

Abstract
Angiogenesis is one of the most important processes for cancer cell survival, tumor growth and metastasis. Vascular endothelial growth factor (VEGF) and its receptor, particularly VEGF receptor-2 (VEGFR-2, or kinase insert domain-containing receptor, KDR), play critical roles in tumor-associated angiogenesis. We developed TTAC-0001, a human monoclonal antibody against VEGFR-2/KDR from a fully human naïve single-chain variable fragment phage library. TTAC-0001 was selected as a lead candidate based on its affinity, ligand binding inhibition and inhibition of VEGFR-2 signal in human umbilical vein endothelial cells (HUVEC). TTAC-0001 inhibited binding of VEGF-C and VEGF-D to VEGFR-2 in addition to VEGF-A. It binds on the N-terminal regions of domain 2 and domain 3 of VEGFR-2. It could inhibit the phosphorylation of VEGFR-2/KDR and ERK induced by VEGF in HUVEC. TTAC-0001 also inhibited VEGF-mediated endothelial cell proliferation, migration and tube formation in vitro, as well as ex vivo vessel sprouting from rat aortic rings and neovascularization in mouse matrigel model in vivo. Our data indicates that TTAC-0001 blocks the binding of VEGFs to VEGFR-2/KDR and inhibits VEGFR-induced signaling pathways and angiogenesis. Therefore, these data strongly support the further development of TTAC-0001 as an anti-cancer agent in the clinic.
Keywords: angiogenesis, cross species reactivity, anti-VEGFR2 monoclonal antibody, TTAC-0001, VEGF, VEGFR-2(KDR)
Abbreviations
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
- KDR
kinase insert domain-containing receptor
- Flk-1
fetal liver kinase 1
- RTK
receptor tyrosine kinase
- ECD
extracellular domain
- mAb
monoclonal antibody
- Kd
dissociation constant
- IgG
immunoglobulin G
- ERK
extracellular signal-regulated kinases
- FBS
fetal bovine serum
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- HUVEC
human umbilical vein endothelial cell
- RT
room temperature
- IHC
immunohistochemistry
- TGI
tumor growth inhibition
- IP
immunoprecipitates
- CHO cells
Chinese hamster ovary cells
- HRP
horseradish peroxidase
- OCT
optimum cutting temperature
- GBM
glioblastoma
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Introduction
Tumor angiogenesis, a process that results in the formation of blood vessels in tumors, is important for tumor growth and the development of distant metastasis.1-3 These vessels can grow either by sprouting from preexisting blood vessels or through the mobilization and differentiation of endothelial precursor cells derived from bone marrow.4-6 Although other growth factors and their cognate receptors have been implicated in tumor angiogenesis,3,7 VEGF is a key regulator of this process. VEGF promotes proliferation, migration, and survival of endothelial cells. In addition, several studies have demonstrated that VEGF is overexpressed in many human tumors, including lung, colon, breast, gastrointestinal tract, renal, and ovarian carcinomas, and there is correlation between increased VEGF expression and tumor progression.8 The 3 VEGF receptors are VEGFR-1, also known as Flt-1; VEGFR-2, also known as Flk-1 (mouse) or KDR (human); and VEGFR-3. VEGFR-2/KDR and Flk-1 are 85% homologous in amino acid sequence.9,10 VEGFR-2/KDR acts as a major regulator of mitogenesis and angiogenesis through its interaction with VEGF. VEGFR-2/KDR is up-regulated in many cancer cells, and it can regulate tumor cell growth and survival through an autocrine pathway.11-13
Several approaches to targeting the VEGF signaling pathway have resulted in potential anticancer therapies. These include a neutralizing anti-VEGF antibody, overexpression of a dominant negative VEGF mutant, soluble VEGF receptors, antisense oligonucleotides targeting VEGF, and small molecule inhibitors of VEGFR signaling.14-21 Results with the humanized anti-VEGF monoclonal antibody (bevacizumab; Avastin®) demonstrated a survival benefit in patients with metastatic colorectal cancer, lung cancer and brain cancer.22-24 A variety of receptor tyrosine kinase (RTK) inhibitors targeting VEGF receptors such as sunitinib (Sutent®) and sorafenib (Nexavar®) have been approved by the US Food and Drug Administration (FDA) for the treatment of renal cell carcinoma (RCC) and imatinib-resistant gastrointestinal stromal tumor (GIST), as well as hepatocellular carcinoma. A therapeutic human IgG1 mAb, ramucirumab (Cyramza®) that was recently approved by FDA binds with high affinity to the extracellular VEGF-binding domain of VEGFR-2/KDR.25-28 Because ramucirumab did not cross-react with mouse VEGFR-2/KDR, the company developed DC101 as a surrogate antibody for use in preclinical studies, DC101 is a rat anti-mouse antibody that inhibits VEGFR-2/Flk-1 (the murine homologues of VEGFR-2/KDR) signaling pathway.29 Furthermore, it has been reported that none of the anti-VEGFR-2/KDR antibodies that have been developed until now have cross-reactivity to mouse VEGFR-2/KDR; thus, studies in appropriate animal models for the evaluation of the drug's efficacy and safety could not be performed.10,30-32 Ramucirumab is currently being investigated in multiple Phase 2 and Phase 3 trials for colorectal cancer, hepatocellular carcinoma, non-small-cell lung cancer, breast cancer, ovarian cancer, prostate cancer, metastatic melanoma, metastatic renal carcinoma, and recurrent glioblastoma.26-28 It was approved by FDA in April 2014 for gastric cancer.
We identified a human anti-VEGFR-2/KDR neutralizing antibody, TTAC-0001, which exhibits potent inhibitory activity in tumor growth and angiogenesis. We report here that TTAC-0001 blocks the binding of VEGF to VEGFR-2/KDR and inhibits VEGFR-2-mediated signaling and angiogenesis. Furthermore, it showed strong anti-angiogenic activity in VEGF-mediated in vivo mouse Matrigel model, as well as ex vivo vessel sprouting in rat. Because TTAC-0001 does cross-react with mouse VEGFR-2 (Flk1), its anti-tumor effect from in vivo models is likely to be due to an inhibition of tumor angiogenesis. Therefore, these data strongly support the further development of TTAC-0001 as an anti-cancer agent. A Phase 1 clinical study of TTAC-0001 is being conducted in Korea.
Results
TTAC-0001 binds to VEGFR-2 specifically and blocks the interaction of VEGF and VEGFR-2
Several anti-VEGFR-2/KDR monoclonal antibodies used in this study were selected from a fully human naïve single-chain variable fragment (scFv) phage library that was generated in-house following the methods described previously.33,34 Based on the binding to purified KDR-ECD(1–3) containing 1–327 amino acids of human VEGFR2, 18 phage clones were selected (data not shown). The phage clones that could inhibit binding of VEGF to human VEGFR2 were selected by ELISA (Fig. 1a). Among them, 3 clones were converted into scFv for E. coli expression and IgG1 format for mammalian expression. Of these, TTAC-0001 inhibited binding of VEGF to its receptor, KDR (Fig 1b) the best. When we added the pre-incubated mixture of antibodies and KDR to coated human VEGF165, the binding of KDR to VEGF was almost completely inhibited at 70 nM of TTAC-0001. In contrast to TTAC-0001, 6C1 and 6G1 inhibited binding only slightly. The complementarity-determining region sequences and affinities of those clones are shown in Figure 1c. The Kd of the TTAC-0001 IgG format was in the sub-nanomolar range (0.23 nM) on immobilized KDR-ECD(1–3)-Fc coating antigen; all other clones had Kd around 10−8 M (Figure S1). TTAC-0001 displayed the strongest inhibition of the binding of VEGF to its receptor, KDR (Fig. 1c).
