

Abstract
The current standard treatment for acute myeloid leukemia (AML) is chemotherapy based on cytarabine and daunorubicine (7 + 3), but it discriminates poorly between malignant and benign cells. Dose-limiting off‑target effects and intrinsic drug resistance result in the inefficient eradication of leukemic blast cells and their survival beyond remission. This minimal residual disease is the major cause of relapse and is responsible for a 5-year survival rate of only 24%. More specific and efficient approaches are therefore required to eradicate malignant cells while leaving healthy cells unaffected. In this study, we generated scFv antibodies that bind specifically to the surface of AML blast cells and AML bone marrow biopsy specimens. We isolated the antibodies by phage display, using subtractive whole-cell panning with AML M2‑derived Kasumi‑1 cells. By selecting for internalizing scFv antibody fragments, we focused on potentially novel agents for intracellular drug delivery and tumor modulation. Two independent methods showed that 4 binders were internalized by Kasumi-1 cells. Furthermore, we observed the AML‑selective inhibition of cell proliferation and the induction of apoptosis by a recombinant immunotoxin comprising one scFv fused to a truncated form of Pseudomonas exotoxin A (ETA'). This method may therefore be useful for the selection of novel disease-specific internalizing antibody fragments, providing a novel immunotherapeutic strategy for the treatment of AML patients.
Keywords: acute myeloid leukemia, anti-AML antibody fragments, phage display technology, primary AML blast cells, AML immunohistochemistry, internalizing phage antibodies, recombinant immunotoxin, in vitro efficacy
Abbreviations
- AML
acute myeloid leukemia
- scFv
single chain variable fragment
- CDR
Complementarity Determining Region
- PBMC
peripheral blood mononuclear cell
- ETA
Pseudomonas exotoxin A
Introduction
Acute myeloid leukemia (AML) is a heterogeneous group of hematologic disorders characterized by the uncontrolled proliferation of haematopoietic precursor cells and the inefficient induction of apoptosis leading to their accumulation in peripheral blood and bone marrow.1 The American Cancer Society estimated there were 14,590 new cases of AML and 10,370 AML-related deaths in 2013.2 The median age at diagnosis is 65 years, which implies there may soon be a dramatic increase in the incidence of AML due to increased life expectancy and the resulting demographic changes.3 The current prognosis is poor, with a 5-year relative survival rate of only 24%.2 The majority of AML patients die because of treatment-associated mortality and relapse caused by blast cells surviving complete remission.1
To control this minimal residual disease, current research focuses on lineage-specific antigen expression on the surface of AML cells, which can be targeted using monoclonal antibodies.4 Several specific binders have been developed against leukemic markers such as the pan‑leukocyte antigen CD45, 5 and the glycoprotein CD66, which is found on mature myeloid cells.6 Because single agents achieve only marginal effects, they are often coupled to radionuclides to create radioimmunoconjugates. Several candidates are currently undergoing clinical Phase 1 and 2 trials, where they are administered prior to allogeneic stem cell transplantation. Apart from these developments, most attention is still focused on a calicheamicin-coupled CD33-targeting antibody, gemtuzumab ozogamicin (Mylotarg®), which was approved by the Food and Drug Administration in 2000 for the treatment of patients 60 years of age and older with recurrent AML, who were not considered candidates for standard chemotherapy.7 The drug was withdrawn in 2010 due to new safety concerns and the product's failure to show clinical benefit to patients enrolled in a confirmatory trial conducted after approval.8
The main drawback of the antigens listed above is that they are not exclusive to AML cells and are also presented on normal cells.9 Drugs targeting these antigens therefore cause toxicity and severe side effects, so there is an urgent need to identify tumor-associated antigens that are overexpressed on AML cells, but not expressed in vital tissues and organs.
We therefore set out to generate highly-specific single-chain variable fragment (scFv) antibodies against internalizing surface molecules predominantly expressed on AML blast cells. We carried out a sequential 3-step panning strategy using viable AML M2‑derived Kasumi-1 cells and the naïve human scFv Tomlinson phage display library J (Geneservice Ltd, UK). Surface-binding and internalizing scFvs were recovered separately during panning. To avoid unwanted cross‑reactivity to common blood cell surfaces, we combined the positive selection with an upstream negative selection (depletion) against healthy peripheral blood mononuclear cells (PBMCs). We screened 108 individual clones for binding to Kasumi‑1 membrane fragments and found that 47% were specific. Sequencing the scFv DNA revealed 4 unique clones with internalizing activity. Two clones showed specific binding to primary cells from AML patients. One of these showed cytolytic activity when fused to a truncated version of Pseudomonas exotoxin A (ETA'). The isolated specific binders will be used to develop new targeted therapies to kill leukemic blast cells and thus prolong survival after AML remission.
Results
Selection of AML-specific antibody fragments on intact cells
To select novel and potentially internalizing scFv antibody fragments binding to AML cells, we screened the Tomlinson phage display library J using viable Kasumi‑1 cells. The library is based on the pIT2 phagemid vector encoding the scFv pIII fusion protein under the transcriptional control of lactose promoter (lacp) and terminator (lact). An upstream bacterial leader sequence (pelB) directs the recombinant protein into the periplasmic space. A myc/His6 tag downstream of the scFv facilitates the detection and purification of the recombinant protein (Fig. 1A). E. coli cells containing the phagemids were infected with either M13KO7 or M13K07ΔpIII helper phage for the production of scFv-presenting phage particles suitable for panning (Fig. 1B). After three rounds of depletion on PBMCs followed by positive selection on intact Kasumi‑1 cells, the scFv library was strongly enriched for Kasumi‑1-specific clones, as determined by visualizing binding activity after each round of selection in 3 independent polyclonal phage ELISAs. Compared with the naïve Tomlinson library J, the absorption value increased 17-fold for the phage pool rescued after cell lysis, without increasing the binding activity on PBMC membrane fragments. The enrichment factor was determined based on the titer of applied and recovered phage suspensions, revealing a 3-fold enrichment for potentially internalizing binders (lysis fraction) after 3 panning rounds.
Figure 1.
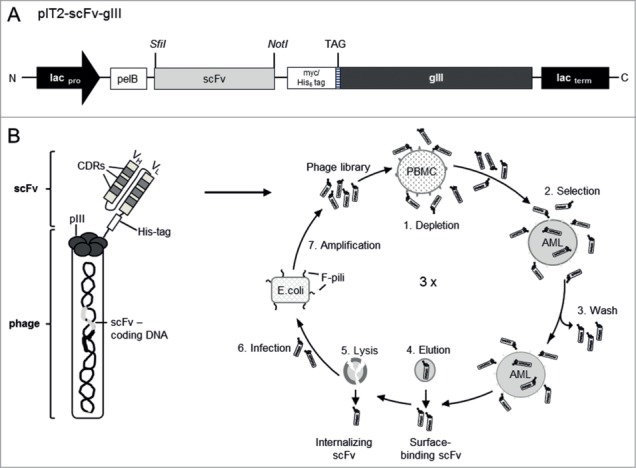
Isolation of AML-specific antibody fragments by phage display. (A) Schematic diagram of the pHEN1-derived pIT2 phagemid expression cassette. Under the transcriptional control of the lactose promoter (lacpro) and terminator (lacterm), the scFv insert is joined in-frame to the gene encoding the pIII phage coat protein (gIII). The phagemid contains a pectate lyase B (pelB) leader sequence upstream of the scFv insert and a myc/His6 tag and amber termination codon (TAG) downstream. Relevant cloning sites are indicated in italics. (B) The cell-based subtractive panning procedure. A phage particle shown on the left, presenting a scFv comprising a heavy chain (VH) and light chain (VL) fused to the pIII minor coat protein, which carries the genetic information. In the panning procedure (shown on the right) the Tomlinson phage library comprising more than 108 different scFv phage particles was first depleted using healthy PBMCs (1) then positively selected on AML-derived cells (2). Non-specific and unbound particles were washed away (3), leaving specific bound phage particles to be eluted from the cell surface (4). Internalized phage particles were recovered by cell lysis (5). The isolated phage particles were used to infect E. coli via their F-pili (6) and amplified for further rounds of selection (7). CDRs, complementarity-determining regions.
Identification of selected scFv clones
Individual binders were identified by randomly picking 108 clones and checking their binding activity on Kasumi‑1 membrane fragments by monoclonal phage ELISA. A total of 51 clones (47%) showed positive binding activity on Kasumi‑1 membrane fragments, 2 thirds of which were recovered from the lysis fraction. The selection criterion for positive binders was a 2.5-fold greater absorption value than negative controls, confirmed in 3 independent experiments. In parallel, we screened the same clones for unwanted cross-reaction to PBMC membrane fragments and found that none of the identified binders showed significant binding activity to PBMC membranes. The cDNAs representing all ELISA-positive binders were sequenced and aligned, revealing 9 sequence-unique scFv clones, 4 recovered after cell lysis. Individual binders were found up to 13 times among the sequenced clones. We confirmed the specific binding activity of scFv‑presenting phage particles on Kasumi‑1 membrane fragments and fixed cells by monoclonal phage ELISA, and on viable Kasumi‑1 cells by flow cytometry to verify native cell surface binding activity (Table 1).
Table 1.
