

Abstract
Lynch syndrome (LS) is a tumor predisposing condition caused by constitutional defects in genes coding for components of the mismatch repair (MMR) apparatus. While hypermethylation of the promoter of the MMR gene MLH1 occurs in about 15% of colorectal cancer samples, it has also been observed as a constitutional alteration, in the absence of DNA sequence mutations, in a small number of LS patients. In order to obtain further insights on the phenotypic characteristics of MLH1 epimutation carriers, we investigated the somatic and constitutional MLH1 methylation status of 14 unrelated subjects with a suspicion of LS who were negative for MMR gene constitutional mutations and whose tumors did not express the MLH1 protein. A novel case of constitutional MLH1 epimutation was identified. This patient was affected with multiple primary tumors, including breast cancer, diagnosed starting from the age of 55 y. Investigation of her offspring by allele specific expression revealed that the epimutation was not stable across generations. We also found MLH1 hypermethylation in cancer samples from 4 additional patients who did not have evidence of constitutional defects. These patients had some characteristics of LS, namely early age at onset and/or positive family history, raising the possibility of genetic influences in the establishment of somatic MLH1 methylation.
Keywords: colorectal cancer, DNA methylation, immunohistochemistry, Lynch syndrome, mismatch repair, microsatellite instability
Abbreviations
- ASE
allele specific expression
- CRC
colorectal cancer
- LS
Lynch syndrome
- MMR
mismatch repair
- MSI
microsatellite instability
- PBLs
peripheral blood leukocytes
- SNuPE
single nucleotide primer extension
Introduction
The molecular alterations of cancer genomes include genetic and epigenetic changes. The best characterized epigenetic modification is hypermethylation of CpG dinucleotides in promoter regions, that typically affects tumor suppressor and DNA repair genes. The causes underlying the development of somatic methylation are poorly understood. Environmental exposures, ageing, inflammation, and genetic factors may variably contribute to the appearance of progressive methylation.1 In addition, in a small fraction of cancer patients, epigenetic changes are detected as constitutional alterations that can be occasionally inherited.2,3
MLH1 is the prototypic human cancer gene associated with inactivating epimutations in colorectal cancer (CRC). Along with MSH2, MSH6 and PMS2, it is one of the 4 DNA mismatch repair (MMR) genes involved in Lynch syndrome (LS), the most common hereditary CRC condition accounting for approximately 1–3% of all CRCs. LS is an autosomal dominant cancer susceptibility syndrome characterized by the early development of CRC and other extracolonic malignancies, namely endometrial, urothelial and small bowel cancers.4,5 Somatic inactivation of the wild type allele encoded by the MMR locus that is constitutionally mutated leads to MMR deficiency and increased mutation rate, which can be demonstrated by the appearance of microsatellite instability (MSI). In addition, since inactivating defects, including promoter DNA methylation, often lead to reduced/absent mRNA synthesis or to extreme mRNA or protein instability, the protein encoded by the defective gene is not detectable by means of immunohistochemical analysis using specific antibodies.
MLH1 promoter methylation is a frequent somatic alteration in CRCs, detected in approximately 15% of CRC samples.6 CRCs showing somatic MLH1 methylation tend to occur at relatively advanced ages, are more common in women and often located in the right colon, and usually also display the somatic activating p.V600E BRAF mutation.7,8 MLH1 promoter methylation also occurs in other sporadic tumors in the clinical spectrum of LS, including gastric, duodenal and endometrial carcinomas.9-11
More rarely, MLH1 hypermethylation occurs as a constitutional defect, detectable in normal cells, such as peripheral leukocytes, in patients presenting with phenotypic characteristics of LS. So far, approximately 60 unrelated patients with MLH1 constitutional epimutations have been reported.3,12 At variance with purely somatic defects, CRCs from patients with LS or constitutional MLH1 epigenetic silencing do not usually harbor somatic BRAF mutations. MLH1 epimutations appear to be reversible in the germline in most cases, but a few instances of intergenerational transmission have been described.13,14 The latter are usually associated in cis with a rare MLH1 haplotype, and are thus considered secondary epimutations, linked to an as yet undefined primary DNA sequence change that induces the aberrant methylation pattern.3,14
Given their apparent rarity, the clinical characteristics associated with constitutional MLH1 epimutations and their relative frequency among patients with intestinal cancers, and with LS in particular, have yet to be fully defined. In this study, we investigated the somatic and constitutional MLH1 methylation status of 14 unrelated subjects with a suspicion of LS but no MMR constitutional mutation detectable and whose tumors did not express MLH1. A novel case of constitutional MLH1 epimutation was identified. In addition, we found MLH1 hypermethylation in cancer samples from 4 additional patients who did not have evidence of constitutional defects, including 2 subjects with early onset cancers.
Results
Constitutional methylation analysis
Constitutional MLH1 hypermethylation was identified by MS-MLPA in 1/14 (7.1%) cases analyzed (case M60; Fig. 1). Patient M60 had developed multiple sporadic cancers: endometrial, urothelial, rectal and breast carcinoma, at the ages of 55, 62, 66 and 67 y, respectively. Family history was negative for LS related cancers (Fig. 2), and a normal MLH1 methylation pattern was detected, by both MS-MLPA and MS-PCR, in both proband's children. The rectal and the breast cancer from patient M60 were investigated for MSI and immunohistochemistry: both were MSI-H and showed loss of MLH1 protein expression (data not shown). The presence of constitutional hypermethylation in case M60 was confirmed by MS-PCR for both the A and the C region 5′ MLH1 fragments tested (data not shown).
