

Abstract
CTLA4-Ig is a highly glycosylated therapeutic fusion protein that contains multiple N- and O-glycosylation sites. Glycosylation plays a vital role in protein solubility, stability, serum half-life, activity, and immunogenicity. For a CTLA4-Ig biosimilar development program, comparative analytical data, especially the glycosylation data, can influence decisions about the type and amount of animal and clinical data needed to establish biosimilarity. Because of the limited clinical experience with biosimilars before approval, a comprehensive level of knowledge about the biosimilar candidates is needed to achieve subsequent development. Liquid chromatography-mass spectrometry (LC–MS) is a versatile technique for characterizing N- and O-glycosylation modification of recombinant therapeutic proteins, including 3 levels: intact protein analysis, peptide mapping analysis, and released glycans analysis. In this report, an in-depth characterization of glycosylation of a candidate biosimilar was carried out using a systematic approach: N- and O-linked glycans were identified and electron-transfer dissociation was then used to pinpoint the 4 occupied O-glycosylation sites for the first time. As the results show, the approach provides a set of routine tools that combine accurate intact mass measurement, peptide mapping, and released glycan profiling. This approach can be used to comprehensively research a candidate biosimilar Fc-fusion protein and provides a basis for future studies addressing the similarity of CTLA4-Ig biosimilars.
Keywords: characterization, CTLA4-Ig fusion protein, glycan, glycosylation modification, intact protein, mass spectrometry, peptide mapping, similarity
Abbreviations
- LC
Liquid chromatography
- UPLC
Ultra-performance liquid chromatography
- Tof
Time of flight
- Q-Tof
quadrupole-time of flight
- MS
mass spectrometry
- ESI
electrospray ionization
- IAM
Iodoacetamide
- DTT
dithiothreitol
- FA
formic acid
- PNGase F
peptide N-glycosidase
- PTMs
post-translational modifications
- 2-AB
2-aminobenzamide
- GFP
[Giu1]-Fibrinopeptide B
- TIC
Total Ion Chromatography
- CTLA-4
cytotoxic T-lymphocyte-associated antigen 4
- RA
Rheumatoid arthritis
- EMA
European Medicines Agency
- FDA
Food and Drug Administration
Introduction
Since 1986, when muromonab-CD3 was approved by the US. Food and Drug Administration (FDA), more than 40 therapeutic monoclonal antibodies (mAbs) and antibody-derivatives, including antigen-binding fragments (Fab), Fc-fusion proteins, radio-immunoconjugates, antibody–drug conjugates, and bispecific antibodies, have received approval for treating many kinds of diseases, including tumors, rheumatoid arthritis (RA), and macular degeneration.1 Unlike small molecule drugs, antibodies are large, heterogeneous proteins that are used as therapeutics due to their controlled properties, specific functions, and long half-life period. However, antibodies and their derivatives are expensive medicines. As increasing healthcare costs are a burden in many countries, reducing the cost of medicines has become a greater economic and public health priority.2 The use of biosimilars offers a solution for healthcare systems facing increasing biologics costs. Biosimilars are defined as biological medicinal products comparable (but not necessarily identical) in quality, efficacy and safety to reference products.3 These qualities have captured the interest of drug companies that are focused on developing less expensive biosimilar antibodies and derivatives, such as TNFR-Ig, VEGFR-Ig, CTLA4-Ig, and PDL1-Ig.4
To expedite the development of biosimilars in Europe, the European Medicines Agency (EMA) has established a series of guidelines.5 Although biosimilar products such as human growth hormone, granulocyte colony-stimulating factor and epoetin have already been approved by regulators, including the EMA, only one biosimilar antibody has been approved because of the complexity of the molecule. Compared to other biosimilar therapeutics, mAb and antibody-derivative biosimilars guidance has experienced delays.6 The main reason is that antibody-derivatives are glycoproteins, which are large, complicated and heterogeneous proteins with the complex post-translational modifications (PTMs). Researchers have shown that regulatory approval of biosimilars of mAb and antibody-derivative is subjected to specific, science-based guidelines. An extensive comparative in vitro characterization to evaluate the biosimilarity of the various functional domains is required.7 The characterization of heterogeneities serves as the basis to research and control them.
Testing the molecular similarity of a biosimilar to the innovator medicine is a challenging process requiring the establishment of rapid and accurate analytical methods that will be accepted by regulators.8 Defining molecular similarity remains a challenge,9 because antibody derivatives can have an average molecular mass of more than 100 kDa and many complex post-translational modifications. Glycosylation is the most important post-translational modification for many reasons. Firstly, glycosylation alters the properties of proteins, including pharmacokinetics,10 pharmacodistribution,11 effector function,12 antigenicity,13 solubility,14 and stability.15 After years of research, we now know that glycosylation can be influenced by cell lines, conformation of proteins, and cell culture conditions. As a result of various cell culture factors, glycosylation variations in mammalian cell bioprocesses can found in the glycan site (macroheterogeneity) and glycan profile (microheterogeneity).16 Glycan structures that are produced from any cell are governed by a network of enzymes that do not always allow for individual reactions. This network is affected by multiple factors, including the availability of precursor, co-factors and enzyme activities levels, all of which give rise to the variable final glycan structure.17
Research on glycosylation variations is important to better understand glycoproteins. In recent developments, mass spectrometry has become an indispensable analytical tool for PTMs analysis due to its superior resolution over other analytical techniques. The development of ESI-TOF-MS technology has transformed the analysis of large heterogeneous biomolecules into a routine task because it has high mass resolution and sensitivity.18 Liquid chromatography-mass spectrometry (LC–MS) is a valuable technique that provides detailed information about glycoproteins via structural analysis of glycans and glycopeptides, including characterizing of N- and O-glycosylation modifications. The development of the electron-transfer dissociation (ETD) technique has made it possible to characterize glycoproteins at the glycopeptide level. ETD can retain the fragile glycosidic linkage with a nonergodic fragmentation process, and at the same time, it can verify the amino acid residue and the glycosylation sites at the peptide level. This is advantageous for O-linked glycosylation analysis, which is a challenging task. Unlike N-linked glycosylation, O-linked glycosylation has a complex sequence for the potential glycosylation site, no common core structure, more heterogeneous structure and high density in Ser/Pro/Thr-rich domains.
Rheumatoid arthritis (RA) is a common inflammatory disease that can be disabling if left untreated.19 It is characterized by chronic joint inflammation and continuous cell infiltration into the synovium. T cells in RA pathophysiology can be mediated through Th1 effector functions, Th17 activity or induction of anti-citrullinated protein antibodies, leading to bone and cartilage destruction.20 It is now thought that 2 signals from the antigen-presenting cell (APC) are needed to activate T cells. The first signal arises when antigenic peptides are presented on the antigen-presenting cell through the major histocompatibility complex (MHC), which is antigen specific. The second co-stimulation signal develops from the interaction between immune molecules on the APC and T cell. Drugs targeting the above signals and current aggressive treatment strategies have greatly improved outcomes for patients with RA over the past decades and slowed the progression of joint damage. In December 2005, abatacept was approved by the US FDA for treatment of patients with RA who have had an inadequate response to other drugs. It was the first selective co-stimulation modulator approved.