Clone characteristics
| scFv phage binder |
scFv SNAP binder |
scFv Fc binder |
Affinity |
Internalization |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Clone | incidence | ELISA | FACS | ELISA | FACS | FACS | IHC | KD ± SD in nM | FACS | Confocal |
| EMI404 | 1x | ++ | ++ | ++ | ++ | + | n.d. | 62.9 ± 6.8 | ++ | + |
| EMI405 | 8x | + | ++ | + | ++ | ++ | ++ | 63.9 ± 8.3 | ++ | ++ |
| EMI406 | 13x | + | ++ | + | ++ | - | n.d. | 159.4 ± 87.5 | ++ | ++ |
| EMI407 | 3x | + | + | + | ++ | - | n.d. | 404.4 ± 183.0 | + | ++ |
Selected binders are categorized as moderate (+) or strong (++) based on the ELISA absorption value (+ < 5x, ++ > 5x higher than background), the percentage of shifted cells identified by FACS (+ < 60%, ++ > 60%), the fluorescence intensity under confocal microscopy (+ high, ++ very high) and the immunohistochemical (IHC) staining intensity (+ moderate, ++ strong). n.d. = not determined. Experiments were carried out at least three times.
Sequence analysis and molecular modeling of selected scFv antibodies
All the identified clones contained a TAG stop codon in the heavy chain CDR2, which we reverse mutated to CAG (glutamine) by site-directed mutagenesis. An average of 5 single colonies was analyzed to find one correctly mutated sequence. The atomic coordinates of the scFv framework region and CDRs were determined automatically using SWISS-MODEL. The comparison of the submitted scFv with and without the (Gly4Ser)3 linker showed that the linker extends from between the VH and VL chains but does not affect scFv folding. The 3D structure of each scFv (Fig. 2A) was constructed by homology modeling based on a template comprising the Staphylococcus aureus domain/human IgM Fab complex (Parent PDB: 1dee; Chain: E). Because of particular residues in key positions, CDRs 1 and 2 of the heavy chain and CDRs 1–3 of the light chain were predicted to be canonical.10,11 The relationship between the selected scFvs was initially investigated by constructing a phenogram using the DNA sequences (Fig. 2B). The greatest lateral distance in the light chain was found between EMI404 and the corresponding sequences of EMI406 and EMI407 (0.048). The greatest lateral distance in the heavy chain was found between EMI404 and EMI406 (0.049) whereas the smallest lateral distance was between EMI406 and EMI407 (0.028).
Figure 2.
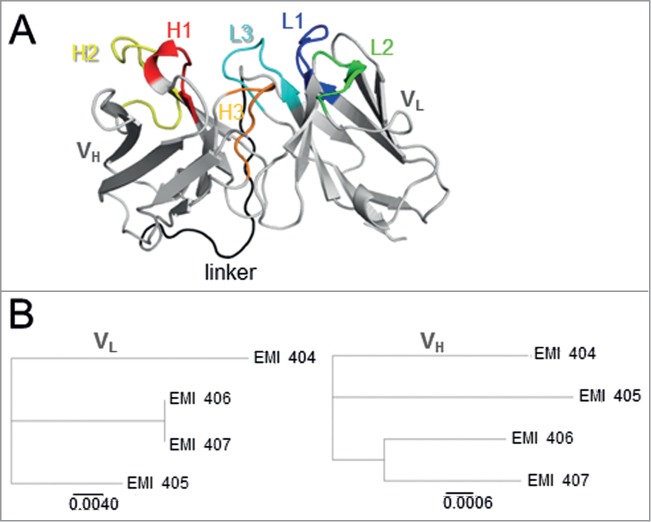
(A) Tertiary structure of scFv EMI405 showing the position of secondary structural elements. The orientation of the scFv is VH–linker–VL. The heavy chain (VH) and light chain (VL) are shown in gray, the VH CDRs are highlighted in warm colors (H1: red, H2: yellow, H3: orange), and the VL CDRs in cold colors (L1: dark blue, L2: green, L3: light blue). The CDRs are defined according to Kabat numbering system. The connecting (Gly4Ser)3 linker is shown in black. (B) Phylogenetic tree based on the neighbor-joining method for the light (VL, left) and heavy (VH, right) chains of the selected scFv antibody fragments. The lateral distance (scale bar) is proportional to their sequence identity.
Binding affinity of soluble scFv-SNAP-tag proteins
The reverse-mutated scFv inserts were transferred to the bicistronic pMS SNAP-tag eukaryotic expression vector to generate scFv-SNAP-tag fusion proteins presenting the selected binders after transfection into HEK293T cells (Fig. 3A). Effective transfection was achieved by selection with zeocin and visual selection (fluorescence microscopy to monitor enhanced green fluorescent protein (eGFP) activity). The scFv-SNAP‑tag fusion proteins were secreted into the supernatant, purified by IMAC and analyzed by SDS-PAGE and western blot. The purified proteins were either used directly or coupled to the fluorophores Vista Green or Alexa Fluor 647 using BG-SNAP substrates.
Figure 3.
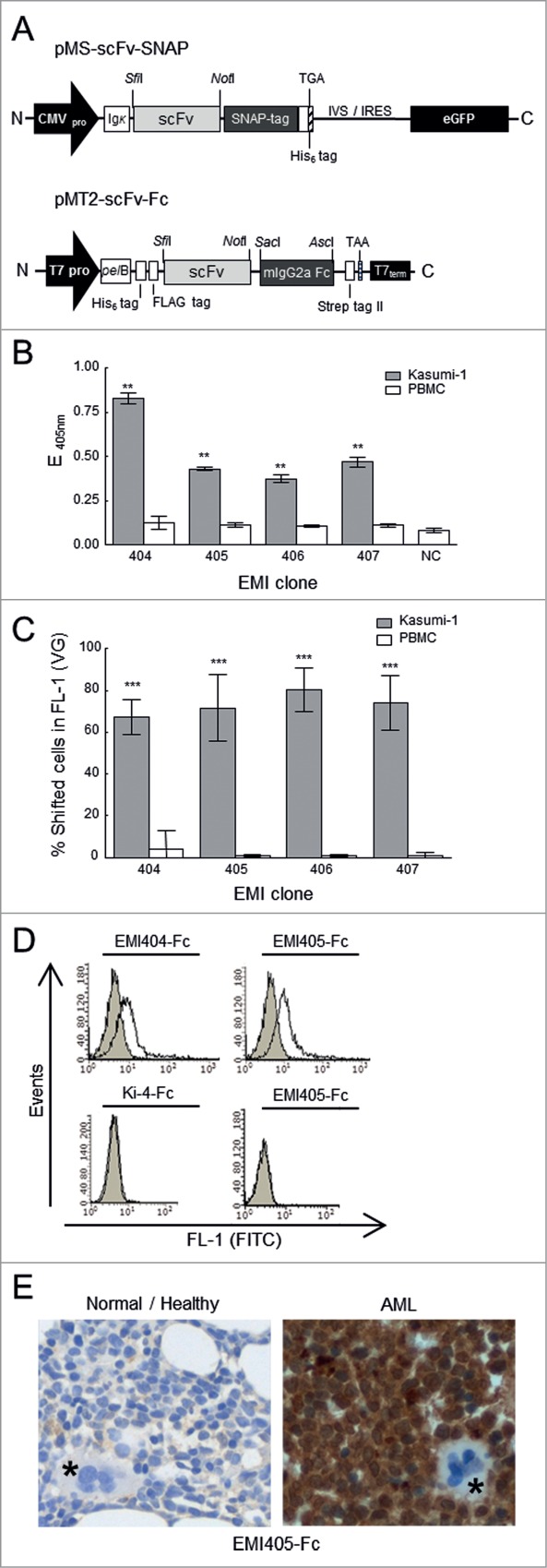
Construction and binding analysis of purified scFv-SNAP-tag and scFv-Fc fusion proteins. (A) Upper diagram: Schematic diagram of the bicistronic eukaryotic expression cassette for recombinant SNAP-tag fusion proteins. Under the control of the cytomegalovirus promoter (CMV pro), the pMS scFv-SNAP vector encodes the antigen binding domain (scFv) joined in-frame to the SNAP-tag. The immunoglobulin kappa leader sequence (Ig κ) upstream of the scFv-SNAP‑tag fusion leads to the secretion of fusion proteins into the supernatant, and a TGA stop codon is placed immediately after the C-terminal His6 tag. The transcribed internal ribosome entry site (IVS-IRES) mediates the cotranslational expression of enhanced fluorescent protein (eGFP). Lower diagram: Schematic diagram of the pET-derived expression cassette. Under the transcriptional control of the T7 promotor (T7pro) and T7 terminator sequence (T7term), the pMT2-scFv-Fc vector encodes a scFv antibody fragment joined in-frame to constant mouse immunoglobulin gamma 2a domain (mIgG2a). IgG2a constant domain enables the use of standard immunohistochemical visualization techniques. The fusion protein is expressed with an N-terminal hexa-histidine tag (His6) followed by a FLAG tag including an enterokinase cleavage site and translocated into the periplasmic space of E. coli by the Erwinia carotovora pectate lyase B (pelB) signal peptide. The stop codon TAA is generated immediately after the C-terminal Strep tag II. His6 tag, FLAG tag and Strep tag II facilitates purification and detection of recombinant proteins. (B) Binding analysis of scFv-SNAP‑tag fusion proteins in a monoclonal scFv ELISA against functional membrane fragments. Tumor binding activity to AML-derived Kasumi‑1 cells (gray bars) was compared to healthy peripheral blood mononuclear cells (PBMCs, white bars). (C) Flow cytometry of fluorophore-labeled scFv-SNAP‑tag proteins on Kasumi‑1 (gray bars) and PBMCs (white bars) cells. After subtraction of background fluorescence, the percentage of fusion protein binding was detected in the FL‑1 channel (488 nm). NC: negative controls. VG: Vista Green. The stars indicate a significant difference relative to the PBMC controls. (D) Flow cytometry analysis showing the binding activity of EMI404-Fc and EMI405-Fc to fresh patient-derived AML cells. Filled gray curves represent untreated cells. Cells were incubated with the fusion protein (black curve). To exclude nonspecific staining of the FITC labeled goat anti-mouse (GAM-FITC) detection antibody, omission of the Fc-tagged fusion proteins served as control (light gray curve). To exclude nonspecific binding of the Fc domain AML blast cells were stained with the irrelevant scFv-Fc fusion protein Ki-4-Fc (left bottom). To exclude unspecific binding, stainings were also performed on healthy donor PBMC (right bottom). (E) Immunohistochemical analysis of bone marrow sections with EMI405-Fc fusion protein, magnification x 400. Right panel (AML bone marrow biopsy sections) shows exemplary the immunological detection of AML blast cells. Left panel (normal/healthy biopsy sections) shows no evidence of blast cell infiltration. Megakaryocytes are marked in asterisks.