Figure 1.
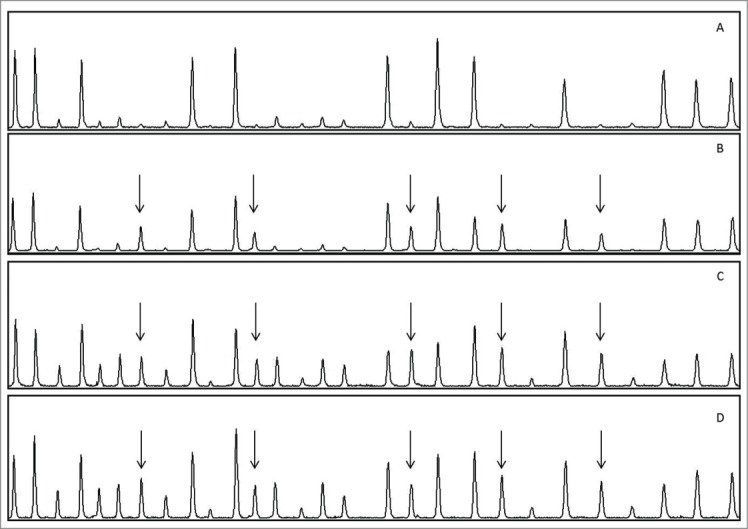
MS-MLPA analysis of samples from patient M60 and an unmethylated control sample. (A) Control (peripheral leukocytes); (B) M60, peripheral leukocytes; (C) M60, rectal cancer; (D) M60, breast cancer. Arrows indicate the 5 MLH1 peaks corresponding to methylated sites. The intensities of these peaks are higher in (C) and (D) compared to (B). In both tumor samples additional peaks appear which are not present or are just barely visible in leukocytes; these correspond to methylated areas in other MMR genes analyzed by the kit.
Figure 2.
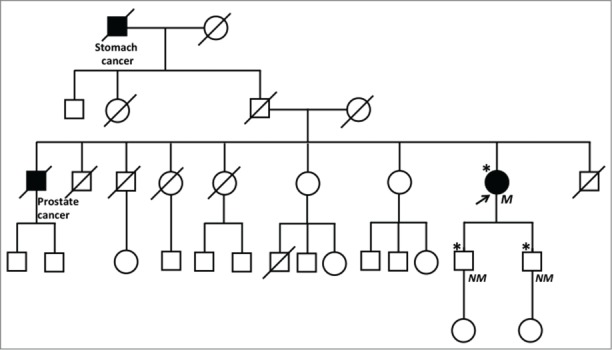
Pedigree of the family of patient M60. The arrow indicates the proband. Asterisks indicate individuals tested for MLH1 methylation. M = MLH1 methylated; NM = MLH1 not methylated.
Somatic methylation and BRAF analysis
The MMR gene methylation status was also investigated by MS-MLPA on 11 tumor samples from 10 patients, 9 of which did not show MLH1 constitutional methylation. MLH1 promoter hypermethylation was detected in 5 cases, including patient M60 (Table 1). Two tumor specimens from patient M60 were analyzed, the rectal and the breast cancer: both samples showed hypermethylated peaks for all 5 MLH1 methylation sensitive probes (Fig. 1). In M60 leukocytes aberrant methylation was limited to the 5 MLH1 probes, while in both tumors methylation was observed also for additional probes corresponding to other genes (MGMT, MSH6, and MSH2). A similar finding was observed for all other tumors showing MLH1 methylation, which also displayed variable degrees of methylation of other genes (data not shown).
Table 1.
Relative methylation levels in samples showing MLH1 hypermethylation
|
MLH1 fragment1 |
||||||
|---|---|---|---|---|---|---|
| Sample ID | Sample type2 | 166 (−13; D) | 196 (−246; C) | 238 (−659; A) | 265 (−383; B) | 292 (206) |
| M60 | PBLs | 0.57 | 0.56 | 0.57 | 0.67 | 0.56 |
| M60 Ka | CRC | 0.55 | 0.61 | 0.69 | 0.73 | 0.63 |
| M60 Kb | BC | 0.67 | 0.63 | 0.65 | 0.57 | 0.65 |
| M49 K | CRC | 0.24 | 0.18 | 0.33 | 0.34 | 0.33 |
| M56 K | CRC | 0.31 | 0.37 | 0.64 | 0.57 | 0.28 |
| M57 K | DC | 0.32 | 0.34 | 0.49 | 0.42 | 0.49 |
| M66 K | CRC | 0.57 | 0.81 | 0.70 | 0.37 | 0.57 |
1Fragments are indicated according to their size in nucleotides; distance in nucleotides from the transcription start site is shown in parentheses, along with the letter indicating the corresponding MLH1 promoter region according to Deng et al.15 The fraction of methylated DNA for each site analyzed was calculated as detailed in the text.
2PBLs: peripheral blood leukocytes; CRC: colorectal carcinoma; BC: breast carcinoma; DC: duodenal carcinoma.
Overall, the BRAF p.V600E mutation was investigated in 7 tumors, including 3 out of the 5 showing somatic hypermethylation. A single CRC, M66, was found to be positive.
