

Abstract
Dihydroartemisinin (DHA) exhibits anticancer activities in a variety of cancer cells, but DHA alone are not effective enough for cancer therapy. In this study we found the stress-regulated protein p8 was obviously increased after DHA treatment in several cancer cells, which further to induce autophagy by the upregulation of endoplasmic reticulum stress-related protein ATF4 and CHOP. Furthermore, when we silenced p8 by siRNA in cancer cells, the apoptosis induced by DHA were notably increased, whereas the overexpression of p8 in cancer cells leaded to the resistance to DHA-induced apoptosis. Moreover, we found the inhibition of autophagy with chloroquine (CQ) can enhance the anticancer effect of DHA both in vitro and in vivo. In conclusion, we found that p8-mediated autophagy attenuates DHA-induced apoptosis in cancer cells, which provides evidence to support the use p8 as a cancer therapeutic target, and suggests that the combination treatment with DHA and autophagy inhibitor might be an effective cancer therapeutic strategy.
Keywords: autophagy, chloroquine, dihydroartemisinin, endoplasmic reticulum stress, p8
Abbreviations
- DHA
dihydroartemisinin
- ART
artemisinin
- CQ
chloroquine
- AVOs
acidic vesicular organelles
- ATF4
activating transcription factor 4
- CHOP
C/EBP homologous protein
- ERs
endoplasmic reticulum stress
- AO
Acridine orange
- MDC
Dansylcadaverine
Introduction
Dihydroartemisinin (DHA), a main active metabolite of artemisinin (ART), which is making a crucial contribution to the therapy of malaria,1 has been demonstrated to exert preferentially cytotoxic effects toward various human cancers, including breast cancer,2 ovarian cancer,3 cervical cancer4 and colorectal carcinoma.5 Recent studies have shown that the anti-cancer activity of ART derivatives including DHA is mainly executed by iron-mediated cleavages of endoperoxide bridges.5,6 Since most cancer cells have higher iron influx than normal cells, DHA exert selective toxicity toward cancer cells.7,8 Additionally, clinical trials have shown that ART derivatives are well tolerated with insignificant side-effects in malaria therapy, indicating DHA have important potential in oncology.
p8, with a theoretical molecular mass of 8872.7Da, also known as nuclear protein 1(Nurp1) or candidate of metastasis-1(Com-1), was first described as being preferentially upregulated in pancreatic acinar cells during the acute phase of pancreatitis.9 P8 participates in a wide range of biological functions in tumorigenesis, including anti-inflammatory,10 cell cycle regulation,11 autophagy,12 and apoptosis inhibition.13 Accumulating studies have shown that p8 is overexpressed in various cancer cells including pancreatic cancer,14 colorectal carcinoma15 and brain tumors.16 Moreover, it has been reported that p8 is involved in the resistance of cancer cells to gemcitabine and adriamycin.14,17 Therefore, p8 might be a new drug-targetable gene in preventing the progression and metastasis of cancer.18
In this study, we discovered DHA upregulates p8 remarkably in several cancer cells, and we sought to find out what role p8 plays in the antiproliferative actions of DHA in cancer cells. It has been reported that endoplasmic reticulum stress (ERs)-associated molecule ATF4 (activating transcription factor 4) and CHOP (C/EBP homologous protein) are downstream targets of p8.19 ER stress occurs when the accumulation of unfolded proteins in the ER is exceeded, and is caused by multiple stimuli including hypoxia, oxidants or reducing agents, glucose deprivation, protein inclusion bodies, etc.20 The major pathway of ER stress is unfolded protein response (UPR), which is identified to recover ER homeostasis and promote cell survival.21 In addition, ER stress has proved to be one of the inducers of autophagy.22 Interestingly, DHA induced-ER stress has been observed in several tumor cells,23,24 and our previous studies have demonstrated that DHA both induce apoptosis and autophagy in various cancer cells.25,26 The Complex relationships among ER stress, autophagy and apoptosis remain unclear, but it's perhaps noteworthy that we found p8 is involved in all of these 3 biological functions induced by DHA.
As shown in our results, when silenced p8 in cancer cells, both ER stress and autophagy induced by DHA were reduced while the apoptosis rates were further increased. Furthermore, when inhibited autophagy in cancer cells through pretreating with chloroquine (CQ), which can arrest degradation of the autolysosome,27 we found the anticancer effect of DHA was notably enhanced. Interestingly, CQ is also widely used as an antimalarial agent, and for the first time, we discovered the 2 classical antimalarial agents, DHA and CQ, have the synergistic effect in cancer suppression.
Results
Dihydroartemisinin upregulates p8, ATF4 and CHOP at transcription level and protein level in HeLa and HCT116 cells
We used human cervical carcinoma cell line HeLa and hunman colon carcinoma cell line HCT116 to investigate the expression changes of p8, ATF4 and CHOP after DHA treatment. As illustrated in Figure 1A, the time-dependent increases in p8, ATF4 and CHOP were observed in both 2 cell lines that were treated with 20 μM DHA. The up-regulation of p8 was observed as early as at 3 h, and the increase of ATF4 was observed at 6 h, whereas the increase of CHOP was observed at least after 9 h treatment under our experimental condition. Likewise, by using real-time quantitative PCR, we confirmed that DHA upregulates the mRNA levels of p8, ATF4 and CHOP both in HeLa and HCT116 cells (Fig. 1B and C).
Figure 1.