Abatacept is a fusion protein consisting of the extracellular domain of the human cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) linked to a modified Fc (hinge, CH2 and CH3 domains) portion of human immunoglobulin 1 (IgG1). It inhibits the activation of T lymphocytes that play an important role in the early stages of pathogenesis of RA and is used to treat RA when other drugs are ineffective. It is licensed for the treatment of RA in combination with methotrexate.21 Abatacept is believed to work by preventing the essential second signal for T cell activation through binding with the extracellular domain of CTLA-4 to immune molecules (CD80 or CD86) on the APC with a higher affinity than CD28. Consequently, T cell activation is prevented and the production of cytokines such as interleukin (IL)-6, IL-8 and tumor necrosis factor are reduced. Use of abatacept is associated with a reduction in joint inflammation, pain, and joint damage in patients with active RA.22 Although there may be some common adverse effects such as headache, hypertension, dizziness, gastrointestinal disorders, and rash, the significant and high therapeutic effect of the fusion protein makes it a potential target for biosimilar development. Production of CTLA4-Ig fusion protein in Chinese hamster ovary (CHO) cells results in a macro- and micro-heterogeneous glycosylation.23
In a study of CTLA4-Ig fusion protein, 3 N-linked sites and one O-linked sites were reported.24 N-glycosylation at N76, N108, and N207 was identified, but detailed information about the glycosylation was not provided. The specific N-glycosylation heterogeneity at each individual site and the O-linked glycosylation sites (and site heterogeneity) of the fusion protein are still ambiguous.24 For definite conclusions to be drawn about the processing and biological function of CTLA4-Ig fusion protein glycosylation, a complete characterization of glycosylation is required. This is also necessary to validate the similarity of a biosimilar to the reference product.
In this study, we successfully used the LC-MS analytical approach to perform in-depth characterization of both N- and O-linked glycosylation of CTLA4-Ig fusion protein. The main analytical tools were liquid chromatography coupled to fluorescence detection and mass spectrometry, combining a subtitle sample preparation strategy. We identified the similarity of a candidate biosimilar CTLA4-Ig fusion protein in the levels of glycosylation: the structures of N-linked glycans, the site heterogeneity of N-linked glycosylation, and the heterogeneity of O-linked glycans. In addition, 4 O-glycosylation sites have been firstly determined using the ETD fragmentation technique. This report represents the first detailed analysis and characterization of the CTLA4-Ig fusion protein and demonstrates the similarity of the CTLA4-Ig fusion protein at the levels of glycosylation patterns.
Results
Ig-fusion proteins are a large group of recombinant glycoproteins in which the carbohydrate can comprise anywhere from less than 1% to more than 80% of the total protein mass. Glycosylation analysis is necessary to meet regulatory requirements for therapeutic proteins, and it is essential to meet emerging challenges: (i) glycosylation may affect the biological function of Fc-fusion proteins, such as complement-dependent cytotoxicity (CDC) and circulatory half-life25; (ii) use of new expression systems such as NS0, SP2/0, CHO or yeast, which had different glycosylation patterns;26 (iii) the differences in glycosylation might be related to structure variants, potency, and safety.27 For example, cetuximab can generate an anaphylactic response due to its heterogeneous glycan forms, such as α-1,3 galactose and hydroxyacetylneuraminic acid (NGNA).28 In our study of CTLA4-Ig glycosylation, we analyzed the biosimilar candidate using 3 different approaches: (1) intact protein level glycan analysis, (2) peptide level glycan analysis, and (3) cleaved glycan level analysis.
Intact protein level glycan analysis
CTLA4-Ig is composed of 2 heavy-chains linked by inter-chain disulfide bonds. It has 2 N-glycan sites in the CTLA4 region and one in the Fc region, all of which are occupied with complex N-linked glycans. There are also some O-linked glycan sites occupied with complex O-linked glycans.24 For this reason, it is very difficult to get detail information about glycosylation heterogeneity at an intact level.29 These oligosaccharides are a complex mixture of different glycans, making them challenging for characterization. However, this characterization is necessary because the glycoforms may potentially be immunogenic and they must be identified, quantified, and limited when the CTLA4-Ig is intended for therapeutic use. When produced in rodent cell lines, nonhuman glycan structure (α-1,3-galactose) may be added to the glycan terminus of the product. Lammerts et al found that the immunogenicity was documented for both the terminal α-Gal on the N-glycan at the cetuximab Fab, while terminal α-Gal on the Fc glycan of other antibodies expressed from mouse-derived cell lines was not immunogenic.30 The Fc fusion proteins have different glycan distributions, therefore researching the glycan at an intact level is necessary.
A combination of high resolution ultra-performance liquid chromatography (UPLC) and ESI-Q-Tof MS was used to measure the molecular masses at the intact protein level. Data processing software (BiopharmaLynx 1.4) was used to automatically deconvolute the collected data to a single peak, which represents average mass for each liquid chromatography peak.31 The deconvoluted results of the intact forms of both the control and biosimilar candidate were shown in Figure 1. From the total ion current (Fig. 1A), we could see the biosimilar and control had the same retention time(RT). To some extent, we may conclude that they had a similar polarity. However, while amplifying the TIC (Fig. 1B-1D), we found that our data processing software BiopharmaLynx 1.4 could not handle the data because the modifications were too complex to analyze at the intact protein level. To some extent, this fusion protein had complex glycosylation modification profiles.
Figure 1.
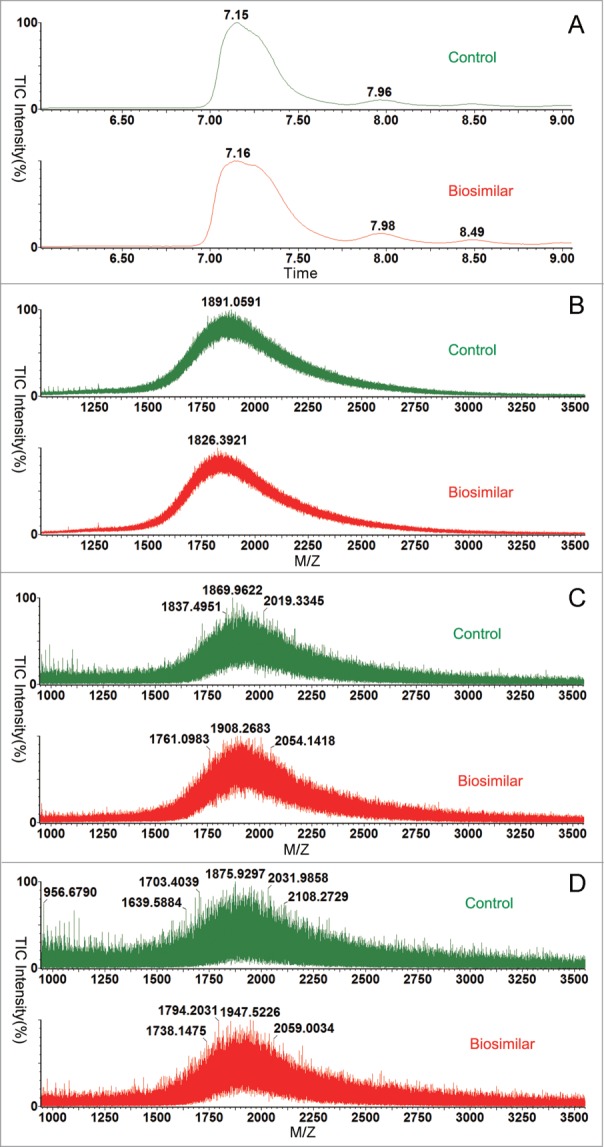
Comparison of biosimilar and control at the intact level. Green line biosimilar; red line control CTLA4-Ig. (A) The total ion current (TIC). (B) The amplification of TIC(7.0–7.6 min). (C) The amplification of TIC(7.8–8.2 min). (D) The amplification of TIC(8.4–8.6 min).
