

Abstract
Background Information: Previous studies have revealed that leptin may be involved in epithelial-mesenchymal transition (EMT), a crucial initiator of cancer progression to facilitate metastatic cascade, increase tumor recurrence, and ultimately cause poor prognosis. However, the underlying mechanism remains unclear. The aim of our present study was to investigate the effect of leptin on EMT of breast cancer cells and the underlying mechanism. Results: Our data demonstrated that leptin significantly increased the phosphorylation of STAT3, Akt, and ERK1/2, elevated the expression of IL-8, and induced breast cancer cells to undergo EMT. The effect of leptin on IL-8 could visibly abolished by the inhibitor of PI3K LY294002. In addition, leptin-induced EMT of breast cancer cells was blocked by anti-IL-8 antibodies. Examination of the expression of ObR, leptin, IL-8 and EMT-related biomarkers in patient specimens demonstrated that malignant breast carcinoma with lymph node metastases (LNM), which represents poor prognosis, expressed higher levels of ObR, leptin, IL-8 than other types of breast cancer, and displayed more obvious EMT transversion. In vivo xenograft experiment revealed that leptin signally promoted tumor growth and metastasis and increased the expressions of IL-8 and EMT-related biomarkers. Conclusions: Our results support that leptin-induced EMT in breast cancer cells requires IL-8 activation via the PI3K/Akt signal pathway.
Keywords: breast cancer, EMT, IL-8, leptin, PI3K/Akt
Abbreviations
- AKT
Protein Kinase B
- COX-2
cyclooxygenase-2
- EMT
epithelial-mesenchymal transition
- ERK
extracellular signal-regulated kinase
- IFN
interferon
- IL-8
Interleukin 8
- JAK
Junas Kinase
- LNM
lymph node metastases
- MAPK
Mitogen-activated protein kinase
- MMP
matrix metalloproteinase
- mTOR
Mammalian Target Of Rapamycin
- NF-κB
Nuclear factor kappa B
- Ob-R
Ob receptor
- PI-3K
phosphatidylinositol-3 kinase
- qRT-PCR
quantify reverse transcription-polymerase chain reaction
- STAT
signal transduction and activators of transcription
- TGF
transforming growth factor
- TNF
tumor necorsis factor
- VEGF
vascular endothelial growth factor
Introduction
Accumulating epidemiologic evidence has shown that obesity, which is characterized by excessive fat accumulation, has rapidly become a major global health issue associated with various metabolic complications. It significantly influences risk, progression and prognosis of different types of hormone-dependent cancers, especially breast cancer in postmenopausal women.1-5 In addition, women with the highest quintile of body mass index (BMI) are more likely to show high breast cancer incidence and mortality.6 Although the association between obesity and breast cancer has been the focus of research for more than 40 years, the cellular and molecular mechanisms underlying the clinical observations are still not fully understood.
As is well known, deregulated secretions of pro-inflammatory chemokines, cytokines, and adipokines as a consequence of adipocytes hypertrophy and hyperplasia, are involved in almost every aspect of tumor pathology.7,8 Among various adipocytokines, leptin, due to its effect on carcinogenesis, has been recognized as an important molecular mediator linking obesity to breast cancer. Leptin is a 16-kDa circulating hormone that is initially thought to be a key factor in controlling immunity, reproduction, and hematopoiesis.9 However, recent studies have demonstrated that leptin could significantly induce survival, migration, invasion of various caners by binding to its specific membrane receptors (Ob-Rs), which are expressed widely in various tissues.10-11 Besides, overexpression of leptin has been observed in the breast tumors examined, while none in the normal cases. In normal mammary epithelial cells, Ob-Rs were not detected by immunohistochemistry, whereas nearly all carcinoma cells showed strongly positive staining.12 Thus, potentially, leptin plays a key role in regulating tumor progression and metastasis; yet, the potential mechanisms underlying leptin-inducedsignaling cascade remain unclear.
Indeed, apart from the effects on tumorigenesis and metastasis, few studies focus on the involvement of leptin in EMT, which is a characteristic phenotypic switch step from adherent eptithelial-like cells to mesenchymal-like cells, thus acting on the processes of embryonic development, wound healing, tissue repair, and various pathological situations, such as invasion and metastasis of malignant cells.13,14 Recent studies show that EMT is a crucial initiator during cancer progression for facilitating metastatic cascade, increasing tumor recurrence, and contributing to poor prognosis.15 Given this, hypotheses connecting leptin to EMT in promoting the metastasis of cancer cells have been established 16, and attempts to elucidate the related signaling pathways are on the way.
Many studies have suggested that cancer cells undergoing EMT can secrete a variety of cytokines, growth factors, and chemokines to potentiate tumor dissemination. Interleukin 8(IL-8), a chemokine produced by macrophages and other cell types such as epithelial cells, is a neutrophil chemotactic factor.17 Despiteacting as animmune reaction mediator, IL-8 also plays an important role in cell proliferation, angiogenesis, migration, and invasion, thereby is involved in the metastatic process of various cancers including breast cancer.18,19 In fact, the metastatic potential of human cancer cells is closely related to the expression of IL-8. Compared with low metastatic tumor cells, highly metastatic tumor cells express a higher level of IL-8 mRNA which is in parallel with the IL-8 protein secretion.20 Intriguingly, in a model of tumor xenograft, tumor cells expressing IL-8 formed dramatically larger tumors with increased microvessel density than the control cells.21 It has been indicated that the essential role of IL-8 secreted by tumor cells in potentiating tumor progression seems to be related to induce adjacent epithelial tumor cells into EMT because blokage of IL-8 signaling could, at least partially, abolish or reverse the EMT.22
Because leptin modulates the expression of multiple genes that are regulated by JAK/STAT, PI3K/AKT, and MAPK/ERK signaling and these signaling pathways also regulate IL-8 expression and potentiate tumor cell migration or invasion, we then postulated that leptin affected EMT via induction of IL-8. Hence, in this current study, we found that leptin promoted epithelial-mesenchymal transition of human breast cancer cells and induced IL-8 expression via PI3K/Akt signaling pathway.