The binding strength was determined by monoclonal scFv-ELISA. We incubated 1 μg of each purified scFv protein with immobilized Kasumi‑1 and PBMC membrane fragments as a negative control. Positive binding was detected using a rabbit anti‑SNAP‑tag primary antibody and a HRP‑labeled goat anti-rabbit secondary antibody, visualized after the addition of ABTS. Selected clones with an absorption value at least 2.5-fold higher than the negative controls were classified as moderate binders, whereas clones with absorption values more than 5-fold higher were classified as strong binders. According to this classification, one of the selected internalizing binders (EMI404) bounds strongly to Kasumi‑1 membrane fragments whereas the others (EMI405, EMI406 and EMI407) were moderate binders (Fig. 3B). We also used flow cytometry to measure scFv binding to viable target cells, and found a minimum of 67.3% for clone EMI404 and a maximum of 80.2 shifted Kasumi‑1 cells in FL-1 for clone EMI406 when incubated with 1 μg Vista Green-labeled protein (Fig. 3C). There was no non-specific binding to PBMCs or other negative control cells such as HEK293T. The KD values were determined by incubating Kasumi‑1 cells with up to 2000 nM of each binder to reach a saturation level. The increasing MFIs of cell-bound scFvs were measured, normalized to background fluorescence and plotted against the applied scFv concentrations in a saturation-binding curve. The calculated KD values of each sample using non‑linear regression ranged from 62.9 ± 26,8 nM for clone EMI404 to 404.4 ± 183.0 nM for clone EMI407 (Table 1). All experiments were carried out at least 3 times.
Binding activity of soluble scFv-Fc-tag proteins toward primary AML cells
The selected scFv's were inserted into the pET-derived expression vector pMT2 (Fig. 3A) and transformed into E. coli BL21(DE3). Recombinant Fc-tag fusion proteins were directed into the periplasmic space after induction with 2 mM IPTG and purified using IMAC on Ni-NTA superflow resins resulting in a single dominant protein band in SDS-PAGE and Western blot at 55 kDa. Protein concentrations were determined colorometrically using the BCA protein assay kit (Pierce Biotechnology, Rockford, IL, USA) and calculated at concentration range of 120-150 μg/ml. Primary tumor cell binding were first determined by flow cytometry using an FITC conjugated goat anti-mouse secondary antibody. 70% of blood cells were determined as CD34+ immature blast cell population. We observed robust cell surface binding of EMI404-Fc (17% increase in MFI) and EMI405-Fc (33% increase in MFI) to the primary AML patient cells, but not to the PBMCs of healthy donors. EMI406-Fc and EMI407-Fc did not show any binding activity. Irrelevant control scFv-Fc fusion protein (Ki-4-Fc) did not show any binding activity to blast cells. (Fig. 3D). Candidate immunotoxin clone EMI405 showed robust immunohistochemical staining for human AML blast cells in the marrow biopsy in contrast to a control (healthy) biopsy. Megakaryocytes were not stained with EMI405-Fc fusion protein indicating tumor-specific binding activity (Fig. 3E).
Internalization behavior
SNAP‑tag scFv fusion proteins labeled with Alexa Fluor 647 were incubated with the target cells at 37°C for 1 h to allow internalization. Having confirmed functional binding activity by flow cytometry, surface-bound scFv proteins were stripped with trypsin and the fluorescent profile was checked again. Clones EMI404, 405, 406 and 407 still generated a significant fluorescent signal in FL‑4 reflecting the presence of endocytosed scFv fusion proteins. In contrast, there was no signal after trypsin elution when incubation was carried out at 4°C or when the target cells were incubated with the non-specific 425(scFv)-SNAP fusion protein (Fig. 4A). Normalizing the signal following trypsin treatment to the signal before treatment revealed that 68.7 ± 3.5% of clone EMI404, 66.2 ± 9.9% of clone EMI405 and 65.5 ± 20.4% of clone EMI406 was internalized after 1 h incubation at 37°C, indicating a quick endocytosis of surface-bound scFv molecules. In comparison, clone EMI407 revealed slow intracellular uptake with an internalization rate of 11.8 ± 6.3% after incubation under the same conditions (Fig. 4B). No surface-bound scFv-SNAP‑tag protein could be detected using a secondary anti-SNAP‑tag antibody after trypsin elution. Using this approach, we confirmed that the surface-bound scFv was efficiently eluted and that the fluorescence signal must represent internalized scFv protein. Each experiment was carried out 3 times. As an example, the internalization pattern of clone EMI405 was monitored by confocal microscopy. The labeled scFv protein bound specifically to Kasumi‑1 cells and was internalized under the same conditions described above, whereas the non-specific 425(scFv)-SNAP protein did not generate a signal. However, incubation at 4°C inhibited internalization (Fig. 5A). Clones that were internalized based on FACS analysis were analyzed to determine their internalization kinetics using the Opera live cell imager, and cellular uptake was observed within the first 15 min of incubation at 37°C (Fig. 5B).
Figure 4.
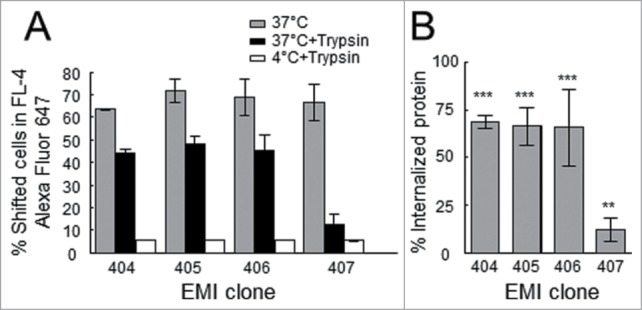
Internalization of scFv-SNAP‑tag fusion proteins. (A) Flow cytometry scFv-SNAP internalization assay. The scFv-SNAP‑tag fusion proteins were labeled with Alexa Fluor 647 and incubated with Kasumi‑1 cells; the fluorescence signal was measured in the FL‑4 channel (gray bars). After treatment with trypsin to remove surface-bound proteins, the fluorescence signal was maintained following incubation at 37°C (black bars), reflecting the internalization of all scFv fusion proteins, but there is no signal following incubation at 4°C (white bars). (B) Relative amount of internalized scFv fusion protein after 60 min incubation at 37°C. The fluorescence signal following the removal of surface-bound scFv was related to the signal before trypsin treatment. The stars indicate a significant difference relative to incubation at 4°C.
Figure 5.
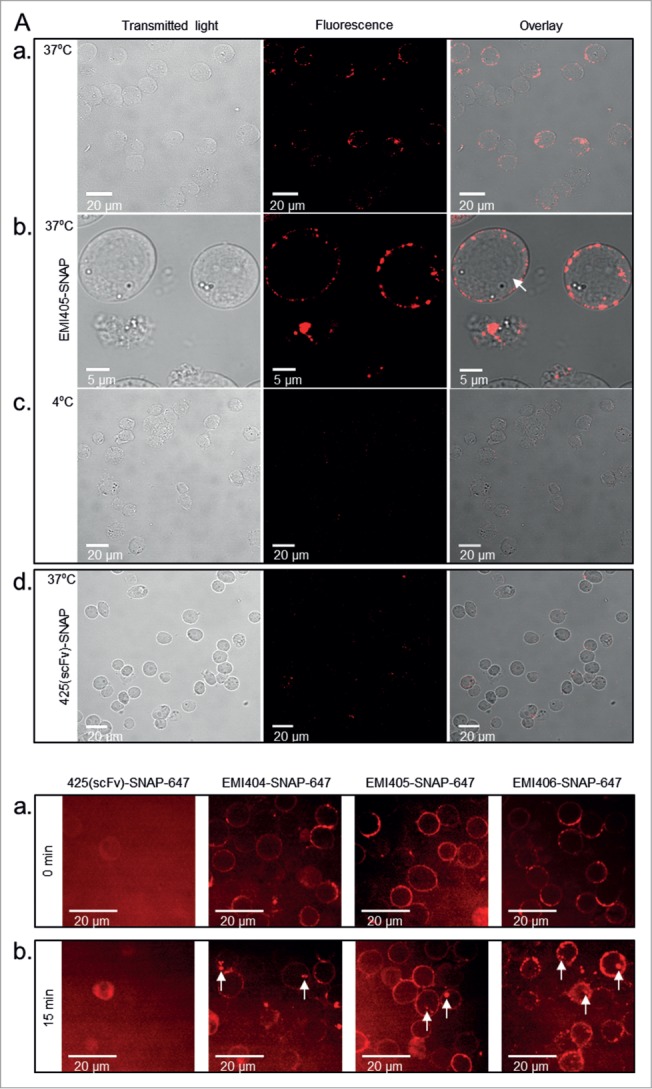
Binding and internalization of scFv-SNAP‑tag fusion proteins analyzed by confocal microscopy. (A) Confocal images of Kasumi‑1 cells incubated with 1 μg EMI405(scFv)-SNAP-647 for 60 min at 37°C (a + b) or at 4°C (c). Surface-bound proteins were removed with trypsin allowing the detection of internalized scFv protein. Kasumi‑1 cells were incubated with the irrelevant 425(scFv)‑SNAP protein at 37°C as a negative control (d). Scale bars show magnification. (B) Internalization kinetics was analyzed using a live cell imager. Kasumi‑1 cells revealed a corona-shaped fluorescent signal when incubated with EMI404, EMI405 and EMI406 at time point 0 min (a). Cellular uptake was observed after 15 min incubation (b) under physiological conditions as shown by the intracellular fluorescent vesicles (white arrows).