MLH1 allele-specific expression assay by SNuPE
To evaluate the effect of MLH1 constitutional hypermethylation on mRNA expression, we performed SNuPE analysis in peripheral blood leukocytes (PBLs) from patient M60 and from a control individual with a normal MLH1 methylation pattern. To this purpose, the exon 8 c.655G>A (p.Ile219Val) polymorphism, for which both the patient and the control were heterozygous, was exploited for primer extension analysis. The calculated ASE value for the control subject was in the normal range (0.83). Unbalanced allelic mRNA expression was detected in M60 PBLs: only the G allele was significantly expressed, with an ASE value of 0.3, indicative of a silencing effect of promoter methylation on the A allele (Fig. 3).
Figure 3.
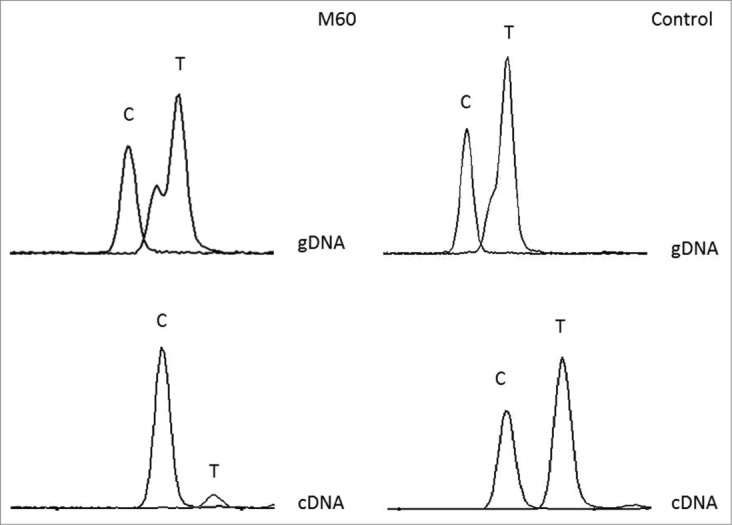
SNuPE analysis of the MLH1 c.655A>G polymorphism (rs1799977) in patient M60 and in a control sample. The complementary sequence is shown, as the experiment was performed using a reverse primer. The T peak (corresponding to the A allele) is clearly underrepresented in cDNA compared to genomic DNA (gDNA) from patient M60.
MLH1 haplotype evaluation
Proband M60 was tested for the presence of the MLH1 single nucleotide variants (c.-27C>A and c.85G>T) previously identified in some individuals with constitutional MLH1 methylation. None of the alleles previously associated with MLH1 hypermethylation was detected.
Discussion
To our knowledge, this is the first systematic analysis of constitutional MLH1 methylation in a series of patients with suspected LS collected at an Italian institution. Our results indicate that MLH1 epimutations are a rare cause of LS also in Italy and further contribute to the definition of the phenotypic characteristics associated with these alterations.
A constitutional MLH1 epigenetic defect was identified in a female proband who presented as a sporadic case affected with multiple primary tumors, which were all diagnosed beginning from 55 y of age. In particular, her rectal carcinoma was detected at 66 y. So far, 62 unrelated patients with confirmed or probable MLH1 epimutations have been reported.3 While most of these cases are sporadic and about 1/3 (22/62) have developed 2 or more tumors, age at diagnosis of the first cancer is usually before 50 y, and, so far, only 7 of the reported probands had their first cancer diagnosis ≥55 y. In addition, the mean age at CRC diagnosis is 39 (±11) years.3
The spectrum of cancers occurring in patients with MLH1 constitutional methylation defects coincides with that of LS. 3 of the tumors (endometrial, urothelial and large bowel) diagnosed in patient M60 are typically associated with LS. The last tumor that she developed is a ductal breast carcinoma. Currently, it is not yet clear whether breast cancer is a component tumor of LS,16 although the most recent studies indicate that MMR gene mutation carriers, may indeed be at increased risk, especially when MLH1 is involved, beginning after the age of 40 y.17-19 There is also evidence that a substantial fraction of breast cancers associated with LS display the characteristic hallmarks of MMR deficiency, that is, MSI-H status and immunohistochemical loss of MMR proteins.20,21 This points to a prominent role of MMR inactivation in the pathogenesis of at least a fraction of the breast carcinomas that arise in LS patients. The only other known breast cancer associated with a constitutional MLH1 epimutation was diagnosed at 63 y, was MSI-H and showed loss of MLH1 protein staining as well as MLH1 loss of heterozygosity in tumor DNA.22 These findings, along with those obtained on patient M60 in this series indicate that breast cancer is likely a manifestation of constitutional MLH1 epimutations. While both cases were diagnosed in the seventh decade, unbiased studies are needed to define age specific risks for breast as well as other tumors in this subset of LS patients.
The 0.07 frequency of MLH1 constitutional epimutations among cases showing MLH1 protein loss in tumors without detectable MLH1 pathogenic variants is in line with previous studies, which observed a range of values from 0.01 to 0.13.23,24 This confirms the rarity of this type of alterations as a cause of LS. However, the estimates obtained in the studies so far conducted may not be representative of the real frequency due to small sample numbers and ascertainment bias. Further investigations on large series of unselected CRCs as well as other LS related tumors will be necessary to define the population frequency of MLH1 epimutations.
