

Abstract
Glucocorticoid (GC) resistance remains a major obstacle to successful treatment of lymphoid malignancies. Till now, the precise mechanism of GC resistance remains unclear. In the present study, dexamethasone (Dex) inhibited cell proliferation, arrested cell cycle in G0/G1-phase, and induced apoptosis in Dex-sensitive acute lymphoblastic leukemia cells. However, Dex failed to cause cell death in Dex-resistant lymphoid malignant cells. Intriguingly, we found that autophagy was induced by Dex in resistant cells, as indicated by autophagosomes formation, LC3-I to LC3-II conversion, p62 degradation, and formation of acidic autophagic vacuoles. Moreover, the results showed that Dex reduced the activity of mTOR pathway, as determined by decreased phosphorylation levels of mTOR, Akt, P70S6K and 4E-BP1 in resistant cells. Inhibition of autophagy by either chloroquine (CQ) or 3-methyladenine (3-MA) overcame Dex-resistance in lymphoid malignant cells by increasing apoptotic cell death in vitro. Consistently, inhibition of autophagy by stably knockdown of Beclin1 sensitized Dex-resistant lymphoid malignant cells to induction of apoptosis in vivo. Thus, inhibition of autophagy has the potential to improve lymphoid malignancy treatment by overcoming GC resistance.
Keywords: autophagy, apoptosis, dexamethasone, glucocorticoid resistance, lymphoid malignancy
Abbreviations
- 3-MA
3-methyladenine
- CQ
chloroquine
- Dex
dexamethasone
- Dox
doxorubicin
- LC3
microtubule-associated protein 1 light chain 3
- MDC
monodansylcadaverine
- mTOR
mammalian target of rapamycin
- OCT
optimum cutting temperature
- Rapa
rapamycin; WST-8, 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2, 4-disulfophenyl)-2H- tetrazolium, monosodium salt
Introduction
Lymphoid malignancies, such as acute/chronic lymphoblastic leukemia, lymphoma and myeloma, are associated with a variety of therapeutic challenges.1 Glucocorticoids (GC) have been wildly used as important therapeutic agents in the treatment of lymphoid malignancies.2 Apoptotic cell death is currently recognized as one of the main mechanisms of GC treatment of lymphoid malignancies for the following reasons: (1) repression of transcription of pro-inflammatory cytokine genes, including NF-κB,3 AP-1,4 and c-Myc;5 (2) other signaling molecules that involved in GC-mediated apoptosis, including calcium,6 RAFTK,7 IL-6, and STAT3.8
Although GC are widely used in clinical therapy, GC resistance on relapse often emerges, which is associated with poor prognosis. In addition, about 30% of the patients are innately resistant to GC. Till now, most studies have revealed that the mechanisms of GC resistance are associated mainly with defective apoptosis machinery, such as over-expression of anti-apoptotic protein Bcl-2 and Mcl-1.9 Recent studies suggested that polymorphisms of GC receptors10 and dysregulated ratio of GC receptor subtypes11 were associated to GC resistance, but the detailed mechanisms remained further elucidated. Thus, exploration of other new mechanisms contributing to GC resistance will promote the optimized design of treatment of lymphoid malignancies.
Autophagy is a dynamic process in which damaged organelles and unfolded proteins are engulfed by autophagosomes, then delivered to lysosomes for degradation.12 As a survival adaptation to tolerate stress and unfavorable conditions, autophagy has been shown to play a key role for therapy resistance during chemotherapy in hepatocarcinoma cancer,13 lung cancer,14 and multiple myeloma.15 For example, Dex induced autophagy by elevating Dig2 expression in murine lymphoma cells. Dig2 knockdown led to increased cell death during Dex treatment.16 Similarly, induction of autophagy contributed to prolonged survival of Bcl-2 positive murine lymphoma cells following Dex treatment. Inhibition of autophagy by 3-MA enhanced cytotoxicity of Dex in Bcl-2-positive cancer cells.17 However, whether autophagy is involved in GC resistance during Dex treatment in human lymphoid malignancies has not been clearly defined.
In this study, we found that autophagic activities were induced by Dex in Dex-resistant lymphoid malignant cells; however, such changes were not observed in Dex-sensitive cells. Dex reduced the activity of mTOR pathway during autophagy induction. Inhibition of autophagy augmented the proliferation inhibition and apoptosis induction effects of Dex both in vitro and in vivo analysis. Thus, our findings suggested a new treatment strategy for GC-resistant lymphoid malignancies.
Results
Dex inhibits cell proliferation in lymphoid malignant cells
To evaluate the effect of Dex on cell proliferation, WST-8 assay was conducted to assess the survival rates of cells treated with increasing concentrations of Dex for 24 and 48 h. We found that the inhibition of cell proliferation induced by Dex was both dose- and time-dependent in CCRF-CEM and Raji cells, while only dose-dependent in U-937 cells (Fig. 1A). We then used trypan blue exclusion assay to enumerate dead cells treated with indicated concentrations of Dex. Interestingly, the increased number of dead cells were consistent with the results of the WST-8 assay in CCRF-CEM cells, but very few dead cells were detected in Raji and U-937 cells (Fig. 1B). The effects of Dex on the induction of apoptosis were determined with Annexin V/PI staining in CCRF-CEM, Raji, and U-937 cells. Flow cytometric analysis displayed significantly increased apoptosis activities in Dex-sensitive CCRF-CEM cells and minor apoptosis in Dex-resistant Raji and U-937 cells (Fig. 1C). Collectively, these results suggested that Dex inhibited the proliferation more significantly in Dex-sensitive CCRF-CEM cells than in Dex-resistant Raji and U-937 cells.
Figure 1.