To keep further analysis of straightforward, the samples were then reduced with DTT (50 mM) to decrease the complexity caused by the pairs of oligosaccharides at the intact protein level. This method can reduce the complexity by half. Individual chain inspections of the reduced CTLA4-Fc fusion protein confirmed that more than one N-linked glycoform and O-linked glycoform were occupied (Fig. 2A), making it is very difficult to analyze in-depth.
Figure 2.
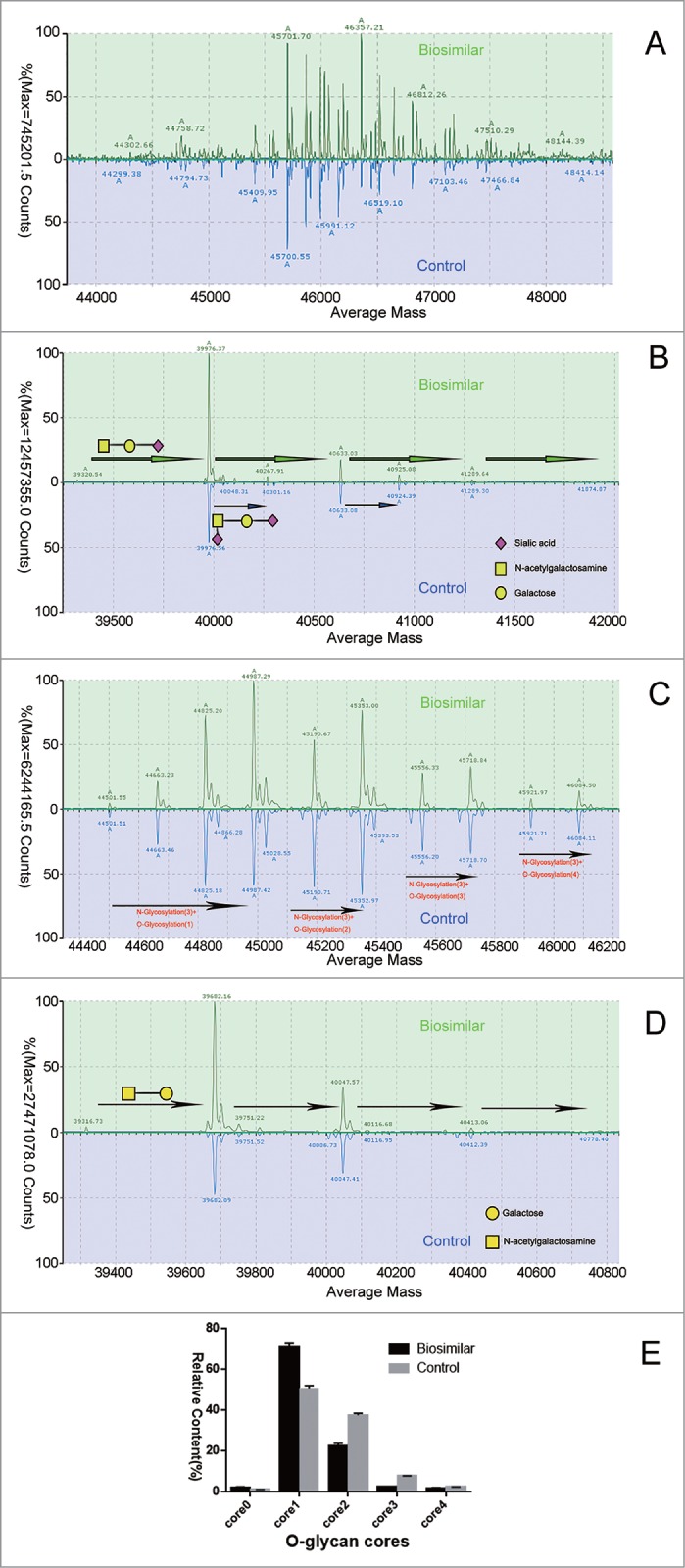
Comparison of biosimilar and control at the subunit level. Green line biosimilar; blue line originator CTLA4-Fc fusion protein. CTLA4-Fc fusion proteins did not deal with enzyme (A), digested by PNGase F (B), by neuraminidase (C), by neuraminidase plus PNGase F, (D); and reduced by DTT, were injected into the C4 UHPLC column. Panel correspond to deconvoluted mass spectra of this fusion protein subunit, respectively (transform algorithm from MassLynx™ 4.1). (E) Relative contents of O-linked glycans quantified by integrated peak intensity of MS (n = 3).
To reduce the glycosylation complexity, many kinds of glycosidases are very useful. We first used PNGase F to remove the N-linked glycans, to further research O-linked glycans at subunit level, as shown in Figure 2B. From Figure 2B, we found that there are more than one O-linked glycan, as the peaks have a different mass with gaps of 656 Da and 291 Da. The mass differences were consistent with the mass increments of sialic acids. As the N-linked and O-linked glycans commonly have more than one sialic acid, we treated the protein with neuraminidase to remove terminal sialic acid, resulting in the reduction of heterogenicity (Fig. 2C). As shown in Figure 2C, results for the biosimilar candidate and control were consistent with 3 N-linked glycans and different O-linked glycanforms. Both have 3 N-linked glycans plus a different number of O-linked glycan forms. The main peaks are G0F, G1F, and G2F with several cores of O-linked glycans, without existing sialic acids as we treated samples with the neuraminidase.
We also used PNGase F and neuraminidase on samples to remove N-linked glycans and the sialic acid, making the samples simpler. Results shown in Figure 2D indicate that both of them were consistent with the number of O-glycan sites. After exposure to the 2 enzymes, there were only cores of O-linked glycans (365 Da). The relative content of O-linked glycans quantified by integrated MS peak intensity was shown in Figure 2E. The biosimilar candidate had a slightly higher level of the core0 and core1, but a little lower level at the core2 and core3 compared with the control.
Overall, we could conclude that the biosimilar and control have complex glycosylation at the intact protein level: first, the main N-linked glycan forms were G0F, G1F, and G2F after removing the sialic acid; second, each chain had 3 N-linked glycans; third, there might exist 4 O-linked glycans; last, the O-linked glycan forms were the core, which consisted of one N-acetylgalactosamine and one Galactose, with one or 2 sialic acids. Additional details on glycosylation were investigated using peptide mapping and free glycan profiling.
Peptide level glycan analysis
Peptide mapping was a useful technique for certifying the proteins amino acid sequences and their PTMs, including glycosylation, oxidation and deamidation.32, 33 The peptide map method could also be used as an alternative or be combined with an oligosaccharide mapping method to acquire site-specific information for proteins with multiple glycosylation sites. For complicated sample preparation, we might treat them with trypsin combined with other proteolytic enzymes to digest the protein into peptide fragments that were amenable to analysis by LC–MS.