Figure 1.

Characterization of binding properties of anti-KDR antibodies. Competitive inhibition of anti-KDR phages (a) or antibodies (b) in binding of KDR(ECD1–3)-Fc to VEGF165. TTAC-0001, closed circle; 6C1, open circle; 6G1, triangle. (c) Complementarity-determining region (CDR) sequences of anti-KDR antibodies and their respective KDR binding affinities (Kd) determined using surface plasmon resonance.
TTAC-0001 binds the N-terminus of domain 2 and domain 3 of extracellular region of VEGFR-2
We also investigated the binding domain of each clone by domain mapping assay. Domain mapping was done using the extracellular domain (ECD) of VEGFR-2/KDR (Fig. 1b) and scFv form of antibodies. All clones showed the highest binding capacity when KDR (ECD 1–3) was used as an antigen. However, the binding pattern of anti-KDR clones with KDR (ECD 1–2, amino acids 1–222 of hVEGFR2) and KDR (ECD 2–3, amino acids 1–327 (Δ 24–116) of hVEGFR2) was different (Fig. 1b). 6C1 scFv and 6G1 scFv showed similar binding affinity to the ECD1–2 and ECD2–3 domains, which suggested that the main binding domain of 6C1 and 6G1 was in Ig domain 2. In contrast, TTAC-0001 scFv had 8-fold higher binding affinity to ECD2–3 compared to ECD1–2 (Fig. 2a). This suggests that the major binding domain of TTAC-0001 looks like in Ig domain 3 that is important for VEGF binding to KDR.9 Thus, the epitope targeted by TTAC-0001 differs from that targeted by 6C1 or 6G1. Based on the results from the above experiments, we selected TTAC-0001 as a lead candidate. 6C1 was used as a negative control. From the domain mapping study, we further investigated the epitopes of TTAC-0001 from the peptide microarray from Abnova (Taipei city, Taiwan). Interestingly, TTAC-0001 has 2 major epitopes,111 ASVIYVY and219 VGYRIYD in KDR (Fig. 2b). The sequence, ASVIYVY, is located in the region between Ig-like domain 1 and 2, and the latter epitope, VGYRIYD, is located in the N-terminus of Ig-like domain 3, which is known to be a critical domain for binding VEGF to VEGFR-2.9 Since the sequence, VGYRIYD, is identical from human to mouse and rat VEGFR-2 and another epitope, ASVIYVY, also showed similarity between species, TTAC-0001 could show cross-species reactivity to rat and mouse VEGFR-2 (Table S1).
Figure 2.
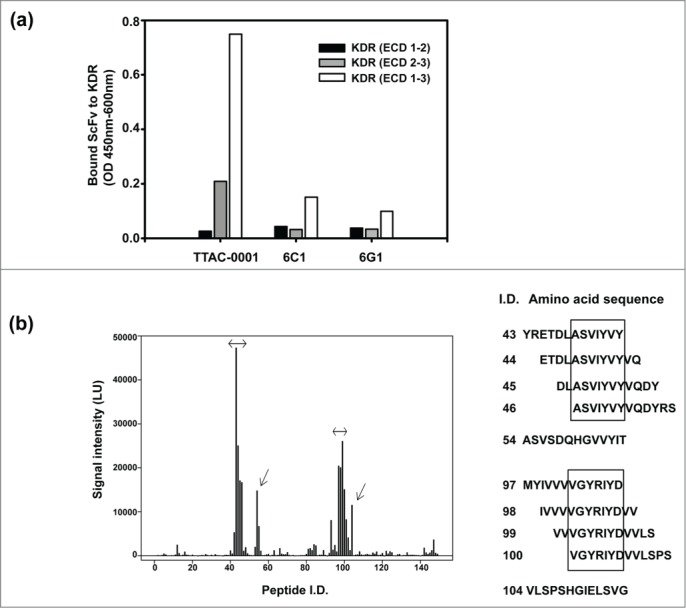
Domain and epitope mapping of anti-KDR antibodies. (a) Domain mapping analysis of anti-KDR antibodies on the extracellular region of KDR. Black bar represents extracellular domain 1 and 2 of KDR (KDR (ECD 1–2)). Gray bar represents extracellular domain 2 and 3 of KDR (KDR (ECD 2–3)). White bar represents extracellular domain 1, 2 and 3 of KDR (KDR (ECD 1–3)). (b) Peptide microarray for epitope mapping. As an antigen, KDR (ECD1–3) was used. The sequence was scanned with a format of 13meric peptides overlapping 11 amino acid residues with the following peptide, resulting in a total of 149 peptides. The binding of primary antibody (TTAC-0001, control human IgG) to the peptide was measured with dylight-649 labeled secondary antibody. Left panel: Fluorescence intensity of peptide. The intensity of fluorescence measured by TTAC-0001 was subtracted the value of control human IgG (Pierce). Arrows indicate peptides showing over 10,000 LU of signal intensity. Right panel: Amino acid sequence (peptide i.d.) showed high intensity of fluorescence (≥ 10,000 LU). The sequences inside the box indicate epitopes for TTAC-0001.
TTAC-0001 inhibits binding of VEGF-C and D to its receptor, VEGFR-2, and it does not bind to VEGFR-1 and VEGFR-3
To validate our assay systems, we investigated binding specificity of TTAC-0001. We first tested the binding of TTAC-0001 to VEGF isoforms to clarify uncertainties of subsequent studies. TTAC-0001 and human IgG did not bind to VEGF-165, VEGF-C and VEGF-D, while bevacizumab bound well to VEGF-165 as expected (Fig. 3a). We also investigated the binding specificity of TTAC-0001 on the family of VEGF receptors by SPR (Fig. 3b). The Fc-fused extracellular domains of hVEGFRs were coated on a CM5 chip and a 50 nM solution of TTAC-0001 was injected as an analyte at a flow rate of 30 μl/min. TTAC-0001 bound well only to VEGFR-2/KDR as expected. It did not bind to VEGFR-1 and VEGFR-3.
Figure 3.

Measurement of the specificity of TTAC-0001. (a) Binding of TTAC-0001 to VEGF isoforms. Black bar represents VEGF-165. Gray bar represents VEGF-C and blank bar represents VEGF-D. hIgG and bevacizumab were used as controls. (b) Specificity measurement of TTAC-0001 on VEGFRs by Biacore. Line, hVEGFR1-Fc coated chip; dashed line (–), hVEGFR2-Fc coated chip; dashed line (—), hVEGFR-3-Fc coated chip. 50 nM of TTAC-0001 was injected at a flow rate of 30 μl/min. (c) Inhibition of TTAC-0001 in binding VEGF isoforms to VEGFR-2. Closed rectangular represents VEGF165. Closed circle represents VEGF-C and Open circle represents VEGF-D. (d) Immunohistochemistry showed positive staining of TTAC-0001 in endothelial cells of mouse aorta and mouse brain bEND.3 cells from Balb/c mice. HUVECs and human GBM tumor tissues were used as controls. Triangle represents stained regions of each cell.
It has been reported that VEGFR-2 can bind fully processed forms of VEGF-C and VEGF-D even though they mostly transmitted their signals through VEGFR-3. Activation of VEGFR-2 is responsible for the angiogenic signal of VEGF-C in many experiments.35-37 Thus, we measured whether TTAC-0001 could inhibit binding of VEGF-C and VEGF-D to VEGFR-2 (Fig. 3c). VEGFR-2 (ECD 1–7)-Fc were used in this experiment. It effectively inhibit binding of VEGF-C and VEGF-D to VEGFR-2 in addition to VEGF-A. It could also inhibit binding of VEGF-E, another VEGFR-2 binding VEGF related protein that was encoded by virus (data not shown). The IC50 values for VEGFs binding were 8.7 nM (VEGF-165), 6.3 nM (VEGF-C) and 7.0 nM (VEGF-D) analyzed by Softmax Pro software. Thus, TTAC-0001 can effectively and specifically inhibit the biological function of VEGFR-2.