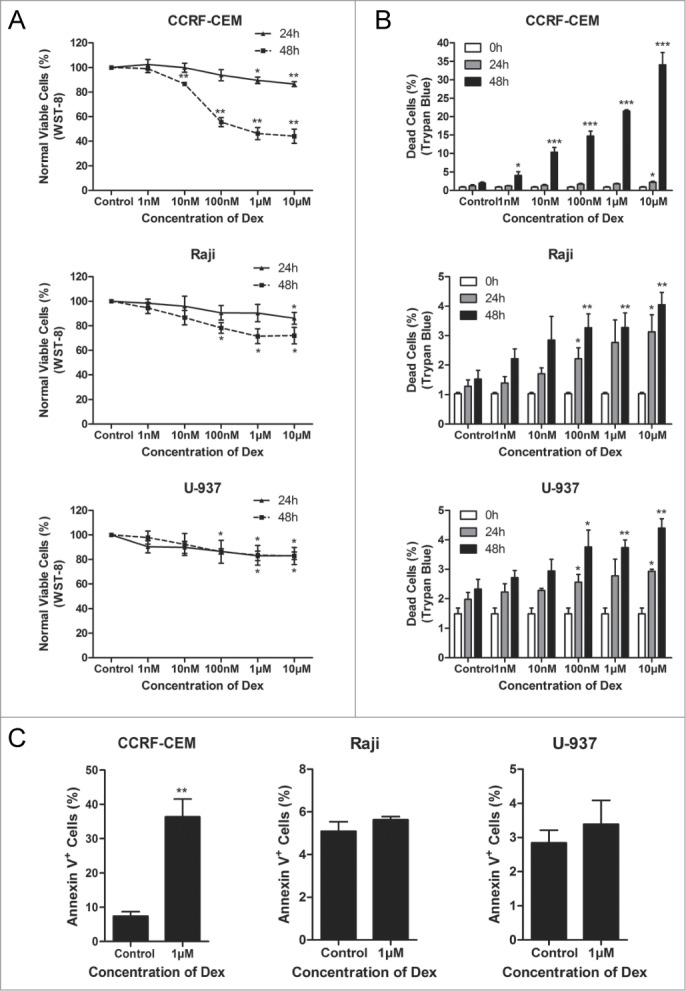
Dex inhibits cell proliferation in CCRF-CEM, Raji and U-937 cells. (A) CCRF-CEM, Raji and U-937 cells were treated with increasing concentrations of Dex for 24 and 48 h, and cell viability was determined by WST-8 assay. Error bars represent the standard errors of 3 independent experiments. (Dex vs. control: *P < 0.05, **P < 0.01) (B) Cells were treated with the indicated concentrations of Dex for the indicated times, and the percentage of dead cells was measured by trypan blue exclusion assay. (Dex versus control: *P < 0.05, **P < 0.01, ***P < 0.001) (C) Cells were treated with or without 1 μM Dex for 48 h. The percentage of apoptotic cells was determined by Annexin V-PI staining followed by FACS analysis. Values represent the means ± SD of 3 independent experiments. (Dex vs. control: **P < 0.01)
Dex arrests cell cycle in G0/G1-phase
We next evaluated whether Dex caused cell cycle arrest in this study. Cells were treated with 1 and 10 μM Dex for 24 h. Flow cytometry was performed to detect the PI-stained tumor cells, which exhibited an increase in G0/G1-phase cells and a decrease in S- and G2/M-phase cells compared with the control group in all cell lines examined (Fig. 2A). The differences were statistically significant in CCRF-CEM and Raji cells, but not in U-937 cells (Fig. 2B). The expression of p-Rb, a pivotal regulator of G1/S transition in the cell cycle process,18 was analyzed by Western blot. We detected a decrease in Ser780 p-Rb expression in CCRF-CEM and Raji cells treated with 1 and 10 μM Dex for 24 h, whereas total Rb levels were not changed (Fig. 2C). In U-937 cells, the decrease of p-Rb was not significant (Fig. 2C). Together, these results revealed that Dex affected the cell cycle distribution more significantly in Raji cells than in CCRF-CEM cells, while there was no statistically significant difference in U-937 cells.
Figure 2.
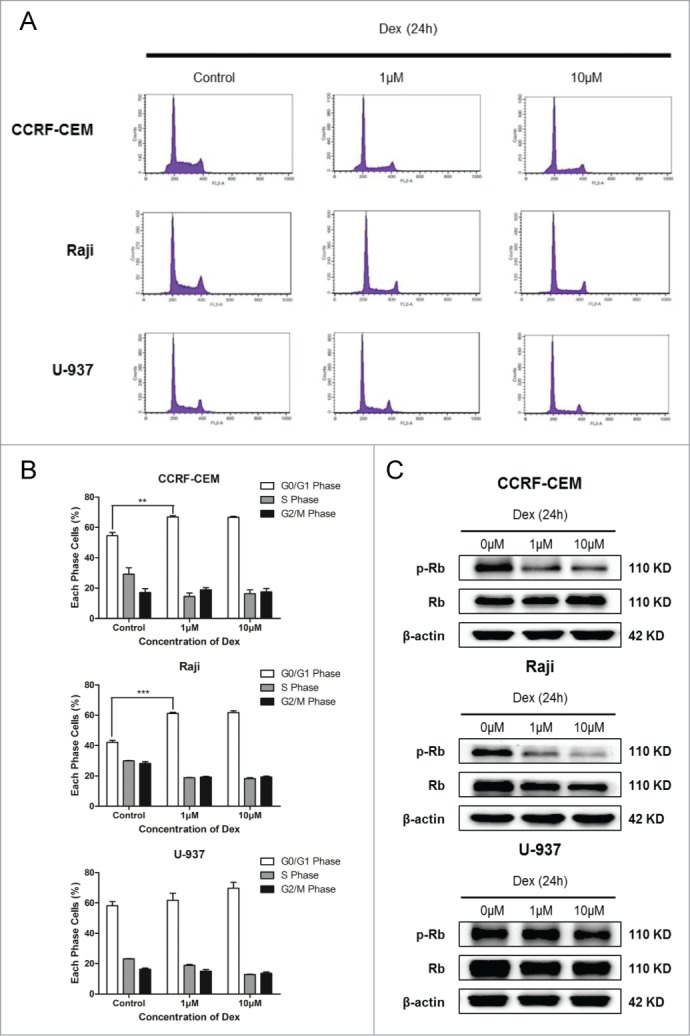
Dex arrests cell cycle in G0/G1-phase. (A, B) CCRF-CEM, Raji, and U-937 cells were treated for 24 h with the indicated concentrations of Dex, fixed, permeabilized, and stained with PI. Flow cytometric analysis of PI-stained cells documented a significant increase in the percentages of CCRF-CEM and Raji cells, but not U-937 cells, arrested in the G0/G1 phase of the cell cycle. Values represent the means ± SD of 3 independent experiments. (Dex versus control: **P < 0.01, ***P < 0.001) (C) Cells were treated with 0, 1, and 10 μM Dex for 24 h, p-Rb and Rb were determined by Western blot.
Dex induces apoptosis in CCRF-CEM cells
To determine whether apoptotic cell death contributed to the decreased cell number treated with Dex, we performed 2 apoptosis assays. CCRF-CEM, Raji, and U-937 cells were treated with Dex at the indicated concentrations for 24 and 48 h, and Western blot was then performed. Levels of cleaved-Caspase 3 and cleaved-PARP were unaffected in Raji and U-937 cells; however, cleaved levels of both proteins were increased significantly in CCRF-CEM cells treated for 48 h (Fig. 3). The results of flow cytometry were consistent with those of Western blot analysis (Fig. 1C). Together, these data indicated that Dex treatment did not elicit significant levels of apoptotic death in Raji and U-937 cells, but instead triggered significant apoptosis in CCRF-CEM cells (Fig. 3, 1C).
Figure 3.
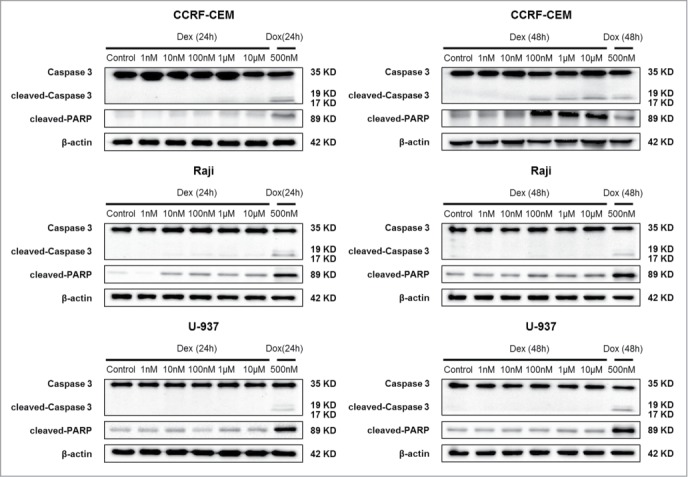
Dex induces apoptosis in CCRF-CEM cells. CCRF-CEM, Raji, and U-937 cells were treated with increasing concentrations of Dex for 24 and 48 h, and cleaved-PARP and cleaved-Caspase 3 were detected by Western blot. Cells were treated with 500 nM doxorubicin for 24 and 48 h as a positive control.