Results
Leptin induced epithelial-mesenchymal transition of breast cancer cells
Binding to the specific receptors (Ob-R) is essential for leptin to execute its biological functions.23 Western blot and Immunofluorescence assay were performed to examine the expression of leptin receptors including long (Ob-Rb) and short receptor (Ob-Rt) isoforms in human breast MCF-7, SK-BR-3, and MD-MB-231 cells. According to the result of Western blot, both Ob-Rb and Ob-Rt were expressed in MCF-7, SK-BR-3 and MD-MB-231 cells (Fig. 1A); fluorescent confocal microscopy also showed that both Ob-Rs were presented on the membrane of all these cancer cells (Fig. 1B). As shown in Fig. 1C, the breast cancer cells treated with 100 ng/ml leptin for 48 h show significant EMT-related morphological changes such as cell-cell junction dissolution, loss of apical-basolateral cell polarity. Besides, to further demonstrate EMT, some of the EMT molecular bio-markers were examined. It was shown that MCF-7 and SK-BR-3 cells exhibited a significant upregulation of mesenchymal markers such as Vimentin and Fibronectin, accompanied by apparent decrease in epithelial markers such as E-cadherin, which indicated a switch from an epthelial to a mesenchymal-like phenotype (Fig. 1D). However, MD-MB-231 cells did not display corresponding EMT changes when treated with leptin.
Figure 1.
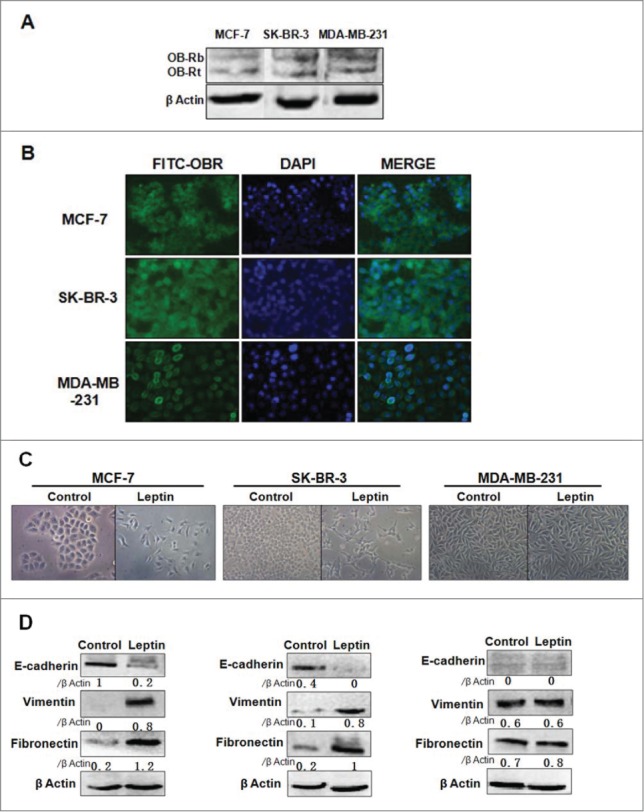
Leptin induces epithelial-mesenchymal transition of breast cancer cells. (A) Proteins were extracted from MCF-7, SK-BR-3 and MD-MB-231 cells. Western blotting analysis using specific antibodies demonstrated that Ob-Rb (125 kDa) and Ob-Rt (100 kDa) were expressed in the breast cancer cells. (B) Immunofluorescence assay with specific antibodies demonstrated leptin receptors were presented on the membrane of breast cancer cells. Original magnification: × 400 (C) The pictures of microscope showed EMT-related morphological changes. The presence of cell-cell junction dissolution, loss of apical-basolateral cell polarity emerged in MCF-7 and SK-BR-3 cells treated with leptin after 16 h serum starvation, while variations of morphological features were not observed in MD-MB-231 cells. Original magnification: ×200 (D) Westren blot analysisdemostrated that leptin downregulated E-cadherin, while upregulated Vimentin and Fibronectin of MCF-7 and SK-BR-3 cells. The EMT-related proteins in MD-MB-231 cells were not investigated.
Leptin-induced activation of IL-8, STAT3, ERK1/2, and AKT was based on Leptin-Leptin ObR axis in breast cancer cells
To investigate whether activation of IL-8 was involved in process of leptin-induced EMT of breast cancer cells, MCF-7 and SK-BR-3 cells were treated with various concentrations (20–200 ng/ml) of leptin in serum-free culture medium for 24 h after 16 h serum starvation or treated with 100 ng/ml leptin for different time from 0h to 48 h. The IL-8 mRNA and protein expression of breast cancer cells was detected by qRT-PCR and western blot, respectively. There was a clearly elevation of IL-8 mRNA and protein expression in a dose (Fig. 2A and C) and time-dependent manner (Fig. 2B and D) in MCF-7 and SK-BR-3 cells. The concentration of leptin at 100ng/ml exerted the maximum effect and thereby being chosen for the further experiments.
Figure 2.

Leptin-Leptin ObR axis was required for activation of IL-8, STAT3, ERK1/2, AKT in breast cancer cells. Serum-starved MCF-7 and SK-BR-3 cells were treated with different concentrations of leptin from 20 ng/ml to 200 ng/ml for 24 h or treated with 100 ng/ml leptin for different time from 0 h to 48 h. Q-PCR and Western blotting analysisdemonstrated leptin facilitated IL8 mRNA or protein of MCF-7 and SK-BR-3 cells with dose (A, C) and time-dependent (B, D) manner. The effect reached to peak level after MCF-7 and SK-BR-3 cells were treated with 100 ng/ml leptin for 24 h.(E) Serum-starved MCF-7 and SK-BR-3 cells were treated with 100 ng/ml leptin for various time intervals from 0h to 24 h. Westren blotting analysis was performed to detect the levels of phosphorylared and total STAT3, ERK1/2, AKT. (F) Antibodies against ObR (4 μg/ml) addition to MCF-7 and SK-BR-3 cells prior to leptin exposure absolutely abolished the expression of phosphorylation STAT3 and AKT (P < 0.05), while no changes was observed in phosphotylation ERK1/2. The specific antibody also down- regulated secretion of IL-8 from breast cancer cells that were treated with leptin (P < 0.05).
Figure 2.

(Continued)
The JAK2/STAT3, phosphatidylinositol 3-kinase (PI3K)/AKT and MAPK/ERK signaling pathways were all reported to be involved in modulating the expression of various gene products that are also regulated by leptin.24,25 To test the effect of leptin on the activation of JAK2/STAT3, PI3K/AKT and MAPK/ERK signaling cascades, the phosphorylated forms of STAT3, ERK1/2, AKT were detected. As shown in Fig. 2E, STAT3 phosphorylation STAT3 was gradually increased within one hour of leptin treatment and then declined. Similarly, leptin induced ERK1/2 phosphorylation, with the maximal induction time at 30 min. Additionally, leptin substantially elevated AKT phosphorylation. However, leptin did not influence the total protein levels of STAT3, ERK1/2 or AKT in MCF-7 and SK-BR-3 cells.