The absence of the 5′ MLH1 variants previously identified in patients with heritable MLH1 epimutations indicates that the establishment of methylation in patient M60 is unlikely to be related to the presence of linked genetic variants.14,25,26 Therefore M60 belongs to the subgroup of primary epimutations, which are not transmitted through generations. While, based on current knowledge, this implies a low recurrence risk for the offspring of primary epimutation carriers, the mechanisms underlying the establishment of MLH1 hypermethylation in these subjects have yet to be defined, and the possibility of tissue specific mosaicism cannot be excluded.
Age at first cancer diagnosis was <50 y in 9 of the total 14 cases with MLH1-negative tumors investigated in the present study, including 2 (M56 and M57) of the 4 cases with methylation confined to tumor DNA. In addition, one of the methylation positive samples with early age at onset, M57, was shown to have a complex phenotype related to the presence of the PTEN mutation c.510T>A (p.Ser170Arg). Although this case had been referred with a suspicion of LS based on the occurrence of duodenal cancer at 37 y, and the results of initial MSI and immunohistochemical testing had supported this hypothesis, no MLH1 mutation was detected and further clinical investigations led to the diagnosis of PTEN hamartoma tumor syndrome (PHTS).27 The latter finding indicates that somatic MLH1 inactivation through promoter methylation may be a molecular event contributing to the pathogenesis of cancers occurring in the context of inherited syndromes other than LS and is not limited to sporadic late onset tumors. The occurrence of MLH1 hypermethylation in CRCs from cases M49 and M66, diagnosed at 68 and 57 y, respectively, is probably an age related event, since the majority of MSI-H CRCs are detected in elderly subjects. Consistent with this hypothesis, the CRC sample of patient M66 was found to harbor the BRAF p.V600E mutation, which is commonly, although not exclusively, associated with somatic MLH1 methylation and CpG island methylator phenotype (CIMP).1 However, case M56 had been diagnosed with a BRAF p.V600E negative CRC at 43 y and had a positive family history for CRC. A few other cases of CRC diagnosed <50 y showing MLH1 hypermethylation in tumor samples in the absence of BRAF p.V600E have been reported.28-30 Due to small numbers and lack of sufficient clinical details, no inference on specific phenotypic characteristics for this category of CRCs can be drawn at the current stage. Interestingly, 3/4 patients with somatic MLH1 hypermethylation had a positive family history. It has been postulated that genetic factors may contribute to the establishment of somatic MLH1 methylation, especially in the context of serrated polyposis31 and, possibly, in a subset of early onset familial cases.28 However, there is currently no clue on the possible identity of the loci influencing MLH1 promoter methylation, and this hypothesis has yet to be proven. Actually, discordance in somatic methylation patterns has been observed in individuals with familial CRC or endometrial cancer not related to constitutional MMR gene mutations.32
The variability in methylation levels as determined by MLPA (e.g., ranging from 0.18 to 0.81 for the 196 nt fragment in the C across samples) could correspond to real differences in the fraction of methylated alleles, with the maximum values indicating biallelic methylation/inactivation (Table 1). The methylated fraction for the 3 samples from patient M60 varied between 0.55 and 0.67 for the C region fragments, with a tendency toward a higher degree of methylation in the breast cancer sample. However, differences were too small to allow any inference. In addition it should be considered that MLPA is a semiquantitative method.
No apparent cause could be identified for the lack of MLH1 staining in tumor cells from 5/10 patients for which methylation analysis in tumor and normal tissue did not reveal any abnormality. A possible explanation, at least for some cases, is the presence of undetected constitutional or somatic MLH1 mutations. It has been shown that somatic biallelic mutations might be induced by an inherited base-excision repair defect in patients with biallelic MUTYH germline mutations.33,34
In conclusion, the investigation of a group of Italian patients with clinical characteristics of LS, MLH1 negative tumors and lack of detectable MLH1 gene mutations has allowed us to identify an additional sporadic case of constitutional MLH1 primary epimutation. This patient displayed some clinical characteristics that are not frequent among MLH1 epimutation carriers, namely relatively late age at tumor diagnosis and occurrence of a breast carcinoma not expressing the MLH1 protein. Further studies are needed in order to define more accurately the clinical spectrum and cancer risks associated with this subset of LS, as well as its frequency in the population. We also found that 36% of the cases had abnormal MLH1 methylation, which was confined to the tumor in the large majority (4/5). The observation that these patients had some characteristics of LS, namely early age at onset and/or positive family history, raises the possibility of genetic influences in the establishment of somatic MLH1 methylation.
Patients and Methods
Patients
The study was performed on 14 subjects with a clinical suspicion of LS in whom previous testing had not identified clearly pathogenetic germline sequence mutations in MMR genes. All probands had been tested by direct sequencing and multiplex ligation dependent probe amplification (MLPA) for the detection of point mutations and large genomic rearrangements, respectively, in MLH1, MSH2 and MSH6. Three of these individuals carried sequence alterations of no or little clinical significance in MLH1 (2 cases) or MSH2 (1 case), corresponding to Class 1 according to the International Agency for Cancer Research (IARC) classification system.35,36 Two other subjects were heterozygous for so far unreported MLH1 gene variants: a silent coding single nucleotide substitution, c.1743G>A (p.Pro581Pro), and a missense change, c.1814A>C (p.Glu605Ala). Both variants should be currently considered of uncertain significance (IARC Class 3), although, based on its nature, c.1743G>A is unlikely to be disease causing. In addition, a PTEN mutation was identified in a patient (M57) who was found to have characteristics of PHTS.27
Patients were selected based on personal and family positive history of LS related cancers, and on absent/reduced MLH1 protein expression in tumor samples (Table 2). In particular, all patients fulfilled the revised Bethesda Criteria.37 Eight probands had a family history of LS related cancers in 1st degree relatives, and 2 were sporadic cases with multiple tumors of the LS spectrum. MSI analysis was performed in tumors from 13 cases.
