

Abstract
DNA methylation is a chemical modification of DNA involved in the regulation of gene expression by controlling the access to the DNA sequence. It is the most stable epigenetic mark and is widely studied for its role in major biological processes. Aberrant DNA methylation is observed in various pathologies, such as cancer. Therefore, there is a great interest in analyzing subtle changes in DNA methylation induced by biological processes or upon drug treatments. Here, we developed an improved methodology based on flow cytometry to measure variations of DNA methylation level in melanoma and leukemia cells. The accuracy of DNA methylation quantification was validated with LC-ESI mass spectrometry analysis. The new protocol was used to detect small variations of cytosine methylation occurring in individual cells during their cell cycle and those induced by the demethylating agent 5-aza-2'-deoxycytidine (5AzadC). Kinetic experiments confirmed that inheritance of DNA methylation occurs efficiently in S phase and revealed a short delay between DNA replication and completion of cytosine methylation. In addition, this study suggests that the uncoupling of 5AzadC effects on DNA demethylation and cell proliferation might be related to the duration of the DNA replication phase.
Keywords: cancer, cell cycle, DNA methylation, flow cytometry, 5-azadeoxycytidine
Abbreviations
- 5AzadC
5-aza-2′-deoxycytidine
- 5mC
5-methylcytosine
- DNMT
DNA methyltransferase
- CGI
CpG island
- MDS
myelodysplastic syndromes
- AML
acute myeloid leukemia
- FACS
fluorescence-activated cell sorting
- LC-ESI MS/MS
liquid chromatography–electrospray ionization tandem mass spectrometry
Introduction
In mammals, DNA methylation participates in the control of the expression of the genetic code and is the most studied epigenetic mark. It consists in the covalent addition of a methyl group on position 5 of the cytosine in the DNA to give 5-methylcytosine (5mC) and is catalyzed by the DNA methyltransferases (DNMTs) in the context of CpG dinucleotides. So far, 3 catalytically active DNMTs, DNMT1, DNMT3A and DNMT3B, have been identified.1,2 In somatic cells, most CpG dinucleotides are methylated, except those (1–2%) located in CpG-rich DNA sequences named CpG islands (CGIs).3-7 CGIs are found mostly in transcriptionally active DNA regions and about half of them coincide with promoters of annotated genes. Their methylation acts as a relatively stable gene silencing event by interfering with the binding of transcription factors or by recruiting additional silencing-associated proteins.8 CGIs are also found within gene bodies (intragenic) and between genes (intergenic) at alternative sites of transcriptional initiation.9,10
Failure in maintaining these epigenetic marks and the establishment of aberrant DNA methylation patterns are associated with under- or over-expression of coding and non-coding RNAs, ultimately leading to diverse pathologies, including neurodegenerative diseases and cancer.11-13 In cancer, the methylome is highly altered, showing global hypomethylation together with localized inappropriate hypermethylation of CGIs located on specific promoters, such as those of tumor suppressor genes.14–16 In addition, CGI shores are hot spots for hyper- and hypo-methylation.17
The reversibility of DNA methylation pointed out DNMTs as targets for anticancer therapies.18–21 To date, 2 cytosine analogs, 5-azacytidine (5AzaC) and 5-aza-2′-deoxycytidine (5AzadC), were shown to be potent DNMT inhibitors and were approved for the treatment of myelodysplastic syndromes (MDS), chronic myelomonocytic leukemia (CMML),22 and acute myeloid leukemia (AML).23 Their ability to reduce global methylation in vivo has been validated in multiple clinical trials24,25 but the link between the demethylation level and the clinical response remains to be understood.26 To this respect, methods combining the analysis of total DNA methylation and cell cycle are of interest for the characterization of the DNA methylation process in tumor cells, as well as the effects induced by DNA methylation inhibitors.
Several experimental strategies exist to follow gene-specific or genome-wide DNA methylation.27-29 However, few methods have been described to quantify the methylation changes in total DNA and to follow small variations.
The development of monoclonal antibodies specific for 5mC resulted in sensitive tools to quantify 5mC in genomic or fragmented DNA samples spotted on nitrocellulose paper or DEAE membranes,30,31 or in fluids of cancer patients, for the dosage of modified nucleosides by immunoassays.32 Commercial kits are now available to measure total DNA methylation by an ELISA-like reaction.33,34 Interestingly, immunolabeling of 5mC can allow the analysis of DNA methylation at the individual cellular level, and, when coupled to fluorescence microscopy,35-37 it gives access to the topology of DNA methylation in the nucleus at the chromosome level. When such information is not required, flow cytometry (FACS) analysis represents an alternative method to measure total DNA methylation in combination with the amount of genomic DNA, in each cell individually.38-44
Here, we developed an improved protocol based on flow cytometry to detect small variations of global DNA methylation in cancer cells, taking into account the concomitant modifications of the cell cycle phases. This new methodology was validated on 2 cell lines from melanoma and leukemia origin, displaying different pharmacological sensitivities to 5AzadC. Parallel quantification by flow cytometry and LC-ESI mass spectrometry (LC-ESI MS/MS) analysis validated the first and showed that flow cytometry can be used to quantify small variations of 5mC. This accurate and reliable approach was used to analyze the coupling between DNA replication and DNA methylation maintenance, by combining the measurement of 5mC content and cell cycle status. This also allowed studying the early kinetics of DNA demethylation after drug treatment.
Results
Analysis of the methylcytosine content by flow cytometry
The experimental conditions of 5mC measurement by flow cytometry were optimized on melanoma cell line WM266–4 (Fig. 1). Cells were labeled with anti-5mC monoclonal antibody followed by a secondary antibody conjugated to a fluorescent probe. The fluorophore Alexa-Fluor 647 was selected for its brightness. Then, DNA was stained with propidium iodide (PI) to measure DNA content and assess cell cycle status of the population. After titration of the commercial antibodies (Supplementary Figure S1A), we routinely used a non-saturating concentration of anti-5mC antibody. Consequently, the intensities of the 5mC labeling varied with the number of cells (Supplementary Fig. S1B). We thus used identical amounts of cells for each sample.
Figure 1.