Given the critical role of Ob-Rs in mediating leptin-induced multiple intracellular signaling pathways by offering an intact intracellular domain, we explored whether relevant leptin-activated signaling molecules were regulated by ObRs. The results showed that the leptin-induced phosphorylation of STAT3 and AKT was abolished by addition of the anti-Ob-Rs antibody (4 μg/ml) to MCF-7 or SK-BR-3 cells before leptin treatment, whereas the phosphorylation of ERK1/2 was not affected (Fig. 2F). Moreover, the addition of the antibody against Ob-Rs could significantly down-regulate secretion of IL-8 from MCF-7 and SK-BR-3 cells.
Leptin-induced epithelial-mesenchymal transition in human breast cancer cells required IL-8 activation via PI3K/AKT-dependent pathway
To determine whether activation of JAK2/STAT or PI3K/AKT signaling pathway is involved in leptin-induced EMT of breast cancer cells, the pharmacologic inhibitor of JAK2/STAT, AG490, or the inhibitor of PI3K, LY294002 was used to pretreat the cells at indicated concentrations, followed by treatment of leptin (100 ng/ml). According to the results, leptin-induced elevation of IL-8 mRNA and protein levels were abolished by PI3K/AKT inhibitor LY294002, whereras blockage of JAK2/STAT3 with AG490 alone did not affect leptin-induced IL-8 expression significantly (Fig. 3A and B). The expressions of the biochemical hallmarks of EMT, Vimentin and Fibronectin, were also inhibited by LY294002 alone and further reduced by LY294002 + AG490, and E-cadherin expression was reversely affected in MCF-7 cells but not in SK-BR-3 cells (Fig. 3B). Next, we silenced Akt in MCF-7 and SK-BR-3 cells using Akt-siRNA. Akt depletion reduced leptin-mediated IL-8 secretion (Fig. 3C). Akt silencing also exhibited the similar level of E-cadherin and vimentin expression even in the presence of leptin treatment (Fig. 3C). These results indicated that leptin-induced IL-8 secretion through PI3K/AKT signal pathway might be essential for EMT in human breast cancer cells.
Figure 3.
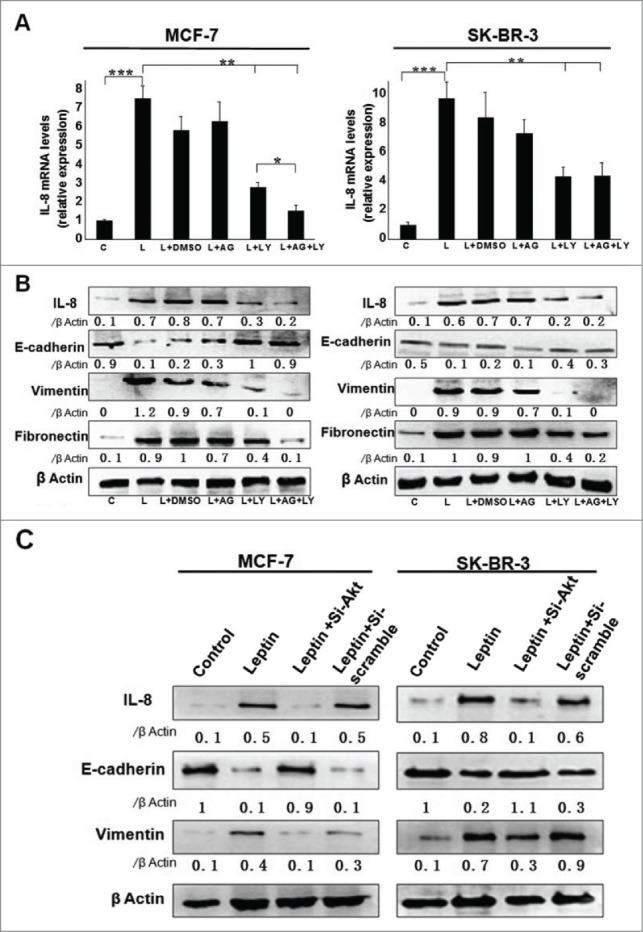
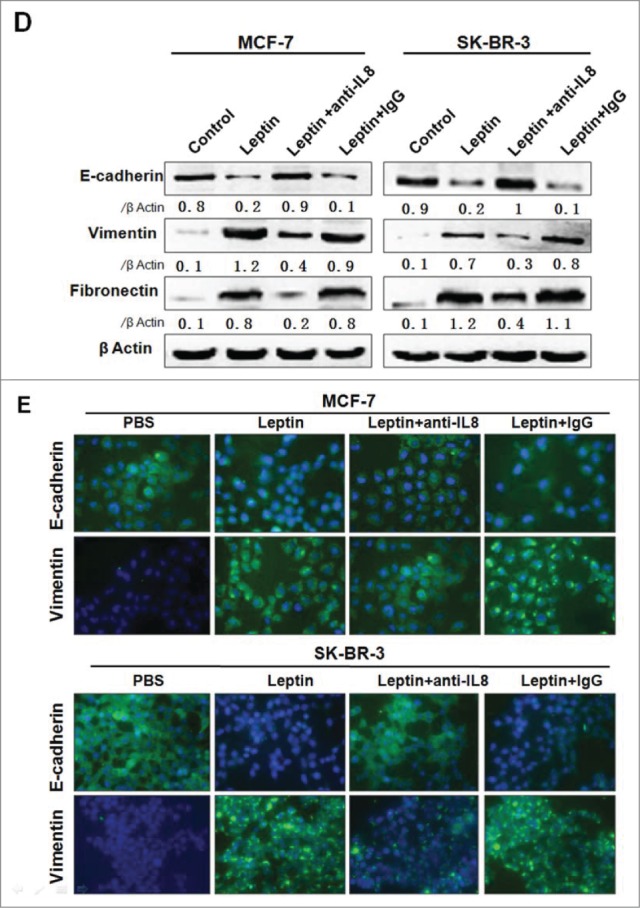
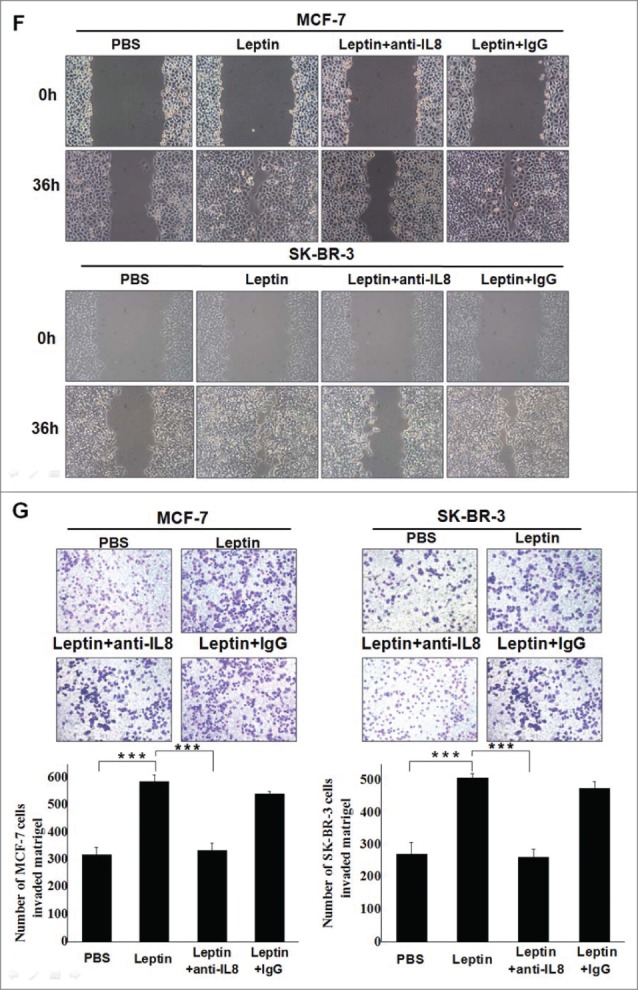
Leptin-induced epithelial-mesenchymal transition of breast cancer cells were abolished in the presence of AKT inhibitor and monoclonal Ab against IL-8. Serum-starved MCF-7 and SK-BR-3 cells were pretreated with 50 μM AG490 or 20 μM LY294002 for 1 h followed by 100 ng/ml leptin treatment for 24 h, DMSO were added as control. For IL-8 blocking experiments, specific antibodies to IL-8 or control IgG were added in serum-starved MCF-7 and SK-BR-3 cells cultures together with leptin and then incubated for 48 h. (A) QPCR and (B) Western blotting analysis indicated that LY294002 could abolish leptin-induced activation of IL-8 of MCF-7, SK-BR-3 cells (P < 0.05). (B) Western blotting analysis indicated that LY294002 could also restore the changes of Vimentin, Fibronectin and E-cadherin achieved by leptin treatment. (C)Western blotting analysis indicated that Akt depletion using Akt-siRNA could also restore the changes of IL-8, Vimentin and E-cadherin achieved by leptin treatment compared with nontargeting control siRNA. (D)Western blotting and (E) Immunofluorescence assay with Fluorescent confocal microscopy showed that comparing with control IgG, IL-8 specific antibody could completely abolish the hallmarks of leptin-induced EMT of MCF-7 and SK-BR-3 cells (P < 0.05).