

Abstract
Remsima® (infliximab) was recently approved as the world's first biosimilar monoclonal antibody (mAb) in both the European Union and Korea. To achieve this, extensive physicochemical characterization of Remsima® in relation to Remicade® was conducted in order to demonstrate the highly similar properties between the two molecules. A multitude of state-of-the-art analyses revealed that Remsima® has identical primary as well as indistinguishable higher order structures compared with the original product. Monomer and aggregate contents of Remsima® were also found to be comparable with those of Remicade®. In terms of charge isoforms, although Remsima® was observed to contain slightly less basic variants than the original antibody, the difference was shown to be largely due to the presence of C-terminal lysine. On the other hand, this lysine was found to be rapidly clipped inside serum in vitro and in vivo, suggesting it has no effect on the biological potency or safety of the drug. Analysis of the glycan contents of the antibodies showed comparable glycan types and distributions. Recent results of clinical studies have further confirmed that the two antibody products are highly similar to each other. Based on this research as well as previous clinical and non-clinical comparability studies, Remsima® can be considered as a highly similar molecule to Remicade® in terms of physicochemical properties, efficacy, and safety for its final approval as a biosimilar product to Remicade®.
Keywords: infliximab, biosimilar, CT-P13, characterization, comparability, Remsima®, Remicade®, reference medicinal product (RMP)
Introduction
Biologic medicinal products (also known as biological drugs or biologics), active drug substances produced by biological processes, are widely used for the treatment of various human diseases (cancer, immune diseases, etc.). Within this class, the recombinant monoclonal antibody (mAb) is a relatively large (about 150K Da) protein that is heterogeneous due to post-translational modifications and carbohydrate attachment. Expiration of patents for biological innovator products, including mAbs, has increased the development of similar versions of the original biopharmaceutical products, termed biosimilars, which can provide affordable biological treatment to patients by increasing consumer access to drugs with lower prices compared with the original product.1
General and specific guidelines for biosimilars have been developed by the European Medicines Agency (EMA). More recently, the EMA has generated guidelines clarifying the non-clinical and clinical requirements for biosimilar mAbs.2 To regulate biosimilars, the Food and Drug Administration (FDA) released a draft guidance regarding biosimilars in early 2012.3 The EMA has approved several biosimilar products according to these guidelines, including biosimilars of human growth hormone (HGH), granulocyte colony-stimulating factor, and erythropoietin,4 although none of these are mAbs. Further, a few biosimilar products have been approved in the US as “follow-on protein products” or “follow-on biologics.”5
Although there are ample guidelines for the regulation of biosimilars, development of a mAb that is highly similar to its originator antibody product is quite difficult, compared with that of a small molecule drug or even small proteins such as HGH, because of its large size and the heterogeneity derived from post-translational modification, carbohydrate attachment. Complexity is added by the multiple therapeutic functions of mAbs, which may be affected by production and development processes (e.g., process changes). Moreover, information on the production of originator products is not publically available.
For this reason, extensive physicochemical characterization of Remsima® in relation to the reference product Remicade® was conducted by analyzing several batches during the manufacturing process at different time points in order to gain as much insight into the originator product as possible. Further, comprehensive comparability studies, including extensive physicochemical and biological characterization, non-clinical studies, clinical trials, and inclusive mechanism of action studies, were performed to demonstrate high similarity with the originator product.
Remsima® was developed according to the latest EMA and FDA guidelines and is thus fully compliant with regulations. Remsima® was granted approval in the European Union (EU)6,7 and in Korea as the first biosimilar mAb of Remicade®. The marketing approvals were for the treatment of all indications of the reference product, including rheumatoid arthritis, adult Crohn's disease, ulcerative colitis, pediatric ulcerative colitis, ankylosing spondylitis, psoriatic arthritis, and psoriasis. In this study, we describe the extensive physicochemical characterization of Remsima® and Remicade® that confirmed comparability between the two products.8-11
Results
Primary structures
Even though both reference and biosimilar mAbs have the same amino sequence, both are heterogeneous due to post-translational modification. To determine sequence variations, the primary structure of each mAb was analyzed.12-15
Primary structures of CT-P13 (Laboratory Code Name of Remsima®) and the reference medicinal product (RMP) were analyzed by amino acid sequence analysis using HCl hydrolysis followed by RP-HPLC. For the most robust amino acids (aspartic acid, glutamic acid, histidine, glycine, threonine, arginine, alanine, phenylalanine, isoleucine, proline, and leucine), molar ratios were similar and at expected levels. All CT-P13 samples tested exhibited comparable results with the RMP (data not shown).
Visual inspection of the tryptic peptide map by HPLC confirmed nearly identical chromatograms between CT-P13 and the RMP without any missing peaks or shifts in retention time (Fig. 1). Amino acid sequences for CT-P13 and RMP were determined by LC-MS peptide mapping (Fig. 2, Table 1). It showed that the detected peptides of the samples match the expected peptides from the amino acid sequence. Peptide sequence coverage was found to be 100% for the heavy chain and 100% for the light chain for all samples tested.
Figure 1.

Expanded chromatogram of peptide mapping.
Figure 2.

Comparison of Total Ion Chromatogram of LC-ESI-MS peptide mapping between (A) CT-P13 and (B) RMP, and peptide peak assignment.