Analysis of 5-methylcytosine (5mC) content in WM266–4 cells by flow cytometry. Asynchronous WM266–4 melanoma cells were labeled with anti-5mC monoclonal antibody prior to DNA staining with propidium iodide (PI). For flow cytometry analysis, cells were selected according to their FSC and SSC parameters (R1 region) (A) and then gated on their PI content (R2 region) (B). 5mC labeling of the R2 cells (gray histogram) and its isotypic control (black histogram) are displayed on a fluorescence histogram with a linear scale (C). Analysis of cell cycle (D) is combined with analysis of DNA methylation, as shown on the dot plot in (E). The graph (F) reports the mean of fluorescence intensities of 5mC (anti-5mC antibody mfi minus isotype control mfi) and PI measured in contiguous intervals (5000 mfi units) on the PI scale.
Flow cytometry analysis was performed on cells selected according to their FSC and SSC parameters (R1 region) to exclude cell debris (Fig. 1A) and then gated on their PI content (R2 region) to exclude cell doublets, aggregates, and apoptotic cells (Fig. 1B). 5mC and PI fluorescence intensities of the gated cells (R2 region) are reported as histograms in Figures 1C and 1D, respectively. Dot-plots display DNA methylation according to the DNA content (Fig. 1E). As shown in Figure 1F, the means of fluorescence intensities (mfis) of 5mC in asynchronous and non-treated cells were linearly related to the mfis of PI. In order to exclude a potential artifact signal due to FRET events between PI and Alexa-Fluor 647 molecules, the same experiment was performed using Alexa-Fluor 405 for the secondary antibody. As expected, labeling with Alexa-Fluor 405 was less intense than with Alexa-Fluor 647 but confirmed the linearity between 5mC and DNA content (Supplementary Fig. S2).
Technical variation in 5mC measurements (means of fluorescence intensities on the R2 region) was estimated in 2 independent experiments performed as triplicates on a single sample and was lower than 8% (data not shown).
Total DNA methylation measurements in melanoma and leukemia cells
This flow cytometry method was used to compare total DNA methylation in 2 cell lines with different DNA content by measuring the 5mC- and DNA-related fluorescence in their respective G0/G1 phases. Melanoma WM266–4 and leukemia KG1 cells were grown as an adherent layer and as a cell suspension, respectively. As shown in Figure 2A, WM266–4 cells contain 1.7-fold more DNA than KG1 cells. In contrast, WM266–4 5mC content is lower than that of KG1 cells (Fig. 2B). In order to compare the density of 5mC on the DNA of each cell line, taking into account their difference in DNA content, 5mC mfis were combined with DNA mfis to calculate a 5mC/DNA index. As shown on Figure 2C, this index is 2.3-fold higher in KG1 cells than in WM266–4 cells. Total 5mC was measured in parallel by LC-ESI MS/MS using genomic DNA from both cell lines (Fig. 2D). The ratio between the percentages of 5mC in each cell line (13.5% and 5.9%, for KG1 and WM266–4 cells, respectively) was also 2.3, in perfect agreement with the flow cytometry analysis (Fig. 2C).
Figure 2.
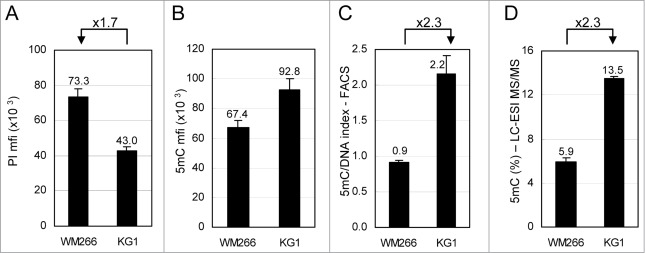
Comparative analysis of total 5-methylcytosine (5mC) content in cell lines by flow cytometry. WM266–4 melanoma and KG1 leukemia cells were labeled with anti-5mC monoclonal antibody prior to DNA staining with propidium iodide (PI). (A). DNA contents in G0/G1 cells were expressed as PI mfis. (B). 5mC contents in G0/G1 cells were expressed as in Figure 1F (C). 5mC/DNA indexes were calculated as ratios of 5mC mfis vs. PI mfis. (D). Percentages of 5mC among total cytosines were measured by LC-ESI MS/MS.
5AzadC-induced DNA demethylation in melanoma and leukemia cells
The FACS analysis was challenged to detect variations in DNA methylation content in cells upon treatment with DNA demethylating agents (Fig. 3). Melanoma WM266–4 cells and leukemia KG1 cells were daily treated with 5AzadC for 3 days. 5mC content was measured as described above and, in parallel, by LC-ESI MS/MS. As shown in Figure 3A, 5AzadC had a moderate effect on the proliferation of WM266–4 melanoma cells at doses ranging from 0.032 μM to 1 μM. These 5AzadC concentrations induced a decrease in 5mC signal that was detected both by FACS (Fig. 3C) and by LC-ESI MS/MS (Fig. 3E), with a significant decrease starting at the lowest 5AzadC concentration (0.032 μM) and a maximal demethylation that plateaued at concentrations above 1 μM. The 5mC content decreased to 75.4% ± 4.4% upon treatment with 3.2 μM 5AzadC, compared to untreated cells, as calculated from data by FACS, and to 50.5% ± 3.3% at 3.2 μM 5AzadC, as calculated by LC-ESI MS/MS.
Figure 3.
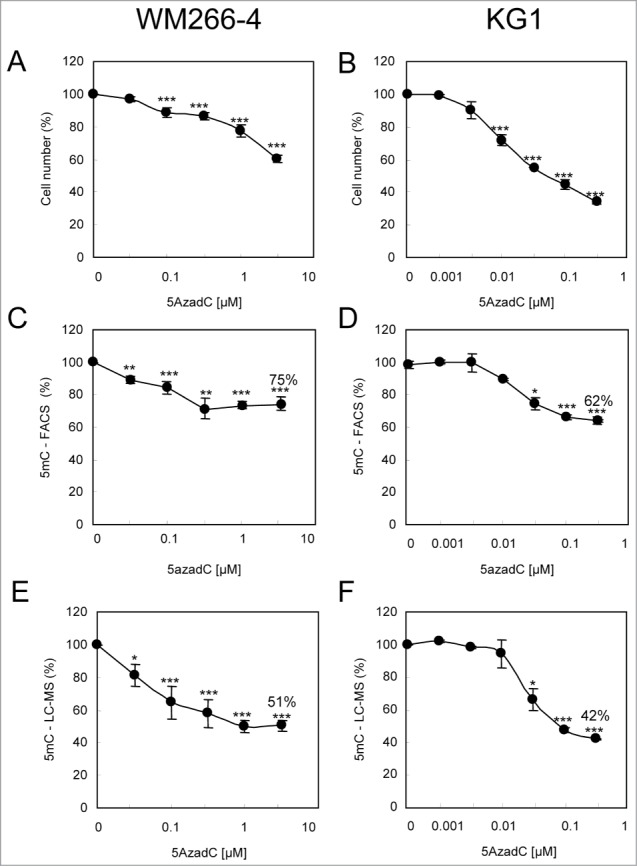
Variations of 5mC content in WM266–4 and KG1 cells upon 5AzadC treatment. WM266–4 cells (A, C, E) and KG1 cells (B, D, F) were seeded at day 0 and treated daily during 3 d with increasing concentrations of 5AzadC. At day 4, cells were collected and numbered (A and B). Samples were analyzed for their 5mC content either by 5mC labeling and FACS analysis (C and D) or by LC-ESI-MS/MS (E and F). 5mC contents, calculated as in Figure 1F are reported as percentages of the 5mC content in untreated cells. * P < 0.05, ** P < 0.01, *** P < 0.005.