Considering the similarity of epitopes between human and mouse VEGFR-2 (Fig. 2b), we examined the binding of TTAC-0001 on mouse tissues. Immunohistochemistry (IHC) showed that TTAC-0001 was stained positively in endothelial cells of aorta from mouse and bEND.3 cells, brain endothelial cells from Balb/c mice (Fig. 3d). Therefore, mouse can be an appropriate species for studying efficacy, safety, and pharmacokinetic and pharmacodynamic properties due to the cross-reactivity of TTAC-0001 even though it was difficult to see direct binding with purified mouse VEGFR-2 by ELISA, surface plasmon resonance (SPR) and fluorescence-activated cell sorting (FACS) (data not shown).
TTAC-0001 inhibits downstream signaling of VEGFR-2 in endothelial cells
The inhibitory activity of TTAC-0001 on VEGF-mediated signaling pathways was investigated. Human umbilical vein endothelial cells (HUVECs) were treated with various concentrations of TTAC-0001 and stimulated with 10 ng/ml of VEGF165 for 10 min. Immunoprecipitation and western blot analysis showed more than 50% inhibition of phosphorylation of VEGFR-2/KDR at TTAC-0001 concentrations of 1 μg/ml and higher, with no change in total VEGFR-2/KDR levels (Fig. 4a). The phosphorylation of downstream signaling molecules such as ERK was also inhibited by TTAC-0001 (Fig. 4b), which was consistent with the inhibition of VEGFR-2/KDR phosphorylation. In contrast to TTAC-0001, 6C1 could not inhibit phosphorylation of KDR and its downstream signal molecule.
Figure 4.

Anti-KDR antibody, TTAC-0001, inhibits VEGF-induced phosphorylation of VEGFR-2 and downstream signaling molecule in HUVECs. HUVECs were pretreated for 30 min with various concentrations of TTAC-0001 anti-KDR neutralizing antibody and stimulated with 10 ng/ml of VEGF for 10 min. (a) Anti-KDR/Flk-1 immunoprecipitates (IP) were analyzed by SDS-PAGE. Immunoblot analysis was performed with anti-phosphotyrosine antibody (anti-PY20). (b) Activation of ERK kinase by VEGF was determined by Western blotting. Various amounts of antibody were pre-treated and stimulated with VEGF.
TTAC-0001 inhibits proliferation, migration and tube formation of endothelial cells
During tumor angiogenesis, angiogenic factors such as VEGF induce endothelial cell proliferation, migration and capillary tube formation, resulting in a dynamic remodeling process of vessel formation. To test the effect of TTAC-0001 on angiogenesis in vitro, we performed an endothelial cell proliferation assay. As shown in Figure 5a, stimulation with hVEGF165 increased HUVEC proliferation compared to un-stimulated control at the extent of 160%. At concentrations over 1 μg/ml, TTAC-0001 inhibited HUVEC proliferation significantly (p-value < 0.01) compared to cells treated with VEGF only. We also performed endothelial cell migration assays with TTAC-0001. HUVECs were treated with TTAC-0001 and hVEGF165 on each chamber and incubated for 3.5 hours and stained with crystal violet to take an image. As shown in Figure 5b, VEGF stimulation increased HUVEC migration compared to un-stimulated control. TTAC-0001 could inhibit HUVEC migration significantly in the VEGF-stimulated conditions (Fig. S2).
Figure 5.

TTAC-0001 inhibits angiogenesis in vitro. (a) TTAC-0001 inhibits VEGF-mediated proliferation of HUVEC. HUVECs were incubated with TTAC-0001 (1, 15 and 20 µg/ml) in the media containing VEGF (10 ng/ml). ** p-value < 0.01 versus VEGF alone treated cells. (b) TTAC-0001 inhibits VEGF-mediated migration of HUVEC. HUVECs were incubated with TTAC-0001 (20 µg/ml) for 0.5 hour and plated onto modified Boyden Chambers, and allowed to migrate in response to 10 ng/ml of VEGF for 3.5 hours at 37 °C. cells were stained with crystal violet. (c) TTAC-0001 inhibits in vitro tube formation. HUVECs were seeded onto matrigels and cultured in the VEGF containing media without TTAC-0001 or with 5, 10 and 20 µg of TTAC-0001. 20 µg of 6C1 was also treated as a negative control.
We also monitored the effect of TTAC-0001 on capillary like tube formation of HUVEC (Fig. 5c). The HUVEC tube formation assay is a simple, but well-established in vitro angiogenesis assay based on the ability of endothelial cells to form 3-dimensional capillary-like tubular structures when the angiogenic growth factor induced endothelial cells were cultured on a Matrigel. During the assay, endothelial cells differentiate, directionally migrate to align, branch, and form the tubular networks of vessels. As shown in Figure 5c, HUVECs cultured in Matrigel containing 10 ng/ml of hVEGF165 formed highly developed and differentiated tube-like structures (VEGF 10 ng/ml) while that cultured without hVEGF165 failed to form the tube-like structures (control). We investigated whether the anti-KDR antibodies could inhibit the tube formation of HUVECs. As the amount of TTAC-0001 increased, the extent of tube formation was decreased and the tubular structures were also more disorganized. 6C1, anti-KDR antibody having lower affinity and VEGF competition activity compared to TTAC-0001 showed lower inhibitory activity of tube formation to TTAC-0001 when we treated with the same amount.
TTAC-0001 inhibits ex vivo vessel sprouting and in vivo neovascularization
Although it was difficult to see binding of TTAC-0001 to Flk-1 using SPR and FACS, we investigated whether it can bind murine vessel system and inhibit angiogenesis based on the positive IHC staining on mouse tissue and similarity of the epitope between human and mouse VEGFR-2. We tested whether TTAC-0001 could inhibit endothelial cell sprouting and microvessel formation in rat aortic ring segments. Segments of rat aorta embedded in Matrigels containing hVEGF165 developed well-formed vascular sprouts (Fig. 6a). The numbers and outgrowth of sprouts from the rat aorta induced by VEGF was inhibited by the treatment of TTAC-0001, and the extent of vessel sprouting inhibition was dependent on the concentration of TTAC-0001.
Figure 6.