Table 1.
Peptide lists identified by LC-ESI-MS peptide mapping of CT-P13 and RMP
| RMP |
CT-P13 |
|||||
|---|---|---|---|---|---|---|
| Peptide No. | Amino Acid No. | Theoretical Mass (Da) | Observed Mass (Da) | RT (min) | Observed Mass (Da) | RT (min) |
| HT01 | 1 - 3 | 374.2 | N/D | N/D | N/D | N/D |
| HT01-HT02 | 1 - 19 | 1901.0 | 1901.0 | 19.8 | 1901.0 | 19.8 |
| HT02 | 4 - 19 | 1544.7 | 1544.8 | 17.0 | 1544.8 | 17.0 |
| HT03 | 20 - 38 | 2310.1 | 2310.1 | 38.0 | 2310.1 | 38.0 |
| HT04 | 39 - 43 | 587.3 | 587.3 | 0.4 | 587.3 | 0.4 |
| HT05 | 44 - 52 | 1071.6 | 1071.6 | 27.1 | 1071.6 | 27.1 |
| HT06* | 53 - 54 | 233.1 | N/D | N/D | N/D | N/D |
| HT07 | 55 - 67 | 1405.7 | 1405.7 | 14.1 | 1405.7 | 14.1 |
| HT08-HT09 | 68 - 74 | 835.5 | 835.5 | 10.8 | 835.5 | 10.8 |
| HT08 | 68 - 69 | 231.1 | N/D | N/D | N/D | N/D |
| HT09 | 70 - 74 | 622.3 | 622.3 | 11.6 | 622.3 | 11.6 |
| HT09-HT10 | 70 - 78 | 1067.5 | 1067.5 | 10.2 | 1067.5 | 10.2 |
| HT10 | 75 - 78 | 463.2 | N/D | N/D | N/D | N/D |
| HT11 | 79 - 89 | 1295.7 | 1295.7 | 24.9 | 1295.7 | 24.9 |
| HT12 | 90 - 100 | 1349.6 | 1349.6 | 12.1 | 1349.6 | 12.1 |
| HT13 | 101 - 124 | 2649.2 | 2649.2 | 27.6 | 2649.2 | 27.6 |
| HT14 | 125 - 136 | 1185.6 | 1185.6 | 22.6 | 1185.6 | 22.6 |
| HT15 | 137 - 150 | 1320.7 | 1320.7 | 20.2 | 1320.7 | 20.1 |
| HT16 | 151 - 213 | 6712.3 | 6712.3 | 43.4 | 6712.3 | 43.4 |
| HT16-HT17 | 151 - 216 | 7054.5 | 7054.5 | 42.7 | 7054.5 | 42.7 |
| HT17 | 214 - 216 | 360.2 | N/D | N/D | N/D | N/D |
| HT18 | 217 - 217 | 488.3 | N/D | N/D | N/D | N/D |
| HT19 | 218 - 221 | 471.3 | 471.3 | 0.6 | 471.3 | 0.6 |
| HT18-HT19 | 217 - 221 | 599.4 | 599.4 | 0.5 | 599.4 | 0.5 |
| HT20* | 222 - 225 | 508.2 | N/D | N/D | N/D | N/D |
| HT21 | 226 - 251 | 2843.4 | 2843.5 | 33.6 | 2843.5 | 33.6 |
| HT22 | 252 - 258 | 834.4 | 834.4 | 14.7 | 834.4 | 14.7 |
| HT23 | 259 - 277 | 2138.0 | 2138.0 | 25.4 | 2138.0 | 25.4 |
| HT24 | 278 - 291 | 1676.8 | 1676.8 | 24.4 | 1676.8 | 24.4 |
| HT25 | 292 - 295 | 500.3 | 500.3 | 0.3 | 500.3 | 0.3 |
| HT26 | 296 - 304 | 1188.5 | 1188.5 | 8.5 | 1188.5 | 8.5 |
| HT27 | 305 - 320 | 1807.0 | 1807.0 | 35.1 | 1807.0 | 35.1 |
| HT27-HT28 | 305 - 323 | 2227.2 | 2227.2 | 33.4 | 2227.2 | 33.4 |
| HT28 | 321 - 323 | 438.2 | N/D | N/D | N/D | N/D |
| HT29* | 324 - 325 | 306.1 | N/D | N/D | N/D | N/D |
| HT30* | 326 - 329 | 446.2 | N/D | N/D | N/D | N/D |
| HT31 | 330 - 337 | 837.5 | 837.5 | 14.9 | 837.5 | 14.9 |
| HT32 | 338 - 341 | 447.3 | 447.3 | 0.6 | 447.3 | 0.6 |
| HT33 | 342 - 343 | 217.1 | N/D | N/D | N/D | N/D |
| HT34 | 344 - 347 | 456.2 | N/D | N/D | N/D | N/D |
| HT33-HT34 | 342 - 347 | 655.4 | 655.4 | 0.4 | 655.4 | 0.4 |
| HT35 | 348 - 358 | 1285.7 | 1285.7 | 17.8 | 1285.7 | 17.8 |
| HT36 | 359 - 363 | 604.3 | 604.3 | 2.3 | 604.3 | 2.3 |
| HT37 | 364 - 373 | 1160.6 | 1160.6 | 22.3 | 1160.6 | 22.3 |
| HT38 | 374 - 395 | 2543.1 | 2543.1 | 30.2 | 2543.1 | 30.2 |
| HT39 | 396 - 412 | 1872.9 | 1872.9 | 31.5 | 1872.9 | 31.4 |
| HT39-HT41 | 396 - 419 | 2672.4 | 2672.4 | 32.1 | 2672.4 | 32.1 |
| HT40 | 413 - 417 | 574.3 | 574.3 | 6.0 | 574.3 | 6.0 |
| HT41 | 418 - 419 | 261.1 | N/D | N/D | N/D | N/D |
| HT42 | 420 - 442 | 2800.3 | 2800.3 | 24.7 | 2800.3 | 24.7 |
| HT43 | 443 - 450 | 787.4 | 787.4 | 12.9 | 787.4 | 12.9 |
| HT43a | 443 - 449 | 659.3 | 659.4 | 15.9 | 659.4 | 15.9 |
| HA01 | 1–74 | 8236.1 | 8236.2 | 43.4 | 8236.2 | 43.4 |
| HA08 | 215 - 223 | 1089.5 | 1089.6 | 1.0 | 1089.5 | 1.1 |
| HA09 | 224 - 251 | 3086.6 | 3086.6 | 33.2 | 3086.6 | 33.2 |
| HA14 | 315 - 358 | 5021.7 | 5021.7 | 24.7 | 5021.7 | 24.9 |
| LT01 | 1 - 18 | 1895.0 | 1895.0 | 31.5 | 1895.0 | 31.5 |
| LT02 | 19 - 24 | 754.3 | 754.3 | 9.9 | 754.3 | 9.9 |
| LT03 | 25 - 39 | 1792.9 | 1792.9 | 22.8 | 1792.9 | 22.8 |
| LT04 | 40 - 45 | 630.3 | 630.3 | 0.5 | 630.3 | 0.5 |
| LT05 | 46 - 49 | 485.4 | 485.4 | 11.4 | 485.4 | 11.5 |
| LT06 | 50 - 61 | 1283.6 | 1283.6 | 15.0 | 1283.6 | 15.0 |
| LT07 | 62 - 107 | 5065.2 | 5065.3 | 41.3 | 5065.3 | 41.3 |
| LT08 | 108 - 108 | 174.1 | N/D | N/D | N/D | N/D |
| LT08-LT09 | 108 - 126 | 2101.1 | 2101.1 | 30.1 | 2101.1 | 30.1 |
| LT09 | 109 - 126 | 1945.0 | 1945.0 | 32.7 | 1945.0 | 32.8 |
| LT10 | 127 - 142 | 1796.9 | 1796.9 | 36.1 | 1796.9 | 36.1 |
| LT11 | 143 - 145 | 346.2 | N/D | N/D | N/D | N/D |
| LT12 | 146 - 149 | 559.3 | 559.3 | 7.0 | 559.3 | 7.0 |
| LT13 | 150 - 169 | 2135.0 | 2135.0 | 13.1 | 2135.0 | 13.1 |
| LT14 | 170 - 183 | 1501.8 | 1501.8 | 24.7 | 1501.8 | 24.7 |
| LT15 | 184 - 188 | 624.3 | 624.3 | 1.0 | 624.3 | 1.1 |
| LT16 | 189 - 190 | 283.2 | N/D | N/D | N/D | N/D |
| LT16-LT17 | 189 - 207 | 2140.1 | 2140.1 | 15.0 | 2140.1 | 15.0 |
| LT17 | 191 - 207 | 1874.9 | 1874.9 | 17.8 | 1874.9 | 17.8 |
| LT18 | 208 - 211 | 522.3 | 522.3 | 1.7 | 522.3 | 1.7 |
| LT19 | 212 - 214 | N/D | N/D | N/D | N/D | N/D |
| LT18-LT19 | 208 - 214 | 868.3 | 868.4 | 5.2 | 868.4 | 5.2 |
H stands for heavy chain; L stands for light chain; T stands for trypsin digested peptides; A stands for Asp-N digested peptides; * The short peptides identified by Asp-N digested peptide map.
The data support the notion that the primary structures of CT-P13 and the RMP are identical, with the exception of C-terminal lysine. Further, modifications such as deamidation, oxidation, and glycosylation were detected at comparable levels between CT-P13 and the RMP. Reduced intact mass analysis yielded a single prominent mass for the light chain along with six possible masses for the heavy chain, corresponding to G0F, G1F, and G2F forms with or without C-terminal lysine (K0/K1) (data not shown). In addition, the content of C-terminal lysine variants was shown to be slightly lower in CT-P13 compared with the RMP. Regarding the light chain, the observed mass was found to be a close match with the expected mass. Moreover, the data confirm that the light chain is not glycosylated. Regarding the heavy chain, the data demonstrate that all six possible combinations of G0F, G1F, and G2F structures with or without C-terminal lysine are present in both CT-P13 and the RMP. In the case of CT-P13, the data show that the G0F and G1F structures lacking lysine are the most commonly occurring heavy chain variants, whereas a greater proportion of K1 species is observed in the RMP. These data are in agreement with the results of LC-MS peptide mapping, as well as the IEF data to be presented later.