In contrast to WM266–4 cells, KG1 leukemia cells proliferation was affected by lower doses of 5AzadC (0.01 μM;Fig. 3B), and a decrease in DNA methylation was observed starting from this concentration. Both techniques detected a maximum demethylation at 0.32 μM 5AzadC with residual 5mC content of 62.5% ± 2.5%, as calculated by FACS analysis (Fig. 3D), and of 42.4% ± 0.3%, as calculated by LC-ESI MS/MS (Fig. 3F), compared to untreated cells.
Effects of 5AzadC on DNA demethylation and cell proliferation.
Concentrations of 5AzadC that efficiently induced DNA demethylation showed very different effects on the proliferation of the 2 studied cell lines (Fig. 3A). Consequently, it was interesting to study the DNA methylation status of cells in each phase of the cell cycle (Fig. 4). WM266–4 cells displayed more than 85% of the cells in G0/G1 phases when untreated (Fig. 4A). Noteworthy, cells were still in an exponential growth phase at the end-point of the experiment, suggesting that this high G0/G1 percentage was not due to a confluence-induced growth arrest (data not shown) but rather to the long duration of the G0/G1 phase in these cells. Demethylating concentrations of 5AzadC (0.032 μM – 1 μM) did not significantly affect this cell cycle phase distribution (Fig. 4A). FACS data showed that, at 3.2 μM 5AzadC, 5mC content was reduced to 74% in G0/G1 cells, while S cells and G2/M cells 5mC content was decreased to 68% and 63%, respectively (Fig. 4C). In contrast, 5AzadC had a strong impact on KG1 cell cycle, with an increasing proportion of cells arrested in G2/M phases at concentrations higher than 0.01 μM 5AzadC (Fig. 4B). The cells in G0/G1 phase underwent greater DNA demethylation (54% of non-treated cells) at the highest 5AzadC concentrations that are cytotoxic. The percentages of total DNA methylation in S cells and G2/M cells were 50% and 56%, respectively, compared to untreated cells (Fig. 4D).
Figure 4.
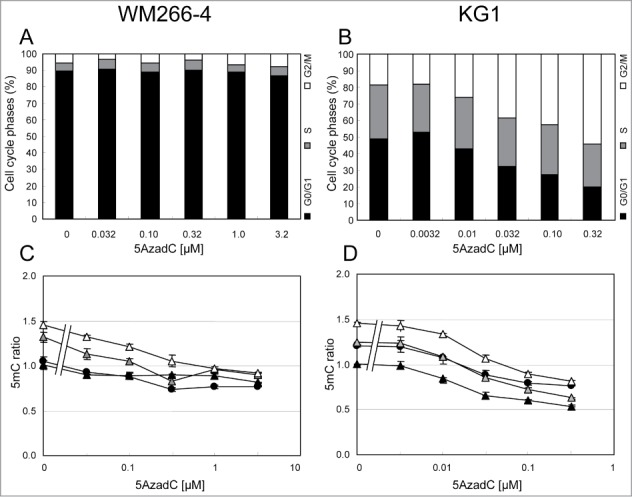
Variations of the 5mC content according to cell cycle status in WM266–4 and KG1 cells upon 5AzadC treatment. WM266–4 cells (A and C) and KG1 cells (B and D) were treated with increasing concentrations of 5AzadC as described in Figure 3. At day 4, cells were analyzed for their 5mC content by FACS analysis according to their cell cycle status. Black circles are for total cells (R2). Black, gray and white triangles are for G0/G1, S and G2/M cells respectively. The 5mC mfis are calculated as in Figure 1F and reported to the 5mC mfi measured on the G0/G1 cells in the control samples.
Early kinetics of cytosine methylation changes in melanoma cells after 5AzadC treatment.
Finally, we explored the kinetics of DNA methylation during the different cell cycle phases and the very early DNA demethylation events induced upon non-toxic but demethylating 5AzadC treatment (Fig. 5). For this experiment, we selected WM266–4 cells, since we previously observed that they are synchronized upon seeding from a confluent culture. Eight hours after plating, cells were adherent and in G0/G1 phase. We then measured total DNA content (Fig. 5A) and total DNA methylation (Fig. 5B) during their first division cycle. Four and a half hours later, most of the cells entered the DNA replication phase. During replication, an increase in 5mC content was detectable only for cells in late S phase. Furthermore, we observed that the first cells in G2/M phase (Fig. 5A and B, 4h30) did not reach the maximum 5mC ratio, measured in G2/M cells later (9h). Clearly, this suggests a lag between DNA replication and completion of cytosine methylation. The 5mC ratio is maximum (1.6) when the whole cell population progressed into the division cycle (6h). Treatment of cells with 5AzadC during their G0/G1 cell cycle phase induced a transient delay in S phase entry (Fig. 5A, 4h30). These cells showed a decrease in 5mC signal (Fig. 5B, 4h30). The maximum difference in 5mC signals was detected 7h30 after treatment, in all cell cycle phases. Noteworthy, 5mC signal was partially restored in treated cells at the end of the experiment (9h).
Figure 5.