(F)Cell scratch assay and (G)Transwell chamber showed that leptin obviously advanced the migration and invasion capability of MCF-7 and SK-BR-3 cells after 36 h incubation, respectively. While IL-8 antibody could reduce the effects compared with control Ab (P < 0.05). Original magnification: ×200.
Figure 3.
(Continued)
Figure 3.
(Continued)
To further determine the role of IL-8 in the process of leptin-induced EMT in breast carcinomas, MCF-7 and SK-BR-3 cells were incubated with the blocking antibodies of IL-8 (1 μg/ml) or the control IgG. Western blot revealed that EMT-related proteins were markedly decreased in the presence of the specific antibodies (Fig. 3D). Besides, immunofluorescence assay showed that MCF-7 and SK-BR-3 cells exhibited significant induction of E-cadherin level along with a decrease in vimentin level when treated with anti-IL-8 antibodies on the basis of leptin, compared with the control IgG treatment group (Fig. 3E). We next analyzed the effect of leptin on migration and invasion of MCF-7 and SK-BR-3 cells. As shown in Fig. 3F and Fig. 3G, leptin (100ng/ml) obviously elevated the migration and invasion of both MCF-7 and SK-BR-3 cells; noteworthily, treatment with the specific antibobies of IL-8 significantly abolished the effect of leptin on migration and invasion of the breast cancer cells.
Expression of ObR, Leptin, IL-8, E-cadherin, and Vimentin in human breast cancer tissues
We next determined the expression of ObR, Leptin, IL-8, E-cadherin and Vimentin in benign breast tissues, invasive breast carcinoma without metastasis, and carcinoma with lymph node metastases (LNM) by immunohistochemical analysis. As shown in Figure 4, the staining intensities of ObR, Leptin and IL-8 in invasive breast carcinoma without axillary lymph node metastases was lower than those in LNM, and higher than those in benign breast tissue. Additionally, high intensity of vimentin and low E-cadherin were observed in LNM.
Figure 4.
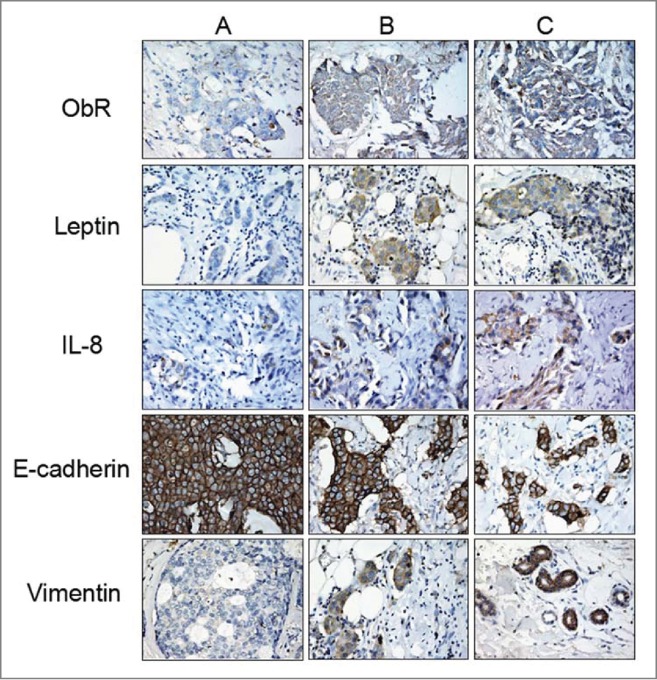
Expression of ObR, Leptin, IL-8, E-cadherin and Vimentin in breast cancer tissues. Clinical pathological specimens of breast tissues were collected and subjected to immunohistochemistry analysis using ObR, Leptin, IL-8, E-cadherin and Vimentin antibodies. A, B and C represented benign hyperplasia breast tissues, invasive breast carcinoma without metastasis and carcinoma with its lymph node metastases (LNM). The staining intensity of ObR, Leptin, IL-8, and Vimentin in invasive breast carcinoma without axillary lymph node metastases was lower than in LNM and stronger compared to that in benign breast tissue, with contrary staining intensity of E-cadherin expression.