Cytotoxicity of the recombinant immunotoxin
The EMI405 scFv sequence was transferred to the pMT ETA' prokaryotic expression vector to generate scFv-ETA' fusion proteins (Fig. 6A). The immunotoxin was successfully produced with 90% purity and specific binding activity was confirmed by flow cytometry after incubating the target cell line Kasumi‑1 with increasing amounts of the immunotoxin (Fig. 6B). Cytotoxicity was evaluated using a colorimetric XTT cell proliferation assay with the target cell line Kasumi‑1 and KG‑1 as negative control, and compared to the effect of the non-specific protein 425(scFv)-ETA'. We observed the dose-dependent inhibition of cell growth with an IC50 value of 265.2 ± 0.2 nM for EMI405(scFv)-ETA' (Fig. 6C), whereas the viability of Kasumi‑1 cells was affected neither by 425(scFv)-ETA' nor by the non-internalizing scFv EMI408. Furthermore, scFv EMI405 had no effect on the viability of the negative control cells KG‑1, confirming its targeted cytotoxicity.
Figure 6.
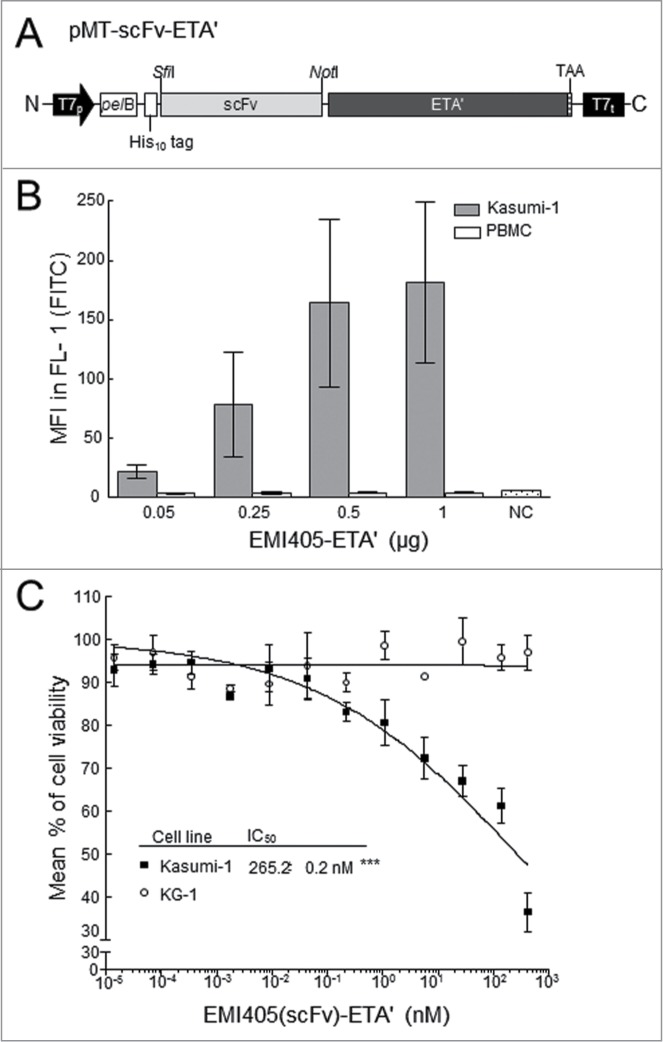
Construction of EMI405(scFv)‑ETA' fusion proteins and their effect on cell viability. (A) Schematic diagram of the pET expression cassette. Under the control of the T7 promoter (T7p) and terminator (T7t), the pMT-scFv-vector encodes the scFv joined in-frame to a truncated variant of Pseudomonas exotoxin A (ETA'). The fusion protein includes an N-terminal His10 tag and protein is translocated into the periplasmic space by the pectate lyase B (pelB) signal peptide. The stop codon TAA is placed immediately after the C-terminal ETA' domain. (B) Flow cytometry reveals dose-dependent binding activity against Kasumi‑1 cells incubated with increasing amounts (14 fM – 420 nM) of EMI405(scFv)-ETA' fusion protein. Surface-bound molecules were detected (FL‑1) using the anti-ETA' monoclonal antibody TC‑1 and mean fluorescence intensity (MFI) was measured in the FL‑1 channel in 3 independent experiments. (C) XTT cell proliferation assays were carried out using EMI405(scFv)-ETA' to determine cytotoxicity against Kasumi‑1 cells (▪) and the negative control line KG‑1 (°). The percentage of viable cells was calculated after incubation with PBS (100% viability) and DMSO (0% viability). The half maximal inhibitory concentration (IC50) was determined by non-linear regression analysis (sigmoidal dose response).
Measurement of apoptosis
Apoptosis was analyzed by labeling the cells with annexin A5-eGFP and propidium iodide, revealing a time-dependent increase in the amount of bound annexin A5 exclusively on Kasumi‑1 cells after 24 h exposure to the immunotoxin EMI405(scFv)-ETA'. The non-specific 425(scFv)-ETA' construct had no effect on Kasumi‑1 cells (Fig. 7A). We also tested for caspase-3/7 activity after incubation with the immunotoxin for 24, 48, 72 and 96 h. The signal representing the metabolized pro-fluorescent caspase substrate in Kasumi‑1 samples was measured and compared to the negative control line KG‑1. After 96 h, we observed apoptosis in 33.0 ± 1.4% of the Kasumi‑1 cells, but only 0.6 ± 0.1% of the KG‑1 cells (P < 0.0001). The percentage of apoptotic Kasumi‑1 cells was 15.9 ± 0.6% after 72 h and 4.2 ± 2.1% after 48 h, indicating a time-dependent increase when incubated with the same amount of immunotoxin (Fig. 7B). The addition of PBS or the non-specific fusion protein 425(scFv)‑ETA' had no effect on apoptosis (data not shown). Each experiment was carried out 3 times.
Figure 7.

Analysis of apoptosis induced by the EMI405(scFv)-ETA' immunotoxin fusion protein. (A) Flow cytometry was used to detect apoptotic cells by annexin A5-FITC/PI double-staining. Kasumi‑1 cells were treated with 1 μg EMI405(scFv)-ETA', non-binding 425(scFv)-ETA' or PBS and analyzed after incubating 4 24 h. Viable cells are shown in the lower left quadrant, early apoptotic cells in the lower right quadrant and late apoptotic/necrotic cells in the upper right quadrant. PtdIns: Propidium Iodide. (B) Histogram shows percent apoptosis based on activation of caspases-3/7 in Kasumi‑1 cells (gray bars) and KG‑1 control cells (white bars) after subtracting background (incubation with PBS). The cells were treated with 1 μg EMI405(scFv)-ETA' for up to 96 h. *** highly significant.
Discussion
There are many subtypes of leukemia, and the 5-year survival rate varies from 15-70% depending on the specific form of the disease and patients age.2 The American Cancer Society predicted a median 5‑year relative survival rate of 25% for AML patients in 2013.2 The likelihood of complete remission depends on a number of prognostic factors, including cytogenetic characteristics and the presence of a mutation in the FLT‑3 transmembrane tyrosine kinase, which promotes the uninhibited proliferation of leukemic blast cells in the absence of a ligand. Several drugs that inhibit FLT‑3 tyrosine kinase are undergoing Phase 1 and 2 clinical studies, but genuine clinical responses are rare.12 Poor prognosis in AML patients mainly reflects the high frequency of relapse due to blast cells surviving complete remission. Post‑remission therapy therefore aims to eradicate these residual blast cells but this is often inefficient, particularly in older patients because of age-related co-morbidities that constrain chemotherapeutic opportunities13 or the intrinsic resistance of leukemic cells to standard chemotherapy.3 More effective and less toxic therapeutic options are therefore required to eliminate minimal residual disease.
Specific molecules targeted to leukemic blast cells would be suitable for this purpose, and although naked monoclonal antibodies have shown only moderate activity, radioactive conjugates and cytotoxic fusion proteins (immunotoxins) have shown promising results.1 Current AML-targeting strategies, however, are limited to tumor-associated antigens such as CD33, CD45 and CD66,4 which, although strongly expressed on blast cells, are also present on healthy cells, resulting in severe off‑target effects.14 To circumvent these drawbacks, we attempted to generate human antibodies against AML-derived tumor cells for the selective destruction of cancer cells. We successfully isolated AML-specific antibody fragments using a whole-cell panning strategy and removed binders to common cell surface antigens by upstream depletion. We then used 2 different internalization assays to confirm the efficiency of panning with strong selection pressure on internalizing scFv antibody fragments and constructed a novel ETA'-based immunotoxin that specifically inhibited cell proliferation and induced apoptosis when incubated with the target cell line Kasumi‑1.