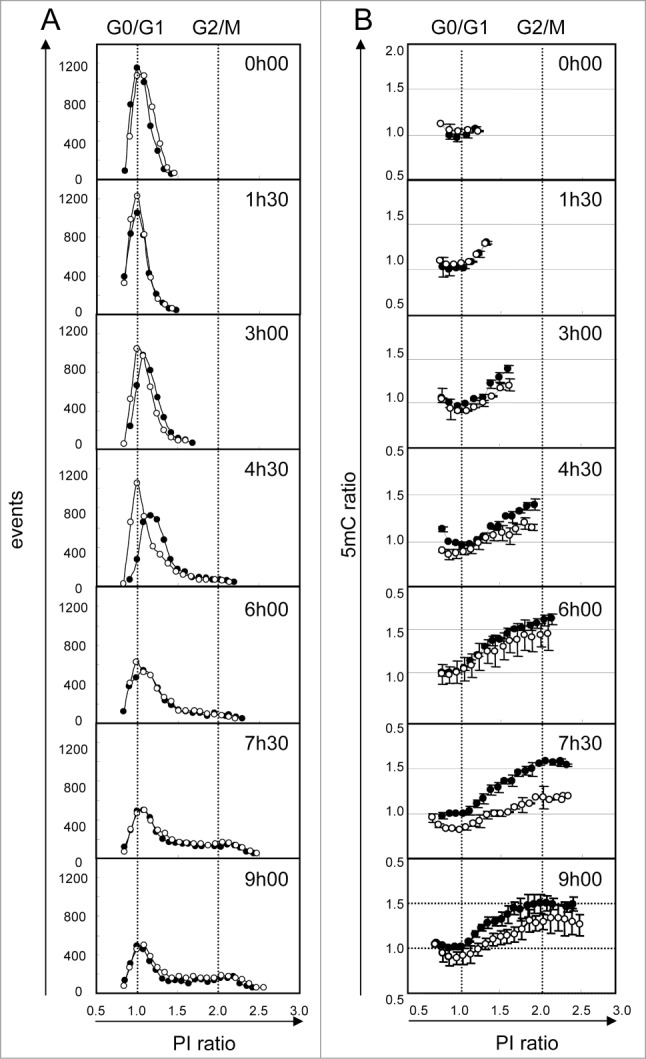
Kinetics of DNA demethylation after 5AzadC treatment in WM266–4 cells. Synchronized WM266–4 cells were treated (white circles) or not (black circles) with 5AzadC (0.32 μM) and collected repetitively at intervals of 90 min. For each sample analysis, the number of cells (column A) and their 5mC labeling (column B) were measured taking 20 contiguous regions according to their PI labeling (intervals of 5000 mfi units). PI and 5mC ratios were calculated using normalized data, as the means of sample fluorescence intensities (mfi) reported to the mfi measured in G0/G1 cells (peak point) in untreated cells at t0. Each circle represents mfis of at least 50 cells. 5mC ratios are reported as means of 2 independent experiments.
Discussion
Modifications of DNA methylation patterns play a major role in physio-pathological situations, such as cancer, by deregulating the “reading” of the genome. Therefore, genome CpG dinucleotide methylation status is extensively studied and different technical approaches have been developed for this purpose. Single CpG modifications are addressed by targeted analysis of specific DNA sequences or genome-wide sequencing after bisulfite conversion on purified genomic DNA. Besides these informative but time-consuming techniques, the measurement of total cytosine methylation levels remains a valuable parameter to follow, in particular, in combination with cell cycle phase.
In order to analyze the relationship between DNA replication and DNA methylation in cancer cells, we developed and validated an improved methodology based on FACS to detect and quantify (1) cellular 5mC content of each single cell population; (2) according to their cell cycle phase; and (3) 5mC content variation during time and upon treatment. Surprisingly, up to now, despite its ability to quantify low variations of fluorescence, flow cytometry was mostly used as a secondary approach to reinforce data obtained by microscopy fluorescence. Here, we have validated it as a quantification tool to compare DNA methylation content in cell lines with different ploidy and to follow variations of global DNA methylation at the cell population scale, with as low as 0.5 x 106 total cells per sample. The sensitivity and accuracy of the approach were challenged by using LC-ESI MS/MS as a direct method for quantification of total 5mC in genomic DNA. We expected some differences in the range of variations detected by the 2 methods, since 5mC immunodetection strongly depends on the specificity and affinity of the anti-5mC antibody for its epitope as well as the local densities and accessibility of 5mC on the DNA. Our data showed that the ratios of 5mC/DNA index, reflecting 5mC density on DNA, were the same with both methods for the 2 cell lines. Cells for which the 5mC contents were ascertained by a direct method, such as LC-ESI MS/MS, can now be included as reference in experiments to accurately estimate absolute 5mC content in other cells.