Dex induces autophagy in both Raji and U-937 cells
We then performed transmission electron microscopy analysis, the golden standard for investigation of autophagy,19 to detect autophagosomes formation treated with Dex. This analysis documented the presence of autophagosomes in the cytoplasm of Dex-treated Raji and U-937 cells (Fig. 4A). We failed to detect autophagosomes in Dex-treated CCRF-CEM cells (Fig. 4A). Acidic vesicular organelles, another morphological characteristic of autophagy, accumulate in the cytoplasm and can be detected by MDC staining.19 Dex significantly increased the accumulation of MDC in Raji and U-937 cells compared with untreated control cells, and this effect was dose-dependent (Fig. 4B). We did not detect autophagic vacuoles in CCRF-CEM cells (Fig. 4B). We then detected the expression of p62 and LC3, which are deemed markers of autophagy,20,21 in Raji and U-937 cells treated with indicated concentrations of Dex using Western blot. The decreasing of p62 and the increasing of LC3-II were Dex dose-dependent (Fig. 4C). This demonstrated that autophagy induced by Dex occurred in Raji and U-937 cells (Fig. 4C). A time-dependent study was also performed, autophagy was increased in a time-dependent manner (Fig. 4D). CCRF-CEM cells did not show autophagy induction (Fig. 4C, D, Supplementary Fig. 1). CCRF-CEM cells did not show autophagy induction (Fig. 4C, D). Taken together, these data showed that Dex induced autophagy in Raji and U-937 cells, but not in CCRF-CEM cells.
Figure 4.
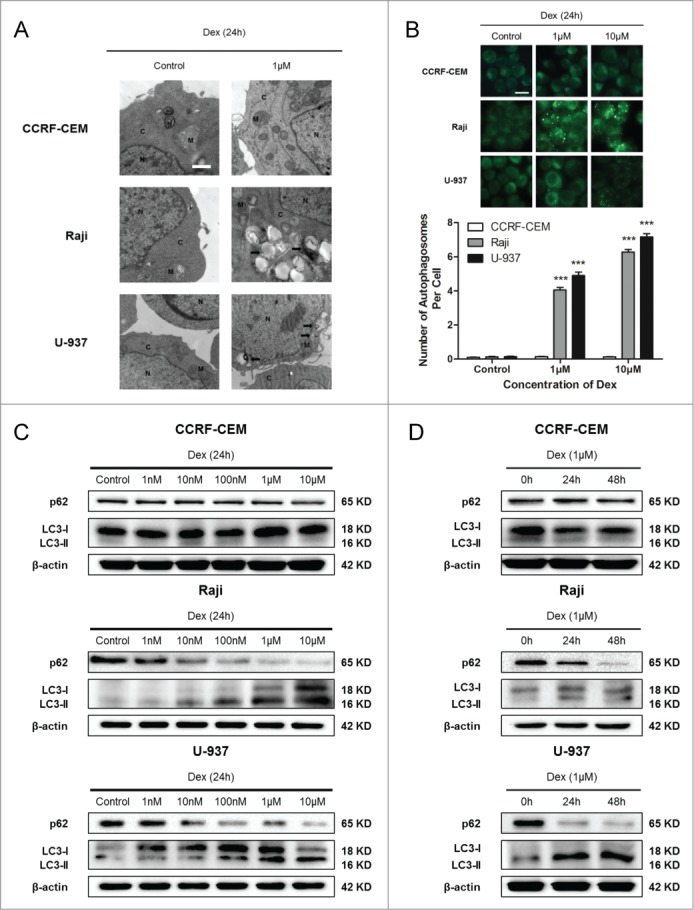
Dex induces autophagy in both Raji and U-937 cells. (A) CCRF-CEM, Raji, and U-937 cells were treated with 0 and 1 μM Dex for 24 h, and autophagosome formation was evaluated by transmission electron microscopy. M indicates mitochondrial structures, N the nucleus, C the cytoplasm, and arrowheads indicate autophagosomes. Scale bar: 500 nm. (B) Cells were treated with 0, 1, and 10 μM Dex for 24 h, then stained with MDC to identify autophagic vacuoles. Scale bar: 50 μm. The number of autophagosomes per cell was quantified by counting the number of autophagosomes in 100 MDC-staining cells. The data were presented as means ± SD of 3 independent experiments. (Dex vs. control: ***P < 0.001) (C) Cells were treated with increasing concentrations of Dex for 24 h, p62 and LC3 were detected by Western blot. (D) Cells were then treated with 1 μM Dex for 0, 24, and 48 h, and p62 and LC3 expression were evaluated by Western blot.
Dex inhibits mTOR pathway in both Raji and U-937 cells
Autophagy is a genetically regulated process, and a variety of signaling pathways participate in its up- or down-stream regulation.22 A number of studies have reported that the mTOR signaling pathway, which is activated in many types of cancer, is important in regulating autophagy.23 We then determined whether Dex affected the mTOR pathway, thereby causing autophagy. Western blot showed a dose-dependent decrease in p-Akt after 24 h of treatment with increasing concentrations of Dex in Raji and U-937 cells (Fig. 5A, B). However, the total Akt levels remained unchanged. The phosphorylation state of the mTORC1 and its downstream substrates p70 S6 kinase and 4E-BP1 (eukaryotic initiation factor 4E binding protein 1) were also decreased by Dex, whereas their total levels were unaffected (Fig. 5A, B). CCRF-CEM cells displayed no change in p-Akt, p-mTOR, p-P70S6K and p-4E-BP1 expression (data not shown). Thus, these data elucidated that Dex reduced the activity of mTOR pathway in Raji and U-937 cells, but not in CCRF-CEM cells.
Figure 5.
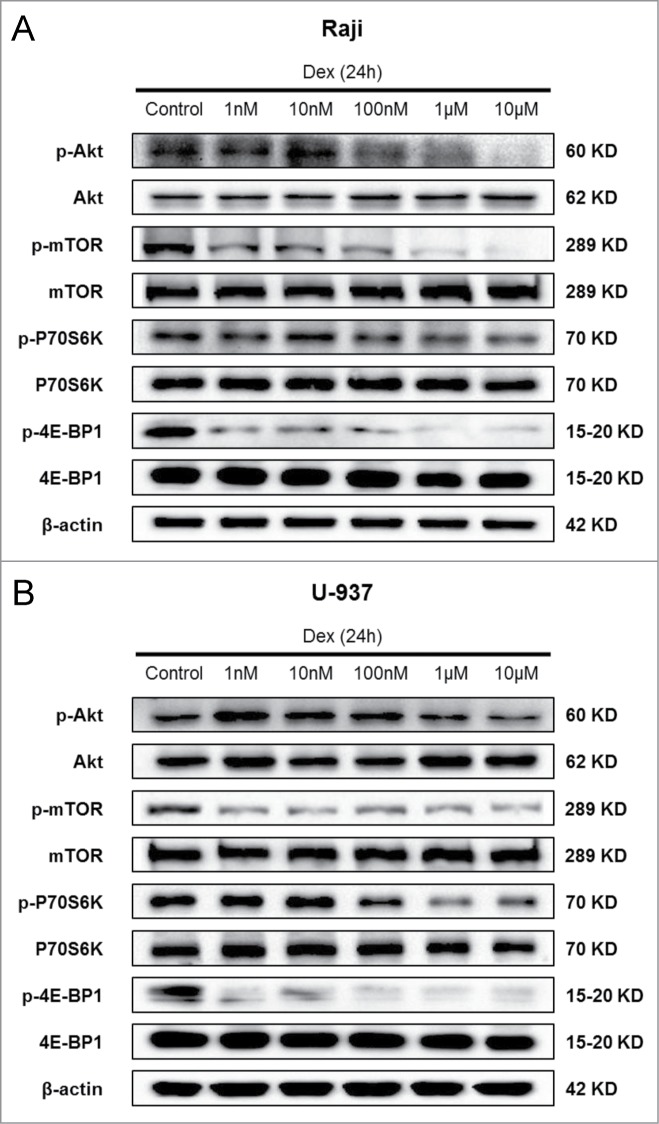
Dex inhibits mTOR pathway in both Raji and U-937 cells. Raji (A) and U-937 (B) cells were treated with increasing concentrations of Dex for 24 h, and cell lysates were subjected to Western blot and probed with the indicated antibodies.