Table 2.
Clinical and molecular characteristics of the patients tested for MLH1 epimutations
| CaseID | Sex | Family history of LS cancers (years at diagnosis) | Type of cancers and age at diagnosis (yrs) | MSI status | BRAF p.V600E** | MLH1 methylation in tumor | MMR gene variants |
|---|---|---|---|---|---|---|---|
| M40 | M | Father: colon (63y) | CRC 33 | MSI-H | nt | − | — |
| M45 | M | — | 1) CRC 33; 2) CRC 40 | MSI-H | WT | − | — |
| M46 | M | Brother: colon (61y) | CRC 43 | nt | nt | nt | — |
| M47 | M | Mother: stomach (71y); maternal uncle: unspecified (35y) | CRC 48 | MSI-H | WT | − | — |
| M49 | F | Daughter: rectum (40y); mother: colon (79y) | CRC 68 | MSI-H | nt | + | MLH1 c.1217G>A; p.Ser406Asn (Class 1) |
| M50 | F | Father: colon 74y; paternal 1st cousin: colon (70y); other paternal relatives with LS-related tumors (>70y) | CRC 52 | MSI-H | nt | nt | — |
| M55 | M | Brother: colon (60y) | 1) CRC 43; 2) CRC 63 | MSI-H | nt | nt | MLH1 c.1039-8T>A (Class 1) |
| M56 | F | Father: colon (81y); paternal uncle: bladder (52y) | CRC 43 | MSI-H | WT | + | — |
| M57* | M | — | DC 37 | MSI-H | WT | + | — |
| M58 | F | — | DC 34 | MSI-H | WT | nt | — |
| M60 | F | — | 1) EC 55; 2) UC 62; 3) CRC 66; 4) BC 76 | MSI-H | nt | + | MSH2 c.1511-9A>T (Class 1) |
| M66 | F | Father: colon (51y) | 1) BC 56; 2) CRC 57 | MSI-H | Mut | + | MLH1 c.1743G>A; p.Pro581Pro (NR; Class 3) |
| M69 | F | — | CRC 35 | MSI-H | nt | − | MLH1 c.1814A>C; p.Glu605Ala (NR; Class 3) |
| M70 | M | — | CRC 41 | MSI-H | WT | − | — |
This patient also had multiple intestinal polyps (mostly hamartomas) and was later found to be heterozygous for the pathogenic PTEN mutation p.Ser170Arg.27
*nt: not tested; WT: wild-type.
DNA was isolated from PBLs using a standard phenol-chloroform protocol, while DNA from formalin-fixed paraffin-embedded microdissected tumor specimens was extracted by QIAamp DNA FFPE Tissue Kit (QIAGEN, Cat. No. 56404). Informed consent for molecular analyses was obtained from all subjects according to institutional procedures.
Molecular analyses
Methylation-specific MLPA (MS-MLPA)
The epigenetic pattern was investigated on PBLs from all patients and on tumor tissues, when available. The MS-MLPA ME011 kit (MRC-Holland) was employed to detect aberrant CpG Island methylation in the promoter regions of MMR genes. The probemix is comprised of 38 probe sequences, 22 of which correspond to MMR genes, containing one or 2 digestion sites for the methylation sensitive HhaI enzyme. In particular, the ME011 kit includes 5 test probes for MLH1. The MLH1 probes that show the best correlation with mRNA expression levels are located at -246 and -13 nt from the transcription initiation site, and correspond to the so called C and D regions, respectively.15 The assay was carried out in duplicate, according to the manufacturer's instructions. Control leukocyte DNA specimens were included in every assay. PCR products were run onto an ABI 310 capillary sequencer and analyzed using GeneMapper v. 4.0 analysis software (Applied Biosystems).
Data were analyzed using the software Coffalyser.NET (MRC-Holland). A dosage ratio of 0.15 or higher between digested (methylated) and undigested (methylated + unmethylated) areas corresponding to the same methylation sensitive site, was assumed to indicate hypermethylation.38
Methylation-specific PCR (MS-PCR)
DNA from PBLs was converted with sodium bisulphite using the EpiTect Bisulphite Kit (Qiagen, Cat. No. 59104) and subjected to PCR amplification with primers specific for methylated (M) and unmethylated (U) sequences.39 2 different fragments of the MLH1 promoter were investigated, encompassing nt −273 to −173 and −711 to −576, which correspond to the C and A regions, respectively.15
MS-PCR was performed using HotStarTaq DNA polymerase (Qiagen, Cat. No. 203203) in a final reaction volume of 20 μl containing 100 ng of bisulphite modified DNA. The PCR reaction consisted of 35 cycles, and the annealing temperature was 50°C and 52°C for the methylated (M) and unmethylated (U) C-region reactions, respectively, while for the M and U A-region reactions the annealing temperature was 59°C and 61°C, respectively. All PCR reactions were performed with positive (M) and negative (U) control DNAs (EpiTect Control DNA, Cat. No. 59655 and 59665, respectively). PCR products were visualized on ethidium bromide stained 2% agarose gels.