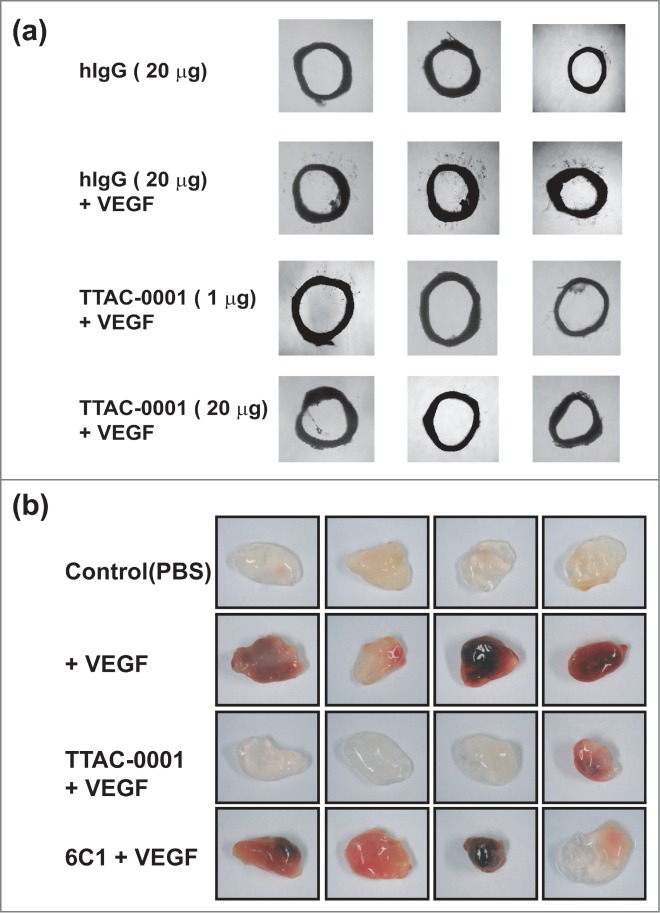
TTAC-0001 inhibits angiogenesis ex vivo and in vivo by targeting murine VEGFR-2. (a) Inhibition of vessel sprouting on hVEGF165-treated rat aortic rings ex vivo by TTAC-0001 (1 and 20 μg) or hIgG antibody (20 μg, negative control). Aortic rings of each group were photographed and representative images are shown. (b) Anti-angiogenic activity of TTAC-0001 was determined using matrigels-implanted in C57/BL6 mice. Matrigels containing PBS alone (control), hVEGF165 (100 ng) or hVEGF165 (100 ng) plus TTAC-0001 or 6C1 antibody (200 µg) were implanted via subcutaneous injection in C57/BL6 mice. Matrigels were removed and photographed after 7 days. 4 replicates are shown.
To examine whether TTAC-0001 could inhibit VEGF-mediated neovascularization in vivo, a matrigel-implanted model in nude mice was used with or without TTAC-0001 in the presence of 100 ng hVEGF165 (Fig. 6b). There was no distinguishable change of color with matrigel alone, whereas the addition of hVEGF165 to matrigel implants showed dark red color, indicating assembly of red blood cells and neovascularization. However, TTAC-0001 treatment resulted in inhibition of neovascularization based on the color of matrigel. The color intensity looks similar to that of the phosphate-buffered saline (PBS)-treatment control in 3 of 4 matrigels. The negative control antibody, 6C1, showed almost no inhibition of VEGF-induced neovascularization. The vessel content inside the plugs was also examined to validate the anti-angiogenic effect of TTAC-0001 (Fig. S3). The sections of matrigel plugs were immunostained with an antibody against the endothelial cell marker CD31. TTAC-0001-treated sample showed a staining pattern that was similar to the VEGF-untreated section. These results indicated that TTAC-0001 could inhibit hVEGF165-induced neovascularization and neutralize Flk-1 in vivo or ex vivo.
Discussion
Various angiogenesis inhibitors targeting the VEGF pathway, such as bevacizumab (Avastin®, Genentech/Roche), sorafenib (Nexavar®, Bayer) and sunitinib (Sutent®, Pfizer) have been developed. In particular, the approval of bevacizumab for first-line metastatic colorectal cancer therapy and other indications has demonstrated the potential benefit of targeting the VEGF/VEGFR signaling pathway. However, even though bevacizumab is clinically validated in various cancers, there may be potential benefits in developing therapeutic antibodies targeting VEGFR. The inhibition of VEGF signaling at the receptor level by a therapeutic antibody may be an efficient approach in many cancers because many solid tumors overexpress VEGF and VEGFR-2 for their survivals. Therefore, blocking VEGFR-2 with a monoclonal antibody may result in both antitumor and anti-angiogenic effects by blocking autocrine, as well as paracrine, signaling through VEGF.5,8 Clinical studies of ramucirumab have demonstrated that it was well tolerated and showed manageable adverse effects.27-28
Here, we demonstrated that TTAC-0001, a monoclonal antibody targeting VEGFR-2/KDR, strongly blocked multiple functional angiogenesis processes in vitro, ex vivo and in vivo. VEGF-mediated proliferation and migration of HUVECs was inhibited by TTAC-0001 at approximately the same concentration that inhibited phosphorylation of VEGFR-2/KDR and ERK. Established functional angiogenesis assays were chosen to demonstrate the potent anti-angiogenic activity of TTAC-0001. The rat aortic ring model recapitulates the entire process of angiogenesis and is known to be regulated by VEGF pathways. We used this model to demonstrate that TTAC-0001 inhibits endothelial cell sprouting and microvessel formation from rat aortic ring segments. In addition, VEGF-mediated vascularization of a subcutaneously implanted matrigel plug in mice was inhibited by TTAC-0001.
We examined the cross-reactivity of TTAC-0001 to Flk-1 to confirm TTAC-0001 targets VEGFR-2 on the host endothelial cells for showing its anti-angiogenic effect. In this study, we observed that TTAC-0001showed positive staining in mouse endothelial cells (bEND.3 cells) and endothelial cells from mouse aorta. This data supports that TTAC-0001 binds to Flk-1 in mouse vessels, and inhibits VEGF-mediated signaling, angiogenesis and tumor growth in mouse xenograft or orthotopic models. Additional IHC showed that endothelial cells of abdominal veins tissues from mouse, rat and cynomolgus were positive by TTAC-0001 (data not shown).
As determined by peptide microarray, the epitope is highly conserved among human, monkey and murine species. The epitope in the critical domain, Ig domain 3, is identical for those species, and another epitope in the N-terminal of Ig domain 2 is highly similar for those species. Domain 3 is known to be an important domain in binding VEGF to KDR. This epitope is conserved from human to mouse. However, there is a substitution of valine (human) to isoleucine (murine) in the epitope of domain 2. Valine and isoleucine has β-branched carbon and aliphatic side chain. Both amino acids have a strong tendency to break α-helix and form β-strand. Thus, substitution of valine to isoleucine is the most frequent homologous substitution in the protein database.38 Even though it can be a homologous substitution, this substitution can cause changes in substrate binding or activity to some extent. Even if it can decrease binding affinity to mouse VEGFR2, the more important aspect is that TTAC-0001 can block the binding of VEGF to mouse VEGFR2, as we can see in the in vivo mouse matrigel study or ex vivo rat aorta ring vessel sprouting assay. This suggests that TTAC-0001 can bind and block mouse VEGFR2 signal even though it binds with lower affinity to mouse VEGFR2 compared to human VEGFR2. Thus, it seems likely that binding to murine VEGFR-2 occurs, and ICH provided evidence of such binding. Direct binding to murine VEGFR-2 was not observed using FACS or SPR, though direct binding to human VEGFR-2 was observed by FACS (data not shown).
Further, we investigated the binding of TTAC-0001 to the assay reagents. It did not bind to hVEGFR-1 or hVEGFR-3 even in the SPR assay (Fig. 3b). We also found that the antibody did not bind to VEGF isoforms, hVEGF-A, hVEGF-C, and hVEGF-D, the exogenous agent in the aorta assay and matrigel assay (Fig. 3a). Thus, this hVEGFR-2 specific antibody (TTAC-0001), which binds to highly similar epitopes from different species, can bind to mouse VEGFR-2 and inhibit angiogenesis.
However, we did not observe a positive binding result from purified Flk-1. Recent reports give some clues regarding this phenomenon. The structure of VEGFR-2 revealed by electron microscope showed that it does not have well-defined structure, suggesting a complex forms when VEGFR-2 binds with VEGF.39 The flexibility of the VEGFR-2 could block the binding of TTAC-0001 to Flk-1 in vitro since conformational changes in the antigen might distort the binding pattern. The patterns of flexibility between mouse and human VEGFR-2 also differ. Globplot, a protein disorder prediction program, predicted that the domain 3 (ECD3), one of major binding regions of KDR by VEGF, was disordered in the N- and C-terminal regions, whereas the ECD3 of Flk-1 was disordered in the central region spanning residues 259–271 (data not shown).40 These regions have been reported to be important for neutralizing VEGFR-2 activity by antibodies.9 Thus, the inconsistency between the in vivo and in vitro assays might be caused by the different flexibility trends of the molecules.