Leptin increased IL-8 expression and induced tumor growth and metastasis in xenografts
To determine the effect of leptin on tumor growth in vivo, MCF-7 cells were transplanted into the mammary fat pads of nude mice. When the tumor diameter reached about 0.5mm on the 15th day, mice were injected respectively with PBS or leptin at 0.1 μg/g biweekly. As shown in Fig. 5A, within 30 days, tumor volume of mice treated with PBS was significantly smaller than that of with leptin injection. On 30th day, partial mice were sacrificed, and the tumor tissue was surgically removed. The results showed that treatment with leptin led to a distinct increase in tumor size compared with PBS group (Fig. 5B and C), besides, none of the mice survived for 60 d In contrast, 40% (2/5) of the mice in PBS group survived for more than 2 months (Fig. 5D). Moreover, H&E staining of the lung and liver metastasis showed that leptin injection resulted in more massive metastasises in lungs and livers compared with that of control (Fig. 5E). To further approve the role of leptin and IL-8 in murine models, the expression of IL-8, EMT associated factors E-cadherin, vimentin, and cell proliferation associated factors Ki67 were analyzed by in situ immunohistochemistry. As revealed in Figure 5F, the expression of IL-8, Ki-67, Vimentin was visibly increased in leptin-treated group, while E-cadherin was strikingly decreased.
Figure 5.

In vivo evidence for Leptin-mediated alterations of EMT markers and IL-8 expression levels. Groups of female nude mice (n = 5) were injected with 1×107 MCF-7 tumor cells in the mammary fat, and 15 d later, intratumor injection of PBS or leptin at 0.1 μg/g was performed biweekly until the 30th day, respectively. The mice were sacrificed. (A, B) Tumor volume and (B) primary weight of mice treated with PBS were significantly smaller than that of leptin injection (P < 0.001). (D) Survival time curve revealed that leptin reduced survival rate of tumor-bearing mice compared with group PBS (P < 0.001). (E) H&E staining demonstrated leptin promoted lung and liver metastasis of breast cancer xenografts. (F) Leptin increased expression of IL-8, Ki67 and Vimentin while decreased expression of E-cadherin evaluated by IHC.
Discussion
EMT, as an essential physiological process for embryogenesis that appears to be reinstated in wound healing and tissue regeneration, has recently been implicated in tumor progression. EMT involves a phenotypic switch that promotes migration and invasion of cancer cell, thus potential resulting in tumor recurrence, aggressiveness, and overall poor prognosis. Leptin, a hormone produced by adipose tissue, has been shown to induce EMT in various human tumor cells, including breast cancer.26,27 We thereby hypothesized that EMT was the likely mechanism that leptin enhanced breast cancer cell migration and invasion. Our findings demonstrated a vital role of leptin in acquisition of mesenchymal characteristics and aggressive behavior in breast cancer. The key mechanism that we expounded to account for the important function of leptin was that it increased IL-8 secretion. We further showed that leptin-mediated secretion of IL-8 was achieved by activation of PI3K/Akt signaling pathway, thus enhancing breast cancer cell migration and invasion.
The biological actions of leptin were executed through interaction with its exclusive receptors, ObR. As is well known, only OB-Rb with full intracellular domain is able to trigger the underlying signal cascade of leptin. Binding of leptin to the extracellular domain of ObRb leads to the activation of broad array of intracellular signals. Many of those have been implicated in carcinogenesis, such as those controlling cell growth and survival (ERK1/2, PI-3K/Akt, mTOR, p38 kinase, cyclin D1), inflammatory response (NF-κB and COX-2), angiogenesis (STAT3, VEGF, FGF, interleukin-1), and differentiation. In contrast to ObRb, ObRt, the shorter isoforms of ObR can bind leptin, but have significantly limited signaling potential.28 Indeed, ObR appears to be frequently expressed in breast cancer tissues (∼80 % of cases). Consistently, in this study, both Ob-Rb and Ob-Rt were expressed in human breast cancer cells MCF-7, SK-BR-3 and MD-MB-231.
Our data also showed that leptin stimulated epithelial- mesenchymal transition of MCF-7 and SK-BR-3 cells, which did not happen in MDA-MB-231 cells. The results were in accord with previous reports which showed that MDA-MB-231 cells were “partial EMT cells.”29 Thus, MDA-MB-231 cell line was dropped in the following study of investigation of leptin-mediated EMT in human breast cancer cells.
To explore the possible molecules that mediated leptin-induced EMT in MCF-7 and SK-BR-3 cells, an array of invasion and metastasis related cytokines and chemokines, such as IL-6, IL-8, IL-10, IL-12, TGF-β, TNF-α, MMP2, MMP7, MMP9, VEGF, et al.30-34 were screened. In this study, we found that leptin significantly upregulated IL-8 expression in both MCF-7 and SK-BR-3 cells in a dose and time-dependent manner. It has been reported that IL-8 secretion could induce tumor proliferation, enhance tumor cell invasion and migration. Besides, positive correlation between elevated serum IL-8 level and diminished survival rate in prostate, breast and pancreatic cancer patients has been reported.35-39 To understand the mechanisms underlying the effect of leptin on EMT and expression of IL-8 in breast cancer cells, we examined leptin-induced activation of JAK/STAT3, PI3K/Akt and MAPK/ERK signaling pathways. Our results showed that leptin stimulated the phosphorylation STAT3, AKT, and ERK1/2. Furthermore, our results showed that anti-ObR antibody blocked leptin-induced IL-8 expression and the phosphorylation of STAT3 and AKT, while had no effect on MAPK/ERK signaling pathway. The effects of leptin on the expression of IL-8, and the EMT biomarkers including vimentin, fibronectin and E-cadherin were abolished by PI3K/AKT inhibitor LY294002 while not by the inhibitor of JAK2/STAT, AG490. Moreover, Akt depletion significantly reduced leptin-mediated IL-8 secretion and exhibited the level of E-cadherin and vimentin expression even in the presence of leptin treatment. Therefore, we hypothesized that IL-8 via the PI3K/AKT signaling pathway might be responsible for leptin-induced EMT in breast cancer cells. The evidence to support the role of IL-8 in leptin-induced EMT was that compared with control IgG, specific antibody against IL-8 not only abolished leptin-induced EMT but also inhibited leptin-induced migration and invasiveness of MCF-7 and SK-BR-3 cells.