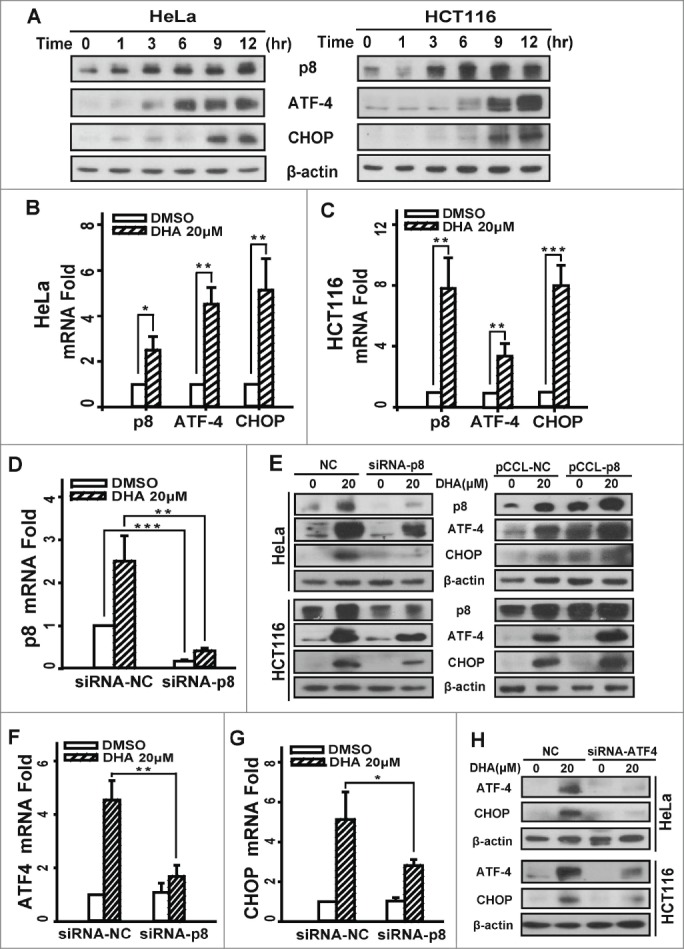
Dihydroartemisinin upregulates p8, ATF4 and CHOP at transcription level and protein level in HeLa and HCT116 cells. (A). HeLa and HCT116 cells were treated with 20 μM DHA for the indicated times, and total cell extracts were probed with antibodies against p8, ATF4, CHOP and β-actin. (B and C).HeLa and HCT116 cells treated with 20 μM DHA for 9 h, then total RNA was extracted and p8, ATF4 and CHOP mRNA expression was analyzed by RT-PCR, using GAPDH as a control gene. Three independent experiments were performed and the values were expressed as the mean ± SD. (E and H). After transfection with pCCL− p8 plasmid or specifically targeted siRNA (p8 or ATF4) for 24 h, HeLa and HCT116 cells were treated with DHA (20 μM) for 24 h, after which p8, ATF4 and CHOP protein levels were measured by protein gel blot analysis. β-actin was measured as the loading control. (D, F-G) After transfection with specifically targeted p8 siRNA for 24 h, HeLa cells treated with DHA (20 μM) for 9 h, after which total RNA was extracted and p8, ATF4 and CHOP mRNA expression was analyzed by RT-PCR, using GAPDH as a control gene. Three independent experiments were performed and the values were expressed as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
To investigate whether ATF4 and CHOP are regulated by p8, which are involved in the response to endoplasmic reticulum stress (ERS), we transfected cells with p8 siRNA, and then we measured the expression levels of ATF4 and CHOP. The transfection efficiency was 84.0%, which detected by real-time quantitative PCR (Fig. 1D). We measured ATF4 and CHOP levels after treatment with DHA for 24 h by western blot. To compare with the negative-knockdown cells, the level of ATF4 and CHOP significantly decreased after DHA exposure in p8-knockdown cells (Fig. 1E). The identical results were obtained in mRNA levels (Fig. 1F and G). Conversely, when cells were transfected with p8-overexpress plasmid, the ATF4 and CHOP express levels further increased than the negative control cells after DHA treatment (Fig. 1D). Taken together, these data demonstrated that DHA-induced increases of ATF4 and CHOP were mediated by p8 upregulation. In addition, the CHOP expression was reduced when ATF4 was knockdown in both HeLa and HCT116 cells (Fig. 1H), indicating that the expression of CHOP is regulated by ATF4 after DHA treatment. The analogous results were validated in human hepatoma HepG2 cells and ovarian carcinoma SKOV3 cells (Fig. S1A and Fig. S2A).
P8 upregulation decreases the sensitivity of tumor cells to dihydroartemisinin
We next investigated the role of p8 in DHA-induced tumor suppression. Cells were transfected with p8 siRNA or p8 overexpressed plasmid and treated with a series of concentration of DHA for 48 h, and after that we figured out the survival functions by SRB assays. As shown in Figure 2A and Figure S1B–C, the survival functions decreased when p8 was silenced by siRNA, whereas the survival functions increased in p8 overexpressed cells compared with the negative control cells.
Figure 2.

p8 upregulation decreases the sensitivity of tumor cells to dihydroartemisinin. After transfection with pCCL− p8 plasmid or specifically targeted p8 siRNA for 24 h, (A) HeLa and HCT116 cells were treated with serial concentrations of DHA for 48 h, and then cell survival was detected by SRB assay. (B) HeLa and HCT116 were treated with 20 μM DHA for 24 h, after which PARP, caspase-3 and cleaved-caspase-3 protein levels were measured by western blot analysis. (C-F) siRNA or plasmid-transfected HeLa cells treated with 20 μM DHA for 24 h, and quantification of apoptosis was performed using Annexin V/PI or Annexin V-PE/7-AAD followed by FACS analysis. Three independent experiments were performed and the values were expressed as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
We further observed the apoptosis after treatment with DHA in p8-knockdown or p8 overexpressed cells. Since activations of caspases are key players in apoptosis execution,28 we investigated the effect of DHA on the protein levels of PARP(a major substrate of caspase3), caspase3 and cleaved-caspase3 in HeLa and HCT116 cells. Compared with in negative-knockdown cells, DHA caused an enhanced cleavage of caspase3 and PARP in p8-knockdown cells (Fig. 2B and Fig. S1A). But in p8 overexpressed cells, DHA caused less cleavage of caspase3 and PARP than in negative control cells (Fig. 2B). The same results were observed when we detected apoptosis rate by flow cytometry analysis. As presented in Figure 2C and D, the apoptosis rate remarkably increased in p8-knockdown HeLa cells treated with DHA (20 μM, 24 h), while the percentage of apoptotic cells caused by DHA was much lower in p8 overexpressed HeLa cells than in negative control HeLa cells (Fig. 2E and F). These results confirmed that the higher expression of p8, the lower effect of DHA performs in tumor inhibition.