The whole-cell selection strategy we developed allows native cell surface proteins and non-protein antigens to be included in the selection process, which minimizes the selection of scFv antibody fragments that bind to the cytosolic face of the cell membrane or denatured epitopes that would not be recognized the native protein in vivo. Furthermore, by carrying out the selection process at 4°C we avoid any changes in the surface antigen pattern or the expression of pre-apoptotic structures. Whole-cell panning also focuses selection on internalizing scFv antibodies, which is beneficial for the development of novel immunotherapeutic agents because receptor-mediated endocytosis plays a crucial role in drug delivery to target cells. Therefore, we increased the incubation temperature to 37°C for 15 min during panning to initiate the endocytosis of scFv-presenting phage particles bound to external receptors. Subsequent interruption of internalization by reducing the temperature to 4°C helped to avoid the intracellular degradation of endocytosed scFv phage particles, allowing them to be recovered from the cytosol in an infectious and completely functional form.15
We initially focused on the characterization of scFv binding activity before carrying out the necessary reverse mutations and transferring the corresponding cDNA into the pMS SNAP‑tag vector for the production of soluble scFv fusion proteins for further analysis.16 The scFv-SNAP‑tag fusion proteins were expressed in HEK293T cells, purified and coupled to Vista Green or Alexa Fluor 647 fluorophores for affinity and internalization assays. We measured the binding affinity and the KD, which is required to determine the potency of an immunotoxin.17,18 The KD of EMI405(scFv)-SNAP was 63.9 ± 8.3 nM, resulting in significant cytotoxicity toward Kasumi-1 cells (IC50 = 265.2 ± 0.2 nM) without affecting other cells. However, a large dose is needed to induce cell death efficiently and this may reflect the relatively small number of target antigens on the cell surface.19
Increasing the avidity of binding could result in a stronger on‑rate but would also enhance the cytotoxicity.20 Furthermore, multivalency would favor internalization because it causes the dimerization of bound receptors.21 The number of antigens presented on the cell surface has a significant effect on cytotoxicity: low-density antigens may require higher concentrations of immunotoxins, in turn causing therapy-limiting off‑target effects.18 Because we carried out selection using whole cells, the antigen recognized by the isolated binders remains unknown. However, the mean fluorescence intensity (MFI) measured during flow cytometry can be used as a surrogate parameter. EMI405, EMI406 and EMI407 each generated a MFI > 25, indicating a moderate density of bound antigens. In contrast, EMI404 generated a MFI < 9, indicating a lower antigen density. Further studies will reveal the identity of the antigens, allowing us to develop targeted fusion proteins with greater cytotoxicity. The addition of valproic acid or mitoxantrone could also improve the efficacy of cell killing because these chemicals have already been shown to enhance the toxicity of specific immunotoxins.7 Because ETA' is prokaryotic in origin, repetitive administration is likely to result in an undesirable immune response, which would limit the therapeutic use of the immunotoxin.22 This could be avoided by replacing the cytotoxic component with human proapoptotic proteins, such as granzyme B/M, angiogenin, DAPK2 and Tau, to form fully human constructs.22-26
Using the Tomlinson phage display library J, we isolated a series of novel scFv antibodies that show specific binding activity against unknown surface antigens on AML‑derived Kasumi‑1 cells. EMI404 and EMI405 showed potent binding activity against human primary AML cells. In addition, candidate immunotoxin clone EMI405 showed strong immunohistochemical staining in bone marrow biopsy specimens from patients with AML. Megakaryocytes were not stained with EMI405-Fc fusion protein indicating tumor-specific binding activity. Cross‑reactivity to common blood cell surface markers was excluded by depletion on healthy PBMCs. Our method could easily be adopted for selection on primary cells, including tumor stem cells if available in sufficient amounts. The promising binding affinity and internalization behavior of scFv EMI405 in particular prompted us to develop immunotoxins combining this scFv with ETA'. The EMI405(scFv)-ETA' fusion protein showed favorable anti-leukemic activity specifically against AML-derived Kasumi‑1 cells, but not other cancer cell lines or healthy PBMCs. EMI405(scFv)-ETA' induced apoptosis in the target cells, but its potency could still be improved either by the addition of toxicity enhancers or by increasing the valency of binding. EMI405(scFv)-ETA' therefore represents a promising candidate for the development of novel therapeutic approaches for AML.
Materials and Methods
Cells and culture methods
The human acute myeloid leukemia M2-derived cell line Kasumi‑1 was purchased from the German Resource Centre for Biological Material (DSMZ, cat. num. ACC-220) and used as the selection antigen. Cells were cultured in 80% (v/v) RPMI 1640 GlutaMAX-I medium (Life Technologies, cat. num. 61870-010) supplemented with 20% (v/v) fetal bovine serum (FBS, Life Technologies, cat. num. 10091-148) at 37°C, 5% CO2 and split every 3–4 days at a ratio of 1:2. Blood samples and bone marrow biopsies were collected from patients with AML after written consent and in strict compliance with applicable ethical, legal and privacy-related conditions in accordance with the Declaration of Helsinki.27 Human PBMCs freshly-isolated from heparinized full blood using Ficoll reagent (GE Healthcare Life Sciences, cat. num. 17-1440-02), the human embryonic kidney cell line HEK293T from the American Type Culture Collection (ATCC, cat. num. CRL-3216) and KG-1 cells (DSMZ, cat. num. ACC-14) were used as negative controls. The cells were grown as above in 90% (v/v) RPMI 1640 GlutaMAX-I medium containing 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin (10,000 U/ml, Life Technologies, cat. num. 15140-122). HEK293T cells were also used for transfection and the expression of scFv-SNAP-tag fusion proteins by seeding 6 × 104 cells/well into 24‑well culture plates and incubating with 1‑2 μg plasmid DNA and 3 μl FuGene HD Transfection Reagent (Promega, cat. num. E2311) for 48 h. Functional protein expression and SNAP-tag activity were tested as previously described.28 Successfully-transfected cells were selected by supplementing the standard medium with 100 μg/ml zeocin (InvivoGen, cat. num. ant-zn-1). For the production of large quantities of protein, transfected cells were cultured in Nunc triple flasks (Sigma-Aldrich, cat. num. F8667) using 200 ml medium renewed every 7‑8 days.
Selection for internalizing scFv antibodies
The human single-fold scFv Tomlinson library J (Medical Research Council Laboratory, Cambridge, UK) was superinfected with either M13KO7 helper phage (NEB, cat. num. N0315S) or M13K07ΔpIII hyperphage (Progen Biotechnik, cat. num. PRHYPE). The scFv‑presenting phage particles from Tomlinson library J were prepared for panning as previously described.16 To remove antibodies binding to common blood-cell surface antigens, the library was pre-incubated with PBMC membrane fragments pre-blocked in 2% skimmed milk powder in PBS (MPBS)29 coated on a Nunc 96‑well microtiter plate (Sigma-Aldrich, cat. num. M9410) shaking at 400 rpm for 1 h at room temperature (depletion)16. Kasumi-1 cells were washed 3 times with 10 ml ice‑cold PBS and blocked for 2 h with 2% MPBS. After depletion, the supernatant containing non‑binding phage particles was transferred to the pre-blocked Kasumi‑1 cells and incubated the rotating platform for 1 h at 4°C. Cells and bound scFv antibodies were pelleted at 1000 x g for 2 min. Non-binders and non-specific binders were washed away with alternating brief and long (5 min) washing steps with pre-chilled PBS. The cells were then re‑suspended in 15 ml pre-warmed PBS and incubated at 37°C for 15 min to allow internalization. The cell membrane-bound scFv phage antibodies were stripped by incubation with 1 ml ice cold elution buffer (76 mM citric acid) and the eluted phage particles were neutralized in 0.5 ml 1 M Tris-HCl (pH 7.5). The cells were washed again with PBS to avoid contaminating the lysis fraction with surface-bound phage, and centrifuged as above. The internalized scFv phage antibodies were recovered by incubating the cell pellet in 1 ml lysis buffer (100 mM triethylamine; TEA) for 10 min and neutralizing in 0.5 ml 1 M Tris-HCl (pH 7.5). The elution and lysate fractions were amplified by infecting mid‑logarithmic growth phase Escherichia coli TG1 cells (Agilent Technologies, cat. num. 200123) for 30 min at 37°C, and were then subjected to the next round of depletion and selection. Three panning rounds were carried out. The enrichment of Kasumi‑1‑specific binders was monitored by polyclonal phage ELISA and the enrichment factor was calculated as previously described.16
Binding analysis: scFv-presenting phage particles and membrane fragments
Polyclonal and monoclonal phage particles were tested by ELISA for binding on immobilized functional membrane fragments29 using an HRP/anti‑M13 monoclonal mouse secondary antibody (GE Healthcare Life Sciences, cat. num. 27-9421-01) and ABTS as the peroxidase substrate (Roche Life Science, cat. num. 11204521001). For polyclonal phage ELISA, phage antibody fragments from each selection fraction were precipitated, adjusted and blocked with 2% MPBS.16 After washing the pre-blocked wells, 100 μl of the phage suspension was applied onto Kasumi‑1 and PBMC membrane fragments and incubated for 1 h at room temperature, shaking at 400 rpm. We then added 100 μl HRP-labeled anti-M13 antibody (1:5000 dilution) and incubated as above. Freshly-prepared ABTS solution was added and the reaction was incubated in the dark for 15–60 min before the colorimetric absorbance was measured at 405 nm using a TECAN reader against a 490 nm reference. Stringent washing with 0.05% PBST was carried out between each step. To determine the binding activity of single clones, dilution series of the elution and lysis fractions were prepared separately, and single clones were transferred to sterile Nunc 96‑well microtiter plates (Sigma-Aldrich). They were checked for binding activity as polyvalent and monovalent scFv‑presenting phage particles after infection with 109 hyperphage and helper phage, respectively.16 The phage-containing supernatant was transferred to the coated wells and binding was detected as described above for the polyclonal phage ELISA. Binders confirmed in 3 rounds of analysis were tested for the presence of a scFv open reading frame by insert PCR. Insert-carrying clones were sequenced to verify their integrity and to determine the number of binders with unique sequences. Premature amber stop codons in positive phage clones were eliminated by site directed mutagenesis as described previously.16
Binding analysis: scFv-presenting phage particles and whole cells
To confirm binding to the outer cell membrane surface, we also carried out whole-cell ELISA on fixed Kasumi‑1 cells. We added 1 × 105 cells to each well and centrifuged the plate for 5 min at 500 x g and then at 4500 x g for 30 min. The cells were air dried for 20 min and fixed with 100 μl/well 1:2 methanol:ethanol at –20°C for 20 min. Binding was detected using the procedure described above for phage ELISA on membrane fragments. We confirmed the ability of the selected binders to bind viable cells by flow cytometry using the scFv‑presenting phage antibodies: 109 monovalent scFv-presenting phage particles representing the unique binders were incubated with 2 × 105 freshly-harvested and washed PBMCs or Kasumi‑1 cells in 2% MPBS containing 0.02% NaN3 for 1 h at 4°C to avoid premature internalization. Unbound phage particles were removed by washing twice with PBS using a cell washer, and the cells were re‑suspended in 1 ml 2% MPBS. We added 1 μl monoclonal anti‑M13 g8 FITC-labeled antibody (ACRIS, Herford, Germany) and incubated the suspension for 30 min at 4°C protected from light. After washing away the unbound antibody, the cell pellet was re‑suspended in 0.5 ml PBS and bound phage particles were detected in FL‑1 using a FACSCalibur flow cytometer (BD Biosciences, Heidelberg, Germany). Binding intensity was evaluated after subtracting the non-specific binding activity of pure helper phage and secondary antibody alone.