Immunohistochemical staining revealed that malignant breast carcinoma with lymph node metastases (LNM) which represents poor prognosis exhibited strong expression of ObR, Leptin, IL-8, and EMT markers. Moreover, xenograft tumor-bearing mice models showed that leptin significantly increased tumor volume and weight, reduced survival rate of tumor-bearing mice, enhanced lung and liver metastases, and increased expression of IL-8 and EMT-related markers.
In summary, the present study showed that leptin induced the EMT in breast cancer cells by promoting IL-8 expression, which may, at least in part, through activation of the PI3K/Akt-dependent signaling pathway. Our findings shed new lights on the specific pathways that are required for leptin-induced EMT and emphasize the critical role of leptin in breast cancer progression.
Materials and Methods
Cell culture and reagents
The human breast cancer cell lines, MCF-7, MDA-MB-231 and SK-BR-3 were obtained from the American Type Culture Collection (ATCC) (Rockville, MD, USA). Cells wre kept in Dulbecco's Modified Eagle Medium containing 10% fetal bovine serum. For treatment, cells were seeded at a density of 1×106 cells/ml in a 100-mm tissue culture dish. After serum starvation for 16 hours, the culture media was changed into serum-free media, which contains leptin (peprotech) treatments as indicated. In other cases, cells were treated with the PtdIns-3 linase inhibitor LY294002 at 10μmol/L or the JAK/STAT inhibitor AG490 (Beyotime, China). Antibodies of ObR (H-300) and IL-8 were purchased from Santa Cruz Biotechnology, Inc. and Abcam respectively. Antibodies of pSTAT-Tyr705, pAKT-Ser473, pERK-Thr202/Tyr204, anti-STAT, anti-AKT, anti-ERK were purchased from Cell Signaling Technologies, USA. Antibodies of E-cadherin, Vimentin and Fibronectin were purchased from Bioword Technology, Inc.
Tissue samples and animals
Breast tissue samples were obtained from patients, who underwent surgery for breast tumor at the department of pathology of the first affiliated hospital of Chongqing Medical University. Tissue samples were classified to breast benign hyperplasia, invasive carcinomas without lymph node metastasis and invasive carcinomas with corresponding lymph node metastases according to histological classification. This study was approved by the Ethics Medical University (Reference Number: CQMU 2010–26). Female nude mice, 5–7 weeks old, were purchased from Center of Laboratory Animals, Chongqing Medical University (Chongqing, China). All procedures were also approved by the Ethics Committee of Chongqing Medical University (Reference Number: CQMU 2010–26).
Western blot analysis
For Western blot, whole protein from cancer cells was subjected to SDS-PAGE and transferred to PVDF membrane, and the antigen-antibody reaction was performed using the previously described antibodies. An enhanced chemiluminescence (ECL) detection system (VIAGENE, USA) was used for immunodetection.
Immunofluorescence and confocal imaging
Breast cancer cells (3 × 105 cells/well) were plated in 6-well chamber slides followed by different treatment as indicted, and then subjected to immunofluorescence staining as described. Anti-ObR, Vimentin or Fibronectin antibodies were diluted in 10% goat serum to a concentration of 1:100. FITC-conjugated anti-rabbit IgG antibodies (Beyotime, China) were diluted to a concentration of 1:1000. Cell nuclei were stained with DAPI. Fluorescence analysis were performed using an Eclipse 80i Microscope (Nikon, Japan).
qRT-PCR analysis
Total cellular RNA was extracted using the TRIzol reagent (Invitrogen, Canada) and was reverse transcribed into cDNA (Invitrogen). Quantitative real-time reverse transcription PCR (qRT-PCR) was performed using the SYBR Premix Ex Taq™ (TaKaRa, Japan), according to the manufacturer's instruction. All reactions were performed in a 25 ul reaction volume in triplicate. Primers for IL8 andβ-actin were as follows: IL-8 sense, 5′-ACTCCAAACCTTTCCACC-3′; IL-8 antisence, 5′-CTTCTCCACAACCCTCTG −3′; β-actin sense, 5′-CACGATGGAGGG GCCGGACTCATC-3′; β-actin antisense, 5′-TAAAGACCTCTATGCCAACACAGT- 3′ ′. PCR generated 155- and 290-bp fragments of the IL-8 and β-actin genes, respectively. Following PCR conditions were an initial denaturation at 95°C for 30 s, 40 cycles of PCR amplification at 95°C for 5 s, 60°C for 20 s and 72°C for 30 s. The relative amount of target gene mRNA was normalized toβ-actin.
RNA Interference
For RNA interference, MCF-7 and SK-BR-3 cells were seeded at a density of 1 × 106 /100-mm tissue culture dish and transfected at 60% confluency with 200 nMof targeted siRNA and control siRNA using Oligofectamine (Invitrogen), the cells were then treated with leptin as indicated for 24 h after transfection. After another 24 h, the cells were harvested and luciferase activities were measured.
Migration assay
Cancer cells were seeded into 24-well culture plates and cultured with DMEM containing 10% FBS to confluence, and then washed with serum-free medium followed by serum starving for 16 h. A sterile pipette tip was used to scratch across the cell layer. After washed with serum-free medium for twice, the culture media were changed to serum-free media which contain treatments as indicated. Plates were photographed after incubation of different time.
Tumor cell invasion assay
Tumor cell invasion assay was performed using Transwell system (Millipore, USA) with 8μm-pore polycarbonate filter membrane coated with Matrigel (Sigma, USA). Each upper chamber was seeded with cancer cells at a density of 5 × 104 cells/ml and placed into the lower chamber contained DMEM with 15% FBS. After 16h of serum starvation, the culture media were changed to serum-free media containing treatments as indicated. After incubation for 18h, the membranes were fixed with 1% paraformaldehyde and stained with 0.1% crystal violet for 10 min. The number of cells passing through the Matrigel was counted in 5 random fields under microscope.