Consequently, peptide mapping was used to study protein sequences and PTMs differences between the biosimilar and the control fusion proteins.34 Using a data acquisition C18 LC-MS with alternate changeable energy full scanning (LC-MSE) was useful for peptide maps.35 Unlike LC-UV/MS peptide mapping methods, MSE acquired exclusive fragments of all precursors. Consequently, all peptide precursors were fragmented in one experiment and getting all information with full-scan fragments. Chakraborty and colleagues had applied LC-MSE for analyzing peptide maps of a protein digest.36 High sequence coverage and good analytical reproducibility were reached in replicated analyses of protein digestion.
Consistent with the previous study, N-glycosylation was only observed in an asparagine (N) residue located in the tryptic peptides T5 (QADSQVTEVCAATYMMGNELTFLDDSICTGTSSG NQVNLTIQGLR), T7 (VELMYPPPYYEGIGNGTQIYVIDPEPCPDSDQEPK) and T14 (EEQYN STYR). For T5, a total of 7 N-linked glycoforms were identified (shown in Fig. 3A for the corresponding glycan structures of the glycoforms). G0F, G1F, G2F, G2FS1, G2FS2, G3FS2 and G4FS2 were automatically assigned by the application manager by their accurate mass and confirmed by examining the MSE spectra. While G2FS1, G2FS2 and G3FS2 were dominant, they were all consist with sialic acids. For T7, we identified 8 N-linked glycoforms, comparable to T5 (shown in Fig. 3B for the corresponding glycan structures of the glycoforms). Compared to T5 and T7, we found major 6 N-linked glycoforms, G0F-N, G0F, G1F, G2F, G2FS1, and G2FS2, in T14 (shown in Fig. 3C for the corresponding glycan structures of the glycoforms). G0F, G1F and G2F were dominant, as expected of recombinant antibodies.
Figure 3.

Mass spectra and MS/MS spectra summed across the 4 boxed-in regions (T5, T7, T8-9, and T14) of the LC–MS tryptic map for the CTLA4-Fc fusion protein, corresponding to the 3 N-linked sites (T5, T7, and T14) and 4 O-linked sites (T8-9) of glycosylation on the CTLA4-Ig fusion protein molecule. Major peaks in the spectra are labeled with oligosaccharide structure assignments. (A) The N-linked glycosylation tryptic peptides T5, (B) the N-linked glycosylation tryptic peptides T7, (C) The N-linked glycosylation tryptic peptides T14, and (D) the O-linked glycosylation tryptic peptides T8-9 respectively. (E-K) The ETD mode to detect T8–9 with 4 O-linked glycosylation sites.
The CTLA4 region is an immunoglobulin- like domain and the N-glycosylation sites at T5 and T7 corresponded to Asn 76 and Asn 108, respectively. The Asn 76 N-glycosylation site was located in β-strand E of the ABED β-sheet, and the Asn 108 was located in the C-terminal G strand of the CC’FG β-sheet on the opposite face of the IgV sandwich. The “MYPPPYY loop” between the 2 β-strands was the key part of the interaction of the CTLA4 homodimer with its ligands.31 Unlike the antibody N-glycans constrained inside the Fc domain cavity, the N-glycans in the CTLA4 domain project out from the surface of the protein into the surrounding medium. Thus, the increase of sialic acids in this area may be very important to the fusion protein's function. At the same time, it was necessary to identify and quantify the glycans, so we separated the fusion protein into 2 parts, the CTLA4 extracellular domain and the Fc domain, with Ides. We compared the 2 parts at the free glycan profiling level. As Figure 4C shows, the result is accordant with the peptide mapping.
Figure 4.

Comparison of LC separation and quantification of 2-AB labeled free glycans released from the control and biosimilar CTLA4-Fc fusion proteins. (A) The fluorescence chromatograms of 2-AB labeled glycans separated by the UPLC HILIC column from intact protein. (B) Integrated peak area of LC-fluorescence chromatograms, relative contents of 2-AB labeled glycans were quantified (n = 3). (C) The fluorescence chromatograms of 2-AB labeled glycans separated by the UPLC HILIC column from the 2 separation part.
The hinge region of CTLA4-Fc fusion protein was rich in serine and threonine. The residues were sites for the attachment to more than one O-linked glycan chains. As previously reported, IgA1 with truncated O-glycans in the hinge region is prone to self-aggregation and forms immune complexes with the IgG antibody against the hinge region of IgA1.37 As such, the O-linked glycans at the hinge region may affect the stability and function of Fc fusion proteins.37 In the CTLA4-Ig fusion proteins, we detected the O-linked glycans on T8-9 (SSDKTHTSPPSPAPELLGGSSVFLF PPKPK). As shown in Figure 3D, at the lower (6v) MS level, we confirmed the peptide sequence utilizing collision-induced dissociation (CID) for glycopeptide fragmentation. However, in the high-energy fragmentation spectra, only neutral loss peaks were observed. O-glycosylation sites were reportedly occupied in the literature, but the specific glycosylation site was not described.31
The ion intensity of the radical anions and selected peptide precursor ions are important factors for effective ETD fragmentation. To confirm this, we removed the N-glycosylations and sialic acids with PNGase F and neuraminidase, and we concentrated the T8-9 with HILIC. As shown in Figure 3E, the deconvoluted ETD spectrum from T8-9 peptide (m/z 841.01343, MH5+) contains 3 core1 O-glycans. A series of fragments were identified, and the annotation for the fragments is shown in Figure 3E. The fragment ion peak at m/z 557.22 (Fig. 3E) corresponds to the c2ions of the peptide with a combined mass of the peptide SS (192.09 Da) and a core 1 tag (365.13 Da). Similarly, the fragment ion peak at 1591.67 matches the mass of peptide SSDKTHTS (861.41 Da) plus 2 core1 tags (2 × 365.13 = 730.26 Da). This ETD information allowed the sites of O-glycosylation to be determined as Ser-2, Ser8 and Ser-11 for the T8-9 glycopeptide eluting at 22.92 min. ETD sequencing was also performed on other O-linked glycan sites (Fig. 3F-3H). As Table 1 shows, the second, eighth, eleventh and twentieth serines each had one O-glycan, which was accordant to the subunit level.
Table 1.
Identified glycosylation sites* from electron-transfer dissociation spectra of glycopeptides from CTLA4-Ig
| O-linked Glycan |
||||
|---|---|---|---|---|
| Tryptic Peptide | Sequence | Core1 | MH+(Da) | RT(min) |
| T8–9 | SSDKTHTSPPSPAPELLGGSSVFLFPPKPK | 3 | 4201.03803 | 22.92 |
| TSPPSPAPELLGGSSVFLFPPKPK | 2 | 3180.60244 | 28.77 | |
| THTSPPSPAPELLGGSS | 1 | 1999.93833 | 16.68 | |
| GSSVFLFPPKPK | 1 | 1668.87273 | 25.41 | |
O-linked glycan sites are indicated in red.
Cleaved glycan level analysis
The released glycans from samples treated with PNGase F were labeled with 2-AB, separated using a hydrophilic interaction chromatography (HILIC) column, and quantified by UPLC with a fluorescence detector. Using ESI Q-Tof mass spectra, the glycan mass profiling and structure elucidation were performed. Thirteen major glycans (G0F, G1F, G2F, G1FS1, G2FS1, G2S2, G2FS2, G3FS1, G3FS2, G3FS3, G4FS2, G4FS3, and G4FS4; Fig. 4A) were identified and quantified for both fusion proteins, and the result was consistent with peptide mapping data. Based on integrated peak areas in Figure 4A, the fluorescence quantification result was shown in Figure 4B. The glycan proportions in the CTLA4-Ig fusion proteins were almost identical, but they were different at some glycan forms. The relative ratios of G2FS1/G2F, G2FS2/G2F, and G3FS1/G2F were increased in the innovator compared to the biosimilar fusion protein. With 3 orthogonal methods applied in this study, we confirmed that the control and biosimilar CTLA4-Ig fusion proteins are comparable in cleaved glycan level.