Combination of Dex and autophagy-inhibitor treatment inhibits the proliferation and increases apoptosis of Raji and U-937 cells
To determine whether autophagy plays a role in the effects of Dex on Dex-resistant cells, we treated cells with Dex and either CQ (an inhibitor of fusion between the autophagosome and lysosome) or 3-MA (a PI3K inhibitor that inhibits the formation of the pre-autophagosomal structure), or the combination of these 3 agents. Western blot showed the autophagy inhibition effects of CQ and 3-MA (Fig. 6A). Treatment with Dex combined with autophagy inhibitors resulted in reduced numbers of cells as measured by WST-8 assay (Fig. 6B), suggesting that autophagy played an important role in cell viability under control conditions. We then evaluated the apoptosis induced by the combination Dex and autophagy inhibitor treatment. Apoptosis was enhanced by Dex after treatment with autophagy inhibitors, while treatment with autophagy inhibitors only had no significant effect (Fig. 6C). This demonstrated that autophagy played a critical role in protecting cancer cells from the apoptotic effects of Dex. Together, these results indicated that combination of Dex and autophagy inhibitor treatment inhibited cell proliferation and increased apoptosis in Dex-resistant cells.
Figure 6.
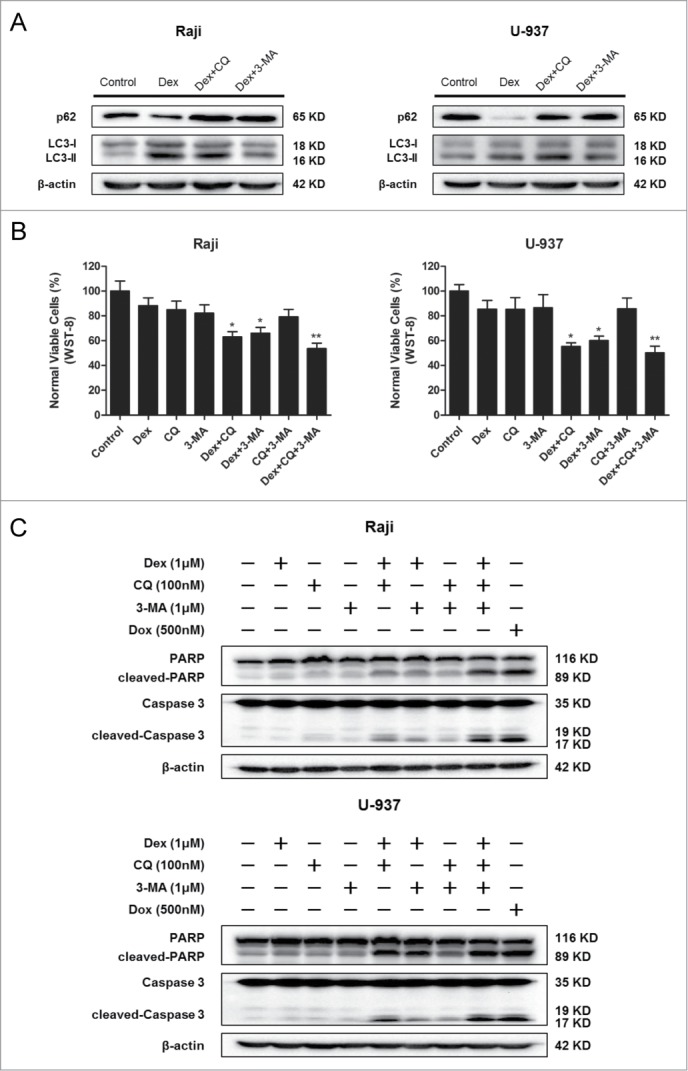
Combination Dex and autophagy-inhibitor treatment inhibits the proliferation and increases apoptosis of Raji and U-937 cells. (A) Raji and U-937 cells were treated with 1 μM Dex for 24 h followed by either 100 nM CQ or 1 μM 3-MA and total cell lysates were subjected to Western blot using anti-p62 and anti-LC3 antibodies. (B) Raji and U-937 cells were treated in the absence or presence of 100 nM CQ, 1 μM 3-MA with 1 μM Dex, or the combination of 2 or all 3 reagents for 24 h. A cell viability assay was performed using WST-8 to assess the inhibition of proliferation. (Treatment versus control: *P < 0.05, **P < 0.01) (C) Cells were treated as described above. Western blot was performed using cell lysates to detect the expression of cleaved-PARP and cleaved-Caspase 3. A doxorubicin (500 nM)-treated sample served as a positive control.
Inhibition of autophagy sensitized Raji cells to Dex-induced apoptosis in vivo
To further corroborate the role of autophagy in GC resistance, we constructed Raji cells stably expressing shBeclin1 and shNC control vectors. The knockdown efficiency of Beclin1 was evaluated by Western blot (Fig. 7A). Compared to shNC control vector, the sh#2 vector showed better knockdown efficiency of Beclin1 than the sh#1 vector. Thus equal numbers of Raji cells (5×106 cells per mouse) stably transfected with sh#2 and shNC vectors were injected into nude mice subcutaneously (n = 5). Dex was intravenously injected from the 12th day. The growth of tumors was measured every 3 days (Fig. 7B). Meanwhile, WST-8 assay was employed to explore whether knockdown of Beclin1 had any effect on proliferation ability of Raji cells in vitro. As shown in Supplementary Fig. 2, both shNC and shBeclin1 (#2) Raji cells proliferated in a similar rate at indicated timepoints (Day 0, Day 1, Day 2, Day 3 and Day 4, respectively). Consistently, both shNC and shBeclin1 (#2) tumors showed similar growth rate till 12th day in vivo. These data indicated that knockdown of Beclin1 did not significantly influence the proliferation ability of Raji cells. However, when subjected to Dex treatment, growth rate of shBeclin1 tumor was significantly slower than that of shNC tumors. Also, the shBeclin1 tumors were significantly smaller than shNC tumors (P < 0.001) (Fig. 7C). Immunofluorescence examination showed much higher immunoreactivity of cleaved-Caspase 3 within shBeclin1 tumors (Fig. 7D). In vitro analysis also showed that Beclin1 knockdown significantly sensitized Raji cells to Dex-induced cytotoxicity (P < 0.05) and apoptosis, as indicated by increased levels of both cleaved PARP and Caspase 3 after Dex treatment (Supplementary Fig. 3). Together, these data demonstrated that inhibition of autophagy sensitized Raji cells to Dex-induced apoptosis in vivo.
