and approved by the institutional review board. Peripheral blood mononuclear cells (PBMC) were purified by density centrifugation as described before.31 PBMC were incubated for 1 h at standard cell culture conditions (i.e., 37°C; 95% (v/v) humidity; 5% (v/v) CO2) in DC medium, consisting of RPMI 1640 (Lonza, Order Nr. BE12–167F), 1% plasma from leukapheresis products (heat-inactivated, 30 minutes, 56°C), 2 mM L-glutamine (Lonza, Order Nr. 17–605E), and 0.04% 20 mg/l gentamycin (BioWhittaker, Order Nr.17–518Z), on plastic cell culture dishes (BD Falcon, BD Bioscience, Order Nr. 353003). The non-adherent fraction (NAF) of the cells was harvested from the supernatant. CD8+ T cells were isolated from the NAF using anti-CD8 MACS-beads (Miltenyi, Order Nr. 130–045–201), according to the manufacturer's instructions. The isolated T cells were cultured in T-cell medium consisting of RPMI 1640, 10% human serum (Sigma, Order Nr. H4522–100ML), 2 mM L-glutamine, 20 mg/l gentamycin, 10 mM HEPES (PAA, Order Nr. S11–001), 1 mM sodium pyruvate (PAA, Order Nr. S11–003), and 1% MEM non-essential amino acids (100x, PAA, Order Nr. M11–003).
Murinisation of the CCT6Am TCR
To murinise the CCT6Am TCR α-chain, we recombined the CCT6Am-specific variable domain and the joining domain with the murine C-domain of a codon-optimised NY-ESO opt vector17 (kindly provided by Wolfgang Uckert), via a restriction site between the variable (V-) domain and the constant (C-) domain. This restriction site was integrated by site directed mutagenesis or PCR.
The murinised CCT6Am TCR β-chain was manufactured by GeneArt® Gene Synthesis, consisting analogous to the D05 CCT6Am α-chain of the Cβ2-domain of the murine TCR and the VDJ-domain specific for CCT6Am.
Production of in-vitro-transcribed mRNA
The sequences encoding the TCRs were cloned into the pGEM4Z-5′UTR-sig-husurvivin-DC.LAMP-3′UTR RNA-production vector32 (kindly provided by Kris Thielemans), replacing the sig-husurvivin-DC.LAMP sequence, and RNA was produced using the mMESSAGE mMACHINE T7 ULTRA Transcription Kit (Life Technologies, Ambion, Order Nr. AM1345–5) according to the manufacturer's instructions. RNA was purified with an RNeasy Kit (Qiagen, Order Nr. 74104) according to manufacturer's protocols.
Electroporation of CD8+ T cells
Electroporations were performed with a Genepulser Xcell system (Biorad, Order Nr. 165–2661). T cells were washed and resuspended in Optimem without phenol red (Life Technologies, Order Nr. 11058–021) at room temperature and pulsed with a 500 V, 5 ms square wave pulse. Cells were transfected with RNA encoding the α- and β-chains of the gp100 TCR, the human (h) CCT6Am TCR, and the murinised (m) CCT6Am TCR. As control, cells were electroporated without RNA. To generate TETARs, different ratios of RNAs encoding the gp100 TCR and the hCCT6Am TCR or the mCCT6Am TCR, i.e. 10 µg or 5 µg of each TCR chain, 10 µg of each gp100 TCR chain and 2 µg of each hCCT6Am TCR chain or mCCT6Am TCR chain, and vice versa, as well as 10 µg of each gp100 TCR chain and 5 µg of each hCCT6Am TCR chain or mCCT6Am TCR chain, and vice versa, were used for electroporation. Finally, 5 µg of each gp100-TCR-chain RNA were chosen for co-electroporation with 10 µg of each hCCT6Am TCR chain or mCCT6Am TCR chain RNA per 100 µl to generate the TETARs. After transfection, T cells were rapidly transferred into T-cell medium. Cells were incubated for 4 h before use in stimulations.