The fusion protein has 2 parts, each serving a different function. The heterogeneity of glycans in the 2 parts may have different effects. We separated the fusion protein into 2 parts, the CTLA4 extracellular domain and the Fc domain, with Ides. After being released from the 2 parts by PNGase F and labeled with 2-AB, the glycans were separated using a HILIC column and quantified with a fluorescence detector. Using ESI Q-Tof mass spectra, the glycan mass profiling and structure elucidation were performed. More than 17 major glycans (G0F, G1F, G2F, G1FS1, G2FS1, G2S2, G2FS2, G3FS1, G3FS2, G3FS3, G4FS2, G4FS3, etc; Fig. 4C) were identified and quantified in the CTLA4 extracellular domain, and about 6 major glycans (G0F, G1F, G2F, G1FS1, G2FS1, G2FS2) were identified and quantified in the Fc domain. In the CTLA4 extracellular domain, the sialic acid represents a greater part and the glycosylation is more complex, compared to the Fc domain. The result was consistent with peptide mapping data.
To some extent, though, it might still be challenging to fully understanding the nature of glycosylation modifications. Under all circumstances, it was important to assess the PTMs with regard to stability, functions and safety, especially among innovator molecules and biosimilars. Glycosylation modifications were likely either key process attributes (KPAs) or critical quality attributes (CQAs). Therefore, information on glycosylation modifications is critical to establishing comparability and similarity, with the ultimate goal of ensuring the efficacy and safety of biosimilar medicines.
Discussion
To date, more than 40 mAbs and antibody-derivatives have been approved and marketed. Data on potency and safety for these products are available. A wealth of experience in the development of antibody derivatives is also available. With the impending patent expiration for commercial antibody derivative medicines, the development of biosimilar antibody derivatives will be less restricted. However, a range of differences between biosimilar products and innovator products may still exist. Developing a standardized series of analytical methods to evaluate the molecular similarity is still needed. With the advancement of technology, a number of physicochemical and biological methods are now available for the characterization of antibody derivatives. It remains challenging to routinely compare biosimiliar antibody derivatives to the corresponding innovator product at the molecular level. In this article, the modern ultra-performance liquid chromatography and LC-MS based methods were successfully applied to research the glycosylation modification of a CTLA4-Ig fusion protein.
Using this analytical technology, we were able to successfully study the glycosylation modification of CTLA4-Ig fusion proteins on 3 levels: intact protein level glycan analysis, peptide level glycan analysis, and released free glycans analysis. As a fast and simple estimation method, intact protein mass measurement was first selected to research obvious differences between the biosimilar and the innovator CTLA4-Ig fusion proteins. Unlike small molecule drugs, antibody fusion proteins are large, heterogeneous proteins with a numerous PTMs. Although they are more similar to antibody medicines than any other medicines, Ig fusion proteins are more complex than antibody medicines. Antibody medicines usually have only 2 glycosylation sites, while Ig-fusion proteins always have several N-linked and more than one O-linked glycosylation sites. As demonstrated in Figure 1A, Ig-fusion proteins cannot be analyzed without any treatment because of the different complex glycosylation modifications. We thus treated the samples with PNGase F and neuraminidase to make the analysis easier, and found that the 2 samples have no evident differences in the intact level glycan analysis. We still consider intact protein mass measurement a useful and fast method to evaluate the similarity between the biosimilar and control Ig-fusion protein medicine with several treatments if the target protein is too complex.
While MS analysis of intact protein is a useful and fast method, it cannot exactly identify the detailed differences. In addition, it has inevitable limitations for detection and characterization of biological medicines, especially for Ig fusion proteins. To further comprehend the detail of modifications and variants, LC-MSE peptide mapping was performed. The peptide mapping of large Ig fusion proteins is a challenging task in general because a large number of peptides must be separated by LC, and because the fusion protein has more than 2 glycosylation sites. The UPLC technology makes separation of the peptides possible.
As the LC-MSE peptide mapping method can identify site-specific modifications, it is useful to research the sites of modifications. In this study, the peptide mapping determined 3 N-linked glycosylation sites on peptides T5 (QADSQVTEVCAATMMGNELTFLDDSICTGTSSGNQVNLTI QGLR), T7 (VELMYPPPYYEGIGNGTQIYVIDP EPCPDSDQEPK), and T14 (EEQYNSTYR) and 4 O-linked glycosylation sites on peptide T8-9 (SSDKTHTSPP SPAPELLGGSSVFLFPPKPK) (Fig. 3A to H). As expected, G0F, G1F, and G2F were the major glycoforms on T14, which correspond to those of the IgG1 antibody. On the contrary, G2FS1 and G2FS2 were the major glycoforms on T5 and T7. This may be very important to the function of the fusion proteins. With the ETD/MS method we identified 4 sites: the second, eighth, 11, and 20 serine in O-linked glycans.
While peptide maps revealed differences in the relative glycosylation patterns for the investigated fusion proteins, the released and labeled glycans with the HILIC LC-fluorescence method was used for accurate quantification of the glycans (Fig. 4A and B). There were small differences in the relative rates of G2FS1 and G2FS2 between the innovator and the biosimilar CTLA4-Ig fusion proteins. The low-abundance glycoforms like Man5 were also quantified. In addition to providing sensitive and accurate quantification, the method has the ability to resolve isomers of glycans such as G1a and G1b, and G1Fa and G1Fb (Fig. 4A). In contrast, the release and recovery of the O-linked glycans have remained a very challenging task due to the lack of available and specific O-glycanases. In this situation, the chemical cleavage method is the only one for the analysis of O-linked glycans in glycoproteins at present. The 2 most commonly used chemical cleavage methods are hydrazinolysis and alkaline β-elimination. Although hydrazinolysis can yield reducing glycans, it can change the sialic acids, like O-Acyl substitutions. Meanwhile alkaline medium can convert glycans to their respective alditols in the alkaline β-elimination process.38 Caution should be taken when dealing with O-linked glycans, because the chemical cleavage and release steps may often cause breakdown of the glycan itself. For this reason, we did not analyze the O-linked glycans at the cleaved glycan level in this article. As the Ig fusion proteins always have 2 parts, and each has different function, the separation and study of the modification of the 2 parts is very important.
In conclusion, intact protein mass measurement can perform a comprehensive elucidation the glycosylation of a biosimilar Ig fusion protein to the control medicine at 3 levels: the intact protein mass measurement, peptide mapping and free glycan quantification. Intact protein mass measurement is a simple and fast method that can both compare molecular mass and reveal heterogeneities. For more complex Ig fusion proteins, it has some fatal weaknesses so we needed to treat the sample to reduce complexity. Peptide mapping is a gold standard method to identify and quantify low-abundance variants and site-specific modifications, such as +15.99 Da of M-oxidation and +0.98 Da of N-deamidation. It can also identify site-specific modifications using ETD, such as glycosylation, oxidation and deamidation sites, which may be very important to their property, potency, and safety. Identification / quantification by LC-MS was perfect for comparison of the biosimilar and innovator product N-glycosylation heterogeneities. With UPLC and ESI-Q-TOF mass spectrometry technologies, we elaborately characterized the biosimilar and innovator CTLA4-Ig fusion proteins at the cleaved glycan level.