Figure 7.
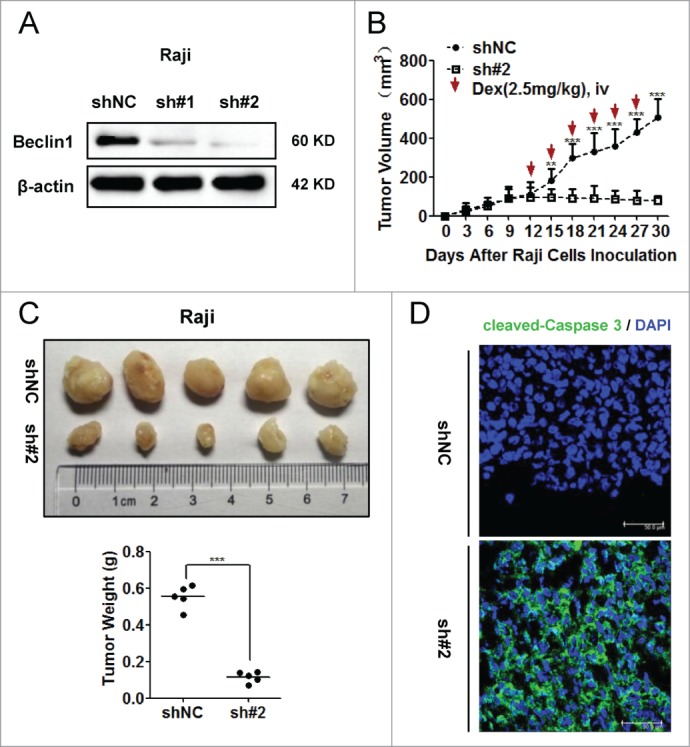
Inhibition of autophagy sensitized Raji cells to Dex-induced apoptosis in vivo. (A) Raji cells stably expressing shBeclin1 (#1, #2) and shNC vectors were constructed by lentivirus infection. The knockdown efficiency of Beclin1 was evaluated by Western blot. (B) Equal number (5 × 106 cells per mouse) of Raji cells stably transfected with shBeclin1 (#2) and shNC were subcutaneously injected into nude mice (n=5). At the beginning of 12th day, Dex (2.5mg/kg) was intravenously injected every 3 days. Tumor volume (mm3) was calculated by the multiplication of (long axis)/2 and (short axis)2. (**P < 0.01, ***P < 0.001) (C) Tumor were excised, photographed and weighed after the mice were sacrificed by cervical dislocation at the end of 30th day. (***P < 0.001) (D) Immunoreactivities of cleaved-Caspase 3 in xenografted tumors were analyzed by the immunofluorescence assay. Nucleus were stained by DAPI. Images were acquired by a confocal microscopy (Leica) and representative data were shown.
Discussion
GC have been used in chemotherapy for lymphoid malignancies (including acute/chronic lymphocytic leukemia, lymphoma, and myeloma) for decades.24 Although they are effective in the early stages, resistance upon relapse often emerges. So far, the molecular mechanisms of resistance to GC have not been fully understood. Here, by conduction of Dex, one representative type of GC for chemotherapy in lymphoid malignancies, several novel findings were made in the present study: (a) Dex inhibited cell proliferation, arrested cell cycle and led to apoptosis in CCRF-CEM cells; (b) Autophagy was significantly induced by Dex in Raji and U-937 cells, both of which showed poor response to Dex with limited apoptosis activation; (c) Inhibition of autophagy overcame Dex-resistance in Raji and U-937 cells by increasing apoptotic cell death both in vitro and in vivo analysis; (d) Dex repressed mTOR signaling activities in Raji and U-937 cells. Accordingly, our findings revealed a novel GC resistance mechanism that induction of autophagy by Dex contributed to GC resistance in lymphoid malignant cells. Inhibition of autophagy showed potential utilization to overcome GC resistance during lymphoid malignancies treatment.
Acquired resistance to chemotherapy remains a major barrier to the successful treatment of lymphoid malignancies. Many mechanisms contributing to drug resistance have been recognized, such as drug export transporters (e.g., permeability-glycoprotein 170),25 resistant haematopoietic stem cells,26 down-regulation of apoptosis, and up-regulation of autophagy.27 Increasing lines of evidence indicated that autophagy protected cancer cells against anticancer treatment by blocking apoptotic pathway.28 An early study illustrated that autophagy induced by Dex had a protective effect on Dex-induced cell death in a mouse T-cell leukemia cell line transfected with Bcl-2.17 Recent studies displayed that Dex caused autophagy in lymphoid malignancies.29 Consistently, in our study, autophagy was significantly induced by Dex in Dex-resistant Raji and U-937 cells (Fig. 4), as indicated by increased LC3 conversion, p62 degradation and autophagosome formation. However, autophagy was not significantly induced in CCRF-CEM cells (Fig. 4), even in the early time points (Supplementary Fig. 1). A previous study proposed that early stages of autophagy were required for, but did not necessarily result in, TNF α-induced apoptosis in CCRF-CEM cells.30 Combined with our data, one possible explanation for the contradictory within the same cell line might be the different types of compounds treatment and diverse autophagic signaling pathways involved. TNF α binds to specific receptors on the cell surface to induce apoptosis via signaling pathway which involves interleukin 1 β-converting enzyme (ICE) and/or ICE-like proteases.31 GC exert their effects through the glucocorticoid receptor (GR, or NR3C1), a member of the nuclear hormone receptor superfamily.32 Moreover, as an evolutionarily conversed catalytic process, autophagy is frequently triggered by stimulus. However, the effect of autophagy on cell survival or cellular death is controversial and context-dependent.33 Thus, it is in a possibility that GC and TNF α might trigger different autophagic signaling, mediating diverse biological effects of autophagy.
As we and others showed that Dex induced autophagy in lymphoid malignant cells, an important question arises: which signaling pathway contributes to the autophagy induced by Dex. Recent studies have shown that mTOR signaling pathway, which is activated in many types of cancer, is involved in autophagy regulation.23 TOR kinase was shown lies upstream of all autophagy-associated genes and plays a critical role in initiating the autophagic signaling in yeast.34 In mammalian cells, mTOR acts as a gatekeeper of the autophagic pathway, regulating autophagy negatively. P70 S6 kinase and 4E-BP1 are downstream targets of mTOR kinase, which reflect the activity of mTOR. Our studies revealed that Dex significantly inhibited the activity of the mTOR signaling pathway (Fig. 5), indicating that the Akt/mTOR pathway was involved in the autophagy induced by Dex. Consistent with our findings, recent reports described that Dex decreased the expression of p-Akt in RS4;11 cells (pre-B ALL cell lines) by PML-dependent manner,29 and in SSC cells (murine squamous cell carcinoma cells) by GR-dependent manner.35 Dex also promoted autophagy by inhibiting IP3-mediated calcium signaling36 or by increasing Dig2 protein.16 Together, based on diverse pharmacological effects of Dex, multiple signaling pathway implicated in Dex-induced autophagy regulation. Combination Dex and inhibitor of specific signaling targets (such as Akt and Dig2) would improve lymphoid malignancy therapy.