In the dose escalation study in mice, the pharmacokinetics of TTAC-0001 showed a linear trend (data not shown). The antitumor activities of TTAC-0001 were also measured with various mouse models such as glioblastoma, lung cancer and colorectal tumors.41 A dose escalation study in the glioblastoma model showed that tumor growth was inhibited in a TTAC-0001 dose-dependent manner.41 These animal studies showed that the tumor growth inhibition by TTAC-0001 is caused by specific inhibition of mouse VEGFR-2 in the mouse vessel.
Based on regulatory guidance, we can use one relevant animal species for the evaluation of safety if only one species is cross-reactive, even though 2 relevant species, murine and non-murine species, are recommended. Thus, lack of direct biochemical binding to murine VEGFR-2 is not a hurdle to starting clinical study. We used mouse and cynomolgus for non-clinical study, even though the cynomolgus study was mainly used to determine the strategy for clinical study in humans. The pharmacokinetic parameters of both species showed similar tendencies. The whole body exposure rate was linearly dependent on the level of dose, and the 2 animal species showed similar safety profiles after TTAC-0001 injection.
The safety of TTAC-0001 was investigated with mouse and cynomolgus (data not shown). Cynomolgus was used as s toxicologically relevant species for TTAC-0001 because TTAC-0001 can positively stain endothelium of abdominal veins of cynomolgus. It can also stain renal endothelium in glomerular and renal epithelial cells, which are known to express VEGFR2.42 (Fig. S4). Furthermore, the amino acid sequences corresponding to epitope of TTAC-0001 are identical between human and monkey (Table S1). The doses for both species were 10 mg/kg, 30 mg/kg and 50 mg/kg at weekly schedule. The toxicological study showed that there was no observed adverse effect level (NOAEL) up to 50 mg/kg at both species. Thus, we have determined NOAEL to be 50 mg/kg. This value corresponds to 16 mg/kg for the human equivalent dose (HED) considering body surface area. With a safety factor of 16, we have set the first human dose level as 1 mg/kg. The safety of TTAC-0001 is currently under investigation in a Phase 1 study in Korea.
Materials and Methods
Cells and reagents
HUVEC was purchased from Lonza. A549 and U87MG cancer cell lines were purchased from ATCC. The Fc fused VEGFRs containing the entire extracellular region were purchased from R&D systems. Recombinant human VEGF121, VEGF165, VEGF-C, VEGF-D and KDR (ECD1–7)-Fc were also purchased from R&D systems. M199 medium for the cultivation of HUVEC was purchased from Invitrogen. The vector used for the expression of KDR (ECD1–3)-Fc, anti-KDR antibodies was pCDNA3.1 (Lifetech) derived pIgG-LD,HD vector, which containing DHFR and IRES elements.34
Protein expression and purification
Cell lines expressing recombinant KDR (ECD 1–3)-Fc were constructed by transfecting the indicated expression vector into dihydrofolate reductase (dhfr)-deficient CHO cells (CHO-DG44) and subsequent gene amplification was performed by the addition of methotrexate as previously described.43 The expressed protein was purified by sequential application of the sample to protein-A, Q-Sepharose, and SP-Sepharose (GE Healthcare) column followed by dialysis against PBS. The expression and purification of the IgG forms of the antibodies were carried out using the same methods applied for KDR (ECD 1–3)-Fc production mentioned above.
Affinity measurement by SPR
The affinities of the anti-KDR antibodies on hVEGFR-2/KDR were measured using a Biacore 3000 (GE Healthcare). KDR (ECD 1–3)-Fc containing 1–327 amino acids of human VEGFR2 (extracellular domain 1–3 of human VEGFR2) was coated to a CM5 chip according to the manufacturer's instruction and anti-KDR antibodies in IgG format were used as the analyte. The TTAC-0001 antibody was diluted and injected to the antigen-coated chip at a concentration of 0.7 nM, 1.4 nM, 2.8 nM, 5.6 nM, 11 nM, 22 nM and 44 nM. The 6C1 was also diluted to a concentration of 25 nM, 50 nM, 100 nM and 200 nM, while 6G1 was diluted to 21.25 nM, 42.5 nM, 85 nM and 170 nM. The flow rate of analyte was 30 μl/min. Kinetic parameters were determined with BIAEvaluation fitting in a condition of less than 10 of Chi square. Langmuir model was used for the binding analysis.
In the case of the hVEGFRs binding tests, hVEGFR1-Fc, hVEGFR2-Fc and hVEGFR3-Fc with entire extracellular domains were coated to CM5 chips at the level of 2,000 RU. 50 nM of TTAC-0001 was injected as an analyte at a flow rate of 30 μl/min.
Competitive inhibition of anti-KDR antibodies in binding VEGFs to VEGFR-2
For competitive VEGF/KDR blocking assays, various amounts of anti-KDR antibodies in the form of phage or IgG were mixed with a fixed amount of KDR (ECD1–3)-Fc (100 ng) or KDR (ECD1–7)-Fc (200 ng, R&D systems) and incubated at RT for 1 hour. The mixture was then transferred to 96-well plates pre-coated with recombinant human VEGFs (hVEGF165, hVEGF-C, hVEGF-D; 200 ng/well, R&D Systems), incubated for an additional 100 min and then washed with DPBS. To block non-specific binding, the plates were washed with PBS containing 3% bovine serum albumin (BSA) for 2 hours at RT. To measure the amount of KDR bound to VEGFs, mouse monoclonal anti-KDR antibody (2 µg/ml, Reliatech) was treated for 1 hour and then, HRP-conjugated anti-mouse antibody (secondary antibody) was added. After 1 hour incubation with secondary antibody, TMB solution was added for color development and absorbance (450–650 nm) was measured using ELISA reader (Tecan).
Domain mapping assay of anti-KDR antibodies
In order to examine which domain of KDR binds to the anti-KDR antibodies, KDR extracellular domain (ECD) fragments were expressed (ECD 1–3, ECD 1–2, ECD 2–3) as a Fc fused form. The Fc fused ECDs were coated into a 96-well plate by incubation at 37 °C for 2 hours. The plate was then washed with PBS and blocked with 2% skim milk/PBS. After washing the plate again with PBS, 330 nM of anti-KDR scFv antibodies were added and allowed it to react at 37 °C for 1.5 hours. Then, the plate was washed again with PBS followed by addition of 1/5000 diluted HRP-conjugated anti-6XHis antibody (abcam, UK). After 1 hour incubation at 37 °C, it was developed with TMB solution and measured absorbance at 450 nm.
Peptide microarray of epitope mapping
Peptide microarray for epitope mapping was done by Abnova (Taipei city, Taiwan). From the amino acid sequence of extracellular domain of VEGFR-2/KDR, 13meric amino acids and overlapping 11 amino acids were synthesized, resulting in a total of 149 peptides. The determination of peptide-antibody binding was performed by PepStar-analysis where the peptide microarray was incubated with TTAC-0001 or control human IgG (Pierce #31154) followed by a Dylight-649 labeled secondary antibody (Pierce #35515) directed against the Fc-part of the primary one. All steps were performed on a TECAN microarray processing station (TECAN HS4800), which enabled highly reliable and reproducible washing and incubation steps. After performing the incubation steps and subsequent to the final washing steps (to remove the secondary antibodies), the microarrays were dried using a nitrogen stream and scanned in a high resolution microarray scanning system with appropriate wavelength settings. After scanning the dry and fluorescently labeled microarray slides, the scanner records a 16-bit numeric image in tagged image file format (*.tif). This tif-image is evaluated using GenepixPro 6.0 software enabling interpretation and quantification of gray scales representing the data of each fluorescent spot on the scanned microarray slide.