Microsatellite instability (MSI), immunohistochemistry and BRAF mutation analysis
MSI analysis was performed using a standard panel of 5 mononucleotides and expression of the MLH1, MSH2 and MSH6 proteins was tested by immunohistochemistry, as previously described.40,41
DNA extracted from formalin-fixed paraffin-embedded tumor specimens was tested for the presence of the p.V600E BRAF variant by direct sequencing of exon 15. BRAF analysis was conducted overall on 7 tumors from which sufficient DNA was available.
Allele–specific expression assay
A semi-quantitative SNuPE assay exploiting the c.655A>G polymorphism in exon 8 of the MLH1 gene (rs1799977) was used for allele-specific expression (ASE) analysis. The assay was performed using the ABI Prism SNaPshot Multiplex Kit (Life Technologies, Cat. No. 4323159). RNA was isolated from blood drawn in PAXgene™ Blood RNA Tubes (BD, Cat. No. 762165) and reverse transcribed. Single nucleotide primer extension-based (SNuPE) analysis was performed on cDNA and genomic DNA (gDNA) samples obtained from PBLs of the same individual. Fragments of gDNA and cDNA encompassing the polymorphic site were PCR-amplified and used as templates for primer extensions. The extension reaction was performed in 10 μL containing 3 μL of template DNA, 5 μL of SNaPshot multiplex ready reaction mix and 0.2 μL of reverse primer. Different length products were obtained for the 2 alleles (A or G).
The products were analyzed on an ABI-PRISM 310 Genetic Analyzer (Life Technologies). ASE was quantified with the GeneMapper Software (Life Technologies) and the allelic ratio was calculated as: (peak area of allele A:peak area of allele G) in cDNA / (peak area of allele A:peak area of allele G) in gDNA.42 The range of balanced allelic ratio was comprised between 0.8 and 1.2, and values below or above the indicated thresholds, respectively, were considered indicative of allelic unbalance.42
Analysis of MLH1 single nucleotide variants
The presence of single nucleotide variants previously associated with constitutional MLH1 promoter hypermethylation was investigated by direct sequencing of the 5′ MLH1 region in PBL genomic DNA.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by a grant from Istituto Toscano Tumori to MG. FC was a recipient of a fellowship from the Associazione Senese per la Prevenzione del Carcinoma Colon-Rettale.
References
- 1.Issa J-P. CpG island methylator phenotype in cancer. Nat Rev Cancer 2004; 4:988-93; PMID:15573120; http://dx.doi.org/ 10.1038/nrc1507 [DOI] [PubMed] [Google Scholar]
- 2.Scott RH, Douglas J, Baskcomb L, Huxter N, Barker K, Hanks S, Craft A, Gerrard M, Kohler JA, Levitt GA, et al. Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet 2008; 40:1329-34; PMID:18836444; http://dx.doi.org/ 10.1038/ng.243 [DOI] [PubMed] [Google Scholar]
- 3.Hitchins MP. The role of epigenetics in Lynch syndrome. Fam Cancer 2013; 12:189-205; PMID:23462881; http://dx.doi.org/ 10.1007/s10689-013-9613-3 [DOI] [PubMed] [Google Scholar]
- 4.Lucci-Cordisco E, Zito I, Gensini F, Genuardi M. Hereditary nonpolyposis colorectal cancer and related conditions. Am J Med Genet 2003; 122A:325-34; PMID:14518071; http://dx.doi.org/ 10.1002/ajmg.a.20475 [DOI] [PubMed] [Google Scholar]
- 5.Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet 2009; 76:1-18; PMID:19659756; http://dx.doi.org/ 10.1111/j.1399-0004.2009.01230.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487:330-37; PMID:22810696; http://dx.doi.org/ 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Meyerhardt JA, Loda M, Giovannucci EL, Fuchs CS. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 2009; 58:90-96; PMID:18832519; http://dx.doi.org/ 10.1136/gut.2008.155473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thiel A, Heinonen M, Kantonen J, Gylling A, Lahtinen L, Korhonen M, Kytölä S, Mecklin JP, Orpana A, Peltomäki P, et al. BRAF mutation in sporadic colorectal cancer and Lynch syndrome. Virchows Arch 2013; 463:613-21; PMID:23963522; http://dx.doi.org/ 10.1007/s00428-013-1470-9 [DOI] [PubMed] [Google Scholar]
- 9.Fleisher AS, Esteller M, Wang S, Tamura G, Suzuki H, Yin J, Zou TT, Abraham JM, Kong D, Smolinski KN, et al. Hypermethylation of the hMLH1 gene promoter in human gastric cancers with microsatellite instability. Cancer Res 1999; 59:1090-5; PMID:10070967 [PubMed] [Google Scholar]