Higher order structures
Numerous state-of-the-art techniques have been employed in order to compare the higher order structures of CT-P13 and the RMP. Such techniques include assessment of disulfide bond positions by native and reduced peptide mapping, free thiol analysis using Ellman's reagent, assessment of secondary structures by FT-IR, assessment of secondary structures by circular dichroism (far-UV), assessment of tertiary structures by circular dichroism (near-UV), assessment of thermal stability by differential scanning calorimetry, and assessment of the general higher order structure of CT-P13 by X-ray crystallography and antibody conformational array.
The disulfide structure of each sample was identified by native and reduced peptide mapping analyses. Eight peaks were identified as disulfide bond-linked peptides in all samples based on MS and MS/MS analyses, indicating identical disulfide structures for CT-P13 and the RMP. The identified disulfide-bonding sites were:
Cys22(HC) – Cys98(HC), Cys147(HC) – Cys203(HC), Cys223(HC) – Cys214(LC), Cys229(HC) – Cys229(HC), Cys232(HC) – Cys232(HC), Cys263(HC) – Cys324(HC), Cys370(HC) – Cys428(HC), Cys23(LC) – Cys88(LC), and Cys134(LC) – Cys194(LC).
The number of moles of free SH groups per mole of IgG, as determined by Ellman's assay, was found to be nearly identical for all CT-P13 and RMP samples tested within the range from 0.11 – 0.12 (free SH mol/IgG mol).
FT-IR spectra were analyzed by comparing the locations and shapes of the amide I and amide II bands, as well as four other bands between 1,000 and 1,800 cm−1. As shown in Figure 3, FT-IR spectra were consistent among all samples in terms of shape and location (amide I band at 1,639 ± 2 cm−1, amide II band at 1,549 ± 3 cm−1, and four characteristic bands between 1,460 and 1,000 cm−1). Far-UV and near-UV CD measurements for CT-P13 and the RMP confirmed highly comparable spectra for all samples. These FT-IR and CD results confirm that the secondary and tertiary structures of CT-P13 and the RMP are highly similar. DSC provides information on the thermal stability of a protein under different solvent conditions and is used in compatibility assays to confirm product stability.16 DSC thermography showed that the transition temperatures of CT-P13 and the RMP correspond to three endothermic transitions at 68°C, 73°C, and 83°C, as shown in Figure 3. These three transition temperatures are related to the CH2, Fab, and CH3 domains of IgG proteins, respectively.7 The second transition point is not well distinguished in the thermogram overlay due to the similar transition temperatures of the CH2 and Fab domains, as well as the relatively low transition temperature of Fab. The similar thermal unfolding profiles and thermal transition midpoint temperatures suggest that the thermal stability and conformation of CT-P13 batches are comparable to those of the RMP. For detailed comparison of higher order structures, antibody conformational array and X-ray crystallography analysis were performed.
Figure 3.

Higher order structure analysis: (A) Far-UV CD; (B) Near-UV CD; (C) FT-IR; (D) DSC.
Antibody conformational array (protein conformational array ELISA) was performed for protein conformation comparison between CT-P13 and the RMP at the molecular level. The result of this analysis is a histogram, which can be likened to a ‘fingerprint’ for any given mAb. Once a ‘fingerprint’ is established, any changes in the mAb during purification or other manipulations will expose a new antigenic site detectable by antibodies on the plate. Conformational antibody array showed that all 14 batches of CT-P13 and the RMP appeared to be very consistent in terms of epitope exposure. Previous research using antibody array ELISA has indicated that new epitope exposure at levels as low as 0.1% within a mAb population can be accurately detected and quantified.17 A less than 50% difference between a biosimilar molecule and its comparator as determined by antibody array ELISA implies a lower than 0.1% difference in terms of epitope exposure, which means the two samples are comparable. Epitope exposure in both the variable and constant regions is illustrated in Figure 4A (Abs 1–12) and Figure 4B (Abs 13–34), respectively. The error bars suggest noisy data rather than real differences between the products. All seven lots of CT-P13 were found to be consistent, as well as within the range of responses defined by the RMP, meaning that the seven CT-P13 and RMP are similar in terms of higher order structure. In contrast, another study on biosimilar mAbs using antibody array ELISA observed significant differences in higher order structure (HOS) among various biosimilar mAbs under development.18,19
Figure 4.

Antibody conformation array of CT-P13 and RMP: (A) variable region (Abs 1–12); (B) constant region (Abs 13–34).
To identify differences in the Fc region between CT-P13 and the RMP, the atomic structures of Fc regions from single batches of CT-P13 and the RMP were determined by X-ray crystallography. Both CT-P13 and RMP Fc crystals were diffracted up to a resolution of 2.4 Å. Ramachandran plot was used for structure validation by checking for any residue located outside of the theoretically favored region. No residue was located in the disallowed region of the Ramachandran plot in both the CT-P13 and RMP Fc structures. Therefore, X-ray crystallography of the Fc domains of CT-P13 and the RMP was able to confirm that both products exhibit almost identical space groups and unit cell dimensions, which suggests that the molecules are packed in the same manner within the crystals. Superposition of the two Fc structures resulted in 0.18 Å rmsd (root mean square deviation) for Cα atoms, suggesting the Fc structures of CT-P13 and the RMP are nearly identical (Fig. 5).
Figure 5.
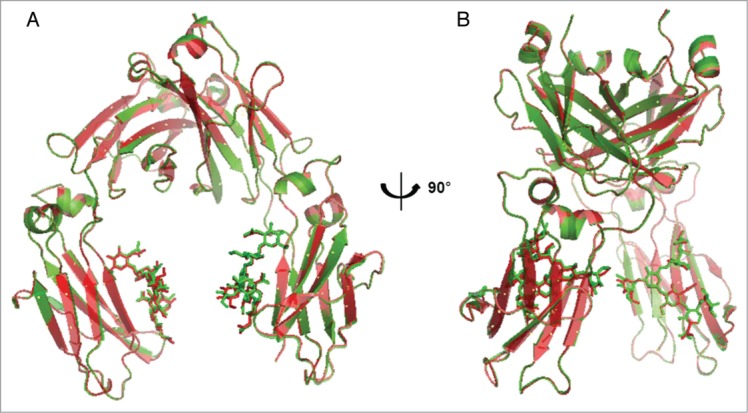
Superimposition of CT-P13 Fc (green) and RMP Fc (red) crystal structures: (A) front view; (B) side view.
Purity and impurity
Product monomer content was determined by three orthogonal methods, including SEC-HPLC, SEC-MALS, and SV-AUC. In SEC-HPLC analysis, the CT-P13 and RMP samples showed prominent monomer peaks within the range from 99.4–99.9% (Fig. 6). Both CT-P13 and the RMP showed a single peak for high molecular weight (HMW) species at a level less than 0.6%. Further, SEC-MALS and AUC analyses were conducted as orthogonal methods of SEC-HPLC. Similar to the results of SEC-HPLC, CT-P13 and the RMP exhibited predominant monomer content (>95%) as determined by both SEC-MALS and AUC analyses. These results confirm that CT-P13 and the RMP are similar with regards to monomeric purity. Amounts of fragments were measured by CE-SDS under reduced conditions, and comparable results were obtained for CT-P13 and the RMP.
Figure 6.