Binding ELISA of TTAC-0001 to VEGFs
VEGF165, VEGF-C and VEGF-D were coated in a 96-well plate at a concentration of 100 ng/well. After washing the plate 3 times with PBS containing 0.05% (v/v) Triton X-100 (PBS-T), the plate was blocked with PBS-T containing 0.5% BSA by incubating at 37 °C for 1 hour. After washing out the blocking solution, 150 μg or 500 μg of hIgG, TTAC-0001 and bevacizumab (anti-VEGF antibody) were added. Then, the plate was washed 3 times with PBS-T, and HRP-conjugated goat anti-hIgG(Fc) was added to the plate. After 1 hour of incubation, TMB solution was added for color development and absorbance (450–650nm) was measured using ELISA reader (Tecan).
VEGFR-2/KDR phosphorylation inhibition by TTAC-0001
Confluent HUVECs were incubated for 6 hours in M199 containing 1% FBS, and cells were stimulated by the addition of hVEGF165 with or without TTAC-0001, anti-KDR neutralizing antibody. After stimulation, cells were lysed in 1 ml of lysis buffer (20 mM of Tris/HCl pH 8.0, 2 mM of EDTA, 137 mM of NaCl, 1 mM of Na3VO4, 1 mM of phenylmethylsulfonyl fluoride, 10% glycerol, and 1% Triton X-100). Lysates were clarified by centrifugation at 15,000×g for 10 min, and the resulting supernatants were immunoprecipitated with 1 µg/ml anti-KDR/Flk-1 antibody, (Santa Cruz Biotechnology) at 4°C for 3 hours, followed by the addition of protein A-agarose beads (Upstate, Lake Placid, NY) at 4°C for 1 hour. Immunoprecipitates were washed 3 times with lysis buffer, and solubilized in SDS-PAGE sample buffer containing β-mercaptoethanol, and further analyzed by Western blotting.
Endothelial cell proliferation assay
HUVECs were seeded at a density of 2 × 104 cells/well in 24-well tissue culture plates (Sarstedt). After 24 hours, cells were washed twice with phenol red-free M199 (Invitrogen) and incubated for 6 hours in phenol red-free M199 containing 1% FBS. Cells were preincubated for 30 min with various concentrations of antibodies and stimulated for 48h by the addition of 10 ng/ml VEGF (R&D systems #293-VE-101). Plates were incubated for 2 hours at 37 °C, and following the addition of 1/10 volume of WST-8 (Dojindo, Japan), absorbance at 450 nm was measured using a microplate reader (Tecan sunrise 96PW).
Endothelial cell migration assays
Chemotactic motility of HUVECs was assayed using Transwell (Corning Costar) with 6.5-mm diameter polycarbonate filters (8-µm pore size). Briefly, the lower surface of the filter was coated with 10 µl of 0.1% gelatin solution (Sigma) and dried at room temperature for 30 min. The 600 µl of serum-starved M199 medium (1% FBS) containing 1 mg/ml BSA (MP Biomedicals) and 10 ng/ml VEGF were replaced in the lower wells. Cells were trypsinized and suspended at a final concentration of 1 × 106 cells/ml in serum-starved M199 medium. 20 μg/ml of TTAC-0001 or 1X PBS were given to the cells for 30 min at room temperature before seeding. One hundred µl of the cell suspension (1 × 105 cells) was loaded into each of the upper wells. After the chamber was incubated at 37°C for 3.5 hours, cells were stained with crystal violet and counted. Briefly, cells were rinsed with PBS, fixed with ethanol, stained with crystal violet solution (0.5% crystal violet, 50% ethanol, 5% formaldehyde, 1xPBS), and then rinsed with distilled water. Non-migrating cells on the upper surface of the filter were removed by wiping with a cotton swab, and dehydrated filters were observed and photographs were taken under a light microscope (Olympus 1×71 microscope with DP71/TH4-200 and DP controller ver.3.11.267). Chemotaxis was quantified by counting the cells that migrated to the lower side of the filter. Ten fields were counted for each assay.
In vitro tube formation in Matrigel
Matrigel matrix (BD Biosciences) was thawed for overnight at 4°C and kept on ice before use. For assaying tube formation of HUVECs in Matrigel, cells were starved for 6 hours and harvested by trypsin treatment. After centrifugation at 1,000 rpm, the precipitated HUVECs were re-suspended with culture medium harboring hVEGF165 (10 ng/ml) at 2×105 cells/ml. Meanwhile, using cooled pipets, 250 μl of the Matrigel was added onto 24-well culture plate on ice and then the plate was incubated at 37°C for 30 min before use. TTAC-0001 (TTAC-0001; 5,10,20 µg) and control antibody (6C1, 20 µg) were added into each well of the plate and then 300 μl of the cell mixtures (6×104 cells of HUVEC) were also added into the same wells. After 20 hours of incubation at 37°C with 5% CO2 atmosphere, the cultures were photographed (× 40).
Ex vivo rat aortic ring vessel sprouting assay
As described previously, aortas were harvested from 6-week-old Sprague Dawley rats.44 Plates (48-well) were coated with 120 µl of Matrigel; after gelling, the rings were placed in the wells and sealed in place with a 50 µl of Matrigel overlay. Recombinant human VEGF165 (10 ng/ml) was suspended in human endothelial serum-free medium (Invitrogen) to a final volume of 200 µ;l and transferred to each well with hIgG (20 μg) or TTAC-0001 (1 and 20 µ;g). On day 6, cells were fixed and stained with Diff-Quick.
In vivo Matrigel plug angiogenesis assay
The Matrigel plug assay was performed as previously described.45 Briefly, 6–8-week-old C57/BL6 mice were injected subcutaneously with 0.6 ml of matrigel containing TTAC-0001 (200 µ;g) or 6C1 (200 μg), hVEGF165 (100 ng), and heparin (10 units). The injected matrigel rapidly formed a single, solid gel plug. After 7 days, the skin of the mouse was easily pulled back to expose the matrigel plug, which remained intact. Matrigel plugs were removed and snap-frozen in liquid nitrogen in the presence of optimum cutting temperature (OCT) compound (Miles, Inc..) and sectioned. Four matrigels were tested for each group. Frozen matrigel sections of 8–12 µ;m from OCT plugs were fixed in 4% (v/v) neutral buffered paraformaldehyde and immunostained with anti-CD31 antibody for the microvessel density measurement.
Immunohistochemistry
To access the potential tissue cross-reactivity of TTAC-0001 (TTAC-0001), IHC study was performed in mouse aorta tissues and bEND.3 cells, brain endothelial cell from Balb/c mice. As a control, HUVECs and human glioblastoma (GBM) tumors were used.
Tissues were fixed in formalin-free IHC zinc fixative (BD Biosciences), embedded in paraffin and cut into 3-µm sections (Wax-it). Slides were deparaffinized in xylene and then hydrated through a graded alcohol series. Endogenous peroxidase in the sections was blocked with 3% (v/v) hydrogen peroxide. Sections were incubated with CAS-BLOCK (Zymed) for 30 min at RT to block non-specific protein binding followed by overnight incubation at 4°C with TTAC-0001. Sections were rinsed in PBS-T and incubated with biotinylated anti-human IgG antibody (1:400 dilution, BD Biosciences) for 1 hour at RT. After washing twice with PBS-T, slides were subsequently incubated with streptavidin-horseradish peroxidase (BD Biosciences). Diaminobenzidine/hydrogen peroxidase (Dako) was used as the chromogen substrate. All slides were counterstained with Meyer's hematoxylin.