ATF4 or CHOP depletion enhances the sensitivity of tumor cells to dihydroartemisinin
In this study we have confirmed ATF4 and CHOP are regulated by p8 after DHA treatment, to test the correlation between the expression levels of ATF4 or CHOP and sensitivity to DHA treatment, we silenced ATF4 or CHOP by transfecting specific siRNAs into HeLa and HCT116 cells before DHA treatment and Annexin V-FITC/PI assays. Notably, ATF4-knockdown increased DHA-driven apoptosis to a greater extent in HeLa cells treated with 20 μM DHA (Fig. 3A and C), and the apoptosis rate of CHOP-knockdown cells rise significantly comparing to negative control cells (Fig. 3B and C). The similar result was also observed in the effect of DHA on the apoptosis execution protein levels. Whether ATF4 depletion or CHOP depletion, the PARP cleavage and caspase3 cleavage driven by DHA were notably increased in both HeLa and HCT116 cells (Fig. 3D), further suggesting that the silencing of ATF4 or CHOP enhances the sensitivity of tumor cells to DHA-driven apoptosis in vitro. The similar results were obtained in HepG2 cells and SKOV3 cells (Fig. S2A-D).
Figure 3.

ATF4 or CHOP depletion enhances the sensitivity of tumor cells to dihydroartemisinin. After transfection with specifically targeted siRNA (ATF4 or CHOP) for 24 h, (A-C) HeLa cells treated with 20 μM DHA for 24 h, and quantification of apoptosis was performed using Annexin V/PtdIns followed by FACS analysis. Three independent experiments were performed and the values were expressed as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001. (D) HeLa and HCT116 were treated with 20 μM DHA for 24 h, after which PARP, caspase-3 and cleaved-caspase-3 protein levels were measured by protein gel blot analysis.
p8-ATF4-CHOP axis is involved in dihydroartemisinin -induced autophagy in HeLa and HCT116 cells
In our early study we have demonstrated that DHA stimulates the induction of autophagy in several cancer cell lines including HeLa and HCT116 cells.26 In this study we found that p8-ATF4-CHOP axis is closely related to DHA-induced autophagy. We determined the effect of DHA on formation of acidic vesicular organelles (AVOs) by acridine orange (AO) staining and MDC staining, to visualize autophagosome formation in the cells. In negative-knockdown HeLa cells, treatment with 20 μM DHA resulted in the formation of red fluorescent AVOs, whereas the DHA-induced AVOs were reduced in p8-knockdown HeLa cells (Fig. 4A). Likewise, when performed by DMC staining after DHA treatment, the fluorescent density was decreased in p8-knockdown HeLa cells compared to the negative-knockdown cells (Fig. 4A), indicating p8 knockdown reverses DHA induced autophagy.
Figure 4.
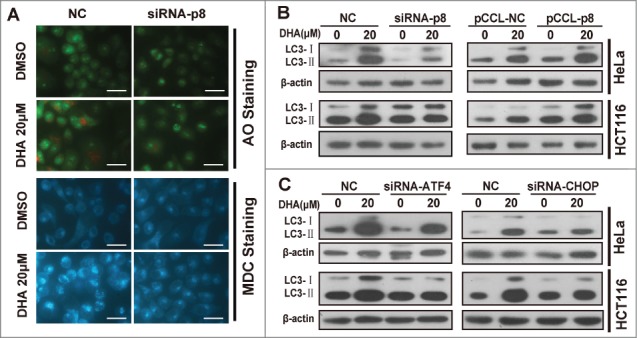
p8-ATF4-CHOP axis is involved in dihydroartemisinin -induced autophagy in HeLa and HCT116 cells. (A) After transfection with specifically targeted p8 siRNA for 24 h, HeLa cells treated with 20 μM DHA for 24 h, and then Acridine orange (AO) staining, MDC staining were performed. Bar, 50 μM. (B) After transfection with pCCL− p8 plasmid or specifically targeted siRNA (p8, ATF4 and CHOP) for 24 h, HeLa and HCT116 were treated with 20 μM DHA for 24 h, after which LC3 protein levels were measured by western blot analysis.
The conversion of LC3-I to LC3-II is a widely accepted autophagy marker.29 As showed in Figure 4B and Figure S1A, the LC3-II accumulations induced by DHA were reduced when p8 was silenced by siRNA. Conversely, the LC3-II accumulations were further increased after DHA treatment when p8 was overexpressed. We discovered similar results when the ATF4 or CHOP was silenced by siRNAs (Fig. 4C and Fig. S2A-B), indicating the p8-ATF4-CHOP axis plays a critical role in DHA induced autophagy in tumor cells.
Inhibition of autophagy increases apoptosis in dihydroartemisinin-treated tumor cells
Autophagy is a lysosomal degradation pathway for the breakdown of damaged or superfluous proteins and organelles, and is a double-edged sword in cancer.30 However, autophagy has a more prominent role in prolonging survival in tumor cells with defects in apoptosis.31 To determine whether autophagy promotes or attenuates apoptosis in DHA-treated tumor cells, we pretreated with 1 mM 3-MA (an inhibitor of an early stage of autophagy) or 10 μM CQ (an inhibitor of which block the downstream steps of autophagy) and then detected the apoptosis rate in HeLa cells that were treated with 20 μM DHA. 3-MA or CQ alone induced low percentages of cells undergoing apoptosis, which were close to the control group. DHA alone induced 20.6% of cells undergoing apoptosis, while combined with 3-MA or CQ, the apoptosis rates rose significantly (Fig. 5A and B). Furthermore, we knockdown autophagy related protein 5(Atg5) to block autophagy in Hela cells, and found that DHA treatment induced higher apoptosis rate in Atg5-knockdown cells than in negative-knockdown cells (Fig. 5C). Taken together, pharmacological inhibition of autophagy at different steps or selective knockdown of Atg5 both strongly increased DHA-induced apoptosis.
Figure 5.