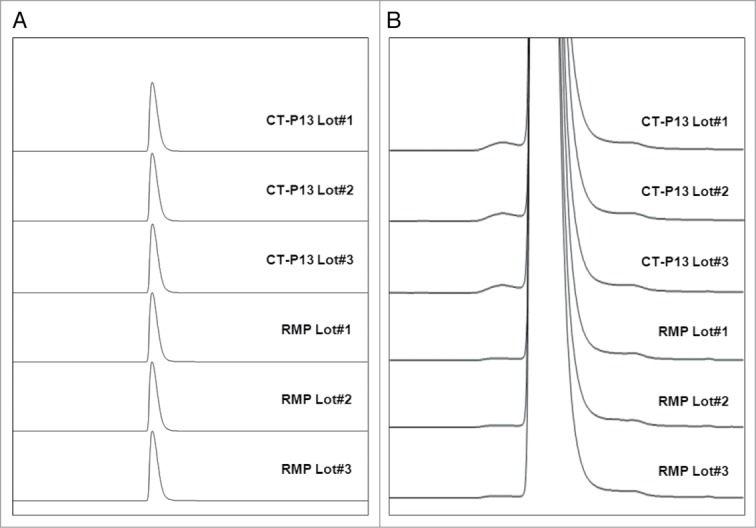
Comparison of Size Exclusion Chromatographies between CT-P13 and RMP: (A) Full scale view of chromatograms; (B) Expanded view of chromatograms.
Charged isoforms
Charge variants in CT-P13 and the RMP were analyzed by isoelectric focusing (IEF) and IEC-HPLC. According to IEF analysis, the calculated pI values of the four bands are comparable (Fig. 7) and fall within similar ranges for both CT-P13 and the RMP. IEC-HPLC analysis of CT-P13 exhibited a distribution of six peaks in the chromatograms (Fig. 8). Although the number and distribution of IEC-HPLC peaks was conserved between CT-P13 and the RMP, the relative proportion (peak ratio) of the six IEC-HPLC peaks varied, especially in the basic variants (Fig. 9). This difference in the relative proportion (peak ratio) of the six IEC-HPLC peaks between CT-P13 and the RMP may be the result of C-terminal lysine variability. To test this hypothesis, single batches of CT-P13 and the RMP were incubated with the enzyme carboxypeptidase B (CPB) in order to eliminate C-terminal lysine variability. Basic bands on the IEF gel were no longer present following CPB treatment (Fig. 7), suggesting that the molecular and charge variants responsible for these bands possess one or two C-terminal lysine residues. The six charge isoforms from IEC-HPLC analysis were then fractionated using a preparative ProPac® WCX-10 column (9 × 250 mm), after which the purity of each collected peak was confirmed by re-injection into an analytical WCX column (Fig. 8A). Figure 10 shows the position of each IEC peak on the IEF gel. The IEC fractions (peaks 1–6) of CT-P13 and the RMP were analyzed by peptide mapping for peak identification. Tryptic peptide mapping identified the structures of the six charged isoforms, as summarized in Table 2. Peak 1 is designated as CT-P13 with no C-terminal lysine, elevation of charged glycans (G1F1SA and G2F1SA), and elevation of deamidation HC Asn57. Peak 2 is designated as CT-P13 with no C-terminal lysine and elevation of charged glycans (G1F1SA and G2F1SA). Peak 3 is designated as CT-P13 with no C-terminal lysine, peak 4 is designated as CT-P13 with one C-terminal lysine, peak 5 is designated as CT-P13 with two C-terminal lysines and elevation of charged glycans (G1F1SA and G2F1SA). Peak 6 is designated as CT-P13 with two C-terminal lysines.
Figure 7.
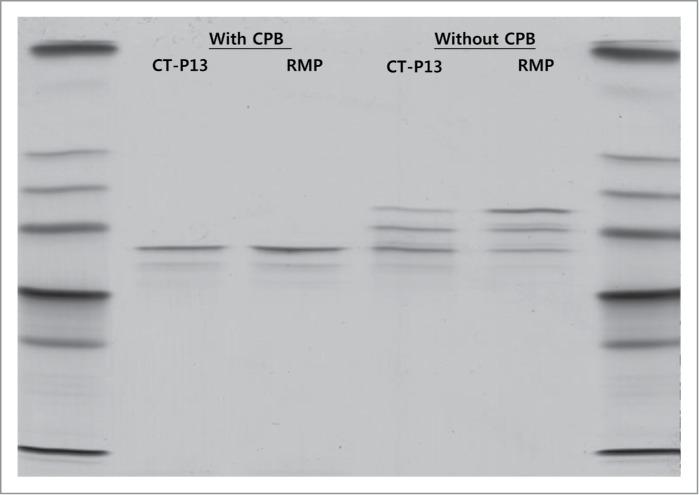
IEF results for CT-P13 and RMP treated with and without CPB.
Figure 8.

Overlaid CEX-HPLC chromatograms: (A) CT-P13 CEX fractions; (B) unfractionated CT-P13; (C) RMP CEC fractions; (D) unfractionated RMP.
Figure 9.
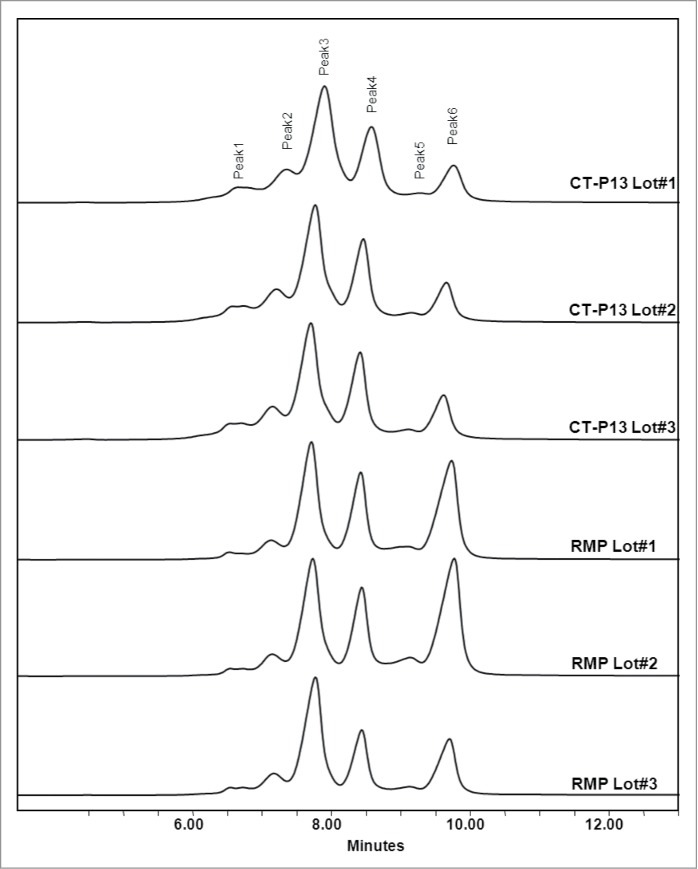
Comparison of charge profiles between RMP and CT-P13 analyzed by Cation Exchange Chromatography.
Figure 10.
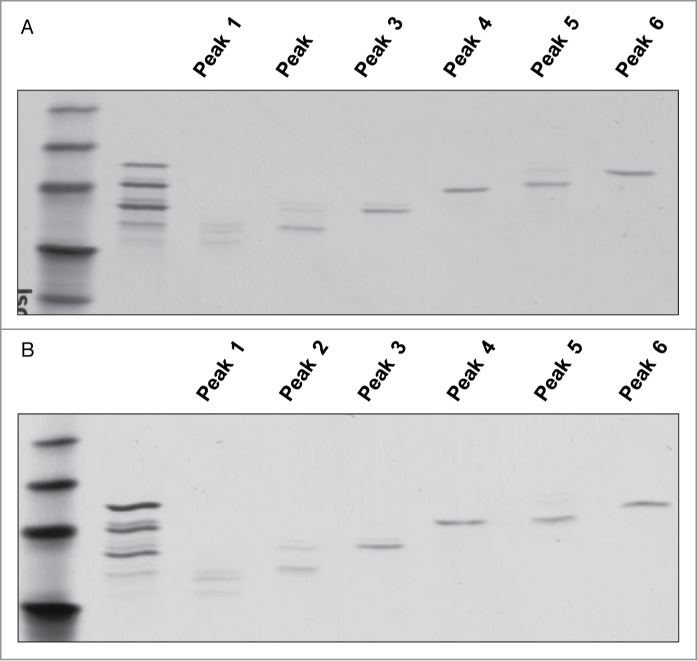
IEF results of each collected peak for (A) CT-P13 and (B) RMP.