Additionally, it was recently shown that Dex caused cell cycle arrest in CCRF-CEM cells.37 We then investigated whether Dex caused cell cycle arrest in Dex-resistant cells. As expected, treatment with Dex resulted in a dramatic accumulation in the G0/G1 phase of the cell cycle and de-phosphorylation of p-Rb at Ser780 (Fig. 2), which is one of the most critical residues for Rb activity during the G1/S transition. These findings demonstrated that Dex-induced cell cycle arrest in Raji cells might contribute to Dex-resistance. Dex failed to induce cell cycle arrest significant in U-937 cells, which is derived from monocyte and called histiocytic lymphoma, together with it failed to cause cell apoptosis might explain why Dex was seldom used in the treatment of these diseases. A recent publication demonstrated that Dex-induced cell cycle arrest was mediated by the p27-Skp2 axis in Dex-sensitive T-lymphoma cells.38 Thus, further investigation is required to clarify the mechanism required to initiate Dex-induced cell cycle arrest in Dex-resistant lymphoid malignant cells.
In conclusion, our findings revealed that autophagy was induced by Dex in Dex-resistant lymphoid malignant cells. Blocking autophagy enhanced the pro-apoptotic effect of Dex. Thus, the current findings may shed light on the future design of optimized therapies in GC-resistant lymphoid malignancies.
Materials and Methods
Cell lines and culture conditions
The human Burkitt lymphoma cell line Raji and histiocytic lymphoma cell line U-937 were purchased from the American Tissue Culture Collection (Manassas, VA, USA). The human acute lymphoblastic leukemia (ALL) cell line CCRF-CEM was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in RPMI 1640 medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone), 2 mM glutamine, and 50 μg/mL penicillin and streptomycin (Solarbio, Beijing, China) at 37ºC in a humidified, 5% CO2 incubator.
Reagents
Antibodies to 4E-BP1 (#9644), phospho-4E-BP1 (Thr37/46) (#2855), Caspase 3 (#9662), PARP (#9532), cleaved-PARP (#9541), Rb (#9309), phospho-Rb (Ser780) (#9307), phospho-Akt (Ser473) (#4060), phospho-P70 S6 kinase (Thr389) (#9205), Beclin1 (#3738), phospho-mTOR (Ser2448) (#2971) and mTOR (#2972) were obtained from Cell Signaling Technology (Danvers, MA, USA). The antibody to P70 S6 kinase (#1494–1) was obtained from Epitomics (Hangzhou, China). The antibody to β-actin (#60008–1) was obtained from Proteintech (Wuhan, China). The antibodies to Akt1 (sc-1618) and p62 (SQSTM1) (sc-28359) were obtained from Santa Cruz Technology (Santa Cruz, Shanghai, China). The antibody to LC3B (L7543) was obtained from Sigma-Aldrich (Shanghai, China). Chloroquine (CQ) (C6628), 3-methyladenine (3-MA) (M9281), puromycin (P9620) and Dexamethasone (Dex) (D1756) were purchased from Sigma-Aldrich. Doxorubicin (Dox) (KGA8181) was purchased from Keygen (Nanjing, China).
Cell viability assay
Cell viability was measured by WST-8 assay. A total of 5 × 103 CCRF-CEM, Raji, or U-937 cells were cultured in 200-μL RPMI1640 medium and treated with different concentrations of Dex for 24 to 48 h. After treatment, cell viability was evaluated using the Cell Counting Kit-8 (Dojindo Laboratories, Tokyo, Japan) according to the manufacturer's instructions. Absorbance of triplicate samples at 450 nm was measured by an EnSpire Multilabel Reader (PerkinElmer).
Trypan blue exclusion assay
Cell death was measured by trypan blue exclusion assay. A total of 2 × 105 CCRF-CEM, Raji, or U-937 cells were cultured in 2-mL RPMI1640 medium and treated with indicated concentrations of Dex for 0, 24, or 48 h. After treatment, the cells were centrifuged and re-suspended with culture media. Then cells were mixed in equal volume with the trypan blue dye solution (CD001; Macgene, Beijing, China) and incubated for 5 min. The cells were then examined under an inverted microscope (CKX31; Olympus, Tokyo, Japan) and scored for the number of dead cells (the blue-stained cells represented those that had taken up the dye). The treated samples were compared with the untreated controls.
Apoptosis detection by flow cytometry
Detection of apoptosis was performed using the Annexin V-FITC/propidium iodide (PI) Apoptosis Detection Kit (KGA107; Keygen). Briefly, 2 × 105 CCRF-CEM, Raji, or U-937 cells were seeded in 6-well plates treated with indicated concentrations of Dex and incubated for 48 h. After treatment, the harvested cells were washed twice in phosphate buffer (PBS), then stained with Annexin V-FITC and PI according to the manufacturer's instructions. The resulting fluorescence was detected by a flow cytometer (FACS Caliber, Becton-Dickinson) with CELL Quest analysis software.
Cell cycle detection by flow cytometry
PI staining was employed to assess the cell cycle distribution. Briefly, 2 × 105 CCRF-CEM, Raji, or U-937 cells were seeded in 6-well plates treated with the indicated concentrations of Dex and incubated for 24 h. After treatment, the harvested cells were washed twice in PBS, then fixed in 75% pre-cold ethanol at 4°C for 4 h. After RNase A (100 μg/mL) (2158; TaKaRa, Dalian, China) digestion at 37°C for 30 min, the cells were stained with 50 μg/mL PI (P4170; Sigma) for 15 min at room temperature before analysis with a flow cytometer using the CELL Quest software.
Western blot analysis
Cells were lysed on ice in RIPA buffer (50 mM Tris [pH 8.0], 150 mM sodium chloride, 0.5% sodium deoxycholate, 0.1% SDS, and 1% NP-40) supplemented with protease inhibitors (1 mM Na3VO4, 1 μg/mL leupeptin, and 1 mM PMSF). The protein concentration was determined by the Coomassie brilliant blue dye method. In all, 20 μg of protein per lane were run in 6% to 12% SDS–PAGE gels and subsequently transferred to a nitrocellulose membrane (Millipore) via submerged transfer. After blocking the membrane at room temperature for 1 h, the membrane was incubated overnight at 4°C with various primary antibodies. After incubation with peroxidase-conjugated secondary antibodies for 1 h at room temperature, the signals were visualized using an enhanced chemiluminescence Western blot detection kit (K-12045-D50; Apgbio, Beijing, China) according to the manufacturer's instructions. The blots were developed using the Bio-Rad Molecular Imager instrument (Bio-Rad, USA).
Detection of autophagic vacuoles with monodansylcadaverine
The autophagic vacuoles were evaluated by monodansylcadaverine (MDC) staining. Briefly, 2 × 105 CCRF-CEM, Raji, or U-937 cells were seeded in 6-well plates treated with indicated concentrations of Dex and incubated for 24 h. After treatment, the cells were incubated with MDC (30432; Sigma) at a concentration of 0.05 mM in PBS at 37°C for 1 h. The cells then were centrifuged onto coverslips that were pre-coated with poly-L-lysine (P8920; Sigma). Fixation with 4% PFA (158127; Sigma) at 4°C for 10 min was performed, and the coverslips were washed with PBS twice and analyzed using a fluorescence microscope (excitation, 380 nm; emission, 525 nm) (DP73; Olympus). The number of autophagosomes per cell was quantified by counting the number of autophagosomes in 100 MDC-staining cells. The data were presented as means ± SD of 3 independent experiments. (Dex versus control: ***P < 0.001).