- 10.Fu T, Pappou EP, Guzzetta AA, Jeschke J, Kwak R, Dave P, Hooker CM, Morgan R, Baylin SB, Iacobuzio-Donahue CA, et al. CpG island methylator phenotype-positive tumors in the absence of MLH1 methylation constitute a distinct subset of duodenal adenocarcinomas and are associated with poor prognosis. Clin Cancer Res 2012; 18:4743-52; PMID:22825585; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-0707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buchanan DD, Tan YY, Walsh MD, Clendenning M, Metcalf AM, Ferguson K, Arnold ST, Thompson BA, Lose FA, Parsons MT, et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J Clin Oncol 2014; 32:90-100; PMID:24323032; http://dx.doi.org/ 10.1200/JCO.2013.51.2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peltomäki P. Epigenetic mechanisms in the pathogenesis of Lynch syndrome. Clin Genet 2014; 85:403-12; http://dx.doi.org/ 10.1111/cge.12349 [DOI] [PubMed] [Google Scholar]
- 13.Morak M, Schackert HK, Rahner N, Betz B, Ebert M, Walldorf C, Royer-Pokora B, Schulmann K, von Knebel-Doeberitz M, Dietmaier W, et al. Further evidence for heritability of an epimutation in one of 12 cases with MLH1 promoter methylation in blood cells clinically displaying HNPCC. Eur J Hum Genet 2008; 16:804-11; PMID:18301449; http://dx.doi.org/ 10.1038/ejhg.2008.25 [DOI] [PubMed] [Google Scholar]
- 14.Hitchins MP, Rapkins RW, Kwok CT, Srivastava S, Wong JJ, Khachigian LM, Polly P, Goldblatt J, Ward RL. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5'UTR. Cancer Cell 2011; 20:200-13; PMID:21840485; http://dx.doi.org/ 10.1016/j.ccr.2011.07.003 [DOI] [PubMed] [Google Scholar]
- 15.Deng G, Chen A, Hong J, Chae HS, Kim YS. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res 1999; 59:2029-33; PMID:10232580 [PubMed] [Google Scholar]
- 16.Vasen HF, Blanco I, Aktan-Collan K, Gopie JP, Alonso A, Aretz S, Bernstein I, Bertario L, Burn J, Capella G, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut 2013; 62:812-23; PMID:23408351; http://dx.doi.org/ 10.1136/gutjnl-2012-304356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrow E, Robinson L, Alduaij W, Shenton A, Clancy T, Lalloo F, Hill J, Evans DG. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet 2009; 75:141-9; PMID:19215248; http://dx.doi.org/ 10.1111/j.1399-0004.2008.01125.x [DOI] [PubMed] [Google Scholar]
- 18.Engel C, Loeffler M, Steinke V, Rahner N, Holinski-Feder E, Dietmaier W, Schackert HK, Goergens H, von Knebel Doeberitz M, Goecke TO, et al. Risks of less common cancers in proven mutation carriers with Lynch syndrome. J Clin Oncol 2012; 30:4409-15; PMID:23091106; http://dx.doi.org/ 10.1200/JCO.2012.43.2278 [DOI] [PubMed] [Google Scholar]
- 19.Win AK, Young JP, Lindor NM, Tucker KM, Ahnen DJ, Young GP, Buchanan DD, Clendenning M, Giles GG, Winship I, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol 2012; 30:958-64; PMID:22331944; http://dx.doi.org/ 10.1200/JCO.2011.39.5590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jensen UB, Sunde L, Timshel S, Halvarsson B, Nissen A, Bernstein I, Nilbert M. Mismatch repair defective breast cancer in the hereditary nonpolyposis colorectal cancer syndrome. Breast Cancer Res Treat 2010; 120:777-82; PMID:19575290; http://dx.doi.org/ 10.1007/s10549-009-0449-3 [DOI] [PubMed] [Google Scholar]
- 21.Buerki N, Gautier L, Kovac M, Marra G, Buser M, Mueller H, Heinimann K. Evidence for breast cancer as an integral part of Lynch syndrome. Genes Chromosomes Cancer 2012; 51:83-91; PMID:22034109; http://dx.doi.org/ 10.1002/gcc.20935 [DOI] [PubMed] [Google Scholar]
- 22.Suter CM, Martin DI, Ward RL. Germline epimutation of MLH1 in individuals with multiple cancers. Nat Genet 2004; 36:497-501; PMID:15064764; http://dx.doi.org/ 10.1038/ng1342 [DOI] [PubMed] [Google Scholar]
- 23.Miyakura Y, Sugano K, Akasu T, Yoshida T, Maekawa M, Saitoh S, Sasaki H, Nomizu T, Konishi F, Fujita S, et al. Extensive but hemiallelic methylation of the hMLH1 promoter region in early-onset sporadic colon cancers with microsatellite instability. Clin Gastroenterol Hepatol 2004; 2:147-56; PMID:15017620; http://dx.doi.org/ 10.1016/S1542-3565(03)00314-8 [DOI] [PubMed] [Google Scholar]
- 24.Crépin M, Dieu MC, Lejeune S, Escande F, Boidin D, Porchet N, Morin G, Manouvrier S, Mathieu M, Buisine MP. Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptibility. Hum Mutat 2012; 33:180-88; http://dx.doi.org/ 10.1002/humu.21617 [DOI] [PubMed] [Google Scholar]
- 25.Ward RL, Dobbins T, Lindor NM, Rapkins RW, Hitchins MP. Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet Med 2013; 15:25-35; PMID:22878509; http://dx.doi.org/ 10.1038/gim.2012.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwok CT, Vogelaar IP, van Zelst-Stams WA, Mensenkamp AR, Ligtenberg MJ, Rapkins RW, Ward RL, Chun N, Ford JM, Ladabaum U, et al. The MLH1 c.-27C>A and c.85G>T variants are linked to dominantly inherited MLH1 epimutation and are borne on a European ancestral haplotype. Eur J Hum Genet 2014; 22:617-24; PMID:24084575; http://dx.doi.org/ 10.1038/ejhg.2013.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ponz de Leon M, Di Gregorio C, Giunti L, Roncucci L, Pedroni M, Tinca AC, Crucianelli F, Tricarico R, Genuardi M. Duodenal carcinoma in a 37-year-old man with Cowden/Bannayan syndrome. Dig Liver Dis 2013; 45:75-8; PMID:23117110; http://dx.doi.org/ 10.1016/j.dld.2012.09.017 [DOI] [PubMed] [Google Scholar]