Table 2.
Peak identification of Infliximab
| Test items | Infliximab |
|---|---|
| Peak 1 | No C-terminal lysine, Charged glycans (G1F1SA and G2F1SA), Deamidation HC Asn57 |
| Peak 2 | No C-terminal lysine, Charged glycans (G1F1SA and G2F1SA) |
| Peak 3 | Main peak (No C-terminal lysine) |
| Peak 4 | One C-terminal lysine |
| Peak 5 | Two C-terminal lysines, Charged glycans (G1F1SA and G2F1SA) |
| Peak 6 | Two C-terminal lysines |
Based on the peak characterizations, peaks 4, 5, and 6 were shown to be C-terminal lysine variants with no differences in biological activities compared with unfractionated RMP. Therefore, peaks 4–6 were confirmed as drug-related substances. In addition, a C-terminal lysine variant was shown to be rapidly clipped both in vitro and in vivo, and thus have no effect on biological potency. None of these properties were attenuated after CPB treatment, suggesting that the increased amount of clipped C-terminal lysine variant in CT-P13 has no effect on biological activities.
Glycosylation
Infliximab, similar to other IgG1 subclass antibodies, is a glycoprotein. To compare glycosylation sites, as well as glycan micro-heterogeneity associated with CT-P13 and Remicade®, a wide range of methods were employed. LC-MS peptide mapping was used to analyze glycan structures displayed by CT-P13 and the RMP at Asn300. These peptide mapping data were employed as a means to determine the range of glycan structures displayed by CT-P13 and the RMP at Asn300. Asn300 was shown to be the only site for N-glycosylation in both CT-P13 and the RMP. Further, no O-linked glycans were detected in CT-P13 and the RMP. The types of detected oligosaccharides were very similar at Asn300, with all lots containing mostly G0F and G1F structures (Fig. 11). Minor species such as Man5, G0F minus N-acetyglucosamine (GlcNAc), G0, G1F minus GlcNAc, G1, G2F, G1F1NeuGc, and G2F1NeuGc were also present in all lots (Fig. 11). The aglycosylation level was below the detection limit. Reduced CE-SDS results also showed that glycosylation occurred at a frequency greater than 99% in both CT-P13 and the RMP (data not shown).
Figure 11.
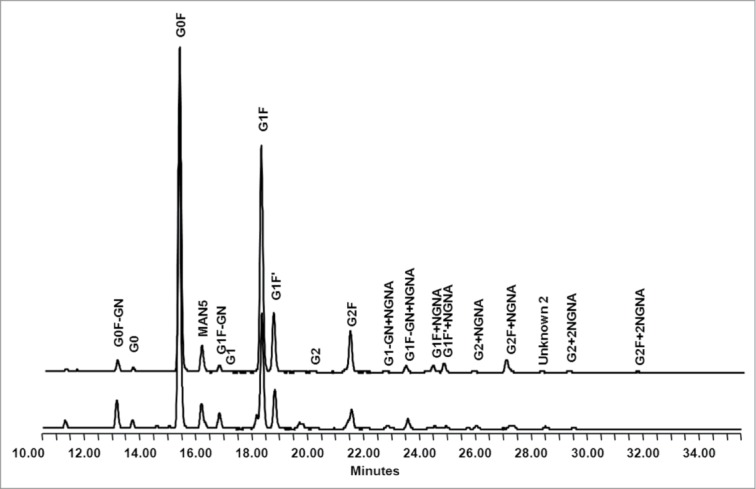
Comparison of oligosaccharide profiles between RMP (up) and CT-P13 (down) analyzed by normal phase chromatography.
To further characterize the glycan micro-heterogeneity associated with Asn300 as a site of N-glycosylation, PNGase was employed for enzymatic cleavage of glycans from molecules, followed by normal phase HPLC with 2AB labeling to resolve and identify oligosaccharide structures (Fig. 11). There were some differences in the levels of sialic acid-containing glycans, including G1FNeuGc and G2F1NeuGc, between CT-P13 and the RMP. However, in general, the amount of glycans was consistent in both CT-P13 and the RMP, with no new glycans detected. The main glycans in both products were G0F and G1F, which is typical of an IgG. Next, the types and amounts of sialic acids capping the glycan structures displayed by CT-P13 and the RMP were assessed. Sialic acid was detected in the form of N-glycolylneuraminic acid (NGNA) in all samples, with comparable levels in CT-P13 and the RMP. Finally, monosaccharide analysis of neutral and amino sugars in CT-P13 and RMP confirmed the presence of fucose (Fuc), GlcNAc, galactose (Gal), and mannose (Man) at similar molar ratios. Taken together, the overall glycosylation levels of CT-P13 and RMP are similar.
Potency
The biological activity of infliximab was determined based on its mechanism of action, including its in vitro tumor necrosis factor (TNF) neutralization activity, TNF-binding affinity (ELISA), and cell-based TNF-binding affinity. TOST (two one-sided t test) was used to confirm that the average values of the two products are similar. In detail, TOST applies a bioequivalence interval of 80–125%. If 90% confidence intervals (90% CI) of the ratios between the two products lie within the range from 80–125%, then the products are considered equivalent. If the corresponding p values are lower than 0.05 (significant level) on both sides, the result is considered equal between CT-P13 and the RMP (Table 3).
Table 3.
Summary of binding affinity and in vitro potency results
| RMP |
||||||
|---|---|---|---|---|---|---|
| Test items | CT-P13Average (%) (Range) | SD | Average (%) (Range) | SD | TOST | Method |
| TNF binding | 99 (97–105) | 2.5 | 100 (94–104) | 2.8 | <0.0001 | ELISA |
| FcRn | 101 (95–109) | 4.2 | 97 (93–103) | 3.3 | <0.0001 | SPR |
| C1q | 100 (91–116) | 6.6 | 98 (87–109) | 8.2 | <0.0001 | ELISA |
| TNF binding | 101 (92–110) | 6.0 | 100 (90–112) | 7.1 | <0.0001 | Cell-based |
| TNF Neutralization | 102 (95–107) | 4.7 | 104 (98–110) | 2.9 | <0.0001 | Cell-based |
| Apoptosis | 101 (91–105) | 5.0 | 101 (92–110) | 2.5 | <0.0001 | Cell-based |
| CDC | 102 (91–116) | 8.0 | 93 (84–115) | 8.0 | 0.0011 | Cell-based |
Discussion
Small biosimilars such as human growth hormone (HGH), granulocyte colony-stimulating factor, and erythropoietin that adhere to guidelines developed by the EMA, FDA, and ICH have been approved in the EU since 2006. Small molecules (i.e., generics) are relatively easy to reproduce with identical quality and properties as the RMP due to their relatively small size and absence of any glycans. However, as is the case with mAbs such as Remsima®, development of complex biosimilars becomes more difficult due to their complicated glycan composition, structural conformation, post-translational modification, and numerous therapeutic functions. Despite this problem, the European Medicines Agency has already provided scientific advice and guideline relating to the development of several biosimilar antibody-based products in development.20
In Europe, by definition a “biosimilar” drug product must share the same amino acid sequence as its reference product.20 Thus, it is necessary to confirm the identity of their amino acid sequence. Extensive physicochemical and biological characterization for Remsima® and its reference product Remicade® was conducted in order to demonstrate their highly similar properties. A series of state-of-the-art analyses showed that Remsima® has 1) identical primary as well as higher order structures as Remicade®; 2) indistinguishable monomer and aggregate contents, overall comparable glycan types, and distributions; and 3) comparable potencies and binding affinities as the RMP. Although Remsima® contains slightly less basic variants in comparison to the RMP, these differences can largely be attributed to C-terminal lysine, which was rapidly clipped in vitro and in vivo and found to have no effect on biological potency or safety. Glycan analysis confirmed similar glycan types and distributions between Remsima® and the RMP.