Transmission electron microscopy
Cells were fixed with 2.5% glutaraldehyde in PBS (pH 7.4), washed with PBS, and fixed by 1% osmium tetroxide (OsO4). They were then washed with PBS and dehydrated with indicated concentrations of ethanol. After gradient dehydration, permeation, and aggregation, thin sections were stained with uranyl acetate and lead citrate for observation under a JEM 1011CX electron microscope (JEOL, Peabody, MA, USA). Images were acquired digitally from a randomly selected pool of 10 to 15 fields under each condition.
Generation of Raji cells stably expressing shRNA vectors
Lentiviral particles containing shBeclin1#1 (131209AZ), shBeclin1#2 (131209BZ) and shNC (131127CZ) control expression vectors were purchased from Shanghai GenePharma Co., Ltd. Raji cells in exponential phase of growth were seeded into wells of a 6-well-plate and exposed to lentivirus particles for 2 h following another 48 h culture. After infection, Raji cells stably expressing shBeclin1 (#1, #2) and shNC vectors were chosen by selection with 2 μg/ml puromycin.
Cell proliferation assay
Cell proliferation rates were measured by WST-8 assay. Briefly, equal numbers (5 × 103) of shNC and shBeclin1 (#2) Raji cells were diluted in 200-μL culture medium and seeded into 96-well plates. Cell viability was evaluated using the Cell Counting Kit-8 (Dojindo Laboratories, Tokyo, Japan) according to the manufacturer's instructions at indicated timepoints. Absorbance (OD) at 450 nm was measured by an EnSpire Multilabel Reader (PerkinElmer). The experiment was triplicatedly performed and statistical comparison was analyzed by the 2-tailed Student's t-test. Differences were considered statistically significant at P < 0.05.
Mouse experiments
Animal experiments were approved by the Institute Animal Care and Use Committee of Dalian Medical University, and carried out in accordance with established institutional guidelines and approved protocols. An equal number (5×106 cells per mouse) of Raji cells stably transfected with shBeclin1 (#2) and shNC were subcutaneously injected into male 6-week-old immune-deficient (non-obese diabetic/severe combined immunodeficient) mice (5 mice for each group). Mice were maintained in the pathogen-free authorized facility where the temperature was maintained at 20–22°C and humidity at 50–60% with a 12 h dark/light cycle. Tumor growth was monitored by the measurement of short axis and long axis of tumor mass with vernier calipers every 3 days. At the beginning of 12th day, Dex (2.5 mg/kg) was intravenously injected every 3 days. Tumor were excised, photographed and weighed after the mice were sacrificed by cervical dislocation at the end of 30th day. Tumor volume (mm3) was calculated by the multiplication of (long axis)/2 and (short axis)2.
Immunofluorescence
For the immunofluorescence analysis of xenografted tumors, 7 μm-thick OCT compound-embedded tissue sections were cut, subsequently fixed with methanol for 5 min at room temperature, and permeabilized (0.25% Triton X-100 in PBS) for 20 min at 4°C. After blocked (3% BSA, 0.1% Triton X-100 in PBS) for 1 h at room temperature, sections were incubated with anti-cleaved-Caspase 3 antibody (diluted in blocking buffer) overnight at 4°C. After three 5 min washes in PBS, sections were incubated in the FITC-conjugated secondary antibody for 1 h at room temperature. Following three further 5 min washes, nucleus were stained by DAPI and images were acquired by a confocal microscopy (Leica).
Statistical analysis
Data are expressed as means ± SD of 3 independent experiments. Statistics were calculated by SPSS software (version 16.0). The significance of differences between experimental variables was determined using the 2-tailed Student's t-test. Differences were considered statistically significant at P < 0.05 (*P < 0.05, **P < 0.01, ***P < 0.001).
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
We thank Quentin Liu lab members for their critical commends and technical support. We also thank for their critical reading of the manuscript.
Funding
This work was supported by the National Basic Research Program of China (973 Program; No. 2012CB967000 to Q. Liu), National Natural Science Foundation of China (No. 81130040 to Q. Liu), the Liaoning (NSF2014029102 to Q. Liu), National Natural Science Foundation of China (81370604 to M. Fang) and Social Development Research Plan of Liaoning Province (2013225002–208 to M. Fang).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Dalla -Favera R. Lymphoid malignancies: many tumor types, many altered genes, many therapeutic challenges. J Clin Invest 2012; 122:3396-7; PMID:23023709; http://dx.doi.org/ 10.1172/JCI66307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sionov RV, Spokoini R, Kfir-Erenfeld S, Cohen O, Yefenof E. Mechanisms regulating the susceptibility of hematopoietic malignancies to glucocorticoid-induced apoptosis. Adv Cancer Res 2008; 101:127-248; PMID:19055945; http://dx.doi.org/ 10.1016/S0065-230X(08)00406-5 [DOI] [PubMed] [Google Scholar]
- 3. De Bosscher K, Vanden Berghe W, Vermeulen L, Plaisance S, Boone E, Haegeman G. Glucocorticoids repress NF-kappaB-driven genes by disturbing the interaction of p65 with the basal transcription machinery, irrespective of coactivator levels in the cell. Proc Nat Acad Sci U S A 2000; 97:3919-24; PMID:10760263; http://dx.doi.org/ 10.1073/pnas.97.8.3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jonat C, Rahmsdorf HJ, Park KK, Cato AC, Gebel S, Ponta H, Herrlich P. Antitumor promotion and antiinflammation: down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell 1990; 62:1189-204; PMID:2169351; http://dx.doi.org/ 10.1016/0092-8674(90)90395-U [DOI] [PubMed] [Google Scholar]
- 5. Thompson EB, Medh RD, Zhou F, Ayala-Torres S, Ansari N, Zhang W, Johnson BH. Glucocorticoids, oxysterols, and cAMP with glucocorticoids each cause apoptosis of CEM cells and suppress c-myc. J Steroid Biochem Mol Biol 1999; 69:453-61; PMID:10419025; http://dx.doi.org/ 10.1016/S0960-0760(99)00063-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chai J, Xiong Q, Zhang P, Zheng R, Peng J, Jiang S. Induction of Ca2+ signal mediated apoptosis and alteration of IP3R1 and SERCA1 expression levels by stress hormone in differentiating C2C12 myoblasts. Gen Comp Endocrinol 2010; 166:241-9; PMID:19723525; http://dx.doi.org/ 10.1016/j.ygcen.2009.08.011 [DOI] [PubMed] [Google Scholar]