We demonstrated that both methods detect DNA methylation variations in melanoma WM266–4 cells and leukemia KG1 cells upon 5AzadC treatment, with similar dose-responses curves. Nevertheless, the extent of variation in absolute 5mC content value is lower, by approximate 1/3, when measured by FACS than by LC-ESI MS/MS. One explanation is that the 5mC antibody did not bind to all 5mC but only to a fraction located in DNA regions with a density of methylation above a threshold. Such a bias in the 5mC immunodetection was indeed shown in experiments using methylated DNA immunoprecipitation techniques.45
We took advantage of this method to study variations of 5mC content induced by repeated 5AzadC treatments in melanoma and leukemia cells at the level of individual cells in a population, according to their cell cycle phase. Interestingly, the FACS method we developed showed, for the first time, that demethylation induced by 5AzadC was associated with different effects on the cell cycle distribution in WM266–4 and KG1 cells. Indeed, these cell lines have similar doubling time (32 h and 36 h, respectively) but showed strong differences in their S phase duration. KG1 cells, which appeared more sensitive to 5AzadC, displayed a longer S phase than WM266–4 cells. Noteworthy, at the end-point of our experiments (96 h), KG1 cells that survived at the highest 5AzadC concentrations showed a higher decrease of 5mC signal than WM266–4 cells. In contrast, DNA demethylation reached a plateau in WM266–4 cells, which might be linked to the shorter S phase duration. These results strongly suggest that 5AzadC demethylating efficiency is related to DNA replication and is in agreement with its described mechanism of action.46,47 We clearly showed that slow DNA replication increased the efficiency of 5AzadC incorporation; inversely, rapid DNA replication with a short S phase limited this process, even at high concentrations of drug. In particular, whereas high 5AzadC concentrations induced cell cycle arrest in G2/M phase and cell death in KG1 cells, they had a limited effect on WM266–4 cells proliferation. These observations correlate with the known limited therapeutic efficiency of demethylating agents on solid tumors.26
Finally, we monitored DNA methylation during the cell cycle in WM266–4 melanoma cells. Our data confirmed that inheritance of DNA methylation efficiently occurs in S phase, as demonstrated earlier.47,48 Interestingly, it also revealed a short delay between DNA replication and completion of cytosine methylation, notably in early S phase cells. In addition, the first cells detected in the G2/M phase (4h30) display lower DNA methylation content compared to G2/M cells at further time-points, suggesting an increase of the global DNA methylation efficiency during progression of the cell cycle. Our data are in agreement with previous results showing that completion of DNA methylation occurs mainly in S phase, but can also be observed in G2/M.48 The ratio of 5mC mfis measured for G2/M cells and G0/G1 cells was 1.6, in agreement with what previously observed.49 Nevertheless, we did not detect a lower amount of DNA methylation in G1 cells than in the S phase, as shown by Brown et al. in HeLa cells.49
Upon a single 5AzadC treatment, we observed loss of 5mC signal in late S cells, which presumably corresponds to the replacement of cytosines by 5AzadC in the DNA strands and DNMT trapping. With time, after the first cell division, this decrease in 5mC was also observed in the G0/G1 cells (at 7h30), suggesting propagation of cytosine methylation defect in daughter cells. Even if we cannot exclude that 5AzadC is also incorporated in G0/G1 phase, as suggested earlier,50 we did not detect this decrease in the early time-points of our experiment (up to 3h00). Finally, 5mC level was partially restored at the end of the first round of cell division. It is well known that, when incorporated into DNA, 5AzadC covalently traps DNMTs, which are then degraded by the proteasome.51 The DNMTs nuclear pool is progressively depleted, leading to loss of DNA methylation. The partial restoration of 5mC signal at 9h is possibly due to incomplete depletion of DNMT at this time.
Taken together, our data bring a refined view of the DNA methylation process throughout the cell cycle in tumor cells, in particular, upon demethylating treatment. To achieve this goal, an improved methodology based on flow cytometry was developed to analyze total DNA methylation. First, we validated this method as a rapid, reliable, and sensitive alternative to LC-ESI-MS/MS technology for routine quantification of 5mC in cells. As such, it can be particularly useful to quickly assess DNA demethylating efficiency of new demethylating agents, before triggering more complex analyses. Moreover, our method might be of interest in the follow-up of clinical efficiency of demethylating drugs when tumor cells from patients are available.52,53,54 Second, in contrast with previous approaches using cell synchronization followed by cell sorting for DNA methylation analysis,49 we were able to analyze simultaneously DNA methylation and cell cycle. This allowed us to detect a delay between DNA replication and completion of DNA methylation during S phase. Subtle events, such as partial restoration of 5mC after a single 5AzadC treatment and the dependence of 5AzadC efficiency upon S phase duration, were shown. Finally, our data further comfort the described mechanism of action of 5AzadC and support the requirement of repeated treatments to induce an extensive total DNA demethylation, as observed in several models.55
Material and Methods
Drugs
The drug 5-Aza-2’-deoxycytidine (5AzadC) was purchased from Sigma-Aldrich (France) and aliquots in water were stored at –20°C.
Cell culture
Metastatic melanoma cell line WM266–4 and leukemia cell line KG1 were obtained from the American Type Culture Collection. WM266–4 cells were grown in DMEM (Invitrogen, France) supplemented with 10% fetal bovine serum (Lonza, France). KG1 cells were grown in RPMI medium (Invitrogen, France) supplemented with 20% fetal bovine serum. Both cell lines were maintained in 5% CO2 atmosphere.
Cell treatments
WM266–4 cells were seeded at 8 x 105 cells/cm2 from an exponential growth culture and set to attach for a 16 h period. In some experiments, in which enrichment in G0/G1 cells was required, cells were seeded from a confluent culture (2 x 106 cells/cm2) and treated 8 h later. In these conditions, more than 90% of the cells were in G0/G1 phase. At the end of the treatment, WM266–4 cells were collected using TrypLE™ Select (Life Technologies, France). KG1 cells were grown in suspension and seeded at 0.2 x 106 cells/mL from an exponential growth culture. Both cell lines were treated with 5AzadC 3 times at 24 h intervals from day 0 to day 3 and harvested for analysis at day 4 after the treatment onset. Proliferation was measured by counting viable cells using an Automated Cell Viability Analyzer (Beckman Coulter Vi-Cell).
5mC labeling
5mC labeling was routinely performed on 0.5 x 106 cells in U-bottom 96 wells microtitration plates and adapted from a previous protocol.38 Incubations were carried out in 100 μL reaction volumes and each step was ended by a 3 min centrifugation at 1200 rpm. Supernatants were discarded by turning vigorously the plates upside down.
Briefly, cells were harvested, washed in PBS and fixed with 4% paraformaldehyde in PBS for 15 min at room temperature. After washing in PBS, cells were permeabilized in 0.5% Triton X-100 in PBS for 15 min at room temperature. Cells were washed in PBS and treated with 2 N HCl for 30 min at 37°C. Neutralization was performed with 100 mM Tris HCl pH 8.8 for 10 min and followed by extensive washes with 0.05% Tween 20 in PBS. After blocking in 1% BSA and 0.05% Tween 20 in PBS for at least 2 h, cells were incubated with a primary monoclonal antibody against 5mC (clone 33D3, AbD Serotec, Germany) at a final concentration of 10 μg/mL or with an isotypic control antibody for 90 min at room temperature. Samples were washed in 1% BSA, 0.05% Tween 20 in PBS and incubated with the secondary antibody (Alexa-Fluor 647- or Alexa-Fluor 405-conjugated goat anti-mouse Ig antibodies; Invitrogen, France) for 45 min at room temperature. Cells were finally washed twice in 1% BSA, 0.05% Tween 20 in PBS and labeled in 5 μg/mL propidium iodide (PI) in PBS for 2 h at 4°C for DNA content analysis.