Statistical analysis
The data were presented as means ± SD and statistical comparisons between groups were performed using 1-way ANOVA followed by the Student's t test.
Funding
This work was supported by a grant from the Korea Health Industry Development Institute/Ministry of Health and Welfare (grant #: A040016) and partly by a grant of the National R&D Program for Cancer Control, Ministry of Health & Welfare, Republic of Korea (grant #: 0720420).
Disclosure of Potential Conflicts of Interest
Weon Sup Lee, Sung-Woo Kim, Sang Ryeol Shim, Ju Ryoung Nam and Jin-San Yoo are employed by and stockholders of PharmAbcine. Do-Hyun Nam is an adviser and stockholder of PharmAbcine. Dong-Sup Lee is a stockholder of PharmAbcine.
Acknowledgments
We thank Dr. Jin-Sang Yoo, Dr. Joongkyu Kim and members of PharmAbcine for helpful scientific discussions and comments on the manuscript. The authors wish to acknowledge members of Samsung Medical Center for their contributions in immunohistochemistry analysis.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995; 1:27-31; PMID:7584949; http://dx.doi.org/ 10.1038/nm0195-27 [DOI] [PubMed] [Google Scholar]
- 2.Kerbel RS. Tumor angiogenesis: past, present and the near future. Carcinogenesis 2000; 21:505-15; PMID:10688871; http://dx.doi.org/ 10.1093/carcin/21.3.505 [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P. Angiogenesis in health and disease. Nat Med 2003; 9:653-60; PMID:12778163; http://dx.doi.org/ 10.1038/nm0603-653 [DOI] [PubMed] [Google Scholar]
- 4.Rafii S, Lyden D, Benezra R, Hattori K, Heissig B. Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy? Nat Rev Cancer 2002; 2:826-35; PMID:12415253; http://dx.doi.org/ 10.1038/nrc925 [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N. VEGF: an update on biological and therapeutic aspects. Curr Opin Biotechnol 2000; 11:617-24; PMID:11102799; http://dx.doi.org/ 10.1016/S0958-1669(00)00153-1 [DOI] [PubMed] [Google Scholar]
- 6.Ferrara N. Vascular endothelial growth factor and the regulation of angiogenesis. Recent Prog Horm Res 2000; 55:15-35; PMID:11036931 [PubMed] [Google Scholar]
- 7.Blume-Jensen P, Hunter T. Oncogenic kinase signaling. Nature 2001; 411:355-65; PMID:11357143; http://dx.doi.org/ 10.1038/35077225 [DOI] [PubMed] [Google Scholar]
- 8.Ferrara N. Vascular endothelial growth factor as a target for anticancer therapy. Oncologist 2004; 9 1:2-10; PMID:15178810; http://dx.doi.org/ 10.1634/theoncologist.9-suppl_1-2 [DOI] [PubMed] [Google Scholar]
- 9.Lu D, Kussie P, Pytowski B, Persaud K, Bohlen P, Witte L, Zhu Z. Identification of the residues in the extracellular region of KDR important for interaction with vascular endothelial growth factor and neutralizing anti-KDR antibodies. J Biol Chem 2000; 275:14321-30; PMID:10799512; http://dx.doi.org/ 10.1074/jbc.275.19.14321 [DOI] [PubMed] [Google Scholar]
- 10.Popkov M, Jendreyko N, Gonzalez-Sapienza G, Mage RG, Rader C, Barbas CF III. Human/mouse cross-reactive anti-VEGF receptor 2 recombinant antibodies selected from an immune b9 allotype rabbit antibody library. J Immunol Methods 2004; 288:149-64; PMID:15183093; http://dx.doi.org/ 10.1016/j.jim.2004.03.005 [DOI] [PubMed] [Google Scholar]
- 11.Dias S, Hattori K, Heissig B, Zhu Z, Wu Y, Witte L, Hicklin DJ, Tateno M, Bohlen P, Moore MA., et al.. Inhibition of both paracrine and autocrine VEGF/VEGFR-2 signaling pathways is essential to induce long-term remission of xenotransplanted human leukemias. Proc Natl Acad Sci U S A 2001; 98:10857-62; PMID:11553814; http://dx.doi.org/ 10.1073/pnas.191117498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aesoy R, Sanchez BC, Norum JH, Lewensohn R, Viktorsson K, Linderholm B. An autocrine VEGF/VEGFR-2 and p38 signaling loop confers resistance to 4-hydroxytamoxifen in MCF-7 breast cancer cells. Mol Cancer Res 2008; 6:1630-8; PMID:18922978; http://dx.doi.org/ 10.1158/1541-7786.MCR-07-2172 [DOI] [PubMed] [Google Scholar]
- 13.Sher I, Adham SA, Petrik J, Coomber BL. Autocrine VEGF-A/KDR loop protects epithelial ovarian carcinoma cells from anoikis. Intl J Cancer 2009; 124:553-61; PMID:9004006; http://dx.doi.org/ 10.1002/ijc.23963 [DOI] [PubMed] [Google Scholar]
- 14.Prewett M, Huber J, Li Y, Santiago A, O'Connor W, King K, Overholser J, Hooper A, Pytowski B, Witte L., et al.. Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res 1999; 59:5209-18; PMID:10537299 [PubMed] [Google Scholar]
- 15.Millauer B, Shawver LK, Plate KH, Risaui W, Ullrich A. Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature 1994; 367:576-9; PMID:8107827; http://dx.doi.org/ 10.1038/367576a0 [DOI] [PubMed] [Google Scholar]
- 16.Kuo CJ, Farnebo F, Yu EY, Christofferson R, Swearingen RA, Carter R, von Recum HA, Yuan J, Kamihara J, Flynn E., et al.. Comparative evaluation of the antitumor activity of antiangiogenic proteins delivered by gene transfer. Proc Natl Acad Sci U S A 2001; 98: 4605-10; PMID:11274374; http://dx.doi.org/ 10.1073/pnas.081615298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saleh M, Stacker SA, Wilks AF. Inhibition of growth of C6 glioma cells in vivo by expression of antisense vascular endothelial growth factor sequence. Cancer Res 1996; 56:393-401; PMID:8542597 [PubMed] [Google Scholar]
- 18.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995; 376:66-70; PMID:7596436; http://dx.doi.org/ 10.1038/376066a0 [DOI] [PubMed] [Google Scholar]
- 19.Ciardiello F, Caputo R, Damiano V, Caputo R, Troiani T, Vitagliano D, Carlomagno F, Veneziani BM, Fontanini G, Bianco AR., et al.. Antitumor effects of ZD6474, a small molecule vascular endothelial growth factor receptor tyrosine kinase inhibitor, with additional activity against epidermal growth factor receptor tyrosine kinase. Clin Cancer Res 2003; 9:1546-56; PMID:12684431 [PubMed] [Google Scholar]
- 20.Ciardiello F, Bianco R, Caputo R, Caputo R, Damiano V, Troiani T, Melisi D, De Vita F, De Placido S, Bianco AR., et al.. Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin Cancer Res 2004; 10: 784-93; PMID:14760102; http://dx.doi.org/ 10.1158/1078-0432.CCR-1100-03 [DOI] [PubMed] [Google Scholar]
- 21.McMahon G. VEGF receptor signaling in tumor angiogenesis. Oncologist 2000; 5 1:3-10; PMID:10804084; http://dx.doi.org/ 10.1634/theoncologist.5-suppl_1-3 [DOI] [PubMed] [Google Scholar]
- 22.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E., et al.. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350:2335-42; PMID:15175435; http://dx.doi.org/ 10.1056/NEJMoa032691 [DOI] [PubMed] [Google Scholar]