- 7. Sharma S, Lichtenstein A. Dexamethasone-induced apoptotic mechanisms in myeloma cells investigated by analysis of mutant glucocorticoid receptors. Blood 2008; 112:1338-45; PMID:18515658; http://dx.doi.org/ 10.1182/blood-2007-11-124156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu T, Fei Z, Gangavarapu KJ, Agbenowu S, Bhushan A, Lai JC, Daniels CK, Cao S. Interleukin-6 and JAK2/STAT3 signaling mediate the reversion of dexamethasone resistance after dexamethasone withdrawal in 7TD1 multiple myeloma cells. Leukemia Res 2013; 37:1322-8; PMID:23871159; http://dx.doi.org/ 10.1016/j.leukres.2013.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Packham G, Stevenson FK. Bodyguards and assassins: Bcl-2 family proteins and apoptosis control in chronic lymphocytic leukaemia. Immunol 2005; 114:441-9; PMID:15804279; http://dx.doi.org/ 10.1111/j.1365-2567.2005.02117.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Russcher H, Smit P, van den Akker EL, van Rossum EF, Brinkmann AO, de Jong FH, Lamberts SW, Koper JW. Two polymorphisms in the glucocorticoid receptor gene directly affect glucocorticoid-regulated gene expression. J Clin Endocrinol Metabol 2005; 90:5804-10; PMID:16030164; http://dx.doi.org/ 10.1210/jc.2005-0646 [DOI] [PubMed] [Google Scholar]
- 11. Ma L, Fang M, Liang Y, Xiang Y, Jia Z, Sun X, Wang Y, Qin J. Low expression of glucocorticoid receptor α isoform in adult immune thrombocytopenia correlates with glucocorticoid resistance. Annal Hematol 2013; 92:953-60; PMID:23435844; http://dx.doi.org/ 10.1007/s00277-013-1705-5 [DOI] [PubMed] [Google Scholar]
- 12. Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463-77; PMID:15068787; http://dx.doi.org/ 10.1016/S1534-5807(04)00099-1 [DOI] [PubMed] [Google Scholar]
- 13. Guo XL, Li D, Hu F, Song JR, Zhang SS, Deng WJ, Sun K, Zhao QD, Xie XQ, Song YJ., et al. Targeting autophagy potentiates chemotherapy-induced apoptosis and proliferation inhibition in hepatocarcinoma cells. Cancer Lett 2012; 320:171-9; PMID:22406827; http://dx.doi.org/ 10.1016/j.canlet.2012.03.002 [DOI] [PubMed] [Google Scholar]
- 14. Xi G, Hu X, Wu B, Jiang H, Young CY, Pang Y, Yuan H. Autophagy inhibition promotes paclitaxel-induced apoptosis in cancer cells. Cancer Lett 2011; 307:141-8; PMID:21511395; http://dx.doi.org/ 10.1016/j.canlet.2011.03.026 [DOI] [PubMed] [Google Scholar]
- 15. Lamy L, Ngo VN, Emre NC, Shaffer AL, 3rd, Yang Y, Tian E, Nair V, Kruhlak MJ, Zingone A, Landgren O., et al. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 2013; 23:435-49; PMID:23541952; http://dx.doi.org/ 10.1016/j.ccr.2013.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Molitoris JK, McColl KS, Swerdlow S, Matsuyama M, Lam M, Finkel TH, Matsuyama S, Distelhorst CW. Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J Biol Chem 2011; 286:30181-9; PMID:21733849; http://dx.doi.org/ 10.1074/jbc.M111.245423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Swerdlow S, McColl K, Rong Y, Lam M, Gupta A, Distelhorst CW. Apoptosis inhibition by Bcl-2 gives way to autophagy in glucocorticoid-treated lymphocytes. Autophagy 2008; 4:612-20; PMID:18362516; http://dx.doi.org/ 10.4161/auto.5920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giacinti C, Giordano A. RB and cell cycle progression. Oncogene 2006; 25:5220-7; PMID:16936740; http://dx.doi.org/ 10.1038/sj.onc.1209615 [DOI] [PubMed] [Google Scholar]
- 19. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/ 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- 21. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726-34; PMID:16148885; http://dx.doi.org/ 10.1038/nrc1692 [DOI] [PubMed] [Google Scholar]
- 23. Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S., et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383-435; PMID:20959619; http://dx.doi.org/ 10.1152/physrev.00030.2009 [DOI] [PubMed] [Google Scholar]
- 24. Greenstein S, Ghias K, Krett NL, Rosen ST. Mechanisms of glucocorticoid-mediated apoptosis in hematological malignancies. Clin Cancer Res 2002; 8:1681-94; PMID:12060604 [PubMed] [Google Scholar]
- 25. Steinbach D, Legrand O. ABC transporters and drug resistance in leukemia: was P-gp nothing but the first head of the Hydra? Leukemia 2007; 21:1172-6; PMID:17429427; http://dx.doi.org/ 10.1038/sj.leu.2404692 [DOI] [PubMed] [Google Scholar]
- 26. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer 2005; 5:275-84; PMID:15803154; http://dx.doi.org/ 10.1038/nrc1590 [DOI] [PubMed] [Google Scholar]
- 27. Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961-7; PMID:17972889; http://dx.doi.org/ 10.1038/nrc2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y., et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51-64; PMID:16843265; http://dx.doi.org/ 10.1016/j.ccr.2006.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Laane E, Tamm KP, Buentke E, Ito K, Kharaziha P, Oscarsson J, Corcoran M, Björklund AC, Hultenby K, Lundin J., et al. Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death Differ 2009; 16:1018-29; PMID:19390558; http://dx.doi.org/ 10.1038/cdd.2009.46 [DOI] [PubMed] [Google Scholar]
- 30. Jia L, Dourmashkin RR, Allen PD, Gray AB, Newland AC, Kelsey SM. Inhibition of autophagy abrogates tumour necrosis factor α induced apoptosis in human T-lymphoblastic leukaemic cells. Br J Haematol 1997; 98:673-85; PMID:9332326; http://dx.doi.org/ 10.1046/j.1365-2141.1997.2623081.x [DOI] [PubMed] [Google Scholar]
- 31. Fraser A, Evan G. A license to kill. Cell 1996; 85:781-4; PMID:8681372; http://dx.doi.org/ 10.1016/S0092-8674(00)81005-3 [DOI] [PubMed] [Google Scholar]
- 32. Niu N, Manickam V, Kalari KR, Moon I, Pelleymounter LL, Eckloff BW, Wieben ED, Schaid DJ, Wang L. Human glucocorticoid receptor α gene (NR3C1) pharmacogenomics: gene resequencing and functional genomics. J Clin Endocrinol Metab 2009; 94:3072-84; PMID:19435830; http://dx.doi.org/ 10.1210/jc.2008-2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Das G, Shravage BV, Baehrecke EH. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol 2012; 4:a008813; PMID:22661635; http://dx.doi.org/ 10.1101/cshperspect.a008813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev 2001; 15:807-26; PMID:11297505; http://dx.doi.org/ 10.1101/gad.887201 [DOI] [PubMed] [Google Scholar]
- 35. Bernardi RJ, Trump DL, Yu WD, McGuire TF, Hershberger PA, Johnson CS. Combination of 1alpha,25-dihydroxyvitamin D(3) with dexamethasone enhances cell cycle arrest and apoptosis: role of nuclear receptor cross-talk and Erk/Akt signaling. Clini Cancer Res 2001; 7:4164-73; PMID:11751517 [PubMed] [Google Scholar]
- 36. Harr MW, McColl KS, Zhong F, Molitoris JK, Distelhorst CW. Glucocorticoids downregulate Fyn and inhibit IP(3)-mediated calcium signaling to promote autophagy in T lymphocytes. Autophagy 2010; 6:912-21; PMID:20814235; http://dx.doi.org/ 10.4161/auto.6.7.13290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang C, Ryu YK, Chen TZ, Hall CP, Webster DR, Kang MH. Synergistic activity of rapamycin and dexamethasone in vitro and in vivo in acute lymphoblastic leukemia via cell-cycle arrest and apoptosis. Leukemia Res 2012; 36:342-9; PMID:22137317; http://dx.doi.org/ 10.1016/j.leukres.2011.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kullmann MK, Grubbauer C, Goetsch K, Jakel H, Podmirseg SR, Trockenbacher A, Ploner C, Cato AC, Weiss C, Kofler R., et al. The p27-Skp2 axis mediates glucocorticoid-induced cell cycle arrest in T-lymphoma cells. Cell Cycle 2013; 12:2625-35; PMID:23907123; http://dx.doi.org/ 10.4161/cc.25622 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.