Flow cytometry analysis
Cell labeling was analyzed on a LSRII cytometer (BD Biosciences) using the BD FACSDiva software. Data acquisition was performed as follows. Cells were displayed using a FSC/SSC dot-plot in order to exclude cellular debris (R1 region). Then they were gated on the basis of their propidium iodide content in order to exclude cell doublets, aggregates, or apoptotic cells (R2 region), according to cell cycle analysis requirements.56 Finally, cells were visualized on a fluorescence histogram. Since the experiments were set up to detect decreases in the signal under DNA demethylating conditions, the voltage applied on the corresponding PMT was adjusted to shift the fluorescence histogram of control (untreated) cells on the right of the scale. Routinely, quantification of fluorescence was based on means of fluorescence intensities (mfis) recorded on at least 5000 events in the R2 region. In some cases, mfis were measured from less cells, 50 cells having been validated as the lower limit allowing an accurate measurement.
For each experiment, the displayed 5mC mfis were calculated by subtracting the isotypic control mfis to the mfis obtained with the anti-5mC antibody. When indicated in the legends, these corrected data were reported to the controls (see also Supplementary Fig. S3).
Genomic DNA purification and hydrolysis
Genomic DNA was prepared using the Blood & Cell Culture DNA Mini Kit (Qiagen, France) and quantified by NanoDrop spectrophotometry (ThermoScientific, USA). DNA hydrolysis was performed as previously described.57 Briefly, 2μg of DNA were digested by nuclease P1 (2 h at 45°C in 0.01 M AcONH4 pH=5.3 buffer), snake venom phosphodiesterase (2 h at 37°C in 0.1 M NH4HCO3 buffer) and Antarctic alkaline phosphatase (1 h at 37°C). Nuclease P1, snake venom phosphodiesterase and Antarctic alkaline phosphatase were bought from US Biological (USA), Sigma-Aldrich (France) and New England Biolabs (France), respectively.
LC-ESI mass spectrometry
Genomic DNA methylation analysis was performed as previously described.57 Nucleotides chromatography and mass spectrometry analysis were performed using a Dionex Ultimate 3000 HPLC (LG3400 micropump and WPS3000 autosampler) coupled to an Applied Biosystems QTrap mass spectrometer through a TurboIonSpray ion source interface.
The four deoxyribonucleosides, 2’-deoxy-5-hydroxymethylcytidine (5hmdC), 2’-deoxy-5-methylcytidine (5mdC), 2’-deoxycytidine (dC), 2’-deoxyguanosine (dG), used to calibrate, were purchased from Sigma-Aldrich (France).
A sample equivalent to 1 μg of digested DNA (10 μL final volume) was injected and chromatographed in a Synergi 4u Polar-RP 80A 1.0 × 150 mm column (Phenomenex) and eluted with buffer A (0.1% formic acid in water) and B (0.1% formic acid in methanol) with a gradient increase of 0 to 50%B in 30 min.
The ESI conditions were as follows: nebulizer gas flow GS1=20, curtain gas flow CUR=20, collision gas flow CAD=high, ion spray voltage IS =+5500V, declustering potential DP = 20V, entrance potential EP = 10V, collision energy CE=10V, collision cell exit potential CXP = 15V. UV chromatograms were performed at λ = 260 nm. LC-ESI-MS/MS chromatograms were acquired in Multiple Reaction Monitoring (MRM) mode by monitoring 4 transition pairs of molecular/fragment positive Ion at m/z 258.1/142.1, 242.1/126.1, 228.2/112.1, 268.1/152.1, for 5hmdC, 5mdC, dC, dG, respectively. The scan time was set to 200 ms for each ion pair. Cytosine methylation levels were calculated using the following formula: 5mdC (%) = (A5mdC / (A5mdC + AdC)) × 100, where A5mdC and AdC are the integrals of the MRM peaks corresponding to 5mdC and dC, respectively.
Statistical analysis
When shown, statistics were obtained from 3 independent experiments, i.e., repeated experiments starting from 3 different cell batches treated with 5AzadC from distinct storage aliquots and using the same experimental design. In each independent experiment, different drug concentrations were tested as one replicate only. Results are expressed as the mean ± standard deviation (SD). Statistical analysis was performed using a paired Student's t-test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Natacha Novosad for providing the KG1 cells and Drs Chantal Etievant and Stephane Vispé for helpful discussions.
Funding
This study was supported by the regional council of Midi-Pyrénées (France), FEDER and ATIP CNRS INC Jeunes Chercheurs to PBA.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Bestor T, Laudano A, Mattaliano R, Ingram V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. the carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J Mol Biol 1988; 203:971-83; PMID:3210246; http://dx.doi.org/ 10.1016/0022-2836(88)90122-2 [DOI] [PubMed] [Google Scholar]
- 2. Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 1998; 19:219-20; PMID:9662389; http://dx.doi.org/ 10.1038/890 [DOI] [PubMed] [Google Scholar]
- 3. Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Pro Nat Acad Sci U S A 2002; 99:3740-5; PMID:11891299; http://dx.doi.org/ 10.1073/pnas.052410099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol 1987; 196:261-82; PMID:3656447; http://dx.doi.org/ 10.1016/0022-2836(87)90689-9 [DOI] [PubMed] [Google Scholar]
- 5. Illingworth RS, Bird AP. CpG islands–'a rough guide'. FEBS Lett 2009; 583:1713-20; PMID:19376112; http://dx.doi.org/ 10.1016/j.febslet.2009.04.012 [DOI] [PubMed] [Google Scholar]
- 6. Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39:457-66; PMID:17334365; http://dx.doi.org/ 10.1038/ng1990 [DOI] [PubMed] [Google Scholar]
- 7. Auclair G, Weber M. Mechanisms of DNA methylation and demethylation in mammals. Biochimie 2012; 94:2202-11; PMID:22634371; http://dx.doi.org/ 10.1016/j.biochi.2012.05.016 [DOI] [PubMed] [Google Scholar]