- 28.van Roon EH, van Puijenbroek M, Middeldorp A, van Eijk R, de Meijer EJ, Erasmus D, Wouters KA, van Engeland M, Oosting J, Hes FJ, et al. Early onset MSI-H colon cancer with MLH1 promoter methylation, is there a genetic predisposition? BMC Cancer 2010; 10:180; PMID:20444249; http://dx.doi.org/ 10.1186/1471-2407-10-180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lubomierski N, Plotz G, Wormek M, Engels K, Kriener S, Trojan J, Jungling B, Zeuzem S, Raedle J. BRAF mutations in colorectal carcinoma suggest two entities of microsatellite-unstable tumors. Cancer 2005; 104:952-961. [DOI] [PubMed] [Google Scholar]
- 30.Thiel A, Heinonen M, Kantonen J, Gylling A, Lahtinen L, Korhonen M, Kytölä S, Mecklin J-P, Orpana A, Peltomäki P, et al. BRAF mutation in sporadic colorectal cancer and Lynch syndrome. Virchows Arch 2013; 463:613-621; PMID:23963522; http://dx.doi.org/ 10.1007/s00428-013-1470-9 [DOI] [PubMed] [Google Scholar]
- 31.Young J, Jass JR. The case for a genetic predisposition to serrated neoplasia in the colorectum: hypothesis and review of the literature. Cancer Epidemiol Biomarkers Prev 2006; 15:1778-84; PMID:1855950417035382; http://dx.doi.org/ 10.1158/1055-9965.EPI-06-0164 [DOI] [PubMed] [Google Scholar]
- 32.Joensuu EI, Abdel-Rahman WM, Ollikainen M, Ruosaari S, Knuutila S, Peltomäki P. Epigenetic signatures of familial cancer are characteristic of tumor type and family category. Cancer Res 2008; 68:4597-605; PMID:18559504; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-6645 [DOI] [PubMed] [Google Scholar]
- 33.Castillejo A, Vargas G, Castillejo MI, Navarro M, Barberá VM, González S, Hernández-Illán E, Brunet J, Ramón Y, Cajal T, et al. Prevalence of germline MUTYH mutations among Lynch-like syndrome patients. Eur J Cancer 2014; 50:2241-2250; PMID:24953332; http://dx.doi.org/ 10.1016/j.ejca.2014.05.022 [DOI] [PubMed] [Google Scholar]
- 34.Morak M, Heidenreich B, Keller G, Hampel H, Laner A, de la Chapelle A, Holinski-Feder E. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur J Hum Genet 2014; Feb 12 http://dx.doi.org/ 10.1038/ejhg.2014.15 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Hogervorst FB, Hoogerbrugge N, Spurdle AB, Tavtigian SV, IARC Unclassified Genetic Variants Working Group . Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 2008; 20:1282-91; http://dx.doi.org/ 10.1002/humu.20880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, Bapat B, Bernstein I, Capellá G, den Dunnen JT, et al. Application of a five-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants lodged on the InSiGHT locus-specific database. Nat Genet 2014; 46:107-15; PMID:24362816; http://dx.doi.org/ 10.1038/ng.2854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96:261-8; PMID:14970275; http://dx.doi.org/ 10.1093/jnci/djh034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gylling A, Abdel-Rahman WM, Juhola M, Nuorva K, Hautala E, Jarvinen HJ, Mecklin JP, Aarnio M, Peltomaki P. Is gastric cancer part of the tumour spectrum of hereditary non-polyposis colorectal cancer? A molecular genetic study. Gut 2007; 56:926-33; PMID:17267619; http://dx.doi.org/ 10.1136/gut.2006.114876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 1998; 95:6870-75; PMID:9618505; http://dx.doi.org/ 10.1073/pnas.95.12.6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roncari B, Pedroni M, Maffei S, Di Gregorio C, Ponti G, Scarselli A, Losi L, Benatti P, Roncucci L, De Gaetani C, et al. Frequency of constitutional MSH6 mutations in a consecutive series of families with clinical suspicion of HNPCC. Clin Genet 2007; 72:230-37; PMID:17718861; http://dx.doi.org/ 10.1111/j.1399-0004.2007.00856.x [DOI] [PubMed] [Google Scholar]
- 41.Giunti L, Cetica V, Ricci U, Giglio S, Sardi I, Paglierani M, Andreucci E, Sanzo M, Forni M, Buccoliero AM, et al. Type A microsatellite instability in pediatric gliomas as an indicator of Turcot sindrome. Eur J Hum Genet 2009; 17:919-27; PMID:19156169; http://dx.doi.org/ 10.1038/ejhg.2008.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Renkonen E, Zhang Y, Lohi H, Salovaara R, Abdel-Rahman WM, Nilbert M, Aittomäki K, Järvinen HJ, Mecklin J-P, Lindblom A, et al. Altered expression of MLH1, MSH2, and MSH6 in predisposition to hereditary nonpolyposis colorectal cancer. J Clin Oncol 2003; 21:3629-37; PMID:14512394; http://dx.doi.org/ 10.1200/JCO.2003.03.181 [DOI] [PubMed] [Google Scholar]