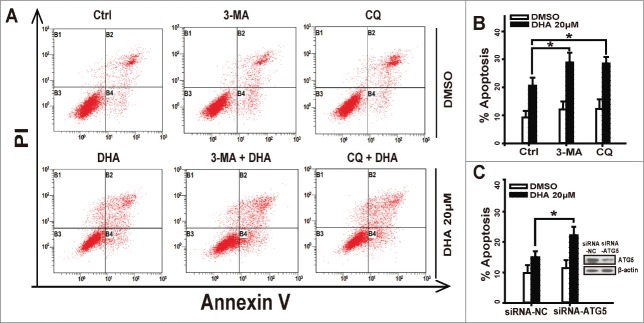
Inhibition of Autophagy increases apoptosis in dihydroartemisinin-treated tumor cells. (A-B) HeLa cells were pretreated with 3-MA(1 mM) or CQ(10 μM)for 1 h, then cotreated with DHA(20 μM) for 24 h and were stained with Annexin V/PI and detected by FACS analysis. (C) After transfection with specifically targeted Atg-5 siRNA for 24 h, HeLa cells were treated with 20 μM DHA and were stained with Annexin V/PtdIns and detected by FACS analysis. Three independent experiments were performed and the values were expressed as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
The combination of dihydroartemisinin and chloroquine exerted synergistic anti-tumor efficacy both in vitro and in vivo
In present study we have observed that CQ, as an inhibitor of autophagy, enhances the apoptosis rate in DHA-treated HeLa cells, so next we sought to further demonstrate the synergistic effects of DHA and CQ on anti-tumor efficacy. First, we employed SRB assay to figure out the survival function and combination index (CI values) of DHA/CQ combination treatment in HeLa and HCT116 cells. Survival fraction curves were displayed in Figure 6A and B, compared with mono-treatment, the combination of DHA and CQ notably decreased the survival function in both HeLa and HCT116 cells. Subsquently, CI values were calculated using Calcucyn at the fixed-ratio concentrations of DHA and CQ. As shown in Table 1, most groups of DHA plus CQ exerted synergy (CI < 0.70) or strong synergy (CI < 0.30) in the tested cancer cells lines. Next, we performed protein gel blot assays. Treatment with DHA (20 μM) combined with CQ (10 μM) for 24 h caused much more significant cleavage of PARP, caspase3 compared with the single agent-treated cells, indicating the involvement of caspases-mediated apoptosis triggered by DHA/CQ combined treatment (Fig. 6C), and further favoring the synergistic efficacy of CQ on DHA-induced apoptosis.
Figure 6.
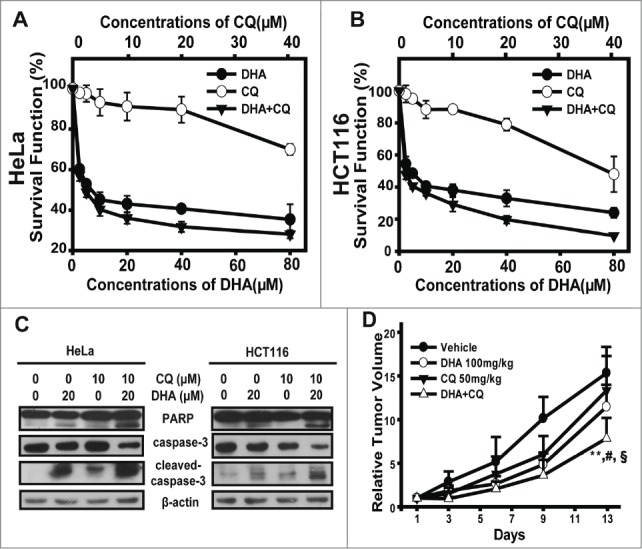
The combination of dihydroartemisinin and chloroquine exerted synergistic anti-tumor efficacy both in vitro and in vivo. (A–B) SRB assays were used to examine the cell proliferation inhibitory activities in HeLa cells (A) and HCT116 cells (B). Cells in 96-well plates were exposed to serial concentrations of DHA, CQ or the constant-ratio mixture of these 2 for 48 h. Dose–response curves of 2 cell lines to DHA, CQ and the combination were presented. Each condition had triplicates, and standard deviation was represented as error bars. The CI values were demonstrated in Table 1. (C) HeLa and HCT116 cells were pretreated with CQ(10 μM)for 1 h, then cotreated with DHA(20 μM) for 48 h and after which PARP, caspase-3 and cleaved-caspase-3 protein levels were measured by protein gel blot analysis. (D) The mice transplanted with HeLa human xenografts were randomly divided into 4 groups and given administration of DHA (100 mg/kg, i.g.), CQ (50 mg/kg, i.p.), the combination or vehicle daily for a period of 13 days. Relative tumor volume are expressed as mean ± SD (n = 8 per group). ** Relative to Vehicle group, P < 0.01; # Relative to DHA group, P < 0.05;§Relative to CQ group, P < 0.05. Tumor weight, inhibition rate, and T/C value (RTV of treatment/RTV of control) were shown in Table 2.
Table 1.
CI values of DHA at concentrations applied in combination with CQ in Hela and HCT116 cells
| Hela |
HCT116 |
||||
|---|---|---|---|---|---|
| DHA (μM) | CQ (μM) | Combination index | DHA (μM) | CQ (μM) | Combination index |
| 2.5 | 1.25 | 0.899 | 2.5 | 1.25 | 0.487 |
| 5 | 2.5 | 0.538 | 5 | 2.5 | 0.446 |
| 10 | 5 | 0.315 | 10 | 5 | 0.544 |
| 20 | 10 | 0.335 | 20 | 10 | 0.485 |
| 40 | 20 | 0.351 | 40 | 20 | 0.289 |
| 80 | 40 | 0.396 | 80 | 40 | 0.126 |
A CI less than 0.90 indicates synergism; 0.10, very strong synergism; 0.10–0.30, strong synergism; 0.30–0.70, synergism; 0.70–0.85, moderate synergism; 0.85–0.9, slight synergism; 0.90–1.10, additive; and more than 1.10, antagonism.
To test whether this synergistic effect could be reproduced in vivo, the combination therapy of DHA and CQ was tested against HeLa xenografts in nude mice. The i.g. administration of DHA at the dose of 100 mg/kg daily or i.p. administration of CQ at dose of 50 mg/kg daily for 13 days only modestly reduced tumor weight; and the combination therapy with DHA and CQ significantly decreased the tumor weight (Table 2). As illustrated in Figure 6D, similar tumor growth inhibitory effect of DHA and CQ were observed on HeLa xenografts models. Both of single agent treatment groups resulted in a moderate therapeutic effect, as the T/C value of DHA-treated group was 74.7%, and T/C value of CQ-treated group was 87.0%. As expected, the combination therapy exhibited distinct tumor growth inhibition (the T/C value 51.2%), with a significant greater extent than DHA-, CQ- or vehicle-treated mice (mean RTV of combination group vs mean RTV of DHA-group: P < 0.05; mean RTV of combination group vs mean RTV of CQ-group: P < 0.05; mean RTV of combination group vs mean RTV of vehicle-group: P < 0.001), but caused no extended body weight loss to the animals. Taken together, these data revealed that the combination of DHA and CQ produced much more potent tumor growth inhibitory effects, without significant toxicities to the animals.