Analysis of scFv-SNAP- and scFv-Fc-tag fusion protein by ELISA and flow cytometry
Successfully-mutated scFv sequences were subcloned in the pMS expression vector for the production of soluble SNAP-tag fusion proteins in eukaryotic cells30 or pET-vector derived pMT2 prokaryotic expression vector for the production of soluble Fc-tag fusion proteins. Cloning, transfection of HEK293T cells and the expression, purification and coupling of scFv-SNAP to fluorophores modified with benzylguanine were carried out as previously described.28 Cloning and expression of scFv-Fc fusion protein were performed in the pET System according to manufactures optimized protocols (NEB, Frankfurt a. M., Germany). His6 tagged fusion proteins were purified by immobilized metal affinity chromatography (IMAC) using Ni-NTA superflow resins according to the manufacturers’ instructions (Qiagen, cat. num. 30410). The functionality of the scFv-SNAP fusion protein was demonstrated by ELISA using the crude cell culture supernatant as well as purified protein. A 96‑well microtiter plate was coated overnight at 4°C with 100 μl of a 1:100 dilution of Kasumi‑1 and PBMC membrane fragments. The plate was washed 3 times with PBS and blocked for 2 h with 2% MPBS, then 100 μl/well of the cell supernatant containing the scFv was added and incubated for 1 h at room temperature, shaking at 400 rpm. Unbound protein was washed away with 0.05% PBST, and bound scFv was detected using 0.2 μg/ml rabbit anti SNAP‑tag polyclonal primary antibody (GenScript, cat. num. A00684) and a polyclonal goat anti-rabbit HRP-labeled secondary antibody diluted 1:5000 (Abcam, cat. num. ab6721). Finally, 100 μl of freshly-prepared ABTS substrate was added to each well and positive binding was evaluated as described above. The binding strength of each scFv-SNAP-tag fusion protein was compared by adding 1 μg of IMAC-purified protein pre-blocked in 2% MPBS to a total volume of 100 μl in each well followed by ELISA as described above. Bound scFv-SNAP‑tag fusion proteins were detected via their SNAP-tag as described above. The binding activity of directly-labeled scFv clones on whole cells was detected qualitatively by incubating 1 μg of the eluted scFv protein for 1 h on ice protected from light with 5 × 105 freshly-harvested PBMCs or Kasumi‑1 cells washed 3 times in blocking buffer (PBS containing 0.5% bovine serum albumin). After two washing steps with PBS in a cell washer, the cells were re-suspended in 300 μl blocking buffer and used directly for binding analysis (flow cytometry) and internalization analysis. Gating was performed according to physical characteristics in forward and sideward scatter modes, or trypan blue staining in FL-3 to exclude dead cells and debris. The binding activity on freshly isolated peripheral mononuclear cells (PBMC) from AML patients of scFv-Fc fusion proteins were analyzed by flow cytometry using a FITC labeled goat anti-mouse (GAM-FITC) secondary antibody (BD Biosciences, cat. num. 554001). Blast cells from AML PBMC samples were analyzed by flow cytometer with monoclonal anti-CD34 antibody (Leica Biosystems, cat. num. END-L-CE), followed by staining with GAM-FITC. Data were acquired with a Guava easyCyte 5HT apparatus (Merck Millipore, cat. num. 0500-4005).
Binding analysis: Immunohistochemistry
Immunohistochemistry was carried out on serial sections (2 μm) of formalin-fixed paraffin embedded routine iliac crest biopsies from AML patients using the Bond-Max automated immunostainer (Leica Biosystems). Tissue sections were deparaffinized with Bond dewax solution (Leica Biosystems, cat. num. AR9222) and antigen retrieval was performed by heating the sections in 10 mM sodium citrate buffer, pH 6.0, at 95°C for 20 min. After PBS washing, unspecific binding was blocked by incubating 5 min in Novocastra peroxidase block (Leica Biosystems, cat. num. RE7101-CE). The slides were incubated first with the recombinant scFv-Fc fusion protein (150 μg/ml) diluted 1:10 in PBS containing 2% milk powder for 20 min at room temperature. After several PBS washes, slides were visualized using the Bond Polymer Refine Detection System (Leica Biosystems, cat. num. DS9800) containing the secondary peroxidase-labeled goat anti-mouse antibody and activated DAB solution according to the manufacturer's instructions and finally counterstained with Mayer's ‘Hämalaun’ solution to visualize cell nuclei.
Functional affinity constant of selected scFv antibodies
The affinity constants of each selected scFv antibody were determined as previously described31 with the following modifications. The incubation of Kasumi‑1 cells with the Vista Green-labeled (NEB, cat. num. S9147S) scFv-SNAP proteins (0.5–2000 nM) was carried out as described above to reach saturation. After subtracting the background signal produced by intrinsic cell fluorescence and the nonspecific binding of scFv-SNAP‑tag proteins, we calculated the geometric mean of the fluorescence intensity for each scFv and applied concentration. The functional affinity to PBMCs was tested in parallel to demonstrate specificity.
Internalization analysis by flow cytometry
Internalization behavior was tested by flow cytometry using scFv-SNAP-tag fusion proteins labeled with Alexa Fluor 647. Initially, 5 × 105 Kasumi‑1 cells were washed 3 times with PBS, re‑suspended in standard medium and incubated with 1 μg scFv protein for 1 h at 37°C to allow internalization. Parallel incubations were carried out at 4°C in 1% NaN3 as a reference for non-internalization. After a further wash, the surface-bound scFv-SNAP proteins were stripped by adding 0.5 ml 0.05% trypsin for 10 min at 37°C. The reaction was stopped by adding 0.5 ml RPMI plus 10% FBS and excess trypsin was removed by washing twice with PBS. The cell pellet was re‑suspended in blocking buffer and the samples were analyzed using a FACSCalibur flow cytometer (BD Biosciences). Each sample was normalized to the non-internalization control (0%) and the positive binding signal before trypsin treatment (100%). To detect any residual scFv fusion protein on the cell surface, we added 0.5 μl rabbit anti SNAP-tag polyclonal primary antibody and 0.5 μl goat anti-rabbit Alexa Fluor 488 polyclonal secondary antibody (Life Technologies, cat. num. A‑11009).
Internalization analysis by confocal microscopy
The internalization behavior of the selected scFvs was confirmed by confocal imaging. The cells were prepared as described above and monitored with a TCS SP5 confocal microscope (LEICA Microsystem). The proteins labeled with Alexa Fluor 647 (NEB, S9136S) were excited using a helium‑neon laser at 633 nm. Images of fluorophore and transmitted light were merged using LEICA software. Cell internalization kinetics was evaluated by introducing the selected scFv-SNAP fusion proteins into the OPERA live cell imaging system after incubating at 4°C with the target cell line Kasumi‑1. For optimal cell density, 50–100 μl of each sample was pipetted into each well of a black 96‑well microtiter plate with a transparent base (μClear, Greiner bio-one, cat. num. 655096)) and the cells were allowed to settle at 4°C. Kinetic measurement was carried out over the course of 12 h in an Opera® microtiter plate imaging reader (Evotec Technologies).