Tumor Xenografts
Female nude mice were bred and kept in strict accordance with the Chongqing Management Approach of Laboratory Animal (Chongqing government order NO.195). All procedures were approved by the Ethics Committee of Chongqing Medical University (Reference Number: CQMU 2010–26). And all surgery was attempted to minimize suffering. Mammary fat pads of the mice were co-injected with 1×107 MCF-7 cells. When the tumor diameter reached about 0.5 mm, intratumor injection of PBS or leptin at 0.1 μg/g was performed biweekly until the 30th day respectively. On the 30th day, partial mice were put to death and the tumor was surgically removed for determining the tumor volumes and weights. In addition, tumor and tissue sections were subjected to histological analysis. The other mice were kept to survey survival rates until 60 d Immunohistochemical staining of IL-8, E-cadherin, Vimentin and Ki67 of tumor tissue sections was carried out according to the manufacturer's protocol. Lung and liver metastasis in mice were measured by HEstaining.
Statistical analysis
Statistical significance was confirmed with the Student's t test and Graph Pad Prism software. P-values <0.05 indicate significant differences.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Lin Wang, Cuiping Tang and Tingmei Chen concepted and designed the research, wrote the article; Lin Wang, Cuiping Tang, Hong Cao, Kuangfa Li, Xueli Pang and Liang Zhong carried out the experimental work; Weiqi Dang, Hao Tang and Yunxiu Huang providied reagents; Lan Wei, Min Su and Tingmei Chen analyzed the data.
Funding
This work is supported by a grant from National Natural Science Foundation of China (no. 81272544); and by a grant from the Natural Science Foundation of Chongqing (no. cstc2012jjA10011).
References
- 1.de Azambuja E, McCaskill-Stevens W, Francis P, Quinaux E, Crown JP, Vicente M, Giuliani R, Nordenskjöld B, Gutiérez J, Andersson M, et al.. The effect of body mass index on overall and disease-free survival in node-positive breast cancer patients treated with docetaxel and doxorubicin-containing adjuvant chemotherapy: the experience of the BIG 02-98 trial. Breast CancerRes Treat. 2010;119:145-53; http://dx.doi.org/ 10.1007/s10549-009-0512-0 [DOI] [PubMed] [Google Scholar]
- 2.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569-78; PMID:18280327; http://dx.doi.org/ 10.1016/S0140-6736(08)60269-X [DOI] [PubMed] [Google Scholar]
- 3.Majed B, Moreau T, Senouci K, Sigal B, Salmon RJ, Fourquet A, Asselain B. Stoutness and prognosis of female non-metastatic breast cancer: results from a French observational cohort study. Bull Cancer. 2009;96:531-41; PMID:19467984 [DOI] [PubMed] [Google Scholar]
- 4.Wolin KY, Carson K, Colditz GA. Obesity and cancer. Oncologist. 2010;15:556-65; PMID:20507889; http://dx.doi.org/ 10.1634/theoncologist.2009-0285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simpson ER, Brown KA. Obesity and breast cancer: role of inflammation and aromatase. J Mol Endocrinol. 2013; 51(3):T51-9; PMID:24163427; http://dx.doi.org/ 10.1530/JME-13-0217 [DOI] [PubMed] [Google Scholar]
- 6.Ray A, Cleary MP. Obesity and breast cancer: a clinical biochemistry perspective. Clin Biochem. 2012;45:189-97; PMID:22178111; http://dx.doi.org/ 10.1016/j.clinbiochem.2011.11.016 [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Bellows CF, Kolonin MG. Adipose tissue-derived progenitor cells andcancer, World J Stem Cells. 2010; 2(5):103-113; PMID:21607127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwaan HC, McMahon B. The role of plasminogen-plasmin system in cancer. Cancer TreatRes. 2009; 148:43-66 [DOI] [PubMed] [Google Scholar]
- 9.Huang L, Li C. Leptin: a multifunctional hormone. Cell Res. 2000;10:81-92; PMID:10896170; http://dx.doi.org/ 10.1038/sj.cr.7290038 [DOI] [PubMed] [Google Scholar]
- 10.Guo S, Gonzalez-Perez RR. Notch, IL-1 and leptin crosstalk outcome (NILCO) is critical for leptin-induced proliferation, migration and VEGF/VEGFR-2 expression in breast cancer. PLoS One. 2011; 6(6):e21467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grossmann ME, Ray A, Nkhata KJ, Malakhov DA, Rogozina OP, Dogan S, Cleary MP. Obesity and breast cancer: status of leptin and adiponectin in pathological processes. Cancer Metastasis Rev. 2010; 29(4): 641-653; PMID:20821253; http://dx.doi.org/ 10.1007/s10555-010-9252-1 [DOI] [PubMed] [Google Scholar]
- 12.Ishikawa M, Kitayama J, Nagawa H. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin Cancer Res. 2004;10:4325-31; PMID:15240518; http://dx.doi.org/ 10.1158/1078-0432.CCR-03-0749 [DOI] [PubMed] [Google Scholar]
- 13.Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011; 27:347-376; PMID:21740232; http://dx.doi.org/ 10.1146/annurev-cellbio-092910-154036 [DOI] [PubMed] [Google Scholar]
- 14.Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002; 2(6):442-454; PMID:12189386; http://dx.doi.org/ 10.1038/nrc822 [DOI] [PubMed] [Google Scholar]
- 15.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119(6):1420-1428; PMID:19487818; http://dx.doi.org/ 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan D, Avtanski D, Saxena NK, Sharma D. Leptin-induced epithelial-mesenchymal transition in breast cancer cells requires β-catenin activation via Akt/GSK3-dependent and MTA1/Wnt1-dependent pathways. J Biol Chem. 2012; 287(11):8598-9612; PMID:22270359; http://dx.doi.org/ 10.1074/jbc.M111.322800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hedges JC, Singer CA, Gerthoffer WT. Mitogen-activated protein kinases regulate cytokine gene expression in human airway myocytes. Am J Respir Cell Mol Biol. 2000,23(1):86-94; PMID:10873157; http://dx.doi.org/ 10.1165/ajrcmb.23.1.4014 [DOI] [PubMed] [Google Scholar]
- 18.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008; 14(21):6735-6741; PMID:18980965; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-4843 [DOI] [PubMed] [Google Scholar]