Table 2.
In vivo efficacy of DHA in combination with CQ against human HeLa xenografts
| No. of animals |
Body Weight |
||||||
|---|---|---|---|---|---|---|---|
| start | end | start | end | Tomur weight (g) | Inhibition (%) | T/C (%) | |
| Vehicle | 8 | 8 | 19.3 | 20.8 | 0.28 ± 0.17 | / | / |
| DHA(100 mg/kg) | 8 | 8 | 20.1 | 22.1 | 0.21 ± 0.08 | 26.9 | 74.7 |
| CQ(50 mg/kg) | 8 | 8 | 19.1 | 20.9 | 0.24 ± 0.08 | 16.6 | 87.0 |
| DHA + CQ | 8 | 8 | 18.9 | 20.7 | 0.14 ± 0.05 *,#, §§ | 51.5 | 51.2 |
Discussion
The cytotoxicity of dihydroartemisinin in multiple kinds of cancer cells have been demonstrated by numerous of researches. Moreover, some clinical trials have also been reported, which provides evidence on the good tolerability of DHA and shows orally administered DHA improved clinical symptoms of advanced cervix carcinoma.32 However, since it has been reported that DHA alone is not effective enough for cancer therapy,33 new effective chemotherapy strategies such as combination of other compounds with DHA are urgently needed. In the present study, we find upregulation of p8 in DHA-treated cancer cells induced autophagy to decrease the anticancer effect, thus we speculate that combination of DHA with p8 inhibitor or autophagy inhibitor may have the synergistic anti-tumor effect. Since we have not found any kinds of p8 inhibitor, we only investigated the combination treatment of DHA and chloroquine (CQ), which is well known as an autophagy inhibitor. Coincidentally, like DHA, CQ is another classical antimalarial agent, and several clinical trials have shown CQ exerts well anticancer effects.34 CQ exerts its effects mainly through blunting the activivty of lysosomal enzymes,35 to arrest degradation of the autolysosome, the latter step of autophagy, which break the energy supply through the autophagy pathway.27 As our expectation, the combination treatment of DHA and CQ reveals notable synergistic anticancer effect both in vitro and in vivo. The mechanism underlying the synergistic effect may be that CQ broke the DHA-induced autophay to enhance apoptosis level in cancer cells. However, it needs to be further investigated because both of 2 agents have complex cellular functions.
During p8 upregulation and autophagy induced by DHA, endoplasmic reticulum stress is also triggered in this process. ER stress is initiated by an imbalance between ER protein folding load and capacity in the ER lumen, and its signal transduction pathway is known as unfolded protein response (UPR). Since ER stress can be caused by oxidative stress, the iron-mediated endoperoxide cleavages of DHA result in the unbalance of redox to induce ER stress.5 To response to ER stress, the translation of ATF4 mRNAs selectively increases, which induces the expression of genes involved in restoring ER homeostasis.36 Among the ATF4 target genes, CHOP is one of the most induced, which is common considered as a mediator of apoptosis. But recent studies have identified that CHOP promotes pro-survival autophagic process through the cooperation with ATF4.37 In this study, we demonstrated that p8 acts upstream of ATF4 and CHOP in DHA-induced ER stress response in tumor cells. Although the exact p8 precise role in these mechanisms has still to be further clarified, the pro-survival autophagy induced by p8-ATF4-CHOP axis should not be ignored in cancer therapies with DHA.
Several studies have shown that ER stress can activate autophay, and even suggested that at least part of autophagosomal membrane originates from enlarged ER membrane.22,38 Like as autophagy, ER stress can activate both prosurvival programs as well as death mechanisms. Carracedo's study identified that ER stress induced by cannabinoid promoted autophagy-mediated cell death via TRB3-dependent inhibition of mTORC1 axis in human glioma cells.12 But another study demonstrated that ER stress induced by salinomycin promoted pro-survival autophagy via inhibiting AkT1 and mTORC1 in human non-small cell lung cancer.39 Therefore, the paradoxical ability of ER stress and autophagy to promote cell survival or death seems to depend on the type of cancer and cytotoxic agents used. In our study, by using RNA interference and autophagy inhibitors, we confirmed that the p8-ATF4-CHOP axis-mediated autophagy contributes to promote cell survival in DHA-treated cancer cells.
In summary, for the first time, we found the stress-inducible protein p8 is upregulated rapidly by DHA treating in tumor cells. Furthermore, we demonstrated that the upregulation of p8 induces ATF4 and CHOP expression, and consequently leads to autophagy, which was confirmed to attenuate apoptosis in DHA treatment. In addition, the combination therapy of DHA and CQ showed well synergistic antitumor effect in both vitro and in vivo. These results provide evidence to support the use p8 as a cancer therapeutic target, and suggest that the combination treatment of DHA and CQ might be an effective cancer therapeutic strategy.
Materials and Methods
Reagents
Dihydroartemisinin (DHA) was kindly provided by Engineer Liuxu of Guiling Pharmaceutical Co. Sulforhodamine B (SRB), Acridine orange (AO), Dansylcadaverine (MDC), Chloroquine diphosphate salt (CQ), and 3-Methyladenine (3-MA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies against ATF4, CHOP, LC3B, Caspase-3, cleaved-caspase3 were purchased from Cell Signaling Technology Inc. (Beverly, MA, USA). Antibodies against PARP and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody against p8 was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
HeLa, HCT116, HepG2 and SKOV3 cell lines were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China) and were cultured in RPMI-1640 (HeLa and SKOV3) or DMEM (HCT116 and HepG2), supplemented with 10% fetal serum in a humidified atmosphere of 5%CO2 at 37°.