- 8. Deaton AM, Bird A. CpG islands and the regulation of transcription. Gen Dev 2011; 25:1010-22; PMID:21576262; http://dx.doi.org/ 10.1101/gad.2037511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ, Smith C, Harrison DJ, Andrews R, Bird AP. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet 2010; 6:e1001134; PMID:20885785; http://dx.doi.org/ 10.1371/journal.pgen.1001134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010; 466:253-7; PMID:20613842; http://dx.doi.org/ 10.1038/nature09165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature 2007; 447:433-40; PMID:17522677; http://dx.doi.org/ 10.1038/nature05919 [DOI] [PubMed] [Google Scholar]
- 12. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol 2010; 28:1057-68; PMID:20944598; http://dx.doi.org/ 10.1038/nbt.1685 [DOI] [PubMed] [Google Scholar]
- 13. Guil S, Esteller M. DNA methylomes, histone codes and miRNAs: tying it all together. Int J Biochem Cell Biol 2009; 41:87-95; PMID:18834952; http://dx.doi.org/ 10.1016/j.biocel.2008.09.005 [DOI] [PubMed] [Google Scholar]
- 14. Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683-92; PMID:17320506; http://dx.doi.org/ 10.1016/j.cell.2007.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 2007; 8:286-98; PMID:17339880; http://dx.doi.org/ 10.1038/nrg2005 [DOI] [PubMed] [Google Scholar]
- 16. Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 2005; 37:853-62; PMID:16007088; http://dx.doi.org/ 10.1038/ng1598 [DOI] [PubMed] [Google Scholar]
- 17. Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178-86; PMID:19151715; http://dx.doi.org/ 10.1038/ng.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang X, Lay F, Han H, Jones PA. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol Sci 2010; 31:536-46; PMID:20846732; http://dx.doi.org/ 10.1016/j.tips.2010.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gros C, Fahy J, Halby L, Dufau I, Erdmann A, Gregoire JM, Ausseil F, Vispe S, Arimondo PB. DNA methylation inhibitors in cancer: recent and future approaches. Biochimie 2012; 94:2280-96; PMID:22967704; http://dx.doi.org/ 10.1016/j.biochi.2012.07.025 [DOI] [PubMed] [Google Scholar]
- 20. Li KK, Li F, Li QS, Yang K, Jin B. DNA methylation as a target of epigenetic therapeutics in cancer. Anticancer Agents Med Chem 2013; 13:242-7; PMID:22934696; http://dx.doi.org/ 10.2174/1871520611313020009 [DOI] [PubMed] [Google Scholar]
- 21. Fahy J, Jeltsch A, Arimondo PB. DNA methyltransferase inhibitors in cancer: a chemical and therapeutic patent overview and selected clinical studies. Expert Opin Ther Pat 2012; 22:1427-42; PMID:23033952; http://dx.doi.org/ 10.1517/13543776.2012.729579 [DOI] [PubMed] [Google Scholar]
- 22. Griffiths EA, Gore SD. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semi Hematol 2008; 45:23-30; PMID:18179966; http://dx.doi.org/ 10.1053/j.seminhematol.2007.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Piekarz RL, Bates SE. Epigenetic modifiers: basic understanding and clinical development. Clin Cancer Res 2009; 15:3918-26; PMID:19509169; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hackanson B, Robbel C, Wijermans P, Lubbert M. In vivo effects of decitabine in myelodysplasia and acute myeloid leukemia: review of cytogenetic and molecular studies. Ann Hematol 2005; 84 <s>1:32-8; PMID:16292549; http://dx.doi.org/ 10.1007/s00277-005-0004-1 [DOI] [PubMed] [Google Scholar]
- 25. Navada SC, Steinmann J, Lubbert M, Silverman LR. Clinical development of demethylating agents in hematology. J Clin Invest 2014; 124:40-6; PMID:24382388; http://dx.doi.org/ 10.1172/JCI69739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res 2009; 15:3938-46; PMID:19509174; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jorda M, Peinado MA. Methods for DNA methylation analysis and applications in colon cancer. Mutat Res 2010; 693:84-93; PMID:20599551; http://dx.doi.org/ 10.1016/j.mrfmmm.2010.06.010 [DOI] [PubMed] [Google Scholar]
- 28. Oakeley EJ. DNA methylation analysis: a review of current methodologies. Pharmacol Ther 1999; 84:389-400; PMID:10665836; http://dx.doi.org/ 10.1016/S0163-7258(99)00043-1 [DOI] [PubMed] [Google Scholar]
- 29. Torano EG, Petrus S, Fernandez AF, Fraga MF. Global DNA hypomethylation in cancer: review of validated methods and clinical significance. Clin Chem Lab Med 2012; 50:1733-42; PMID:23089701; http://dx.doi.org/ 10.1515/cclm-2011-0902 [DOI] [PubMed] [Google Scholar]
- 30. Achwal CW, Chandra HS. A sensitive immunochemical method for detecting 5mC in DNA fragments. FEBS Lett 1982; 150:469-72; PMID:6819163; http://dx.doi.org/ 10.1016/0014-5793(82)80791-6 [DOI] [PubMed] [Google Scholar]
- 31. Oakeley EJ, Podesta A, Jost JP. Developmental changes in DNA methylation of the two tobacco pollen nuclei during maturation. Proc Nat Acad Sci U S A 1997; 94:11721-5; PMID:9326677; http://dx.doi.org/ 10.1073/pnas.94.21.11721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reynaud C, Bruno C, Boullanger P, Grange J, Barbesti S, Niveleau A. Monitoring of urinary excretion of modified nucleosides in cancer patients using a set of six monoclonal antibodies. Cancer Lett 1992; 61:255-62; PMID:1739950; http://dx.doi.org/ 10.1016/0304-3835(92)90296-8 [DOI] [PubMed] [Google Scholar]
- 33. Kremer D, Metzger S, Kolb-Bachofen V. Quantitative measurement of genome-wide DNA methylation by a reliable and cost-efficient enzyme-linked immunosorbent assay technique. Anal Biochem 2012; 422:74-8; PMID:22197418; http://dx.doi.org/ 10.1016/j.ab.2011.11.033 [DOI] [PubMed] [Google Scholar]