Construction, expression and purification of scFv-ETA' immunotoxin
Recombinant immunotoxins were constructed using the scFv genes from the ELISA-positive phagemid clones. The reverse-mutated scFv inserts were cloned into the pMT ETA' prokaryotic expression vector32 and introduced into E. coli Rosetta 2 (DE3) (Merck Millipore, cat. num. 70954) for the high-level expression of scFv-ETA' fusion proteins. The scFv-ETA' immunotoxin was expressed under stress in the presence of compatible solutes as previously described.33 The periplasmic fraction was recovered by pelleting the bacteria and re‑suspending in buffer (75 mM Tris‑HCl, 300 mM NaCl, 10% glycerol, one tablet protease inhibitor per 50 ml) then sonicating for 9 × 1 min at 70% intensity. The periplasmic fraction was recovered after centrifugation at 30,000 x g for 30 min and the recombinant immunotoxin was purified in a batch procedure using Ni‑NTA Sepharose (QIAGEN, Hilden, Germany). The flow-through was collected after 2 rounds of incubation. The resin was washed once with buffer 1 (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole), twice with buffer 2 (50 mM NaH2PO4, 300 mM NaCl, 40 mM imidazole) and bound protein was stripped with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole). The eluted protein was finally purified using size exclusion chromatography as described33 and analyzed quantitatively by SDS-PAGE. Functional binding activity was confirmed by flow cytometry using the anti-ETA' monoclonal antibody TC-1 (kindly provided by Dr Galloway, Ohio, USA).
Cytotoxicity of scFv-ETA' constructs
Inhibition of cell proliferation was tested as previously described.30,34,35 A serial dilution of the recombinant immunotoxin (initially 30 ng/μl) was distributed in a 96‑well microtiter plate, and after adding 4 × 104 Kasumi‑1 cells in complete medium, the plates were incubated for 72 h under standard conditions. Cell proliferation was determined by measuring the conversion of tetrazolium salt (XTT) to an orange formazan dye (Cell proliferation kit II, Roche Life Science, cat. num. 11465015001), whose absorbance was measured at 450 nm in an ELISA plate reader. All samples were measured in triplicate. Each sample was normalized to DMSO-treated positive controls (0%) and untreated cells (100%) to determine IC50 values.
The induction of apoptosis was detected using a caspase-3/7 activity detection kit (Apo-One Homogeneous Caspase-3/7 Assay, Promega, cat. num. G7792) according to the manufacturer's instructions. We seeded 0.5 ml aliquots of 5 × 105 Kasumi‑1 cells in 24-well microtiter plates and incubated them with 1 μg immunotoxin for 24 h under standard conditions. Samples were analyzed in duplicate in an ELISA plate reader. Apoptosis was also analyzed by annexin A5 staining as previously described.25 Cells were treated as described above and apoptotic cells were stained with HEK293T cell supernatant containing an annexin A5 eGFP fusion protein.25 After counterstaining necrotic cells with propidium iodide, samples were measured by flow cytometry in a FACSCalibur device.
Bioinformatic analysis of antibody variable regions
The complementarity-determining regions (CDRs) in the anti-AML scFv antibodies were identified using standard bioinformatics tools and techniques. First, positive binders identified by monoclonal phage ELISA were prepared for sequencing by extracting the plasmid vectors using the NucleoSpin Plasmid kit (Macherey-Nagel, 740588.10) and subsequent insert PCR using the primers LMB3 (5'-CAG GAA ACA GCT ATG AC-3') and fdSEQ1 (5'-GAA TTT TCT GTA TGA GG-3') as described.16 The cDNA and amino acid sequences were analyzed using the Vector NTI Advance 11 Sequence Analysis Software and Vector NTI AlignX algorithm (Invitrogen), respectively. Unique sequences were then analyzed with V-BASE (http://vbase.mrc-cpe.cam.ac.uk/) and IgBLAST (www.ncbi.nlm.nih.gov/blast/Blast.cgi) using the Kabat numbering system36 to determine the framework regions and CDRs of the variable heavy (VH) and variable light (VL) chains. SWISS-MODEL (http://swissmodel.expasy.org/)37-39 was used to model the VH and VL chains and PyMOL v1.5 was used for computer graphic modeling. Homology among the selected scFv antibodies was investigated by phylogenetic analysis using the Neighbor-Joining algorithm.
Data analysis
Quantitative analysis of soluble scFv proteins was carried out using AIDA image analyzer software v4.27 (Raytest, Straubenhardt, Germany) after digital scanning of Coomassie-stained SDS polyacrylamide gels. Fluorophore-labeled scFvs were detected in the VersaDoc MP System (BIO-Rad, Offenbach, Germany) using QuantityOne Basic 1-D analysis software v4.2.1 (BIO-Rad). Data from flow cytometry were evaluated using CellQuest software (Becton Dickinson, Heidelberg, Germany) and the Windows Multiple Document Interface for Flow Cytometry v2.8 (WinMDI, Joseph Trotter, USA). Statistical analysis was carried out with GraphPad Prism software (GraphPad, La Jolla, CA, USA). Data were presented as mean ± standard deviation (SD). A two-tailed t-test was used to determine the significance of independent experiments. The criterion p < 0.05 was considered significant (*), p < 0.01 very significant (**) and p < 0.001 highly significant (***). Saturation binding curves were generated by non‑linear regression using the Levenberg-Marquardt algorithm. Immunotoxin IC50 values were calculated by non‑linear regression using a sigmoidal dose–response equation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We like to thank Dr. Stefano Di Fiore for his support in confocal microscopy and Dr. Richard Twyman for critical reading of the document.
Funding
This publication is based upon work that was partly supported by the Deutsche Krebshilfe (109383 and 109127).
References
- 1. Abutalib SA, Tallman MS. Monoclonal antibodies for the treatment of acute myeloid leukemia. Curr Pharm Biotechnol 2006; 7:343-69; PMID:17076651; http://dx.doi.org/ 10.2174/138920106778521578 [DOI] [PubMed] [Google Scholar]
- 2.American Cancer Society Cancer Facts & Figures 2013. Atlanta: American Cancer Society, 2013. Available from: http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-036845.pdf . [Google Scholar]
- 3. Stone RM. The difficult problem of acute myeloid leukemia in the older adult. CA Cancer J Clin 2002; 52:363-71; PMID:12469764; http://dx.doi.org/ 10.3322/canjclin.52.6.363 [DOI] [PubMed] [Google Scholar]
- 4. Mulford D. Antibody therapy for acute myeloid leukemia. Semin Hematol 2008; 45:104-9; PMID:18381105; http://dx.doi.org/ 10.1053/j.seminhematol.2008.02.008 [DOI] [PubMed] [Google Scholar]
- 5. Pagel JM, Gooley TA, Rajendran J, Fisher DR, Wilson WA, Sandmaier BM, Matthews DC, Deeg HJ, Gopal AK, Martin PJ, et al. Allogeneic hematopoietic cell transplantation after conditioning with 131I-anti-CD45 antibody plus fludarabine and low-dose total body irradiation for elderly patients with advanced acute myeloid leukemia or high-risk myelodysplastic syndrome. Blood 2009; 114:5444-53; PMID:19786617; http://dx.doi.org/ 10.1182/blood-2009-03-213298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bunjes D, Buchmann I, Duncker C, Seitz U, Kotzerke J, Wiesneth M, Dohr D, Stefanic M, Buck A, Harsdorf SV, et al. Rhenium 188-labeled anti-CD66 (a, b, c, e) monoclonal antibody to intensify the conditioning regimen prior to stem cell transplantation for patients with high-risk acute myeloid leukemia or myelodysplastic syndrome: results of a phase I-II study. Blood 2001; 98:565-72; PMID:11468151; http://dx.doi.org/ 10.1182/blood.V98.3.565 [DOI] [PubMed] [Google Scholar]
- 7. ten Cate B, Bremer E, de Bruyn M, Bijma T, Samplonius D, Schwemmlein M, Huls G, Fey G, Helfrich W. A novel AML-selective TRAIL fusion protein that is superior to Gemtuzumab Ozogamicin in terms of in vitro selectivity, activity and stability. Leukemia 2009; 23:1389-97; PMID:19262596; http://dx.doi.org/ 10.1038/leu.2009.34 [DOI] [PubMed] [Google Scholar]
- 8. US Food and Drug Administration Pfizer Voluntarily Withdraws Cancer Treatment Mylotarg from U.S. Market; 2010. http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm216448.htm [Google Scholar]
- 9. Frankel AE, Sievers EL, Scheinberg DA. Cell surface receptor-targeted therapy of acute myeloid leukemia: a review. Cancer Biother Radiopharm 2000; 15:459-76; PMID:11155818; http://dx.doi.org/ 10.1089/cbr.2000.15.459 [DOI] [PubMed] [Google Scholar]