Although we analyzed the N-glycans and the sites of O-glycans in detail, we have yet to illustrate the relationship between the modifications with the function, half-life, and stability to the Ig-fusion protein. In recent years, researchers have found that a CHO ortholog of N-acetyllactosaminide 3-α-galactosyltransferase-1 is active, and can affect glycosylated protein products produced in CHO. 39 They also observed α 1,3-Gal glycan form in a CTLA4-Ig product, which had an abundance of ∼0.2% of the mass of total N-linked glycans.39 No data on this immunogenic glycan variant is provided in this study because we found that the N-linked glycans had an abundance below 0.1% (data not shown). Further study of the Ig fusion proteins using additional analytical methods and preclinical and clinical trials of them need to be done in the future.
Materials and Methods
Samples and materials
The control CTLA4-Ig was the commercial therapeutic protein abatacept (Bristol-Myers Squibb, East Syracuse, NY). The biosimilar CTLA4-Ig was expressed in our lab. It was manufactured in CHO cell lines, purified with several chromatographic techniques and maintained at 4 °C. NaBHCN, iodoacetamide (IAM), dithiothereitol (DTT), guanidine hydrochloride (GuHCl), ammonium bicarbonate (NH4HCO3), trypsin, peptide N-glycosidase (PNGase F) and 2-AB- labeling Kit were purchased from Sigma, formic acid (FA) from EM sciences, Optima-grade acetonitrile (ACN) from Fisher Scientific, and HILIC solid phase extraction (SPE) posts were obtained from Waters Corp.
Instrumentation
Separations were performed on Waters ACQUITY UPLCTM system equipped with fluorescence detector. All MS measurements were implemented on a Waters Xevo G2-S system equipped with an ESI source and a Thermo Orbitrap Fusion system equipped with ETD. The analytic softwares are the Biopharmalynx 1.3 (Waters) with MaxEnt and Proteome Discoverer 1.4 (ThermoFish er Scientific) with SequestHT.
Intact protein mass measurements
The fusion proteins were reduced with DTT. The intact and reduced samples were analyzed by reverse-phase LC-MS. The reverse-phase desalting separations of intact samples were performed on a Waters MassPREPTM Micro Desalting Column (2.1 × 5 mm) using a gradient (3–10 min, 5–90% B). The reverse-phase separations of reduced samples were performed on a Waters C4 Column (2.1 × 50 mm, 1.7 μm) using a gradient (8–18 min, 5–50% B). The mobile phase B was acetonitrile with 0.1% formic acid, while mobile phase A was water with 0.1% formic acid. The flow rate was maintained at 0.400 m L/min, and column temperature was maintained at 80°C to Desalting Column and 40°C to C4 Column respectively. Mass spectrometric analysis was performed on a Waters Xevo G2-S Q-TOF MS system with a positive ion mode. The desolvation gas and source temperature were set to 400°C and 120°C. The capillary and cone voltages were set at 3,000 and 35V, respectively. Transfer collision energies was set at 0V. The m/z scan range was set to 500–3500. The deconvolution of ESI mass spectra of intact and reduced samples was performed by Biopharmalynx 1.3 using MaxEnt 1 algorithm.
Peptide mapping analysis for N-linked Glycans
After denaturing with guanidine hydrochloride at 37°C for 1 hour and reducing with 50 mM DTT and alkylated with IAM for 45 minutes, exchanging the buffer to 50 mM NH4HCO3 solution. Enzyme (Trypsin) digests of fusion proteins were prepared using trypsin (1:25 w/w) at 37°C for 2 hours. The digests were terminated at 0.1% FA. Two ug protein digests were injected for each run. Peptide mixture was separated on a BEH300 C18 column (2.1 × 150 mm, 1.7 μm) at 45°C using a 80-min gradient (1-36% B). The LC mobile phases was the same as in above intact protein mass analyses and the flow rate was maintained at 0.200 mL/min. The eluted peptides were detected by MS with a changing low collision energy (6 V) and high collision energy (ramping from 25 to 45V) ESI acquisition mode to obtain the precursor ions (MS) and their fragmentation data (MSE), respectively. Scan time was 0.5 second. A capillary voltage of 3000V, source temperature of 120°C, cone voltage of 35 V, cone gas flow of 50 L/h were set during the analyses, respectively. An auxiliary pump was used to spray a solution of 100 fmol/ ml Glu1-fibrinopeptide B (GFP) for mass accuracy (lockmass channel), with a flow rate of 10 ul/min.
The collected LC-MS/MS data were processed by BiopharmaLynx 1.3 software, using fully tryptic cleavage rules for the sequences of the control and biosimilar candidate CTLA4-Ig fusion proteins. We set the fixed modification and variable modification, while searching the protein digest library. Glycan structures were also set as variable modifications. The mass tolerance for both precursors and fragments was set to less than 30 ppm. The identified peptides were confirmed by MSE spectra with at least 5 fragment ions.
Peptide mapping analysis for O-linked Glycans
To reduce the complexity of the sample and get the in-depth information of the fusion protein, we used 20 mg sample (deglycosylated by adding PNGase F and incubating at 37°C overnight) and digested with trypsin like above. Separating the samples with HILIC column, we got about 200 ug peptides of the fusion protein with O-linked Glycans. 2 ug peptide mixture was separated by an Easy-nLC system (ThermoFisher Scientific) equipped with an Easy-Spary column (C18, 75 um × 15 cm, 3 um, Thermofisher Scientific). Peptides were eluted with an acetonitrile gradient and 0.1% formic acid in 40 minutes gradient at a flow rate of 350 nl/min. The effluents were analyzed by Orbitrap Fusion (ThermoFisher Scientific) using a data-dependent scan mode. Precursor ions were scanned by Orbitrap at a resolution of 60K. Then the most intense ions were selected by Quadrupole and dissociated by ETD with a supplemental activation. Product ions were scanned by ion trap at a rapid scan mode. Raw Files generated by Orbitrap Fusion were processed by Proteome Discoverer 1.4 (ThermoFisher Scientific) using the search engine of SequestHT. All MS2 spectrums were matched against the sequence of T8-9. Precursor ion tolerance was set to 20 ppm and 1.2 Da for product ion, for 2 charged precursor ions, weight of c, y and z ions were set to 0.25, 0.75, and 1; for precursor ions with more than 2 charges, weight of c, y and z ions were set to 1, 0.25, and 1. The O-linked Glycosylation (+365.131 Da) was set to dynamic modification. Searching results were all manually checked to ensure each identified peptide had a consecutive series of product ions.
Free glycan profiling
N-linked glycans of the fusion proteins were deglycosylated by adding PNGase F and incubating at 37°C overnight, under denaturation conditions. After precipitating deglycoslated samples with cold ethanol and centrifuging the mixture at 14,000 × g for 30 min, the supernatant was transferred to a new sample tube and evaporated to dryness. Released and dried glycans were labeled with 2-aminonbensamide (2-AB) solution for 4 hours at 65°C. HILIC SPE post was used to remove excess labeling agent again. The flow-through containing the labeled oligosaccharides was transferred to a new sample tube and evaporated to dryness. The dried oligosaccharides were diluted to 80% ACN in water prior to analysis.