A recently reported clinical outcome along with the above physicochemical and biological results clearly demonstrate that Remsima® is a highly similar molecule to the RMP.21,22 Thorough comparability analysis has also showed that CT-P13 possesses highly similar properties in terms of primary/higher order structures and purity/impurity. Regarding charge isoforms, the number and distribution of charge variants were shown to be conserved between CT-P13 and Remicade® despite noticeable differences in the relative proportion of basic variants. However, the amount of basic variants derived from C-terminal lysine was shown to have no effect on biological potency due to rapid clipping by CPB in vivo.
Remsima®, as the biosimilar of Remicade®, received approval in the EU and South Korea for indications such as rheumatoid arthritis, adult Crohn's disease, ulcerative colitis, pediatric ulcerative colitis, ankylosing spondylitis, psoriatic arthritis, and psoriasis following comprehensive product development and demonstration of RMP similarity, which was based on extensive physicochemical and biological properties, comparative clinical and non-clinical studies, and comprehensive mechanism of action studies. This outcome confirms that it is possible to develop a large biosimilar such as a mAb and gain approval for marketing upon expiration of the original patent drug.
Materials and Methods
Materials
Lots of CT-P13 (Remsima®), a Remicade® biosimilar, were manufactured at Celltrion Inc., Korea. Remicade® lots were purchased from a pharmacy located in the EU.
Amino acid analysis
Amino acid analysis was performed via hydrolysis of peptide bonds with 6 M HCl, followed by pre-column derivatization using o-phthalaldehyde (OPA) and 9-fluorenyl-methylchloroformate (FMOC-Cl), separation by RP-HPLC, and fluorescence detection. Samples were first de-salted using PD-10 columns (GE Healthcare) and then aliquoted into hydrolysis tubes containing internal standards. The sample/internal standard mixtures were then hydrolyzed using 6 M HCl for 24 h at 110°C under reduced pressure. The hydrolysates were dried under reduced pressure, reconstituted in 50 μl of 0.1 M HCl and transferred to HPLC vials. The HPLC vials were then loaded onto the sample tray of an Agilent 1200 HPLC system for automated derivatization of the liberated amino acids. Amino acids containing a primary amino group (all except proline) were derivatized with OPA in the presence of 3-mercaptopropionic acid. Proline was derivatized by reaction with FMOC-Cl. The internal standard used for amino acid derivatizations was norvaline, with the exception of proline derivatization in which sarcosine was utilized. Amino acid analysis was conducted by RP-HPLC with fluorescence detection. Retention times and fluorescence responses were calibrated using a standard mixture of amino acids. Amino acid concentrations of samples were calculated in units of pmol/μl from respective calibration curves showing plots of the response ratio vs. the amount ratio. The molar ratio relative to leucine was then calculated.
HPLC peptide mapping analysis
Samples were analyzed by HPLC peptide mapping after reduction with DTT (Sigma-Aldrich), alkylation with iodoacetamide (Sigma-Aldrich), and digestion with trypsin (Promega) at 37°C. The resulting peptides were separated by RP-HPLC using a C18 column (5 μm, 250 × 4.6 mm; Vydac, Hesperia) and acetonitrile gradient (Burdick and Jackson) containing trifluoroacetic acid (Sigma-Aldrich). Absorbance was monitored at 214 and 280 nm using a Waters-2695 Alliance HPLC system equipped with a UV detector (Waters).
Reduced intact mass analysis
Samples were reduced with DTT, followed by LC-ES-MS analysis using an Agilent 1200 HPLC coupled online to an Agilent 6530 Q-TOF mass spectrometer (Agilent Technologies). The m/z (mass/charge) data were collected from 900 to 4000 m/z at a scan rate of 1 spectrum per second, followed by deconvolution of the mass spectra to intensity vs. molecular mass.
LC-MS-ESI peptide mapping analysis
Samples were analyzed by LC-MS peptide mapping after reduction, alkylation, and three enzyme digestions with trypsin, Lys-C (Roche; Nutley), and Asp-N (Roche). The resulting peptides were separated by reversed-phase UPLC using a C18 column (1.7 μm, 2.1 × 50 mm; Waters) and acetonitrile gradient containing formic acid (Fluka). Chromatography was performed on a Waters Acquity UPLC. An online AB SCIEX Triple TOF 5600 mass spectrometer with an electrospray source (AB SCIEX) was used to collect mass spectra of the intact peptide, as well as to fragment the peptides for sequencing (MS/MS analysis).
Free thiol analysis by Ellman assay
The free thiol (SH) groups in the samples were determined by the 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB, Ellman's reagent) method. Briefly, l-cysteine standard samples were mixed with DTNB in 6 M guanidine HCl, 100 mM sodium phosphate pH 8.0, and 1 mM EDTA, followed by measurement of absorbance at 412 nm.
Fourier transform infrared (FT-IR) spectroscopy
FT-IR spectra were recorded on a Nicolet 6700 (Thermo-Nicolet) with a smart iTR ATR accessory and diamond crystal at a resolution of 4 nm, no. of scan 16, and spectral range from 4000–600 cm−1. All spectra were baseline and ATR-corrected with instrument software. To generate the difference spectra, buffer spectra were recorded as a blank and subsequently subtracted. Sample solutions were transferred onto the crystal and allowed to dry. FT-IR spectra were analyzed by comparing the locations and shapes of the amide I and amide II bands, as well as four other bands between 1000 and 1800 cm−1.
Circular dichroism (CD) spectroscopy
CD experiment was performed using a Chirascan-plus CD spectrometer (Applied Photophysics) equipped with a Pelltier controlled temperature regulator at 293K. Cells of quartz glass along with optical path lengths of 1.0 and 0.10 cm were used for near-UV and far-UV CD measurements, respectively. Protein concentrations used for the far-UV spectra and near-UV CD spectra were 0.2 and 1.0 mg/ml, respectively. The formulation buffer was measured as a blank and subsequently subtracted. Noise reduction was applied to the baseline-corrected protein spectra using the smoothing option of the device software for the spectrometer. Conversion of the measured CD signals to mean residue molar ellipticities [(θ)MRE] was also performed using this software.
Differential scanning calorimetry (DSC)
Thermal stability of the samples was evaluated by measuring their Tm values using a Microcal VP-DSC microcalorimeter (MicroCal). The thermogram was obtained with a scan rate of 1°C/min. DSC data were analyzed using a non-two state model using the Origin software package (OriginLab Corporation) to determine three thermal transition temperatures.
Antibody conformational array
A RemiBridge antibody array ELISA kit (ArrayBridge) was used for protein conformational array. This assay is in sandwich ELISA format wherein the plate is coated with a panel of antibodies raised against peptides derived from the full-length Remicade® sequence for recognition of 12 and 22 conformational epitopes in the variable and constant regions of infliximab, respectively. After incubating 5 μg/ml of CT-P13 or the RMP in the plate to allow protein capture by the panel of antibodies, biotin-conjugated anti-human IgG antibody was added and incubated for 1 h to allow binding to any captured proteins. Streptavidin-HRP (horseradish peroxidase) conjugate and TMB (3,3′,5,5′-tetramethylbenzidine) substrate were added subsequently to detect colored product by captured HRP. The intensity of the generated color was detected using a microtiter plate reader capable at a wavelength of 450 nm. CT-P13 and RMP samples were tested pairwise in triplicate on seven sets of RemiBridge ELISA plates. All plates were handled identically, enabling side-by-side comparison of all 14 samples. Data was shown graphically grouped both pairwise and in total.