- 34. Niveleau A, Drouet E, Reynaud C, Fares F, Bruno C, Pain C. Polymerase chain reaction products containing 5-methyldeoxycytidine: a microplate immunoquantitation method. Nucleic Acids Res 1994; 22:5508-9; PMID:7816646; http://dx.doi.org/ 10.1093/nar/22.24.5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li Y, Miyanari Y, Shirane K, Nitta H, Kubota T, Ohashi H, Okamoto A, Sasaki H. Sequence-specific microscopic visualization of DNA methylation status at satellite repeats in individual cell nuclei and chromosomes. Nucleic Acids Res 2013; 41:e186; PMID:23990328; http://dx.doi.org/ 10.1093/nar/gkt766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pfarr W, Webersinke G, Paar C, Wechselberger C. Immunodetection of 5'-methylcytosine on Giemsa-stained chromosomes. Biotechniques 2005; 38:527-8, 30; PMID:15884667; http://dx.doi.org/ 10.2144/05384BM01 [DOI] [PubMed] [Google Scholar]
- 37. Gertych A, Farkas DL, Tajbakhsh J. Measuring topology of low-intensity DNA methylation sites for high-throughput assessment of epigenetic drug-induced effects in cancer cells. Exp Cell Res 2010; 316:3150-60; PMID:20813111; http://dx.doi.org/ 10.1016/j.yexcr.2010.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Habib M, Fares F, Bourgeois CA, Bella C, Bernardino J, Hernandez-Blazquez F, de Capoa A, Niveleau A. DNA global hypomethylation in EBV-transformed interphase nuclei. Exp Cell Res 1999; 249:46-53; PMID:10328952; http://dx.doi.org/ 10.1006/excr.1999.4434 [DOI] [PubMed] [Google Scholar]
- 39. Karouzakis E, Gay RE, Michel BA, Gay S, Neidhart M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis and Rheum 2009; 60:3613-22; PMID:19950268; http://dx.doi.org/ 10.1002/art.25018 [DOI] [PubMed] [Google Scholar]
- 40. Bian Y, Alberio R, Allegrucci C, Campbell KH, Johnson AD. Epigenetic marks in somatic chromatin are remodelled to resemble pluripotent nuclei by amphibian oocyte extracts. Epigenetics 2009; 4:194-202; PMID:19440040; http://dx.doi.org/ 10.4161/epi.4.3.8787 [DOI] [PubMed] [Google Scholar]
- 41. Li C, Chen Z, Liu Z, Huang J, Zhang W, Zhou L, Keefe DL, Liu L. Correlation of expression and methylation of imprinted genes with pluripotency of parthenogenetic embryonic stem cells. Hum Mol Gen 2009; 18:2177-87; PMID:19324901; http://dx.doi.org/ 10.1093/hmg/ddp150 [DOI] [PubMed] [Google Scholar]
- 42. Santos F, Dean W. Using immunofluorescence to observe methylation changes in mammalian preimplantation embryos. Methods Mol Biol 2006; 325:129-37; PMID:16761724. [DOI] [PubMed] [Google Scholar]
- 43. Santos F, Hendrich B, Reik W, Dean W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol 2002; 241:172-82; PMID:11784103; http://dx.doi.org/ 10.1006/dbio.2001.0501 [DOI] [PubMed] [Google Scholar]
- 44. MacKay AB, Mhanni AA, McGowan RA, Krone PH. Immunological detection of changes in genomic DNA methylation during early zebrafish development. Genome 2007; 50:778-85; PMID:17893737; http://dx.doi.org/ 10.1139/G07-055 [DOI] [PubMed] [Google Scholar]
- 45. Nair SS, Coolen MW, Stirzaker C, Song JZ, Statham AL, Strbenac D, Robinson MD, Clark SJ. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics 2011; 6:34-44; PMID:20818161; http://dx.doi.org/ 10.4161/epi.6.1.13313 [DOI] [PubMed] [Google Scholar]
- 46. Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980; 20:85-93; PMID:6156004; http://dx.doi.org/ 10.1016/0092-8674(80)90237-8 [DOI] [PubMed] [Google Scholar]
- 47. Leonhardt H, Page AW, Weier HU, Bestor TH. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 1992; 71:865-73; PMID:1423634; http://dx.doi.org/ 10.1016/0092-8674(92)90561-P [DOI] [PubMed] [Google Scholar]
- 48. Hervouet E, Nadaradjane A, Gueguen M, Vallette FM, Cartron PF. Kinetics of DNA methylation inheritance by the Dnmt1-including complexes during the cell cycle. Cell division 2012; 7:5; PMID:22348533; http://dx.doi.org/ 10.1186/1747-1028-7-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brown SE, Fraga MF, Weaver IC, Berdasco M, Szyf M. Variations in DNA methylation patterns during the cell cycle of HeLa cells. Epigenetics 2007; 2:54-65; PMID:17965591; http://dx.doi.org/ 10.4161/epi.2.1.3880 [DOI] [PubMed] [Google Scholar]
- 50. Yamagata Y, Szabo P, Szuts D, Bacquet C, Aranyi T, Paldi A. Rapid turnover of DNA methylation in human cells. Epigenetics 2012; 7:141-5; PMID:22395463; http://dx.doi.org/ 10.4161/epi.7.2.18906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Patel K, Dickson J, Din S, Macleod K, Jodrell D, Ramsahoye B. Targeting of 5-aza-2'-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme. Nucleic Acids Res 2010; 38:4313-24; PMID:20348135; http://dx.doi.org/ 10.1093/nar/gkq187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Claus R, Pfeifer D, Almstedt M, Zucknick M, Hackanson B, Plass C, Lubbert M. Decitabine induces very early in vivo DNA methylation changes in blasts from patients with acute myeloid leukemia. Leuk Res 2013; 37:190-6; PMID:23158571; http://dx.doi.org/ 10.1016/j.leukres.2012.10.015 [DOI] [PubMed] [Google Scholar]
- 53. Oki Y, Jelinek J, Shen L, Kantarjian HM, Issa JP. Induction of hypomethylation and molecular response after decitabine therapy in patients with chronic myelomonocytic leukemia. Blood 2008; 111:2382-4; PMID:18055864; http://dx.doi.org/ 10.1182/blood-2007-07-103960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Aparicio A, North B, Barske L, Wang X, Bollati V, Weisenberger D, Yoo C, Tannir N, Horne E, Groshen S, et al. LINE-1 methylation in plasma DNA as a biomarker of activity of DNA methylation inhibitors in patients with solid tumors. Epigenetics 2009; 4:176-84; PMID:19421002; http://dx.doi.org/ 10.4161/epi.4.3.8694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer cell 2012; 21:430-46; PMID:22439938; http://dx.doi.org/ 10.1016/j.ccr.2011.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wersto RP, Chrest FJ, Leary JF, Morris C, Stetler-Stevenson MA, Gabrielson E. Doublet discrimination in DNA cell-cycle analysis. Cytometry 2001; 46:296-306; PMID:11746105; http://dx.doi.org/ 10.1002/cyto.1171 [DOI] [PubMed] [Google Scholar]
- 57. Song L, James SR, Kazim L, Karpf AR. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal Chem 2005; 77:504-10; PMID:15649046; http://dx.doi.org/ 10.1021/ac0489420 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.