SRB assay
The effects of DHA and CQ on cell proliferation inhibition were determined by the sulforhodamine B (SRB) assay. Briefly, cells were seeded into 96-well plates, cultured overnight, and treated with the compounds for the indicated time. The cells were then fixed with 10% trichloroacetic acid and stained with SRB. SRB in the cells was dissolved in 10 mM Tris–HCl and measured at 515 nm using a multiwell spectrophotometer (Thermo Electron Corp., Marietta, OH, USA). The rate of inhibition of cell proliferation was calculated for each well as [(A515 control cells-A515 treated cells)/A515 control cells] × 100% (whereA515 is the OD value at 515 nm).
siRNA transfection
The siRNA sequence was duplexes produced by Genepharma, Co. (Shanghai, China). The sequences of siRNAs used were as follows (sense): si-p8#1 (for HeLa) 5′-GGAUGAAUCUGACCUCUAUTT-3′, si-p8#2 (for HCT116,SKOV3 and HepG2) 5′-GACUCCUGCCUUGGUUACATT-3′, si-ATF4#1 (for HeLa, SKOV3 and HepG2) 5′-TCCCTCAGTGCATAAAGGATT-3′, si-ATF4#2 (for HCT116) 5′-GCCTAGGTCTCTTAGATGATT-3′, si-CHOP 5′-GAGCUCUGAUUGACCG-AAUTT-3′, siAtg5 5′-GGAAUAUCCUGCAGAAGAAUU-3′, negative control siRNA 5′-UUCUCCGAACGUGUCACGUTT-3. The transfection was performed using Opti-MEM (Invitrogen), siRNA and Oligofectamine (Invitrogen) according to the manufacturer's recommendations, 24 h prior to the drug treatment.
Quantitative real-time reverse transcription PCR assay
Total RNA was prepared using Trizol reagent (Sangon Biotech Co., Ltd), precipitated by isopropyl alcohol, and rinsed with 70% ethanol. Single-stranded cDNA was prepared from the purified RNA using an oligo(dT) primer (Thermoscript RT kit; Invitrogen) followed by SYBR(Takara) green real-time PCR. The primers were as follows(forward primer and reverse primer): p8, 5′-GAAGAGAGGCAGGGAAGACA-3′ and 5′-CTGCCGTGCGTGTCTATTTA-3′; ATF4, 5′-GGCCACCATGGCGTATTA-3′ and 5′-TGCTGAATGCCGTGAGAA-3; CHOP, 5′-GCACCTCCCAGAGCCCTCACTCTCC-3′ and 5′-GTCTACTCCAAGCCTTCCCCCTGCG-3′; GAPDH, 5′-TGCACCACCAACTGCTTAGC-3′ and 5′-GGCATGGACTGTGGTCATGAG-3′. Relative expression levels of the target genes were normalized with the control gene, GAPDH.
AO staining and MDC staining
Acridine orange (AO) and Dansylcadaverine (MDC) were used to evaluate the abundance of autophagic vacuoles in the cells. Following treatment, the cells were stained with AO (1 μg/ml) or MDC (50 μM) for 15 min at 37° and subsequently examined by fluorescence microscopy.
Annexin V-FITC/PtdIns and Annexin V-PE/7-AAD assay
Cells undergoing apoptosis were detected using the Annexin V-FITC/PI apoptosis detection kit (Invitrogen) or Annexin V-PE/7-AAD apoptosis detection kit (MultiSciences Biotech Co, Ltd).Briefly, cells were resuspended in cold binding buffer and incubated for 15 min in dark at room temperature; addition of Annexin V–FITC(or Annexin V-PE) and PtdIns (or 7-AAD) solutions following the manufacturer's instructions. Flow cytometry analysis was performed using a FACSCalibur cytometer (Beckman Coulter, FC500MCL).
Western blotting
The cells were collected and lysed in 2× SDS gelloading buffer [24 mM Tris-HCl (pH 6.8), 0.02% mercaptoethanol, 4% SDS, 0.4% bromphenol blue, 20% glycerol] and then boiled for 10–15 minutes. Equal volumes of cell lysates were resolved on 8%–15% SDS-PAGE gels, and the proteins were transferred to PVDF membranes (Pierce Chemical). The blots were incubated with the indicated primary antibodies and then the appropriate horseradish peroxidase-conjugated secondary antibodies. The signals were visualized by the ECL Plus western blotting detection system (Abbkine, Inc., Redlands, CA, USA).
Measurement of in vivo activity
Human cervical cancer HeLa xenografts were established by 5 × 106 cells subcutaneously into the armpit of the nude mice. When the tumor reached a mean group size of˜50 mm3, the mice were randomized to vehicle and treated groups, and received vehicle (0.1%CMC-Na in physiological saline solution, i.g. administration and physiological saline solution i.p. administration) daily, DHA (100 mg/kg, i.g. administration) daily, CQ (50 mg/kg, i.p. administration) daily, and combination group (DHA, 100 mg/kg, i.g. administration and CQ, 50 mg/kg, i.p. administration) daily for 13 days. Tumor volume was calculated as V = (length × width2)/2. The individual relative tumor volume (RTV) was calculated according to the following formula: RTV = Vn/V0, where Vn is the tumor volume on day n and V0 is the tumor volume on day of initial treatment. The T/C% was determined by calculating relative tumor volume (RTV) as T/C% = 100 × (mean RTV of treated group)/(mean RTV of vehicle group). The tumor growth inhibition rate was calculated by using the following formula: inhibition rate (%) = (1−TWt/TWv) × 100, where TWt and TWv are the mean tumor weight of the treatment and vehicle groups, respectively. The animal study was approved by the Animal Research Committee at Zhejiang University (log number Zju2011101065 and Zju2012101016), and animal care was provided in accordance with institutional guidelines.
Statistical analysis
For all parameters measured, the values for all samples in the different experimental conditions were averaged, and the standard error or standard deviation of the mean was calculated. Analysis of variance followed by Student's t-test was used for the statistical analyses. Significance was defined as P < 0.05.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by research grants from the National Natural Science Foundation of China (No. 81072659), the Zhejiang Provincial Science and Technology Program (No. 2008C23067) and the Health Bureau of Zhejiang Province, China (No. 2008W10923).