Subsequently, the labeled glycans were separated on a HILIC column (2.1 × 150 mm, glycan, 1.7 um) at 60°C and detected by an ACQUITY UPLC fluorescence detector plus Q-TOF MS. Separations were performed at a 45 min gradient (72–62% B) and 0.4 ml/min flow rate using 30 mM ammonium formate, pH 4.75 as mobile phase A and 100% ACN as mobile phase B. The desolvation gas and source temperature was set to 350°C and 120°C. The capillary and cone voltages were set at 3,000 and 45V, respectively. Transfer collision energies was set at 6V. The m/z scan range was set to 500–3000.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from Natural Science Foundation of China (81330061), Ministry of Science and Technology of China (973 projects 2010CB833605 and 863 projects 2014AA021004), State Key Project for New Drug Development (2013ZX09101021; 2013ZX09401303), Shanghai Commission of Science and Technology (Key Laboratory and Projects 13DZ1930100), and Shanghai Excellent technical leader (13XD1424000).
References
- 1.Reichert JM. Marketed therapeutic antibodies compendium. mAbs 2012; 4:413-5; PMID:22531442; http://dx.doi.org/ 10.4161/mabs.19931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Unützer J, Schoenbaum M, Katon WJ, Fan MY, Pincus HA, Hogan D, Taylor J. Healthcare costs associated with depression in medically Ill fee-for-service medicare participants. J Am Geriatr Soc 2009; 57:506-10; PMID:19175438; http://dx.doi.org/ 10.1111/j.1532-5415.2008.02134.x [DOI] [PubMed] [Google Scholar]
- 3.Belsey MJ, Harris LM, Das RR, Chertkow J. Biosimilars: initial excitement gives way to reality. Nat Rev Drug Discov 2006; 5:535-6; PMID:16883647; http://dx.doi.org/ 10.1038/nrd2093 [DOI] [PubMed] [Google Scholar]
- 4.Sockolosky JT, Tiffany MR, Szoka FC. Engineering neonatal Fc receptor-mediated recycling and transcytosis in recombinant proteins by short terminal peptide extensions. Proc Natl Acad Sci USA 2012; 109:16095-100; PMID:22991460; http://dx.doi.org/ 10.1073/pnas.1208857109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie H, Chakraborty A, Ahn J, Yu YQ, Dakshinamoorthy DP, Gilar M, Chen W, Mazzeo JR. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry technologies. mAbs 2010; 2:379-94; PMID:20458189; http://dx.doi.org/ 10.4161/mabs.11986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen RC, Naiyanetr P, Shu SA, Wang J, Yang GX, Kenny TP, Guggenheim KC, Butler JD, Bowlus C, Tao MH. Antimitochondrial antibody heterogeneity and the xenobiotic etiology of primary biliary cirrhosis. Hepatology 2013; 57:1498-508; PMID:23184636; http://dx.doi.org/ 10.1002/hep.26157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Declerck PJ. Biosimilar monoclonal antibodies: a science-based regulatory challenge. Expert Opin Biol Ther 2013; 13:153-6; PMID:23286777; http://dx.doi.org/ 10.1517/14712598.2012.758710 [DOI] [PubMed] [Google Scholar]
- 8.Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov 2012; 11:527-40; PMID:22743980; http://dx.doi.org/ 10.1038/nrd3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jez J, Antes B, Castilho A, Kainer M, Wiederkum S, Grass J, Rüker F, Woisetschläger M, Steinkellner H. Significant impact of single N-glycan residues on the biological activity of Fc-based antibody-like fragments. J Biol Chem 2012; 287:24313-9; PMID:22589538; http://dx.doi.org/ 10.1074/jbc.M112.360701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bumbaca D, Boswell CA, Fielder PJ, Khawli LA. Physiochemical and biochemical factors influencing the pharmacokinetics of antibody therapeutics. AAPS J 2012; 14:554-8; PMID:22610647; http://dx.doi.org/ 10.1208/s12248-012-9369-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Werner RG, Kopp K, Schlueter M. Glycosylation of therapeutic proteins in different production systems. Acta Paediatr Suppl 2007; 96:17-22; PMID:17391433; http://dx.doi.org/ 10.1111/j.1651-2227.2007.00199.x [DOI] [PubMed] [Google Scholar]
- 12.Jefferis R. Isotype and glycoform selection for antibody therapeutics. Arch Biochem Biophys 2012; 526:159-66; PMID:22465822; http://dx.doi.org/ 10.1016/j.abb.2012.03.021 [DOI] [PubMed] [Google Scholar]
- 13.Zhu D, McCarthy H, Ottensmeier CH, Johnson P, Hamblin TJ, Stevenson FK. Acquisition of potential N-glycosylation sites in the immunoglobulin variable region by somatic mutation is a distinctive feature of follicular lymphoma. Blood 2002; 99:3562-8; PMID:11986208; http://dx.doi.org/ 10.1182/blood.V99.7.2562 [DOI] [PubMed] [Google Scholar]
- 14.Lee JW, Forciniti D. Effect of glycosylation on the partition behavior of a human antibody in aqueous two-phase systems. Biotechnol Prog 2013; 29:943-50; PMID:23657984; http://dx.doi.org/ 10.1002/btpr.1741 [DOI] [PubMed] [Google Scholar]
- 15.Zheng K, Bantog C, Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. mAbs 2011; 3:568-76; PMID:22123061; http://dx.doi.org/ 10.4161/mabs.3.6.17922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andersen DC, Bridges T, Gawlitzek M, Hoy C. Multiple cell culture factors can affect the glycosylation of Asn-184 in CHO-produced tissue-type plasminogen activator. Biotechnol Bioeng 2000; 70:25-31; PMID:10940860; http://dx.doi.org/ 10.1002/1097-0290(20001005)70:1%3c25::AID-BIT4%3e3.0.CO;2-Q [DOI] [PubMed] [Google Scholar]
- 17.Butler M. Optimisation of the cellular metabolism of glycosylation for recombinant proteins produced by Mammalian cell systems. Cytotechnology 2006; 50:57-76; PMID:19003071; http://dx.doi.org/ 10.1007/s10616-005-4537-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gadgil HS, Pipes GD, Dillon TM, Treuheit MJ, Bondarenko PV. Improving mass accuracy of high performance liquid chromatographyelectrospray ionization time-of-flight mass spectrometry of intact antibodies. J Am Soc Mass Spectrom 2006; 17:867-72; PMID:16631376; http://dx.doi.org/ 10.1016/j.jasms.2006.02.023 [DOI] [PubMed] [Google Scholar]