- 19.Xie K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 2001; 12(4):375-391; PMID:11544106; http://dx.doi.org/ 10.1016/S1359-6101(01)00016-8 [DOI] [PubMed] [Google Scholar]
- 20.Singh RK, Gutman M, Radinsky R, Bucana CD, Fidler IJ. Expression of interleukin 8 correlates with the metastatic potential of human melanoma cells in nude mice. Cancer Res 1994;54:3242-3247; PMID:8205546 [PubMed] [Google Scholar]
- 21.Ning Y, Manegold PC, Hong YK, Zhang W, Pohl A, Lurje G, Winder T, Yang D, LaBonte MJ, Wilson PM, et al.. Interleukin-8 is associated with proliferation, migration, angiogenesis and chemosensitivity in vitro and in vivo in colon cancer cell line models. Int J Cancer. 2011;128:2038-2049; PMID:20648559; http://dx.doi.org/ 10.1002/ijc.25562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Claudia P, Duane HH, Romaine IF. Influence of IL-8 on the epithelial–mesenchymal transition and the tumor microenvironment. Future Oncol. 2012,8(6): 713-722; PMID:22764769; http://dx.doi.org/ 10.2217/fon.12.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo S, Liu M, Wang G, Torroella-Kouri M, Gonzalez-Perez RR. Oncogenic role and therapeutic target of leptinsignaling in breast cancer and cancer stem cells. Biochim Biophys Acta. 2012; 1825(2): 207-222; PMID:22289780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo S, Liu M, Wang G, Torroella-Kouri M, Gonzalez-Perez RR, et al.. Oncogenic role and therapeutic target of leptin signaling in breast cancer and cancer stem cells. Biochim Biophys Acta. 2012; 1825(2): 207-222; PMID:22289780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Perez RR, Lanier V, Newman G. Leptin's Pro-Angiogenic Signature in Breast Cancer. Cancers (Basel). 2013; 5(3):1140-1162; PMID:24202338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nath AK, Brown RM, Michaud M, Sierra-Honigmann MR, Snyder M, Madri JA. Leptin affects endocardial cushion formation by modulating EMT and migration via Akt signaling cascades. J Cell Biol. 2008; 181(2):367-380; PMID:18411306; http://dx.doi.org/ 10.1083/jcb.200708197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kushiro K, Núñez NP. Ob/ob serum promotes a mesenchymal cell phenotype in B16BL6 melanoma cells. Clin Exp Metastasis. 2011; 28(8):877-886; PMID:21879359; http://dx.doi.org/ 10.1007/s10585-011-9418-4 [DOI] [PubMed] [Google Scholar]
- 28.Jarde T, Perrier S, Vasson MP, Caldefie-Chezet F. Molecular mechanisms of leptin and adiponectin in breast cancer. Eur J Cancer. 2011;47(1):33-43; PMID:20889333; http://dx.doi.org/ 10.1016/j.ejca.2010.09.005 [DOI] [PubMed] [Google Scholar]
- 29.Karnoub AE1, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007.449(7162):557-63; PMID:17914389; http://dx.doi.org/ 10.1038/nature06188 [DOI] [PubMed] [Google Scholar]
- 30.Luboshit SG, Shina S, Kaplan O, Engelberg S, Nass D, Lifshitz-Mercer B, Chaitchik S, Keydar I, Ben-Baruch A. Elevated expression of the CC chemokine regulated on activation, normal T cell expressed and secreted (RANTES) in advanced breast carcinoma. Cancer Res. 1999; 59(18):4681-1687; PMID:10493525 [PubMed] [Google Scholar]
- 31.Germano G, Allavena P, Mantovani A. Cytokines as a key component of cancer-related inflammation. Cytokine. 2008; 43(3):374-379; PMID:18701317; http://dx.doi.org/ 10.1016/j.cyto.2008.07.014 [DOI] [PubMed] [Google Scholar]
- 32.Eiró N, González L, González LO, Fernandez-Garcia B, Lamelas ML, Marín L, González-Reyes S, del Casar JM, Vizoso FJ. Relationship between the inflammatory molecular profile of breast carcinomas and distantmetastasis development. PLoS One. 2012,7(11):e49047; PMID:Can't; http://dx.doi.org/ 10.1371/journal.pone.0049047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slattery ML, John E, Torres-Mejia G, Stern M, Lundgreen A, Hines L, Giuliano A, Baumgartner K, Herrick J, Wolff RK. Matrix metalloproteinase genes are associated with breast cancer risk and survival: the breast cancer health disparities study. PloS One. 2013; 8(5): e63165; PMID:23696797; http://dx.doi.org/ 10.1371/journal.pone.0063165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z, Bandyopadhyay A, Nichols RW, Wang L, Hinck AP, Wang S, Sun LZ. Blockade of autocrine TGF-βsignaling inhibits stem cell phenotype, suivival, and metastasis of murine breast cancer cells. J Stem Cell Res Ther, 2012,2(1):1-8; http://dx.doi.org/ 10.4172/2157-7633.1000116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schadendorf D, Möller Algermissen , Worm M, Sticherling M, Czarnetzki BM, et al.. IL-8 produced by human malignant melanoma cells in vitro is an essential autocrine growth factor. J Immunol. 1993; 151(5):2667-2675; PMID:8360485 [PubMed] [Google Scholar]
- 36.Yuan A, Chen JJ, Yao PL, Yang PC. The role of interleukin-8 in cancer cells and microenvironment interaction. Front Biosci. 2005; 10: 853-865; PMID:15569594; http://dx.doi.org/ 10.2741/1579 [DOI] [PubMed] [Google Scholar]
- 37.Varney ML, Li A, Dave BJ, Bucana CD, Johansson SL, Singh RK. Expression of CXCR1 and CXCR2 receptors in malignant melanoma with different metastatic potential and their role in interleukin-8 (CXC L-8)-mediated modulation of metastatic phenotype. Clin Exp Metastasis. 2003; 20(8):723-731; PMID:14713106; http://dx.doi.org/ 10.1023/B:CLIN.0000006814.48627.bd [DOI] [PubMed] [Google Scholar]
- 38.Lehrer S, Diamond EJ, Mamkine B, Stone NN, Stock RG. Serum interleukin-8 is elevated in men with prostate cancer and bone metastases. Technol Cancer Res Treat. 2004; 3(5):411; PMID:15453805; http://dx.doi.org/ 10.1177/153303460400300501 [DOI] [PubMed] [Google Scholar]
- 39.Benoy IH, Salgado R, Van Dam P, Geboers K, Van Marck E, Scharpé S, Vermeulen PB, Dirix LY. Increased serum interleukin-8 in patients with early and metastatic breast cancer correlates with early dissemination and survival. Clin Cancer Res. 2004; 10(21):7157-7162; PMID:15534087; http://dx.doi.org/ 10.1158/1078-0432.CCR-04-0812 [DOI] [PubMed] [Google Scholar]