X-ray crystallography for Fc region
Fc regions of CT-P13 and the RMP were obtained by papain digestion, followed by recombinant protein A chromatography and gel filtration purification. Next, 10–15 mg/ml of each Fc sample was crystallized by the hanging-drop vapor diffusion method in 10–15% PEG4000, 4–6% PEG400, 50 mM LiSO4, and 0.1 M MES (pH 6.0) at room temperature. Single crystal was mounted in a stream of gaseous nitrogen at 100 K with 25% ethylene glycol as a cryo-protectant. Diffraction data were collected with an X-ray diffractometer (MicroMaxTM-007 HF microfocus X-ray generator and R-AXIS IV++ imaging plate area detector) (Rigaku). Diffraction data sets were indexed, integrated, and scaled using the HKL2000 software package.23 Immunoglobulin gamma Fc structure (PDB ID: 2WAH) was used as an initial search model in molecular replacement using the CNS software suite,24 and model building and visualization were performed using the ‘Coot’ molecular graphic software package.25 Structure refinement utilized the CNS software suite, with 5% of data in the reflection files set aside for free R calculation. PROCHECK was used to check stereochemical quality of the structures.26
Size-exclusion chromatography (SEC-HPLC)
SEC-HPLC was performed under non-denaturing conditions with the Waters-2695 Alliance HPLC system on a TSK G3000SWXL column (Tosoh, Japan) with aqueous-buffered mobile phase. The isocratic elution profile was monitored using UV detection at 214 nm.
SEC-multi angle light scattering (SEC-MALS)
Aliquots of each sample were diluted in PBS buffer to a final concentration of 12 mg/ml. A Waters HPLC system equipped with a Superdex 200 10/300GL column (Amersham Biosciences) was used, and detection was monitored using Optilab rEX, UV 280 nm for RI, as well as a DAWN® HELEOS™ II (Wyatt Technology) for MALS detection.
Sedimentation velocity analytical ultracentrifugation (SV-AUC)
SV-AUC was performed on a Beckman Coulter XL-A AUC instrument at 20°C. A rotor speed of 28,000 rpm with 150 scans at 6 min intervals was selected for each SV-AUC run. Data were assessed using the SEDFIT program to obtain the c(s) profile of the sedimentation coefficient (s) values, reported in Svedberg units (S).
Capillary sodium dodecyl sulfate gel electrophoresis (CE-SDS)
CE-SDS was performed under reducing conditions for analysis of purity/impurities. A Beckman Coulter, PA 800 capillary electrophoresis system was used with a 57 cm, 50 μm I.D. bare-fused silica capillary. Reducing CE-SDS was performed for determination of purity by sum of heavy and light chains.
Isoelectric focusing (IEF)
IEF was used to determine pI values of charge variants in the CT-P13 and RMP samples. Electrophoresis was performed on IsoGel agarose IEF plates in the pH range from 3–10 using a flatbed electrophoresis system (Multiphor II, Amersham Biosciences). pI values were calculated against IEF pI markers (pI range: 9.45–6.00) and compared with the pI values of the reference standard. pI values were calculated using Quantity One software (Bio-Rad).
Ion exchange chromatography
The IEC-HPLC method was used to evaluate the distribution of charge variants by cation exchange chromatography. The HPLC system (Waters) was equipped with a Propac WCX 10 analytical column and guard column set (Dionex) at ambient temperature. Gradient NaCl elution was performed, and UV signals were obtained at 214 nm. Peaks in the IEC HPLC chromatogram were integrated, and percentage peak areas of each peak were calculated.
Monosaccharide analysis
Monosaccharide analysis of neutral and amino sugars was performed by hydrolyzing the samples with 2 N trifluroacetic acid at 100°C, followed by anion exchange chromatography with the pulsed amperometric detection (HPAEC-PAD) system (Dionex) with a CarboPac PA-20 analytical column (Dionex) and isocratic NaOH elution. Monosaccharide content was measured based on relative response of monosaccharide standards against an internal standard (2-deoxy-D-glucose). Monosaccharide content was determined as relative mole ratios (monosaccharide/protein, mole/mole).
Sialic acid analysis
To analyze sialic acid species (e.g., N-acetylneuraminic acid (NANA) and N-glyconeuraminic acid (NGNA)), sialic acids were released from antibodies by mild acid hydrolysis (0.1 M HCl) at 80°C. Anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD) was used for the analysis. A CarboPac PA-20 analytical column with an amino trap column was combined with a sodium hydroxide/acetate gradient elution to elute sialic acid species. Triple waveform and integration detection mode with an AgCl reference electrode were selected as PAD detection parameters. The sialic acid content was quantified based on the responses of sialic acid standards (NGNA) relative to an internal standard, 2-keto-3-deoxy-D-glycero-D-glacto-nononic acid (KDN). The results were reported as molar ratios (sialic acid/protein, mole/mole).
Oligosaccharide profile analysis
For oligosaccharide profile analysis, N-linked glycans were released from the antibody using PNGase F treatment at 37°C. PNGase F-cleaved glycans were analyzed by HPLC with a FLD detector (Waters). Released N-linked glycans by PNGase F treatment were extracted from deglycosylated protein solution using a MassPREP HILIC μElution plate (Waters). Extracted glycans were labeled with 2-AB labeling reagent, followed by removal of excess labeling reagent using a MassPREP HILIC μElution plate. Finally, 2-AB-labeled N-linked glycans were analyzed by normal phase chromatography with a GlycoSep N column (Glyko) and fluorescence detector.
N-glycan analysis by LC-MS
For structural analysis of N-linked oligosaccharide at Asn300 by LC-MS (AB SCIEX), trypsin-digested peptides prepared for peptide mapping were evaluated. Extracted ion chromatograms were used to quantify each oligosaccharide species. The percentage calculation was based on each glycosylation site. For each site, all detectable oligosaccharide structures were counted.
Enzyme-linked immunosorbent assay (ELISA)
To assess the binding affinity of antibody to TNF, ELISA was performed. TNF was coated onto 96-well plates overnight. After washing, the plates were blocked with diluent buffer (Teknova) at RT, and diluted antibodies were loaded and incubated for 1 h at room temperature. After washing, anti-kappa light chain HRP conjugate (Sigma) was added and incubated for 30 min at RT and then washed. The plate was incubated with TMB substrate solution (Sigma), and the reaction was stopped by sulfuric acid. For C1q ELISA, TNF was used to coat the plate. After binding to TNF, the samples were incubated with C1q (Quidel) and then treated with secondary antibody conjugate (anti-C1q HRP, Thermo) for 30 min. All other procedures were repeated in the same manner as those for the TNF ELISA. Data were evaluated with GraphPad Prism.
Surface plasmon resonance
The binding affinity against FcRn was evaluated by surface plasmon resonance using BiaCore 3000. FcRn was immobilized on a CM5 chip with an amine coupling kit (GE healthcare). The samples were serially diluted and injected onto the ligand-immobilized CM5 chip. The regeneration step was performed with glycine (GE healthcare). The KD value was evaluated using Bia Evaluation software.
TNF neutralization in vitro assay
Serially diluted samples in RPMI 1640 medium (Gibco) were prepared in 96-well plates, after which human TNF was added and incubated for 1 h at 37°C in a CO2 incubator. WEHI 164 cells (ATCC) were seeded onto the 96-well plates pre-incubated with samples and TNF. After incubation for 20–24 h, cell viability was analyzed using CCK-8 (Dojindo). The EC50 value was calculated using SoftMax software (Molecular Devices).