- 19.Emery P, Kavanaugh A, Bao Y, Ganguli A, Mulani P. Comprehensive disease control (CDC): what does achieving CDC mean for patients with rheumatoid arthritis? Ann Rheum Dis 2014; 0:1-10; PMID:25139667; http://dx.doi.org/ 10.1136/annrheumdis-2014-205302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hot A, Miossec P. Effects of interleukin (IL)-17A and IL-17F in human rheumatoid arthritis synoviocytes. Ann Rheum Dis 2011; 70:727-32; PMID:21345813; http://dx.doi.org/ 10.1136/ard.2010.143768 [DOI] [PubMed] [Google Scholar]
- 21.Maxwell LJ, Singh JA. Abatacept for rheumatoid arthritis: a Cochrane systematic review. J Rheumatol 2010; 37:234-45; PMID:20080922; http://dx.doi.org/ 10.3899/jrheum.091066 [DOI] [PubMed] [Google Scholar]
- 22.Maxwell L, Singh JA. Abatacept for rheumatoid arthritis. Cochrane Database Syst Rev 2009; 4:7277-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang C. Receptor-Fc fusion therapeutics, traps, and MIMETIBODY technology. Curr Opin Biotechnol 2009; 20:692-9; PMID:19889530; http://dx.doi.org/ 10.1016/j.copbio.2009.10.010 [DOI] [PubMed] [Google Scholar]
- 24.Bongers J, Devincentis J, Fu J, Huang P, Kirkley DH, Leister K, Liu P, Ludwig R, Rumney K, Tao L. Characterization of glycosylation sites for a recombinant IgG1 monoclonal antibody and a CTLA4-Ig fusion protein by liquid chromatography–mass spectrometry peptide mapping. J Chromatogr A 2011; 1218:8140-9; PMID:21978954; http://dx.doi.org/ 10.1016/j.chroma.2011.08.089 [DOI] [PubMed] [Google Scholar]
- 25.Satoh M, Iida S, Shitara K. Non-fucosylated therapeutic antibodies as next-generation therapeutic antibodies. Expert Opin Biol Ther 2006; 6:1163-73; ; http://dx.doi.org/ 10.1517/14712598.6.11.1161 [DOI] [PubMed] [Google Scholar]
- 26.Li H, Sethuraman N, Stadheim TA, Zha D, Prinz B, Ballew N, Bobrowicz P, Choi B-K, Cook WJ, Cukan M. Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat Biotechnol 2006; 24:210-5; PMID:16429149; http://dx.doi.org/ 10.1038/nbt1178 [DOI] [PubMed] [Google Scholar]
- 27.Eon-Duval A, Broly H, Gleixner R. Quality attributes of recombinant therapeutic proteins: an assessment of impact on safety and efficacy as part of a quality by design development approach. Biotechnol Prog 2012; 28:608-22; PMID:22473974; http://dx.doi.org/ 10.1002/btpr.1548 [DOI] [PubMed] [Google Scholar]
- 28.Commins SP, Kelly LA, Rönmark E, James HR, Pochan SL, Peters EJ, Lundbäck B, Nganga LW, Cooper PJ, Hoskins JM. Galactose-α-1, 3-galactose–specific IgE is associated with anaphylaxis but not asthma. Am J Respir Crit Care Med 2012; 185:723-30; PMID:22281828; http://dx.doi.org/ 10.1164/rccm.201111-2017OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beck A, Wagner-Rousset E, Bussat M-C, Lokteff M, Klinguer-Hamour C, Haeuw J-F, Goetsch L, Wurch T, Dorsselaer AV, Corvaïa N. Trends in glycosylation, glycoanalysis and glycoengineering of therapeutic antibodies and Fc-fusion proteins. Curr Pharm Biotechnol 2008; 9:482-501; PMID:19075687; http://dx.doi.org/ 10.2174/138920108786786411 [DOI] [PubMed] [Google Scholar]
- 30. Lammerts van Bueren JJ, Rispens T, Verploegen S, van der Palen-Merkus T, Stapel S, Workman LJ, James H, van Berkel PH, van de Winkel JG, Platts-Mills TA, et al. Anti-galactose-alpha-1,3-galactose IgE from allergic patients does not bind alpha-galactosylated glycans on intact therapeutic antibody Fc domains. Nat Biotechnol 2011; 29:574-6; PMID:21747378; http://dx.doi.org/ 10.1038/nbt.1912 [DOI] [PubMed] [Google Scholar]
- 31.Xie H, Gilar M, Chakraborty A, Chen W, Berger S. Monitoring deamidation progression in an antibody tryptic digest using UPLCMSE with BiopharmaLynx and a Xevo QTof MS system. Waters Appl Note 2009. (Milford, Massachusetts); 720002879en [Google Scholar]
- 32.Dick LW Jr, Mahon D, Qiu D, Cheng K-C. Peptide mapping of therapeutic monoclonal antibodies: improvements for increased speed and fewer artifacts. J Chromatogr B Technol Biomed Life Sci 2009; 877:230-6; PMID:19112052; http://dx.doi.org/ 10.1016/j.jchromb.2008.12.009 [DOI] [PubMed] [Google Scholar]
- 33.Chelius D, Xiao G, Nichols AC, Vizel A, He B, Dillon TM, Rehder DS, Pipes GD, Kraft E, Oroska A. Automated tryptic digestion procedure for HPLCMSMS peptide mapping of immunoglobulin gamma antibodies in pharmaceutics. J Pharm Biomed Anal 2008; 47:285-94; PMID:18313251; http://dx.doi.org/ 10.1016/j.jpba.2008.01.018 [DOI] [PubMed] [Google Scholar]
- 34.Xie H, Gilar M, Gebler JC. Characterization of protein impurities and site-specific modifications using peptide mapping with liquid chromatography and data independent acquisition mass spectrometry. Anal Chem 2009; 81:5699-708; PMID:19518054; http://dx.doi.org/ 10.1021/ac900468j [DOI] [PubMed] [Google Scholar]
- 35.Castro-Perez JM, Kamphorst J, DeGroot J, Lafeber F, Goshawk J, Yu K, Shockcor JP, Vreeken RJ, Hankemeier T. Comprehensive LC− MSE lipidomic analysis using a shotgun approach and its application to biomarker detection and identification in osteoarthritis patients. J Proteome Res 2010; 9:2377-89; PMID:20355720; http://dx.doi.org/ 10.1021/pr901094j [DOI] [PubMed] [Google Scholar]
- 36.Chakraborty AB, Berger SJ, Gebler JC. Use of an integrated MS-multiplexed MSMS data acquisition strategy for high-coverage peptide mapping studies. Rapid Commun Mass Spectrom 2007; 21:730-44; PMID:17279597; http://dx.doi.org/ 10.1002/rcm.2888 [DOI] [PubMed] [Google Scholar]
- 37.Royle L, Roos A, Harvey DJ, Wormald MR, van Gijlswijk-Janssen D, Redwan el RM, Wilson IA, Daha MR, Dwek RA, Rudd PM. Secretory IgA N- and O-glycans provide a link between the innate and adaptive immune systems. J Biol Chem 2003; 278:20140-53; PMID:12637583; http://dx.doi.org/ 10.1074/jbc.M301436200 [DOI] [PubMed] [Google Scholar]
- 38.Huang Y, Mechref Y, Novotny MV. Microscale nonreductive release of O-linked glycans for subsequent analysis through MALDI mass spectrometry and capillary electrophoresis. Anal Chem 2001; 73:6063-9; PMID:11791581; http://dx.doi.org/ 10.1021/ac015534c [DOI] [PubMed] [Google Scholar]
- 39.Bosques CJ, Collins BE, Meador JW III, Sarvaiya H, Murphy JL, DelloRusso G, Bulik DA, Hsu I-H, Washburn N, Sipsey SF, et al. Chinese hamster ovary cells can produce galactose-α-1,3-galactose antigens on proteins. Nat Biotechnol 2010; 28:1153-6; PMID:21057479; http://dx.doi.org/ 10.1038/nbt1110-1153 [DOI] [PMC free article] [PubMed] [Google Scholar]