- 23.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003; 9:669-76; PMID:12778165; http://dx.doi.org/ 10.1038/nm0603-669 [DOI] [PubMed] [Google Scholar]
- 24.Ferrara N, Hillan K, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 2004; 3:391-400; PMID:15136787; http://dx.doi.org/ 10.1038/nrd1381 [DOI] [PubMed] [Google Scholar]
- 25.Keren P, Zhu Z. Development of angiogenesis inhibitors to vascular endothelial growth factor receptor 2. current status and future perspective. Front Biosci 2005; 10:1415-39; PMID:15769635; http://dx.doi.org/ 10.2741/1629 [DOI] [PubMed] [Google Scholar]
- 26.Lee SH. TTAC-0001 (TTAC-0001): a fully human monoclonal antibody targets vascular endothelial growth factor receptor 2 (VEGFR-2). Arch Pharm Res 2011; 34:1223-6; PMID:21910042; http://dx.doi.org/ 10.1007/s12272-011-0821-9 [DOI] [PubMed] [Google Scholar]
- 27.Spratlin JL. Ramucirumab (IMC-1121B): monoclonal antibody inhibition of vascular endothelial growth factor receptor-2. Curr Oncol Rep 2011; 13:97-102; PMID:21222245; http://dx.doi.org/ 10.1007/s11912-010-0149-5 [DOI] [PubMed] [Google Scholar]
- 28.Spratlin JL, Mulder KE, Mackey JR. Ramucirumab (IMC-1121B): a novel attack on angiogenesis. Future Oncol 2010; 6:1085-94; PMID:20624120; http://dx.doi.org/ 10.2217/fon.10.75 [DOI] [PubMed] [Google Scholar]
- 29.Kunkel P, Ulbricht U, Bohlen P, Brockmann MA, Fillbrandt R, Stavrou D, Westphal M, Lamszus K. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res 2001; 61:6624-8; PMID:11559524 [PubMed] [Google Scholar]
- 30.Witte L, Hicklin DJ, Zhu Z, Pytowski B, Kotanides H, Rockwell P, Böhlen P. Monoclonal antibodies targeting the VEGF receptor-2 (Flk1/KDR) as an anti-angiogenic therapeutic strategy. Cancer Metastases Rev 1998; 17:155-61; PMID:9770111; http://dx.doi.org/ 10.1023/A:1006094117427 [DOI] [PubMed] [Google Scholar]
- 31.Zhu Z, Hattori K, Zhang H, Jimenez X, Ludwig DL, Dias S, Kussie P, Koo H, Kim HJ, Lu D., et al.. Inhibition of human leukemia in an animal model with human antibodies directed against vascular endothelial growth factor receptor 2. correlation between antibody affinity and biological activity. Leukemia 2003; 17:604-11; PMID:12646950; http://dx.doi.org/ 10.1038/sj.leu.2402831 [DOI] [PubMed] [Google Scholar]
- 32.Lu D, Jimenez X, Zhang H, Bohlen P, Witte L, Zhu Z. Selection of high affinity human neutralizing antibodies to VEGFR-2 from a large antibody phage display library for antiangiogenesis therapy. Intl J Cancer 2002; 97:393-9; PMID:11774295; http://dx.doi.org/ 10.1002/ijc.1634 [DOI] [PubMed] [Google Scholar]
- 33.Burmester J. Pluckthu A Construction of scFv Fragments from Hybridoma or Spleen Cells by PCR Assembly. In: Kontermann R., Dubel S (eds), Antibody Engineering. Berlin: Springer; 2001; 19-40 [Google Scholar]
- 34.Yoo JS, Lee WS, Shim SR, Park MI, Kang JE, Kim DY, Lee JC, Lee DH, Cho DY, Sul SS et al.. Korea Research Institute of Bioscience and Biotechnology Human Monoclonal Antibody Neutralizing Vascular Endothelial Growth Factor Receptor and Use Thereof. Korea Patent, KR, 10-2007-00057719, 2007, 13 [Google Scholar]
- 35.Benest AV, Harper SJ, Ylä-Herttuala SY, Alitalo K, Bates DO. VEGF-C induced angiogenesis preferentially occurs at a distance from lymphangiogenesis. Cardiovasc Res 2008; 78:315-23; PMID:18065770; http://dx.doi.org/ 10.1093/cvr/cvm094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lohela M, Heloterä H, Haiko P, Dumont DJ, Alitalo K. Transgenic induction of vascular endothelial growth factor-C is strongly angiogenic in mouse embryos but leads to persistent lymphatic hyperplasia in adult tissues. Am J Pathol, 2008; 173:1891-901; PMID:18988807; http://dx.doi.org/ 10.2353/ajpath.2008.080378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anisimov A, Alitalo A, Korpisalo P, Soronen J, Kaijalainen S, Leppanen VM, Jeltsch M, Yla-Herttuala S, Alitalo K. Activated forms of VEGF-C and VEGF-D provide improved vascular function in skeletal muscle. Circ Res, 2009; 104:1302-12; PMID:19443835; http://dx.doi.org/ 10.1161/CIRCRESAHA.109.197830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ladunga I., Smith R.F. Amino acid substitutions preserve protein folding by conserving steric and hydrophobicity properties. Protein Eng 1997; 10:187-96; PMID:9153083; http://dx.doi.org/ 10.1093/protein/10.3.187 [DOI] [PubMed] [Google Scholar]
- 39.Ruch C, Skiniotis G, Steinmetz MO, Walz T, Ballmer-Hofer K. Structure of a VEGF-VEGF receptor complex determined by electron microscopy. Nature Struct Mol Biol 2007; 14:249-50; PMID:17293873; http://dx.doi.org/ 10.1038/nsmb1202 [DOI] [PubMed] [Google Scholar]
- 40.Linding R, Russell RB, Neduva V, Gibson TJ. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res 2003; 31:3701-8; PMID:12824398; http://dx.doi.org/ 10.1093/nar/gkg519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim DG, Jin YG, Jin JY, Yang HY, Joo KM, Lee WS, Shim SR, Kim SW, Yoo JS, Lee SH, et al., Anticancer activity of TTAC-0001, a fully human anti-vascular endothelial growth factor receptor 2 (VEGFR-2/KDR) monoclonal antibody, is associated with inhibition of tumor angiogenesis. 2015. Unpublished manuscript. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanellis J, Fraser S, Katerelos M, Power DA. Vascular endothelial growth factor is a survival factor for renal tubular epithelial cells. Am J Physiol Renal Physiol 2000; 278:F905-915; PMID:10836978 [DOI] [PubMed] [Google Scholar]
- 43.Hwang SJ, Choi HH, Kim KT, Hong HJ, Koh GY, Lee GM. Expression and purification of recombinant human angiopoietin-2 produced in chinese hamster ovary cells. Protein Exp Purif 2005; 39:175-83; PMID:15642468; http://dx.doi.org/ 10.1016/j.pep.2004.09.005 [DOI] [PubMed] [Google Scholar]
- 44.Nicosia RF, Ottinetti A. Modulation of microvascular growth and morphogenesis by reconstituted basement membrane gel in three-dimensional cultures of rat aorta: a comparative study of angiogenesis in matrigel, collagen, fibrin, and plasma clot. In Vitro Cell Dev Biol 1990; 26:119-28; PMID:1690206; http://dx.doi.org/ 10.1007/BF02624102 [DOI] [PubMed] [Google Scholar]
- 45.Min JK, Han KY, Kim EC, Kim YM, Lee SW, Kim OH, Kim KW, Gho YS, Kwon YG. Capsaicin inhibits in vitro and in vivo angiogenesis. Cancer Res 2004; 64:644-51; PMID:14744780; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-3250 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.