References
- 1.Krishna S, Bustamante L, Haynes RK, Staines HM.. Artemisinins: their growing importance in medicine. Trends Pharmacol Sci 2008; 29:520-7; PMID: 18752857; http://dx.doi.org/ 10.1016/j.tips.2008.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh NP, Lai H.. Selective toxicity of dihydroartemisinin and holotransferrin toward human breast cancer cells. Life Sci 2001; 70:49-56; PMID: 11764006; http://dx.doi.org/ 10.1016/S0024-3205(01)01372-8 [DOI] [PubMed] [Google Scholar]
- 3.Chen T, Li MA, Zhang RW, Wang H.. Dihydroartemisinin induces apoptosis and sensitizes human ovarian cancer cells to carboplatin therapy. J Cell Mol Med 2009; 13:1358-70; PMID: 18466355; http://dx.doi.org/ 10.1111/j.1582-4934.2008.00360.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu CJ, Zhou L, Cai Y.. Dihydroartemisinin induces apoptosis of cervical cancer cells via upregulation of RKIP and downregulation of bcl-2. Cancer Biol Ther 2014; 15:279-88; PMID: 24335512; http://dx.doi.org/ 10.4161/cbt.27223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu JJ, Chen SM, Zhang XW, Ding J, Meng LH.. The anti-cancer activity of dihydroartemisinin is associated with induction of iron-dependent endoplasmic reticulum stress in colorectal carcinoma HCT116 cells. Invest New Drugs 2011; 29:1276-83; PMID: 20607588; http://dx.doi.org/ 10.1007/s10637-010-9481-8 [DOI] [PubMed] [Google Scholar]
- 6.Mercer AE, Maggs JL, Sun XM, Cohen GM, Chadwick J, O'Neill PM, Park BK.. Evidence for the involvement of carbon-centered radicals in the induction of apoptotic cell death by artemisinin compounds. J Biol Chem 2007; 282:9372-82; PMID: 17227762; http://dx.doi.org/ 10.1074/jbc.M610375200 [DOI] [PubMed] [Google Scholar]
- 7.Shterman N, Kupfer B, Moroz C.. Comparison of transferrin receptors, iron content and isoferritin profile in normal and malignant human breast cell-lines. Pathobiology 1991; 59:19-25; PMID: 2043266; http://dx.doi.org/ 10.1159/000163611 [DOI] [PubMed] [Google Scholar]
- 8.Lai H, Singh NP.. Selective cancer cell cytotoxicity from exposure to dihydroartemisinin and holotransferrin. Cancer Lett 1995; 91:41-6; PMID: 7750093; http://dx.doi.org/ 10.1016/0304-3835(94)03716-V [DOI] [PubMed] [Google Scholar]
- 9.Mallo GV, Fiedler F, Calvo EL, Ortiz EM, Vasseur S, Keim V, Morisset J, Iovanna JL.. Cloning and expression of the rat p8 cDNA, a new gene activated in pancreas during the acute phase of pancreatitis, pancreatic development, and regeneration, and which promotes cellular growth. J Biol Chem 1997; 272:32360-9; PMID: 9405444; http://dx.doi.org/ 10.1074/jbc.272.51.32360 [DOI] [PubMed] [Google Scholar]
- 10.Vasseur S, Folch-Puy E, Hlouschek V, Garcia S, Fiedler F, Lerch MM, Dagorn JC, Closa D, Iovanna JL.. p8 improves pancreatic response to acute pancreatitis by enhancing the expression of the anti-inflammatory protein pancreatitis-associated protein I. J Biol Chem 2004; 279:7199-207; PMID: 14660681; http://dx.doi.org/ 10.1074/jbc.M309152200 [DOI] [PubMed] [Google Scholar]
- 11.Brannon KM, Million Passe CM, White CR, Bade NA, King MW, Quirk CC.. Expression of the high mobility group A family member p8 is essential to maintaining tumorigenic potential by promoting cell cycle dysregulation in LbetaT2 cells. Cancer Lett 2007; 254:146-55; PMID: 17451874; http://dx.doi.org/ 10.1016/j.canlet.2007.03.011 [DOI] [PubMed] [Google Scholar]
- 12.Salazar M, Carracedo A, Salanueva IJ, Hernandez-Tiedra S, Lorente M, Egia A, Vázquez P, Blázquez C, Torres S, García S, et al.. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest 2009; 119:1359-72; PMID: 19425170; http://dx.doi.org/ 10.1172/JCI37948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamidi T, Algul H, Cano CE, Sandi MJ, Molejon MI, Riemann M, Calvo EL, Lomberk G, Dagorn JC, Weih F, et al.. Nuclear protein 1 promotes pancreatic cancer development and protects cells from stress by inhibiting apoptosis. J Clin Invest 2012; 122:2092-103; PMID: 22565310; http://dx.doi.org/ 10.1172/JCI60144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giroux V, Malicet C, Barthet M, Gironella M, Archange C, Dagorn JC, Vasseur S, Iovanna JL.. p8 is a new target of gemcitabine in pancreatic cancer cells. Clin Cancer Res 2006; 12:235-41; PMID: 16397047; http://dx.doi.org/ 10.1158/1078-0432.CCR-05-1700 [DOI] [PubMed] [Google Scholar]
- 15.Davies ML, Parr C, Sanders AJ, Fodstad O, Jiang WG.. The transcript expression and protein distribution pattern in human colorectal carcinoma reveal a pivotal role of COM-1/p8 as a tumour suppressor. Cancer Genomics Proteomics 2010; 7:75-80; PMID: 20335521 [PubMed] [Google Scholar]
- 16.Ree AH, Bratland A, Kroes RA, Aasheim HC, Florenes VA, Moskal JR, Fodstad O, Bruland OS, Maelandsmo GM.. Clinical and cell line specific expression profiles of a human gene identified in experimental central nervous system metastases. Anticancer Res 2002; 22:1949-57; PMID: 12174869 [PubMed] [Google Scholar]
- 17.Vasseur S, Hoffmeister A, Garcia-Montero A, Mallo GV, Feil R, Kuhbandner S, Dagorn JC, Iovanna JL.. p8-deficient fibroblasts grow more rapidly and are more resistant to adriamycin-induced apoptosis. Oncogene 2002; 21:1685-94; PMID: 11896600; http://dx.doi.org/ 10.1038/sj.onc.1205222 [DOI] [PubMed] [Google Scholar]