Apoptosis
Jurkat cells, stably transfected with tm-TNF, were purchased from Kyushu University, seeded onto 96-well plates, and incubated with samples for 18 h. The cells were then stained with an Annexin V apoptosis kit (BD PharMingen). The fraction of apoptotic cells was determined through the bivariate dot-plot presentation of FACS analysis. The apoptotic effect of each sample was assessed by calculation of the gated percentage.
Complement-dependent cytotoxicity assay
CDC effect was measured by absorbance using tmTNF-Jurkat cell lines as a target cell. Serially diluted samples, target cells, and complement (Quidel) were plated onto 96-well plates sequentially. After incubation, each well was treated with CCK-8 (Dojindo), followed by incubation for more than 6–8 h at 37°C in a humidified 5% CO2 incubator. EC50 values were calculated using SoftMax software.
Charge variants characterization study
To identify the charge isoforms, each peak in the IEC was fractionated using a preparative ProPac® WCX-10 column (9 × 250 mm), and the purity of each collected peak was confirmed by re-injection into an analytical weak cationic chromatography (WCX) column. Each IEC peak was then fractioned using a WCX column and subjected to analysis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Dr. Hye Yeon Kim and Dr. Woo Cheol Lee at the Korea Basic Science Institute for use of their X-ray diffractor and facilities. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (No. NRF-2013M3A9B6075887).
References
- 1.Cornes P. The economic pressures for biosimilar drug use in cancer medicine. Target Oncol 2012; 7(Suppl 1):S57-67; PMID:22249658; http://dx.doi.org/ 10.1007/s11523-011-0196-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues (revision 1). London: European Medicines Agency 2014; Available from: http://www.ema.europa.eu/">www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/05/WC500127960.pdf. [Google Scholar]
- 3.Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry. Scientific considerations in demonstrating biosimilarity to a reference product. Rockville: Food and Drug Administration; 2012; Available from: www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf">http://www.fda.gov/">www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf. [Google Scholar]
- 4.GaBi online. Biosimilars approved in Europe. 2011; July 8. Available from: http://gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe. [Google Scholar]
- 5.Woodcock J, Griffin J, Behrman R, Cherney B, Crescenzi T, Fraser B, Hixon D, Joneckis C, Kozlowski S, Rosenberg A, et al. . The FDA's assessment of follow-on protein products: a historical perspective. Nat Rev Drug Discov 2007; 6:437-42; PMID:17633790; http://dx.doi.org/ 10.1038/nrd2307 [DOI] [PubMed] [Google Scholar]
- 6.European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Remsima - European public assessment report (EPAR). 2013; Available from: www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002576/WC500151486.pdf">http://www.ema.europa.eu/">www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002576/WC500151486.pdf. [Google Scholar]
- 7.Beck A, Reichert JM. Approval of the first biosimilar antibodies in Europe: a major landmark for the biopharmaceutical industry. MAbs 2013; 5:621-3; PMID:23924791; http://dx.doi.org/ 10.4161/mabs.25864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beck A, Dimer H, Ayoub D, Debaene F, Wagner-Rousset E, Carapito D, Van Dorsselaer A, Sanglier-Cianférani S. Analytical characterization of biosimilar antibodies and Fc-fusion proteins. Trends Analyt Chem 2013; 48:81-95; http://dx.doi.org/ 10.1016/j.trac.2013.02.014 [DOI] [Google Scholar]
- 9.Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianférani S. Characterization of therapeutic antibodies and related products. Anal Chem 2013; 85:715-36; PMID:23134362; http://dx.doi.org/ 10.1021/ac3032355 [DOI] [PubMed] [Google Scholar]
- 10.Beck A, Sanglier-Cianférani S, Van Dorsselaer A. Biosimilar, biobetter, and next generation antibody characterization by mass spectrometry. Anal Chem 2012; 84:4637-46; PMID:22510259; http://dx.doi.org/ 10.1021/ac3002885 [DOI] [PubMed] [Google Scholar]
- 11.Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov 2012; 11:527-40; PMID:22743980; http://dx.doi.org/ 10.1038/nrd3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie H, Chakraborty A, Ahn J, Yu YQ, Dakshinamoorthy DP, Gilar M, Chen W, Skilton SJ, Mazzeo JR. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry technologies. MAbs 2010; 2:11986; PMID:20458189; http://dx.doi.org/ 10.4161/mabs.11986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gahoual R, Burr A, Busnel JM, Kuhn L, Hammann P, Beck A, François YN, Leize-Wagner E. Rapid and multi-level characterization of trastuzumab using sheathless capillary electrophoresis-tandem mass spectrometry. MAbs 2013; 5:479-90; PMID:23563524; http://dx.doi.org/ 10.4161/mabs.23995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Visser J, Feuerstein I, Stangler T, Schmiederer T, Fritsch C, Schiestl M. Physicochemical and functional comparability between the proposed biosimilar rituximab GP2013 and originator rituximab. BioDrugs 2013; 27:495-507; PMID:23649935; http://dx.doi.org/ 10.1007/s40259-013-0036-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen SL, Wu SL, Huang LJ, Huang JB, Chen SH. A global comparability approach for biosimilar monoclonal antibodies using LC-tandem MS based proteomics. J Pharm Biomed Anal 2013; 80:126-35; PMID:23563225; http://dx.doi.org/ 10.1016/j.jpba.2013.02.040 [DOI] [PubMed] [Google Scholar]
- 16.Lubiniecki A, Volkin DB, Federici M, Bond MD, Nedved ML, Hendricks L, Mehndiratta P, Bruner M, Burman S, Dalmonte P, et al. . Comparability assessments of process and product changes made during development of two different monoclonal antibodies. Biologicals 2011; 39:9-22; PMID:20888784; http://dx.doi.org/ 10.1016/j.biologicals.2010.08.004 [DOI] [PubMed] [Google Scholar]
- 17.Wen J, Jiang Y, Nahri L. Effect of carbohydrate on thermal stability of antibodies. Am Pharm Rev 2008:1-6. [Google Scholar]
- 18.Wang X, Li Q, Davies M. Development of antibody arrays for monoclonal antibody Higher Order Structure analysis. Front Pharmacol 2013; 4:103; PMID:23970865; http://dx.doi.org/ 10.3389/fphar.2013.00103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Li Q, Davies D. Higher order structure comparability: Case studies of biosimilar monoclonal antibodies. Bioprocess Int 2014; 12:32-7. [Google Scholar]
- 20.Reichert JM, Beck A, Iyer H. European Medicines Agency workshop on biosimilar monoclonal antibodies: July 2, 2009, London, UK. MAbs 2009; 1:394-416; PMID:20065643; http://dx.doi.org/ 10.4161/mabs.1.5.9630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park W, Hrycaj P, Jeka S, Kovalenko V, Lysenko G, Miranda P, Mikazane H, Gutierrez-Ureña S, Lim M, Lee Y-A, et al. . A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis 2013; 72:1605-12; PMID:23687259; http://dx.doi.org/ 10.1136/annrheumdis-2012-203091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoo DH, Hrycaj P, Miranda P, Ramiterre E, Piotrowski M, Shevchuk S, Kovalenko V, Prodanovic N, Abello-Banfi M, Gutierrez-Ureña S, et al. . A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA. Ann Rheum Dis 2013; 72:1613-20; PMID:23687260; http://dx.doi.org/ 10.1136/annrheumdis-2012-203090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997; 276:307-26; http://dx.doi.org/ 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 24.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc 2007; 2:2728-33; PMID:18007608; http://dx.doi.org/ 10.1038/nprot.2007.406 [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004; 60:2126-32; PMID:15572765; http://dx.doi.org/ 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 26.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK - a program to check the stereochemical quality of protein structures. J Appl Cryst 1993; 26:283-91; http://dx.doi.org/ 10.1107/S0021889892009944 [DOI] [Google Scholar]