Surface-expression analysis of CD25 and Vβ14-TCR-chains
For surface staining of Vβ14-TCR-chains or the CD25 activation marker, 50,000–100,000 cells (transfected as described above) per condition were harvested 4 h after electroporation or taken from an overnight stimulation with peptide-loaded D05-Mel#6 cells in a 1:1 ratio in 96-well round-bottom plates in a total volume of 200 µl per well. The T cells were washed once in FACS solution, consisting of PBS (LONZA; Order Nr. BE17–512F) supplemented with 1% FCS (PAA, Order Nr. A15–151) and 0.02% sodium azide (Merck, Order Nr. 822335), and incubated with anti-Vβ14-PE antibody (Immunotech, Order Nr. 2047) or anti-CD25-FITC antibody (BD Biosciences, Order Nr. 555431) for 30 minutes at 4°C in FACS solution. Immunofluorescence was detected using a FACScan cytofluorometer (BD Biosciences, Heidelberg, Germany) equipped with CellQuest software (BD Biosciences, Heidelberg, Germany).
Peptide-loading of D05-EBV and D05-Mel#6 cell lines
EBV-transformed B cells (D05-EBV) or cells from a melanoma cell line (D05-Mel#6) were washed once in RPMI 1640 and loaded with peptide at 10 µg/ml for 1 h at 37°C / 5% CO2 in DC medium. Cells were harvested, washed once in RPMI 1640 and used in stimulations. Peptides used in this study were: the gp100-derived HLA-A2-binding peptide gp100280–288 (YLEPGPVTA) and an HLA-B27-binding peptide from CCT6A bearing an individual mutation in the melanoma cell line D05-Mel#6 (manuscript in preparation).
Cytokine analysis
Cells were transfected as described above and rested for 4, 24, and 48 h after electroporation. Then, the T cells were stimulated with D05-EBV cells, which were UV-inactivated (0.005 J/cm2) and afterwards peptide-loaded as described above, in a 1:1 ratio (50,000 cells each) in 96-well round-bottom plates in a total volume of 200 µl per well for 20 h. Cytokine concentrations in the supernatants were analyzed using a Th1/Th2 Cytometric Bead Array Kit II (BD Biosciences, Order Nr. 551809) following the manufacturer's instructions. Immunofluorescence was detected using a FACScan cytofluorometer (BD Biosciences, Heidelberg, Germany) equipped with CellQuest software (BD Biosciences, Heidelberg, Germany).
Cytotoxicity assay
Cytotoxicity was tested with a standard 4–6 h 51chromium-release assay: EBV-transformed B cells (D05-EBV) were labeled with 100 µCi of Na251CrO4/106 (PerkinElmer, Order Nr. NEZ030001MC) for 1 h, washed once, loaded with peptides as described above, and washed twice before being used in co-incubations with effector T cells. Target cells were added to 96-well plates at 1,000 cells / well. Effector cells were added at indicated T:E ratios. The chromium-release was measured with a Wallac 1450 MicroBeta plus Scintillation Counter (Wallac, Turku, Finnland). The percentage of cytolysis was calculated from the 51Cr release as follows: [(measured release – background release)] / [(maximum release – background release)] × 100%.
Statistical analysis
Statistical analysis was performed using the paired Student's t-test. A Gaussian distribution was assumed. P-values are indicated as follows: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
We thank Wolfgang Uckert for the murine TCR constant domains, Kris Thielemans for the pGEM4Z-5_UTR-sig-huSurvivin-DC.LAMP-3_UTR vector, and Christian Hofmann for preparatory work. We also thank Stefanie Baumann and Verena Wellner for excellent technical assistance, and the medical staff for acquisition of donor material.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was partially financed by the BMBF (project DCmutaVacc, Förderkennzeichen 01GU1107A to GS) and by the IZKF of the Medical Faculty of the FAU Erlangen-Nürnberg.