- 10. Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol 1987; 196:901-17; PMID:3681981; http://dx.doi.org/ 10.1016/0022-2836(87)90412-8 [DOI] [PubMed] [Google Scholar]
- 11. Chothia C, Lesk AM, Tramontano A, Levitt M, Smith-Gill SJ, Air G, Air G, Sheriff S, Padlan EA, Davies D, et al. Conformations of immunoglobulin hypervariable regions. Nature 1989; 342:877-83; PMID:2687698; http://dx.doi.org/ 10.1038/342877a0 [DOI] [PubMed] [Google Scholar]
- 12. Stone RM, O'Donnell MR, Sekeres MA. Acute myeloid leukemia. Hematology Am Soc Hematol Educ Program 2004:98-117; PMID:15561679; http://dx.doi.org/ 10.1182/asheducation-2004.1.98 [DOI] [PubMed] [Google Scholar]
- 13. Tallman MS. New strategies for the treatment of acute myeloid leukemia including antibodies and other novel agents. Hematology Am Soc Hematol Educ Program 2005:143-50; PMID:16304372; http://dx.doi.org/ 10.1182/asheducation-2005.1.143 [DOI] [PubMed] [Google Scholar]
- 14. Khandare JJ, Minko T. Antibodies and peptides in cancer therapy. Crit Rev Ther Drug Carrier Syst 2006; 23:401-35; PMID:17425513; http://dx.doi.org/ 10.1615/CritRevTherDrugCarrierSyst.v23.i5.20 [DOI] [PubMed] [Google Scholar]
- 15. Becerril B, Poul MA, Marks JD. Toward selection of internalizing antibodies from phage libraries. Biochem Biophys Res Commun 1999; 255:386-93; PMID:10049718; http://dx.doi.org/ 10.1006/bbrc.1999.0177 [DOI] [PubMed] [Google Scholar]
- 16. Fitting J, Killian D, Junghanss C, Willenbrock S, Murua Escobar H, Lange S, Nolte I, Barth S, Tur MK. Generation of recombinant antibody fragments that target canine dendritic cells by phage display technology. Vet Comp Oncol 2011; 9:183-95; PMID:21848621; http://dx.doi.org/ 10.1111/j.1476-5829.2010.00246.x [DOI] [PubMed] [Google Scholar]
- 17. Barth S, Winkler U, Diehl V, Engert A. [Immunotoxins. Mechanism of action and applications in malignant diseases]. Internist (Berl) 1997; 38:1063-9; PMID:9453955; http://dx.doi.org/ 10.1007/s001080050118 [DOI] [PubMed] [Google Scholar]
- 18. Hetzel C, Bachran C, Tur MK, Fuchs H, Stocker M. Improved immunotoxins with novel functional elements. Curr Pharm Des 2009; 15:2700-11; PMID:19689340; http://dx.doi.org/ 10.2174/138161209788923930 [DOI] [PubMed] [Google Scholar]
- 19. Chiron M, Jaffrezou JP, Carayon P, Bordier C, Roubinet F, Xavier C, Brandely M, Laurent G. Induction and increase of HLA-DR antigen expression by immune interferon on ML-3 cell line enhances the anti-HLA-DR immunotoxin activity. Clin Exp Immunol 1990; 82:214-20; PMID:2122930; http://dx.doi.org/ 10.1111/j.1365-2249.1990.tb05429.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stish BJ, Chen H, Shu Y, Panoskaltsis-Mortari A, Vallera DA. Increasing anticarcinoma activity of an anti-erbB2 recombinant immunotoxin by the addition of an anti-EpCAM sFv. Clin Cancer Res 2007; 13:3058-67; PMID:17505009; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-2454 [DOI] [PubMed] [Google Scholar]
- 21. Schmidt MM, Thurber GM, Wittrup KD. Kinetics of anti-carcinoembryonic antigen antibody internalization: effects of affinity, bivalency, and stability. Cancer Immunol Immunother 2008; 57:1879-90; PMID:18408925; http://dx.doi.org/ 10.1007/s00262-008-0518-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hetzel C, Bachran C, Fischer R, Fuchs H, Barth S, Stocker M. Small cleavable adapters enhance the specific cytotoxicity of a humanized immunotoxin directed against CD64-positive cells. J Immunother 2008; 31:370-6; PMID:18391759; http://dx.doi.org/ 10.1097/CJI.0b013e31816a2d23 [DOI] [PubMed] [Google Scholar]
- 23. Stahnke B, Thepen T, Stocker M, Rosinke R, Jost E, Fischer R, Tur MK, Barth S. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol Cancer Ther 2008; 7:2924-32; PMID:18790773; http://dx.doi.org/ 10.1158/1535-7163.MCT-08-0554 [DOI] [PubMed] [Google Scholar]
- 24. Schiffer S, Letzian S, Jost E, Mladenov R, Hristodorov D, Huhn M, Fischer R, Barth S, Thepen T. Granzyme M as a novel effector molecule for human cytolytic fusion proteins: CD64-specific cytotoxicity of Gm-H22(scFv) against leukemic cells. Cancer Lett 2013; 341:178-85; PMID:23973499; http://dx.doi.org/ 10.1016/j.canlet.2013.08.005 [DOI] [PubMed] [Google Scholar]
- 25. Tur MK, Neef I, Jager G, Teubner A, Stocker M, Melmer G, Barth S. Immunokinases, a novel class of immunotherapeutics for targeted cancer therapy. Current pharmaceutical design 2009; 15:2693-9; PMID:19689339; http://dx.doi.org/ 10.2174/138161209788923877 [DOI] [PubMed] [Google Scholar]
- 26. Hristodorov D, Nordlohne J, Mladenov R, Huhn M, Fischer R, Thepen T, Thepen T, Barth S. Human microtubule-associated protein tau mediates targeted killing of CD30(+) lymphoma cells in vitro and inhibits tumour growth in vivo. Br J Haematol 2014; 164:251-7; PMID:24164493; http://dx.doi.org/ 10.1111/bjh.12626 [DOI] [PubMed] [Google Scholar]
- 27. World Medical A. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 2013; 310:2191-4; PMID:24141714; http://dx.doi.org/ 10.1001/jama.2013.281053 [DOI] [PubMed] [Google Scholar]
- 28. Kampmeier F, Ribbert M, Nachreiner T, Dembski S, Beaufils F, Brecht A, Barth S. Site-Specific, Covalent Labeling of Recombinant Antibody Fragments via Fusion to an Engineered Version of 6-O-Alkylguanine DNA Alkyltransferase. Bioconjug Chem 2009; 20:1010-5; PMID:19388673; http://dx.doi.org/ 10.1021/bc9000257 [DOI] [PubMed] [Google Scholar]
- 29. Tur MK, Rothe A, Huhn M, Goerres U, Klimka A, Stocker M, Engert A, Fischer R, Finner R, Barth S. A novel approach for immunization, screening and characterization of selected scFv libraries using membrane fractions of tumor cells. Int J Mol Med 2003; 11:523-7; PMID:12632108 [PubMed] [Google Scholar]
- 30. Stocker M, Tur MK, Sasse S, Krussmann A, Barth S, Engert A. Secretion of functional anti-CD30-angiogenin immunotoxins into the supernatant of transfected 293T-cells. Protein Expr Purif 2003; 28:211-9; PMID:12699683; http://dx.doi.org/ 10.1016/S1046-5928(02)00709-X [DOI] [PubMed] [Google Scholar]
- 31. Benedict CA, MacKrell AJ, Anderson WF. Determination of the binding affinity of an anti-CD34 single-chain antibody using a novel, flow cytometry based assay. J Immunol Methods 1997; 201:223-31; PMID:9050944; http://dx.doi.org/ 10.1016/S0022-1759(96)00227-X [DOI] [PubMed] [Google Scholar]
- 32. Tur MK, Huhn M, Thepen T, Stocker M, Krohn R, Vogel S, Jost E, Osieka R, van de Winkel JG, Fischer R, et al. Recombinant CD64-specific single chain immunotoxin exhibits specific cytotoxicity against acute myeloid leukemia cells. Cancer Res 2003; 63:8414-9; PMID:14679004 [PubMed] [Google Scholar]
- 33. Tur MK, Huhn M, Jost E, Thepen T, Brummendorf TH, Barth S. In vivo efficacy of the recombinant anti-CD64 immunotoxin H22(scFv)-ETA' in a human acute myeloid leukemia xenograft tumor model. Int J Cancer 2011; 129:1277-82; PMID:21077160; http://dx.doi.org/ 10.1002/ijc.25766 [DOI] [PubMed] [Google Scholar]
- 34. Barth S, Huhn M, Matthey B, Klimka A, Galinski EA, Engert A. Compatible-solute-supported periplasmic expression of functional recombinant proteins under stress conditions. Appl Environ Microbiol 2000; 66:1572-9; PMID:10742244; http://dx.doi.org/ 10.1128/AEM.66.4.1572-1579.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barth S, Huhn M, Matthey B, Tawadros S, Schnell R, Schinkothe T, Diehl V, Engert A. Ki-4(scFv)-ETA', a new recombinant anti-CD30 immunotoxin with highly specific cytotoxic activity against disseminated Hodgkin tumors in SCID mice. Blood 2000; 95:3909-14; PMID:10845927 [PubMed] [Google Scholar]
- 36. Kabat EA, Wu TT. Identical V region amino acid sequences and segments of sequences in antibodies of different specificities. Relative contributions of VH and VL genes, minigenes, and complementarity-determining regions to binding of antibody-combining sites. J Immunol 1991; 147:1709-19; PMID:1908882 [PubMed] [Google Scholar]
- 37. Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2006; 22:195-201; PMID:16301204; http://dx.doi.org/ 10.1093/bioinformatics/bti770 [DOI] [PubMed] [Google Scholar]
- 38. Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res 2003; 31:3381-5; PMID:12824332; http://dx.doi.org/ 10.1093/nar/gkg520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997; 18:2714-23; PMID:9504803; http://dx.doi.org/ 10.1002/elps.1150181505 [DOI] [PubMed] [Google Scholar]