- 18.Cano CE, Hamidi T, Sandi MJ, Iovanna JL.. Nupr1: the Swiss-knife of cancer. J Cell Physiol 2011; 226:1439-43; PMID: 20658514; http://dx.doi.org/ 10.1002/jcp.22324 [DOI] [PubMed] [Google Scholar]
- 19.Carracedo A, Lorente M, Egia A, Blazquez C, Garcia S, Giroux V, Malicet C, Villuendas R, Gironella M, González-Feria L, et al.. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell 2006; 9:301-12; PMID: 16616335; http://dx.doi.org/ 10.1016/j.ccr.2006.03.005 [DOI] [PubMed] [Google Scholar]
- 20.Kim I, Xu W, Reed JC.. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 2008; 7:1013-30; PMID: 19043451; http://dx.doi.org/ 10.1038/nrd2755 [DOI] [PubMed] [Google Scholar]
- 21.Zhou Y, Liang X, Chang H, Shu F, Wu Y, Zhang T, Fu Y, Zhang Q, Zhu JD, Mi M.. Ampelopsin-induced autophagy protects breast cancer cells from apoptosis through Akt-mTOR pathway via endoplasmic reticulum stress. Cancer Sci 2014; 105:1279-87; PMID: 25088800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, et al.. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest 2012; 122:4621-34; PMID: 23143306; http://dx.doi.org/ 10.1172/JCI62973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu JJ, Chen SM, Zhang XW, Ding J, Meng LH.. The anti-cancer activity of dihydroartemisinin is associated with induction of iron-dependent endoplasmic reticulum stress in colorectal carcinoma HCT116 cells. Invest New Drugs 2011; 29:1276-83; PMID: 20607588; http://dx.doi.org/ 10.1007/s10637-010-9481-8 [DOI] [PubMed] [Google Scholar]
- 24.Gao X, Luo Z, Xiang T, Wang K, Li J, Wang P.. Dihydroartemisinin induces endoplasmic reticulum stress-mediated apoptosis in HepG2 human hepatoma cells. Tumori 2011; 97:771-80; PMID: 22322845 [DOI] [PubMed] [Google Scholar]
- 25.Zhou HJ, Wang Z, Li A.. Dihydroartemisinin induces apoptosis in human leukemia cells HL60 via downregulation of transferrin receptor expression. Anticancer Drugs 2008; 19:247-55; PMID: 18510170; http://dx.doi.org/ 10.1097/CAD.0b013e3282f3f152 [DOI] [PubMed] [Google Scholar]
- 26.Hu W, Chen SS, Zhang JL, Lou XE, Zhou HJ.. Dihydroartemisinin induces autophagy by suppressing NF-kappaB activation. Cancer Lett 2014; 343:239-48; PMID: 24099910; http://dx.doi.org/ 10.1016/j.canlet.2013.09.035 [DOI] [PubMed] [Google Scholar]
- 27.Kimura T, Takabatake Y, Takahashi A, Isaka Y.. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res 2013; 73:3-7; PMID: 23288916; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-2464 [DOI] [PubMed] [Google Scholar]
- 28.Green DR. Apoptotic pathways: the roads to ruin. Cell 1998; 94:695-8; PMID: 9753316; http://dx.doi.org/ 10.1016/S0092-8674(00)81728-6 [DOI] [PubMed] [Google Scholar]
- 29.Mizushima N, Yoshimori T.. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID: 17611390; http://dx.doi.org/ 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- 30.White E, DiPaola RS.. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res 2009; 15:5308-16; PMID: 19706824; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathew R, Karantza-Wadsworth V, White E.. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961-7; PMID: 17972889; http://dx.doi.org/ 10.1038/nrc2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jansen FH, Adoubi I, J C KC, DE Cnodder T, Jansen N, Tschulakow A, Efferth T.. First study of oral Artenimol-R in advanced cervical cancer: clinical benefit, tolerability and tumor markers. Anticancer Res 2011; 31:4417-22; PMID: 22199309 [PubMed] [Google Scholar]
- 33.Lai HC, Singh NP, Sasaki T.. Development of artemisinin compounds for cancer treatment. Invest New Drugs 2013; 31:230-46; PMID: 22935909; http://dx.doi.org/ 10.1007/s10637-012-9873-z [DOI] [PubMed] [Google Scholar]
- 34.Sotelo J, Briceno E, Lopez-Gonzalez MA.. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med 2006; 144:337-43; PMID: 16520474; http://dx.doi.org/ 10.7326/0003-4819-144-5-200603070-00008 [DOI] [PubMed] [Google Scholar]
- 35.Sharma N, Thomas S, Golden EB, Hofman FM, Chen TC, Petasis NA, Schönthal AH, Louie SG.. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett 2012; 326:143-54; PMID: 22863539; http://dx.doi.org/ 10.1016/j.canlet.2012.07.029 [DOI] [PubMed] [Google Scholar]
- 36.Verfaillie T, Salazar M, Velasco G, Agostinis P.. Linking ER stress to autophagy: potential implications for cancer therapy. Int J Cell Biol 2010; 2010:930509; PMID: 20145727; http://dx.doi.org/ 10.1155/2010/930509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.B'Chir W, Chaveroux C, Carraro V, Averous J, Maurin AC, Jousse C, Muranishi Y, Parry L, Fafournoux P, Bruhat A.. Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell Signal 2014; 26:1385-91; PMID: 24657471; http://dx.doi.org/ 10.1016/j.cellsig.2014.03.009 [DOI] [PubMed] [Google Scholar]
- 38.Bernales S, Schuck S, Walter P.. ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy 2007; 3:285-7; PMID: 17351330; http://dx.doi.org/ 10.4161/auto.3930 [DOI] [PubMed] [Google Scholar]
- 39.Li T, Su L, Zhong N, Hao X, Zhong D, Singhal S, Liu X.. Salinomycin induces cell death with autophagy through activation of endoplasmic reticulum stress in human cancer cells. Autophagy 2013; 9:1057-68; PMID: 23670030; http://dx.doi.org/ 10.4161/auto.24632 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.