References
- 1.Xue S, Gillmore R, Downs A, Tsallios A, Holler A, Gao L, Wong V, Morris E, Stauss HJ. Exploiting T cell receptor genes for cancer immunotherapy. Clin Exp Immunol 2005; 139:167-72; PMID:15654813; http://dx.doi.org/ 10.1111/j.1365-2249.2005.02715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolhuis RL, Willemsen RA, Gratama JW. Clinical applications of redirected cytotoxicity. In Sitkovsky MV, Henkart PA, editors. Cytotoxic cells. Philadelphia: Lippincott, Williams & Wilkins; 2000; 423-40. [Google Scholar]
- 3.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM et al.. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002; 298:850-4; PMID:12242449; http://dx.doi.org/ 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 2012; 12:269-81; PMID:22437939; http://dx.doi.org/ 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hawkins RE, Gilham DE, Debets R, Eshhar Z, Taylor N, Abken H, Schumacher TN, ATTACK Consortium . Development of adoptive cell therapy for cancer: a clinical perspective. Hum Gene Ther 2010; 21:665-72; PMID:20408760; http://dx.doi.org/ 10.1089/hum.2010.086. [DOI] [PubMed] [Google Scholar]
- 6.Schumacher TN. T-cell-receptor gene therapy. Nat Rev Immunol 2002; 2:512-9; PMID:12094225; http://dx.doi.org/ 10.1038/nri841. [DOI] [PubMed] [Google Scholar]
- 7.Schaft N, Dorrie J, Muller I, Beck V, Baumann S, Schunder T, Kampgen E, Schuler G. A new way to generate cytolytic tumor-specific T cells: electroporation of RNA coding for a T cell receptor into T lymphocytes. Cancer Immunol Immunother 2006; 55:1132-41; PMID:16344988; http://dx.doi.org/ 10.1007/s00262-005-0098-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, Rosenberg SA, Morgan RA. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther 2006; 13:151-9; PMID:16140584; http://dx.doi.org/ 10.1016/j.ymthe.2005.07.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krug C, Wiesinger M, Abken H, Schuler-Thurner B, Schuler G, Dorrie J, Schaft N. A GMP-compliant protocol to expand and transfect cancer patient T cells with mRNA encoding a tumor-specific chimeric antigen receptor. Cancer Immunol Immunother 2014; 63:999-1008; PMID:24938475; http://dx.doi.org/ 10.1007/s00262-014-1572-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 2002; 3:991-8; PMID:12407406; http://dx.doi.org/ 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 11.Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, Wolfel C, Huber C, Wolfel T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci U S A 2005; 102:16013-8; PMID:16247014; http://dx.doi.org/ 10.1073/pnas.0500090102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hadrup S, Donia M, thor SP. Effector CD4 and CD8 T cells and their role in the tumor microenvironment. Cancer Microenviron 2013; 6:123-33; PMID:23242673; http://dx.doi.org/ 10.1007/s12307-012-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Restifo NP, Wunderlich JR. Principles of tumor immunity: biology of cellular immune responses. In DeVita VT, Hellman S, Rosenberg SA, editors. Biologic Therapy of Cancer. Philadelphia: Lippincott Co 1996; 3-21. [Google Scholar]
- 14.Hofmann C, Hofflin S, Huckelhoven A, Bergmann S, Harrer E, Schuler G, Dorrie J, Schaft N, Harrer T. Human T cells expressing two additional receptors (TETARs) specific for HIV-1 recognize both epitopes. Blood 2011; 118:5174-7; PMID:21926350; http://dx.doi.org/ 10.1182/blood-2011-04-347005. [DOI] [PubMed] [Google Scholar]
- 15.Bialer G, Horovitz-Fried M, Ya'acobi S, Morgan RA, Cohen CJ. Selected murine residues endow human TCR with enhanced tumor recognition. J Immunol 2010; 184:6232-41; PMID:20427762; http://dx.doi.org/ 10.4049/jimmunol.0902047. [DOI] [PubMed] [Google Scholar]
- 16.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res 2006; 66:8878-86; PMID:16951205; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sommermeyer D, Uckert W. Minimal amino acid exchange in human TCR constant regions fosters improved function of TCR gene-modified T cells. J Immunol 2010; 184:6223-31; PMID:20483785; http://dx.doi.org/ 10.4049/jimmunol.0902055. [DOI] [PubMed] [Google Scholar]
- 18.Tanic N, Brkic G, Dimitrijevic B, Dedovic-Tanic N, Gefen N, Benharroch D, Gopas J. Identification of differentially expressed mRNA transcripts in drug-resistant versus parental human melanoma cell lines. Anticancer Res 2006; 26:2137-42; PMID:16827156. [PubMed] [Google Scholar]
- 19.Svane IM, Verdegaal EM. Achievements and challenges of adoptive T cell therapy with tumor-infiltrating or blood-derived lymphocytes for metastatic melanoma: what is needed to achieve standard of care? Cancer Immunol Immunother 2014; 63:1081-91; PMID:25099366; http://dx.doi.org/ 10.1007/s00262-014-1580-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slingluff CL, Jr., Colella TA, Thompson L, Graham DD, Skipper JC, Caldwell J, Brinckerhoff L, Kittlesen DJ, Deacon DH, Oei C et al.. Melanomas with concordant loss of multiple melanocytic differentiation proteins: immune escape that may be overcome by targeting unique or undefined antigens. Cancer Immunol Immunother 2000; 48:661-72; PMID:10752474; http://dx.doi.org/ 10.1007/s002620050015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marchand M, Brichard V, van Baren N, Coulie PG. Biological and clinical developments in melanoma vaccines. Expert Opin Biol Ther 2001; 1:497-510; PMID:11727521; http://dx.doi.org/ 10.1517/14712598.1.3.497. [DOI] [PubMed] [Google Scholar]
- 22.Kaluza KM, Thompson JM, Kottke TJ, Flynn Gilmer HC, Knutson DL, Vile RG. Adoptive T cell therapy promotes the emergence of genomically altered tumor escape variants. Int J Cancer 2012; 131:844-54; PMID:21935923; http://dx.doi.org/ 10.1002/ijc.26447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wolfel T, Holzel M et al.. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012; 490:412-6; PMID:23051752; http://dx.doi.org/ 10.1038/nature11538. [DOI] [PubMed] [Google Scholar]
- 24.Liu K, Caldwell SA, Greeneltch KM, Yang D, Abrams SI. CTL adoptive immunotherapy concurrently mediates tumor regression and tumor escape. J Immunol 2006; 176:3374-82; PMID:16517705; http://dx.doi.org/ 10.4049/jimmunol.176.6.3374. [DOI] [PubMed] [Google Scholar]
- 25.Carretero R, Romero JM, Ruiz-Cabello F, Maleno I, Rodriguez F, Camacho FM, Real LM, Garrido F, Cabrera T. Analysis of HLA class I expression in progressing and regressing metastatic melanoma lesions after immunotherapy. Immunogenetics 2008; 60:439-47; PMID:18545995; http://dx.doi.org/ 10.1007/s00251-008-0303-5. [DOI] [PubMed] [Google Scholar]
- 26.Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med 2014; 65:333-47; PMID:24274181; http://dx.doi.org/ 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hart DP, Xue SA, Thomas S, Cesco-Gaspere M, Tranter A, Willcox B, Lee SP, Steven N, Morris EC, Stauss HJ. Retroviral transfer of a dominant TCR prevents surface expression of a large proportion of the endogenous TCR repertoire in human T cells. Gene Ther 2008; 15:625-31; PMID:18305579; http://dx.doi.org/ 10.1038/sj.gt.3303078. [DOI] [PubMed] [Google Scholar]
- 28.Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther 2013; 24:717-27; PMID:23883116; http://dx.doi.org/ 10.1089/hum.2013.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, Chew A, Carroll RG, Scholler J, Levine BL et al.. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res 2010; 70:9053-61; PMID:20926399; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM et al.. Mesothelin-specific Chimeric Antigen Receptor mRNA-Engineered T cells Induce Anti-Tumor Activity in Solid Malignancies. Cancer Immunol Res 2014; 2:112-20; PMID:24579088; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schaft N, Dorrie J, Thumann P, Beck VE, Muller I, Schultz ES, Kampgen E, Dieckmann D, Schuler G. Generation of an optimized polyvalent monocyte-derived dendritic cell vaccine by transfecting defined RNAs after rather than before maturation. J Immunol 2005; 174:3087-97; PMID:15728524; http://dx.doi.org/ 10.4049/jimmunol.174.5.3087. [DOI] [PubMed] [Google Scholar]
- 32.Bonehill A, Heirman C, Tuyaerts S, Michiels A, Breckpot K, Brasseur F, Zhang Y, Van Der BP, Thielemans K. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J Immunol 2004; 172:6649-57; PMID:15153480; http://dx.doi.org/ 10.4049/jimmunol.172.11.6649. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.